

Abstract
We have studied Ca2+/calmodulin-dependent protein kinase II (CaMKII) isoform distribution and activity in embryonic hippocampal neurons developing in culture. We have found a strong correlation between the expression of the α subunit of the enzyme and the ability to undergo depolarization-dependent phosphorylation, which in young neurons is limited to the somatodendritic pool of the kinase. The lack of responsiveness of the axons of young αCaMKII-positive neurons is not caused by a lower Ca2+ influx but rather by a differential balance between kinase and phosphatase activities in this compartment. After the establishment of synaptic contacts, the presynaptic pool of the kinase displays an increasing level of activity and acquires the parallel ability to phosphorylate synapsin I, which represents one of the major CaMKII presynaptic targets in mature nerve terminals. In contrast, the activity of the postsynaptic pool of the kinase remains constant throughout synaptogenesis. In the presence of a nearly homogeneous subcellular distribution, this highly regionalized regulation of activity may reflect the multifunctional roles of CaMKII in both developing and mature neurons.
Keywords: protein phosphorylation, neuronal differentiation, synapse, phosphospecific antibodies, hippocampal neurons, synapsin
The multifunctional serine/threonine kinase Ca2+/calmodulin-dependent protein kinase II (CaMKII) is highly concentrated in the brain, where it accounts for 1–2% of total protein (Kennedy et al., 1983; Kelly et al., 1984; McGuinness et al., 1985) and phosphorylates several substrates that are involved in the modulation of neuronal excitability and neurotransmission (Soderling, 2000). The elucidation of the biochemical properties of CaMKII has provided evidence that brief [Ca2+]i signals can activate the kinase and stimulate an autophosphorylation reaction, resulting in the generation of long-lasting, Ca2+-independent autonomous activity (Lai et al., 1986; Miller et al., 1986). These features support a role for CaMKII as a memory molecule coupled to Ca2+ signaling in neurons (Lisman, 1994;Braun and Schulman, 1995; Soderling, 2000).
At the synapse, CaMKII has been localized both presynaptically and postsynaptically and has been implicated in specific functions related to plasticity phenomena (Stevens et al., 1994; Mayford et al., 1995;Lisman et al., 1997). At the postsynaptic level, it has been involved in the stabilization of the dendritic arbor structure (Wu and Cline, 1998), in modulation of dendritic exocytosis (Maletic-Savatic et al., 1998), in Ca2+-dependent facilitation of L-type Ca2+ channels (Dzhura et al., 2000), and in the regulatory phosphorylation of AMPA- and NMDA-type glutamate receptors (Barria et al., 1997; Gardoni et al., 2001). Furthermore, a functional role of the kinase in this subcellular compartment was suggested by the observation that exogenous αCaMKII translocates from the dendritic shaft to the postsynaptic density after NMDA receptor stimulation (Shen and Meyer, 1999).
At the presynaptic level, a fraction of CaMKII was shown to be present on the synaptic vesicle (SV) membrane (Benfenati et al., 1992, 1996), and its activity was proposed to regulate the efficiency of neurotransmitter release, via phosphorylation of the SV-associated protein synapsin I (Greengard et al., 1993).
Much less is known about CaMKII function during neuronal differentiation. Expression of the kinase is developmentally regulated (Hanson and Schulman, 1992), and the enzyme has been implicated in the control of neuronal growth and synaptogenesis (Zou and Cline, 1996). Furthermore, results obtained by expressing constitutively active forms of the kinase indicate that CaMKII plays a role in the developmental program of central glutamatergic synapses, as well as in determining their density (Wu et al., 1996; Rongo and Kaplan, 1999).
The observation that neuronal [Ca2+]i signals can be extremely localized and that CaMKII activation has specific functions in distinct subcellular compartments prompted us to investigate the processes underlying the targeting and activation of CaMKII during the development of hippocampal neurons in culture.
To monitor the topographic activation of CaMKII in developing neurons, we took advantage of phosphorylation state-specific antibodies, recognizing either the autophosphorylated kinase or synapsin I specifically phosphorylated by CaMKII (Czernik et al., 1991; Menegon et al., 2000). Our results show that CaMKII can be subdivided into distinct functional pools that are differentially activated during neuronal development.
MATERIALS AND METHODS
Materials. The anti-synapsin I/II (clone 19.11) mouse monoclonal antibody and anti-α/βCaMKII (RU16), anti-phosphoCaMKII (RU48), and phosphosite-3-specific anti-synapsin I (RU19) rabbit polyclonal antibodies were prepared and characterized at The Rockefeller University (New York, NY) (Czernik et al., 1991; Benfenati et al., 1992, 1996; Vaccaro et al., 1997; Menegon et al., 2000). Conjugation of the RU19 antibody with the fluorescent dye Cy3 and purification of the labeled antibodies were performed with the FluoroLink Cy3 kit and the Hitrap affinity column (Amersham Biosciences, Buckinghamshire, UK) according to the manufacturer's instructions.
The following antibodies were purchased from the indicated sources: anti-αCaMKII-specific (clone 6G9) mouse monoclonal antibody (Affinity Bioreagents, Golden, CA); anti-βCaMKII-specific (clone CBβ-1) mouse monoclonal antibody (Zymed, San Francisco, CA); anti-tau-1 antibody (Boehringer Mannheim, Mannheim, Germany); peroxidase-conjugated goat anti-rabbit and goat anti-mouse antibodies (Bio-Rad, Hercules, CA); FITC-conjugated goat anti-mouse, tetramethylrhodamine isothiocyanate-conjugated goat anti-rabbit, FITC-conjugated donkey anti-rabbit, 7-amino-4-methylcoumarin-3-acetic acid-conjugated donkey anti-mouse, and pure rabbit IgG antibodies (Jackson ImmunoResearch, West Grove, PA). The enhanced chemiluminescence detection system was from Amersham Biosciences, and the BCA protein assay reagent was from Pierce (Rockford, IL). Ionomycin, norokadaone, okadaic acid (OA), and cyclosporin A were from Calbiochem (La Jolla, CA). All chemicals were of the highest grade available.
Cell cultures. Low-density primary cultures of hippocampal neurons were prepared from embryonic day 18 rat embryos (Charles River, Calco, Italy), essentially as described previously (Banker and Cowan, 1977; Verderio et al., 1999a). Hippocampi were dissociated by a 15 min incubation with 0.25% trypsin at 37°C, and cells were plated at a density of 20,000–30,000/cm2 on poly-l-lysine (1 mg/ml)-treated glass coverslips in MEM (Invitrogen, San Giuliano Milanese, Italy), supplemented with 10% horse serum (Hyclone, Logan, UT), 2 mmglutamine (Biowhittaker, Ververs, Belgium), and 3.3 mm glucose. After allowing neurons to adhere to the substrate for 3–4 hr, coverslips were transferred to dishes containing a monolayer of cortical astrocytes (Booher and Sensenbrenner, 1972), without physical contact between neurons and glial cells. Cells were then maintained in serum-free MEM supplemented with 1% N2 supplement (Invitrogen), 2 mmglutamine (Biowhittaker), 0.1% ovalbumin (Sigma, St. Louis, MO), 4 mm glucose, and 1 mm sodium pyruvate.
Primary cultures of cerebellar granule neurons were prepared from postnatal day 5 rats (Charles River) as described previously (Gallo et al., 1987; Menegon et al., 1997). To favor neuronal survival and prevent glial cell proliferation, the medium was supplemented 3 d after plating with 10 mm KCl and 7.5 μmcytosine-1-β-d-arabinofuranoside (Sigma).
Immunoblot analysis. Cerebellar granule neurons at 12 din vitro (DIV) or hippocampal pyramidal neurons at 14 DIV were rapidly washed with Krebs–Ringer's solution buffered with HEPES (KRH; in mm: 150 NaCl, 5 KCl, 1.2 MgSO4, 1.2 KH2PO4, 2 CaCl2, 10 glucose, and 10 HEPES/Na, pH 7.4); supplemented with 2 mm EGTA; solubilized by scraping with solubilization buffer (1% SDS, 2 mm EDTA, and 10 mm HEPES/Na, pH 7.4); and immediately frozen in liquid nitrogen. After thawing, lysates were boiled for 3 min and sonicated. Equal amounts of protein were subjected to SDS-PAGE (Laemmli, 1970) and transferred to nitrocellulose as described previously (Towbin et al., 1979). Filters were blocked for 1 hr at room temperature in TBS (in mm: 150 NaCl and 100 Tris-Na, pH 7.5), supplemented with 5% nonfat dry milk, incubated for 2 hr with either anti-α/βCaMKII (RU16; 1:10,000) or phosphospecific anti-CaMKII (RU48; 1:5000) antibodies in TBS/milk, washed five times for 5 min with TBS/0.1% Triton X-100, incubated for 1 hr with a peroxidase-conjugated goat anti-rabbit antibody (1:10,000) in TBS/milk, washed five times for 5 min with TBS/0.1% Triton X-100, and finally developed by chemiluminescence. Digital images were obtained with a Speedy II scanner (Umax, Willich, Germany) and processed with the PhotoShop 5.0 program (Adobe Systems, San Jose CA).
Immunofluorescence analysis. Cells were quickly washed with KRH/EGTA and fixed for 30 min at 37°C in 4% formaldehyde (freshly prepared from paraformaldehyde) dissolved in 120 mm sodium phosphate buffer (pH 7.4) supplemented with 4% sucrose and 4 mm EGTA. After washing three times for 10 min in PBS (in mm: 180 NaCl and 10 Na-phosphate buffer, pH 7.4), cells were incubated with antibodies at the appropriate concentrations in goat serum dilution buffer (in mm: 450 NaCl and 20 sodium phosphate buffer, pH 7.4, 15% goat serum, and 0.3% Triton X-100). Between successive incubations with antibodies, cells were washed three times for 10 min in a high salt buffer (in mm: 500 NaCl and 20 mm sodium phosphate buffer, pH 7.4). After the standard double-labeling protocol, in some instances, Cy3-conjugated phosphosite-3-specific anti-synapsin I antibody (RU19) was added. Incubation with the conjugated antibody was performed in the presence of 10 μg/ml rabbit IgG and was preceded by a 20 min blocking reaction in the same solution.
After the final incubation, cells were washed three times in high salt buffer, once in PBS, and finally once in 5 mm sodium phosphate buffer, pH 7.4. Coverslips were mounted with 70% glycerol in PBS supplemented with phenylenediamine (1 mg/ml; Sigma) as an anti-bleaching agent and viewed with a Zeiss (Oberkochen, Germany) Axiovert 135 inverted microscope equipped with epifluorescence and differential interference contrast (DIC) optics. Images were taken with a high-resolution Hamamatsu (Shizuoka, Japan) Orca-II digital camera and processed with the Adobe PhotoShop 5.0 program; DIC images were filtered with the Image Pro plus program (Media Cybernetics, Silver Spring, MD).
fura-2 videomicroscopy. Neurons were loaded for 1 hr with 5 μm fura-2 pentacetoxy-methylester in KRH at 37°C, washed three times for 10 min in the same solution without fura-2 to allow de-esterification of the dye, and transferred to the recording chamber of a Zeiss Axiovert 100 inverted microscope equipped with a Ca2+ imaging unit using a modified CAM-230 dual-wavelength Jasco (Tokyo, Japan) microfluorimeter as a light source. Experiments were performed at room temperature using the Axon Imaging Workbench 2.2 equipped with a Personal Computer Optics (Computer Optics GmBH, Kelheim, Germany) Super Video Graphics Array SensiCam (Axon Instruments, Foster City, CA). Images were acquired at 0.5–1 340/380 ratios/sec, and ratio values in discrete areas of interest were calculated from sequences of images to obtain temporal analyses. At the end of the experiments, the imaged fields were marked using a diamond-tipped objective, and cells were fixed and processed for immunofluorescence (Pravettoni et al., 2000).
RESULTS
Characterization of the phosphospecific anti-CaMKII antibody
To characterize the phosphospecific anti-CaMKII antibody (RU48), rat cerebellar granule neurons were lysed either under control conditions or after exposure to KCl (55 mm) for 1 min in the absence or presence of EGTA (2 mm). Immunoblot analysis with an anti-α/βCaMKII (RU16) antibody revealed the presence of two prominent 58–60 kDa bands, corresponding to the β/β′ isoforms of CaMKII and of a minor 50 kDa band corresponding to the α isoform of the kinase (Fig. 1A). These results are consistent with the low levels of αCaMKII expression observed in homogenates of rat cerebellum (McGuinness et al., 1985). Both the α and the β/β′ isoforms underwent phosphorylation after depolarization of the cells in the presence of extracellular Ca2+, as revealed by labeling with the phosphospecific anti-CaMKII antibody (RU48).
Fig. 1.
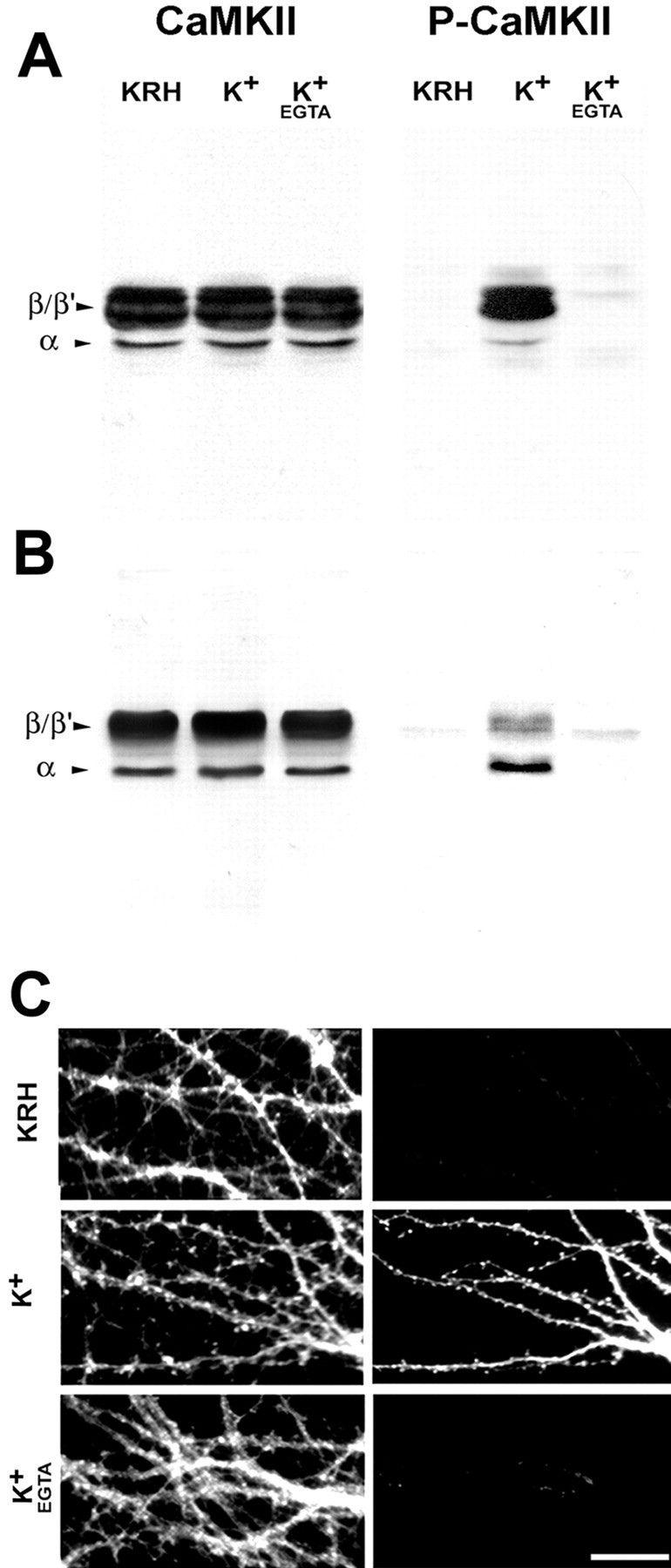
Characterization of the anti-phosphoCaMKII antibody. A, B, Western blot analysis of the specificity of the anti-phosphoCaMKII antibody. Cerebellar granule neurons at 12 DIV (A) or embryonic hippocampal neurons at 14 DIV (B) were lysed under control conditions (KRH) or after exposure to KCl (55 mm) for 1 min either in the absence or in the presence of EGTA (2 mm). Equal amounts of protein were loaded into each lane. Parallel samples were probed with either an anti-α/βCaMKII (CaMKII) or the phosphospecific anti-CaMKII (P-CaMKII) antibody. C, Immunocytochemical analysis of the specificity of the anti-phosphoCaMKII antibody. Embryonic hippocampal neurons at 21 DIV, treated as in B, were fixed and processed by double immunofluorescence with anti-αCaMKII (left) and phosphospecific anti-CaMKII (right) antibody. Note the Ca2+-dependent labeling of phosphorylated CaMKII after depolarization. Scale bar, 20 μm.
In hippocampal neurons at 14 DIV, the α isoform was relatively more abundant, although it represented a minor fraction of the total kinase (Fig. 1B) and appeared to be preferentially phosphorylated after depolarization. Consistent results were obtained by immunofluorescence analysis of rat cerebellar granule (data not shown) or hippocampal (Fig. 1C) neurons after fixation of the cells under the same conditions.
Developmental analysis of CaMKII expression and phosphorylation in embryonic hippocampal neurons in culture
CaMKII is characterized by regional and developmental variations in concentration and isoform ratio in the brain, consistent with a highly regulated role in neuronal function (Hanson and Schulman, 1992). Therefore, we analyzed the cellular distribution of the α and β CaMKII isoforms in rat embryonic hippocampal neurons developing in culture, using isoform-specific antibodies (Fig.2A,B). Although βCaMKII was already present at early stages of development and was ubiquitously and homogeneously distributed in all cells, αCaMKII was characterized by a regulated developmental expression. Indeed, it was not expressed by stage I-II cells (i.e., neurons with short, unpolarized processes), starting to be expressed by a fraction (13%) of stage III neurons (i.e., neurons with an identifiable axon). The proportion of αCaMKII-positive neurons progressively increased during development (40% of stage IV neurons; i.e., neurons in the phase of dendritic outgrowth) to approach the entire population (97%) of mature, stage V neurons. As observed in the case of βCaMKII, in αCaMKII-positive stage III neurons, the kinase displayed an apparently homogeneous distribution, with no particular enrichment in any subcellular compartment.
Fig. 2.
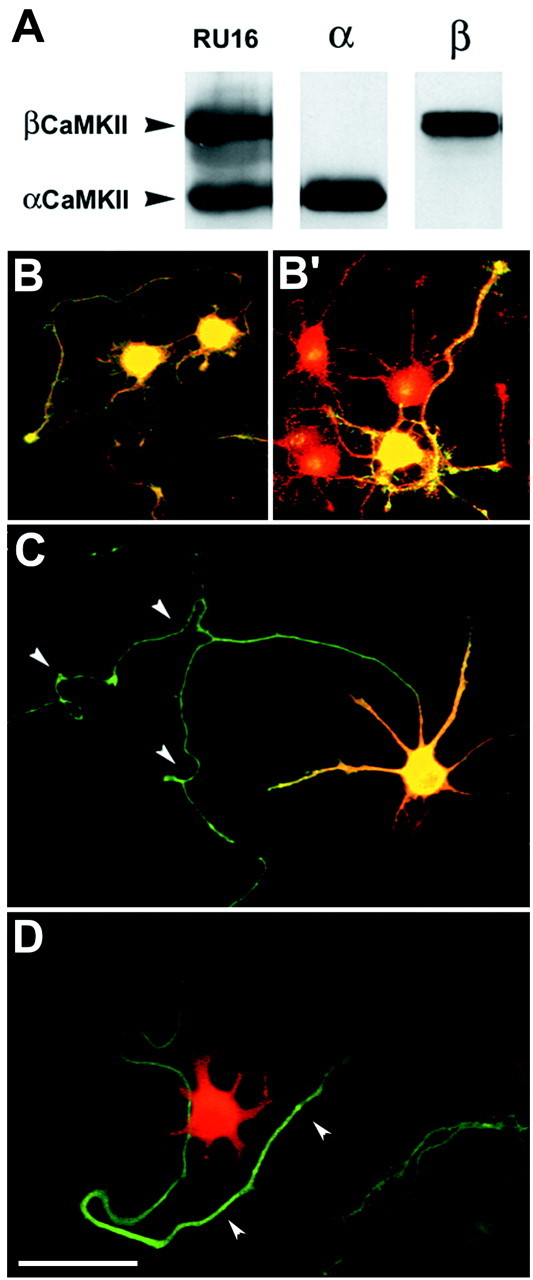
Morphological analysis of CaMKII isoform expression and phosphorylation in stage III embryonic hippocampal neurons in culture. A, Hippocampal slices were lysed under control conditions and probed by immunoblotting with either anti-α/β (RU16) or α- or β-specific anti-CaMKII antibodies.B, B′, Stage III embryonic hippocampal neurons were fixed under control conditions and processed by double immunofluorescence with anti-α/βCaMKII antibody (red) and either anti-βCaMKII (B) or anti-αCaMKII (B′) antibodies (green). C, Stage III embryonic hippocampal neurons were exposed for 1 min to KCl (55 mm), fixed, and processed for double immunofluorescence with anti-αCaMKII (green) and anti-phosphoCaMKII (red) antibodies. D, Neurons treated as in C were processed for double immunofluorescence with anti-tau-1 (green) and anti-phosphoCaMKII (red) antibodies. Thearrowheads in C and Dpoint to the axon, which is virtually devoid of labeling for phosphoCaMKII. Scale bar, 20 μm.
After exposure to depolarizing agents, a prominent phosphorylation of the kinase was apparent in the fraction of cells that corresponded at all stages exactly to the αCaMKII-positive neurons. Phosphorylation of the kinase was confined to the somatodendritic compartment, whereas the axon (identified by staining with antibodies to the axonal protein tau-1) was negative, despite the presence of the protein (Fig. 2C,D).
Analysis of the role of Ca2+ in CaMKII activation
Depolarization-induced Ca2+ influx and CaMKII activation in stage III hippocampal neurons were monitored by fura-2 Ca2+ imaging, followed by retrospective double immunofluorescence with anti-αCaMKII and phosphospecific anti-CaMKII antibodies. Figure3 shows an example of these experiments. The temporal analysis of depolarization-induced [Ca2+]i variations in the neuronal cell bodies showed similar kinetics but different peak levels in the various cells. CaMKII phosphorylation was apparent only in those cells expressing the α isoform of CaMKII, regardless of the [Ca2+]i levels reached.
Fig. 3.

Analysis of Ca2+ influx in phosphoCaMKII-positive and -negative stage III neurons. A, B, Time-resolved (A) and space-resolved (B) analysis of [Ca2+]i dynamics in stage III embryonic hippocampal neurons loaded with fura-2 and treated with KCl (55 mm) for 1 min. The image shown in Bcorresponds to the peak of [Ca2+]i inA. C, Retrospective double immunofluorescence with anti-αCaMKII (green) and phosphospecific anti-CaMKII (red) antibodies of the cells in the field shown in B. Scale bar, 10 μm. Note that the [Ca2+]i levels in one of the phosphoCaMKII-negative cells (1) are higher than those reached in the phosphoCaMKII-positive, αCaMKII-expressing cell (2).
The lack of CaMKII phosphorylation in the axons of αCaMKII-positive neurons was not caused by lower [Ca2+] levels reached after depolarization in this compartment. Figure4 shows a time-resolved analysis of [Ca2+]i variations in single neurons. The [Ca2+]i levels reached in the axon after exposure to KCl were similar to those reached in the dendritic compartment of the same cell, whereas phosphorylation of the kinase was apparent only in the cell body and proximal dendrites.
Fig. 4.
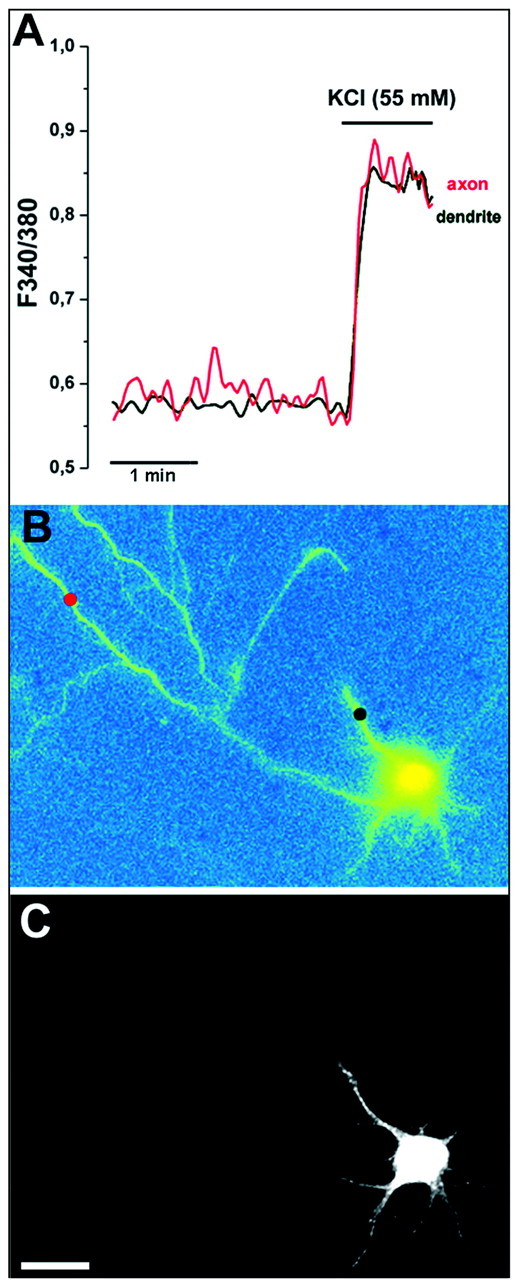
Analysis of Ca2+ influx in the axonal and dendritic compartments of αCaMKII-expressing stage III neurons. A, B, Time-resolved (A) and space-resolved (B) analysis of [Ca2+]i dynamics in the processes of a neuron treated and analyzed as described in the legend to Figure 3.Traces in A refer to the color-codedspots marked in B. C, Retrospective immunofluorescence with phosphospecific anti-CaMKII antibody of the cell shown in B. Note the similar [Ca2+]i levels reached after depolarization in the phosphoCaMKII-positive dendrites and in the phosphoCaMKII-negative axon. Scale bar, 10 μm.
To determine whether the observed pattern of CaMKII activation was specifically coupled to Ca2+ influx through voltage-gated channels, stage III neurons were exposed to ionomycin (1 μm) for 2 min in the presence of either physiological (2 mm) or high (10 mm) [Ca2+]o. Although virtually no CaMKII phosphorylation could be observed in the presence of 2 mm[Ca2+]o, regardless of the expression of the α isoform, a prominent and diffuse phosphorylation of CaMKII was apparent in the presence of 10 mm[Ca2+]o, in both αCaMKII-positive and -negative cells. Furthermore, using the high [Ca2+]o protocol, the kinase also underwent phosphorylation in the axonal compartment (Fig. 5A, top andmiddle panels).
Fig. 5.
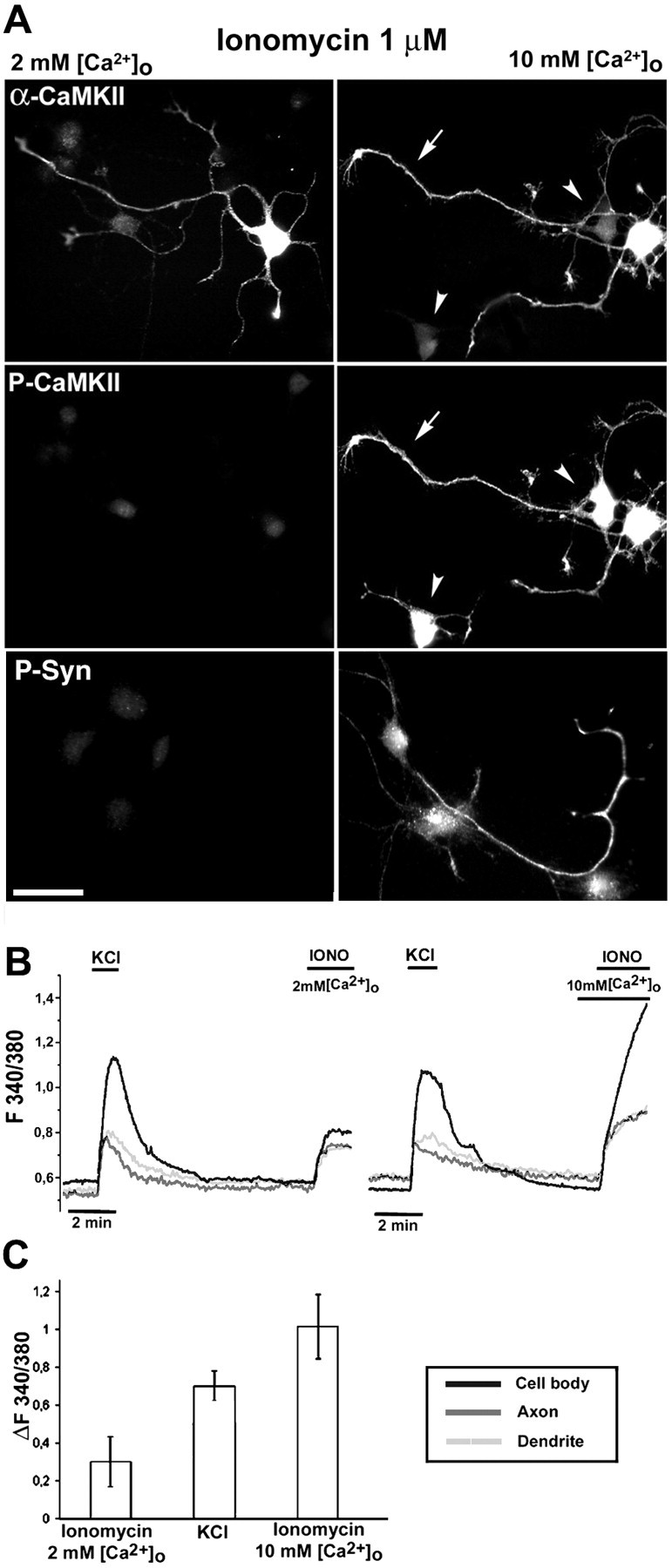
Analysis of CaMKII and synapsin I phosphorylation in stage III neurons treated with ionomycin in either physiological or high [Ca2+]o. A, Stage III neurons were exposed to ionomycin (1 μm) in either physiological (2 mm) or high (10 mm) [Ca2+]o for 2 min, fixed, and either double-labeled with anti-αCaMKII (top panels) and phosphospecific anti-CaMKII (P-CaMKII; middle panels) antibodies or labeled with a phosphospecific anti-synapsin I antibody (P-Syn; bottom panels). The phosphosynapsin staining is superimposed to the corresponding DIC image to visualize the axonal compartment. Note the prominent CaMKII phosphorylation in the high [Ca2+]o protocol, where phosphorylation of the kinase can be seen also in the axonal compartment (arrows) and in αCaMKII-negative cells (arrowheads). Under this condition, the CaMKII presynaptic substrate synapsin I also undergoes phosphorylation. Scale bar, 20 μm. B, Temporal analysis of fura-2 F340:380 ratio values measured in the cell body, dendrite, and axon of representative cells exposed sequentially to KCl (55 mm) in 2 mm[Ca2+]o and then to ionomycin (IONO; 1 μm) in either physiological (2 mm, left) or high (10 mm,right) [Ca2+]o.C, Analysis of peak fura-2 F340:380 ratio values (difference from basal ratio values, ΔF340:380) measured in cell bodies of neurons after exposure to the following stimulating protocols: ionomycin (1 μm) in physiological (2 mm) [Ca2+]o for 2 min, KCl (55 mm) for 1 min, and ionomycin (1 μm) in high (10 mm) [Ca2+]o for 2 min. Bars represent means ± SD with n = 10 cells/condition. For the KCl-treated sample, the mean value is calculated on α-positive, responsive neurons, whereas the average value calculated on the entire population is 0.51 ± 0.22 (n = 46 cells).
This observation prompted us to analyze phosphorylation of the CaMKII substrate synapsin I in the axon, using a previously characterized phosphosite-3-specific antibody directed against Ser603 of synapsin I, phosphorylated by CaMKII (Menegon et al., 2000). Exposure of neurons to ionomycin (1 μm) for 2 min in the presence of 10 mm[Ca2+]o caused a strong and specific phosphorylation of synapsin I (Fig. 5A,bottom panels). In contrast, labeling for phosphosynapsin I was not observed after exposure to ionomycin in the presence of 2 mm[Ca2+]o or after exposure to depolarizing agents (data not shown), as expected by the lack of axonal CaMKII activation.
The distinct responses in terms of CaMKII phosphorylation to the various experimental protocols (depolarizing agents, ionomycin in the presence of physiological or high [Ca2+]o) were related to the different peak levels of [Ca2+]i, as shown by measuring the mean difference between basal and peak ratio values (ΔF340/380) of the fura-2 signal in the cell bodies. The results showed that the cells treated with ionomycin in high [Ca2+]o reached a peak [Ca2+]i, which was approximately threefold higher than that reached in physiological [Ca2+]o. Intermediate levels of [Ca2+]i were reached in the case of neurons treated with KCl, with higher levels in the case of the responsive α-positive cells (Fig. 5C).
Single-cell temporal analyses were performed to compare the relative [Ca2+]i levels reached after depolarization in the various subcellular compartments (soma, axon, and dendrites) with those reached after exposure to ionomycin in either physiological or high [Ca2+]o. The traces indicated that, in the case of the high [Ca2+]o protocol, the axonal compartment reached higher [Ca2+]i levels than under the other experimental conditions both in terms of peak levels (Fig. 5B) and in terms of the of total amount of Ca2+ influx, as evaluated by comparison of the integrals under the curves (data not shown).
Effect of phosphatase inhibitors on the state of phosphorylation of axonal CaMKII
In principle, the failure to reveal a depolarization-dependent increase in the state of phosphorylation of CaMKII in the axons of stage III neurons might be attributable either to a lack of activation of the kinase or to a high level of phosphatase activity in this compartment. To distinguish between these possibilities, neurons were treated with serine/threonine phosphatase inhibitors.
Virtually no immunolabeling for phosphoCaMKII could be detected in the axons of neurons stimulated with KCl after exposure to cyclosporin, a selective inhibitor of calcineurin (Fig.6). In contrast, in the axons of neurons stimulated after exposure to OA, an inhibitor of protein phosphatase 2A (PP2A) and PP1, an intense labeling for phosphoCaMKII was evident. Under these conditions, in the same compartment, site 3 phosphorylation of synapsin I could also be detected. In neurons exposed to the OA-related, inactive compound norokadaone, depolarization failed to induce labeling for either phosphoCaMKII or site 3-phosphorylated synapsin I.
Fig. 6.

Effect of phosphatase inhibitors on the KCl-induced activation of CaMKII in stage III hippocampal neurons. Neurons were incubated for 10 min with KRH in the presence or absence of either OA (500 nm) or cyclosporin (10 nm) and successively stimulated for 2 min with 55 mm KCl. Double immunofluorescence was performed with either phosphospecific anti-CaMKII (P-CaMKII) and anti-αCaMKII (top panels) or site 3 phosphospecific anti-synapsin I (P-Syn I) and anti-total synapsin (bottom panels). Arrowheads point to axons negative for phosphoCaMKII or phosphosynapsin I in cells stimulated with KCl in the presence or absence of cyclosporin;arrows point to axons positive for phosphoCaMKII or phosphosynapsin I in cells stimulated in the presence of OA. Theinset in the top panel shows an αCaMKII-negative cell in which labeling for phosphoCaMKII was undetectable after KCl stimulation in the presence of OA. Scale bar, 20 μm.
The effect of OA was specific for the axons of αCaMKII-positive cells: after KCl stimulation in the presence of OA, αCaMKII-negative cells remained negative for phosphoCaMKII in all neuronal compartments.
Developmental organization of the presynaptic CaMKII/synapsin I complex
A preformed complex between αCaMKII and synapsin I on the SV membrane has been proposed to participate in the regulation of neurotransmitter release from mature nerve terminals (Benfenati et al., 1992; Greengard et al., 1993).
In our model system, when neurons start to establish intercellular contacts (stage IV), the presynaptic pool of CaMKII and its phosphorylation were generally undetectable, because of the abundance of the adjacent postsynaptic pool. The low percentage of stage IV neurons responsive to K+ depolarization allowed us to directly assess phosphorylation of the presynaptic pool of the kinase, which could be visualized in neurons making synapses on αCaMKII-negative cells. In those cases, presynaptic CaMKII appeared to be concentrated in varicosities and undergo phosphorylation (Fig.7A). In the same synaptic boutons, phosphorylation of synapsin I, which becomes progressively enriched in the terminals undergoing maturation, could also be detected. Although synapsin I underwent phosphorylation in virtually all of the terminals after exposure of the cells to ionomycin in the presence of high [Ca2+]o, only a fraction of the terminals appeared to be labeled with either ionomycin applied in physiological [Ca2+]o or after K+ depolarization, suggesting heterogeneity in the state of maturation of the boutons (Fig.7B). Interestingly, synapsin I phosphorylation could be detected only in presynaptic terminals possessing a postsynaptic counterpart, as assessed by DIC microscopy. Quantitative analysis performed on KCl-treated neurons demonstrated that the percentage of phosphosynapsin I-positive terminals, initially low, steadily increased over development, with the recruitment of virtually the entire population of mature synaptic boutons at 21 DIV (Fig.8). However, the apparent stoichiometry of synapsin I phosphorylation varied markedly in the various synaptic terminals.
Fig. 7.

Distribution and phosphorylation of presynaptic αCaMKII and synapsin I in stage IV neurons.A, Stage IV embryonic hippocampal neurons were exposed for 1 min to KCl (55 mm) in the presence of 2 mm [Ca2+]o. Cells were then fixed and processed for triple immunofluorescence with phosphospecific anti-CaMKII (P-CaMKII;green), site 3 phosphospecific anti-synapsin (P-Syn I; red), and anti-vesicle-associated membrane protein-2 (VAMP-2; blue) antibodies. The presynaptic pool of the kinase can be visualized in axons of αCaMKII-positive cells making contacts on αCaMKII-negative neurons (asterisks). Note the white spots(arrowheads), corresponding to structures that stain positively for the three antibodies, identifying them as synaptic boutons in which phosphorylation of both CaMKII and synapsin I has occurred. B, Stage IV embryonic hippocampal neurons were exposed to either KCl (55 mm) for 1 min or ionomycin (1 μm) for 2 min in the presence of either physiological (2 mm) or high (10 mm) [Ca2+]o. Cells were then fixed and processed for double immunofluorescence with anti-synapsin (green, top panels) and phosphospecific anti-synapsin I (P-Syn I;red, bottom panels) antibodies. At this stage, synapsin labeling starts to be concentrated in varicosities. Although only a fraction of the synapsin pool appears to be phosphorylated after depolarization or exposure to ionomycin in physiological [Ca2+]o, the entire synapsin pool undergoes phosphorylation with the high [Ca2+]o protocol. The fluorescence labeling in B is superimposed to the corresponding DIC image. Scale bars: A, left panel, 20 μm; right panel, 1 μm; B, 12 μm.
Fig. 8.
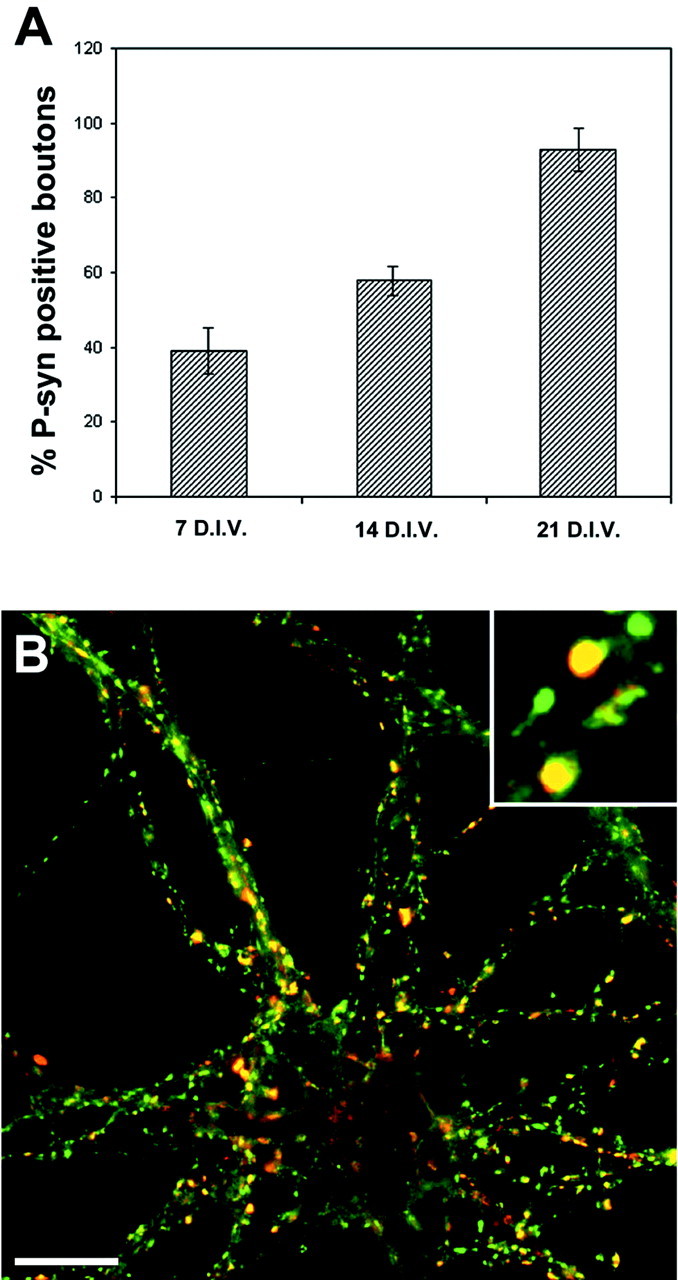
CaMKII-dependent synapsin I phosphorylation in stage V neurons. A, Stage V embryonic hippocampal neurons at 7, 14, or 21 DIV were exposed to KCl (55 mm) for 1 min, fixed, and processed for double immunofluorescence with anti-synapsin and site 3 phosphospecific anti-synapsin I (P-syn) antibodies. The histograms show the percentage of phosphosynapsin I-positive boutons (mean ± SD) at each stage. The number of phosphosynapsin I-positive boutons was normalized for the number of synapsin-positive presynaptic terminals present in each field. B, Representative immunofluorescence image of a 21 DIV neuron double-stained with anti-synapsin (green) and site 3 phosphospecific anti-synapsin I (red) antibodies. Scale bars: B, 10 μm; inset in B, 2 μm.
Exposure of the neurons to OA (but not to the related inactive compound norokadaone) before the depolarizing pulse strongly increased synapsin I phosphorylation at immature presynaptic terminals. Indeed, under these conditions, >80% of the synaptic boutons were labeled by phosphospecific antibodies to synapsin I already at 7 DIV (Fig.9). In addition, in the phosphosynapsin I-positive boutons, the apparent stoichiometry of synapsin I phosphorylation was increased.
Fig. 9.

Effect of phosphatase inhibitors on synapsin I phosphorylation in stage V neurons. A, Embryonic hippocampal neurons at 7 DIV were exposed for 10 min to KRH in the absence or presence of either OA (O.A.; 500 nm) or norokadaone (Nok; 500 nm). In the stimulated samples (KCl), 55 mm KCl was present during the last minute of the incubation. The samples were then fixed and processed for double immunofluorescence with anti-synapsin (Syn; left panels) and phosphospecific anti-synapsin I (P-syn I; right panels) antibodies. Scale bar, 10 μm. B, The percentage of phosphosynapsin I-positive boutons (mean ± SD) was calculated for all of the samples in A (n = 260–280,gray bars). In phosphosynapsin I-positive boutons, the average fluorescence intensity ratio between the signals for phosphosynapsin I and for total synapsins was also calculated (white bars).
Spatially heterogeneous activation of CaMKII in the somatodendritic compartment of mature neurons
Depolarization-induced phosphorylation of CaMKII in the somatodendritic compartment was apparently homogeneous in younger neurons. In contrast, in stage V neurons, two distinct and physically separated domains of activation of the kinase became apparent: a domain strongly and uniformly activated by depolarizing agents, represented by the neuronal cell body and proximal dendrites, and a distal domain (>20–30 μm from the cell body) characterized by the absence of activation in the dendritic shaft and by the presence of isolated spots of activation (Fig. 10). These spots were identified as postsynaptic spines, because they appeared to be always juxtaposed to synapsin-positive presynaptic terminals and were negative for MAP2 immunostaining.
Fig. 10.
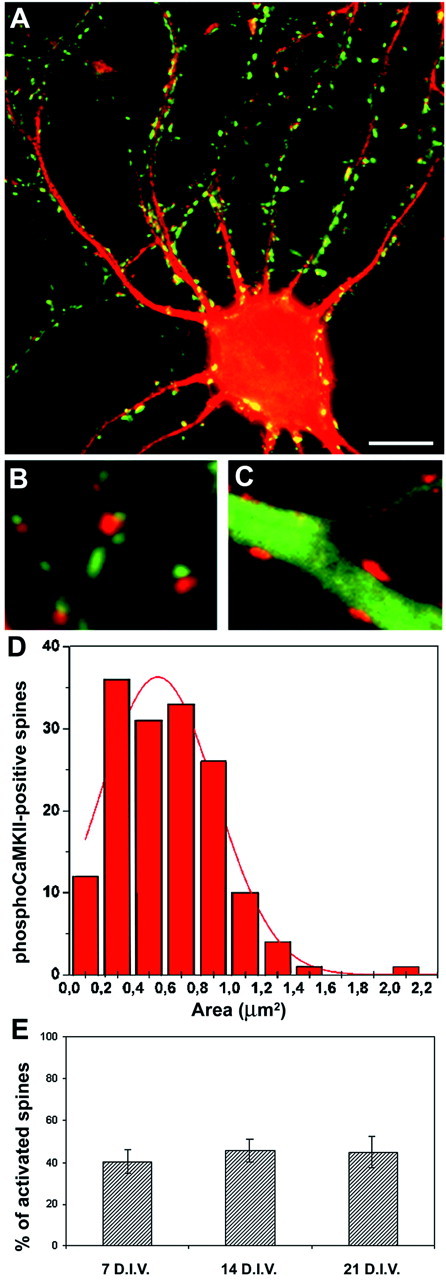
Postsynaptic CaMKII phosphorylation in stage V neurons. A, Stage V embryonic hippocampal neurons at 21 DIV were exposed to KCl (55 mm) for 1 min, fixed, and processed for double immunofluorescence with anti-synapsin (green) and phosphospecific anti-CaMKII (red) antibodies. B, C, High-magnification images of distal dendrites treated as inA and processed for double immunofluorescence with phosphospecific anti-CaMKII (red) and either anti-synapsin (B) or anti-MAP2 (C) antibodies (green). Note the absence of labeling for phosphoCaMKII in the dendritic shaft. The phosphoCaMKII-positive spots are always juxtaposed to synapsin-positive, presynaptic terminals and confined to MAP2-negative areas, defining them as spines. D, Size distribution of the phosphoCaMKII-positive areas in distal dendrites. The continuous red line represents the Gaussian fitting of the curve. E, Stage V embryonic hippocampal neurons at 7, 14, and 21 DIV were processed as in A. The histograms show the percentage of phosphoCaMKII-positive spines (mean ± SD) at each stage. The number of activated spines was normalized for the number of synapsin-positive presynaptic boutons present in each field. Scale bars: A, 6 μm; B, C, 2 μm.
Quantitative analysis of the surface area of the spots labeled in the distal region demonstrated that CaMKII activation remained confined to spines. Indeed, the histogram representing the classes of phosphoCaMKII-labeled areas was consistent with the reported size distribution of dendritic spines (Harris, 1999; Kennedy, 2000). The percentage of activated spines remained constant during development in culture, once normalized for the number of presynaptic boutons present in the field.
DISCUSSION
CaMKII is a multifunctional protein kinase that represents a central element in neuronal signaling and a likely candidate as a Ca2+-sensing memory molecule. Several functions of the kinase are associated with specific subcellular compartments in mature neurons.
We investigated the expression and activity of the different functional pools of the kinase during neuronal development, taking advantage of phosphorylation state- and isoform-specific antibodies directed toward CaMKII or its major presynaptic target, synapsin I.
Four distinct CaMKII isoforms (α, β, γ, and δ) encoded by distinct genes are known. The α and β isoforms are restricted to the nervous tissue, whereas the γ and δ isoforms are expressed in a variety of tissues, including the brain (Braun and Schulman, 1995). The various isoforms can coassemble to form multisubunit holoenzymes whose composition varies in different brain regions (Hanson and Schulman, 1992). We have found that in embryonic hippocampal neurons in culture, the α and β isoforms display differential regulation during development. βCaMKII is expressed by all cells as early as 12 hr after plating (corresponding to stage I neurons), and its level of expression remains constant throughout differentiation. In contrast, αCaMKII is not present at early developmental stages, starts to be expressed by a fraction of stage III neurons, and becomes expressed by the entire population of stage V neurons. An increase in the expression of this isoform has also been observed during brain development (Hanson and Schulman, 1992).
In our model system, we have detected a strict correlation between αCaMKII expression and depolarization-induced activation of CaMKII, which, before synaptogenesis, is confined to the somatodendritic pool of the kinase. The apparent requirement for αCaMKII expression is consistent with the reported low levels of Ca2+/calmodulin-stimulated protein phosphorylation in 2 DIV cultures of hippocampal neurons (Scholz et al., 1988).
Although the [Ca2+]i levels reached after depolarization are generally higher in αCaMKII-expressing stage III neurons than in neurons of the same developmental stage negative for this isoform of the kinase, the unresponsiveness of αCaMKII-negative neurons is not strictly linked to a lower Ca2+ influx, as demonstrated by fura-2 videomicroscopy.
In αCaMKII-expressing young neurons, this isoform of the kinase is present also in the axon, although in this compartment, it does not undergo depolarization-dependent phosphorylation. The lack of activation of the kinase in the axons of the αCaMKII-expressing neurons is not attributable to a reduced Ca2+ influx, because the [Ca2+]i levels reached in this compartment after depolarization are generally comparable with those reached in the dendritic tree of the same cell. However, the selectivity of activation of the kinase in the somatodendritic region of the αCaMKII-expressing neurons is lost using stimulating protocols that induce supraphysiological increases in [Ca2+]i levels (i.e., ionomycin in the presence of 10 mm[Ca2+]o), indicating that both βCaMKII in αCaMKII-negative cells and the axonal pool of the kinase in the αCaMKII-expressing cells are not completely refractory to phosphorylation. In addition, this observation indicates that Ca2+ influx through voltage-gated channels is not strictly required.
Okadaic acid, which has been shown previously to increase the state of phosphorylation of CaMKII in rat hippocampal neurons (Kasahara et al., 1999), is able to determine a marked increase in the state of phosphorylation of axonal CaMKII, suggesting that a differential balance between kinase and phosphatase activities plays a role in selectively silencing CaMKII activity in the axonal compartment of stage III neurons. Okadaic acid is known to inhibit PP2A and PP1. However, the phosphatase involved in silencing axonal CaMKII is likely to be PP2A, because the experimental conditions that we have used conform to established protocols for the selective inhibition of PP2A in intact cells (Favre et al., 1997).
PP2A activity is known to be modulated by various regulatory subunits, which determine the catalytic activity, substrate specificity, and subcellular localization of the enzyme (Janssens and Goris, 2001). Interestingly, one of these subunits, β′α, is selectively localized in the soma and proximal dendrites, thus providing a possible explanation for our observations. The actual involvement of this or other subunits in the observed differential behavior between axons and somatodendrites in terms of PP2A activity, as well as the mechanisms of their targeting to selected neuronal compartments, will be an interesting subject for future investigations.
Interestingly, OA was not able to rescue CaMKII phosphorylation in neurons not expressing the α subunit of the kinase, suggesting that the absence of detectable CaMKII phosphorylation cannot be ascribed to the activity of PP2A in these neurons.
In neurons that have already established synaptic contacts, treatment with OA increased the percentage of synapses in which CaMKII-mediated synapsin phosphorylation can be detected after depolarization, indicating that the kinase-silencing mechanism is active also within the immature presynaptic compartment. In contrast, at the postsynaptic level, the percentage of phosphoCaMKII-positive spines was not altered by OA, consistent with the notion that in this compartment, CaMKII is dephosphorylated by PP1 and not by PP2A (Strack et al., 1997).
In mature nerve terminals, a complex of synapsin I and CaMKII on the SV membrane has been implicated in the regulation of SV availability for neurotransmitter release (Benfenati et al., 1992, 1996; Greengard et al., 1993). Although both synapsin I and CaMKII are expressed starting from early developmental stages, with an overlapping pattern of expression in the axonal compartment, their functional coupling can be seen only in mature terminals. Thus, activation of the presynaptic pool of CaMKII, coupled to synapsin I phosphorylation, increases with development in culture in parallel with synaptogenesis and synaptic maturation. Together, our data support the hypothesis that, during development, dephosphorylated synapsin I plays a role in the interaction of SVs with the cytomatrix of nascent synapses, thus acting as a molecular switch that operates to assemble SV clusters before and during synapse formation and that dynamically modulates SV availability for exocytosis in mature nerve terminals (Greengard et al., 1993;Valtorta et al., 1995).
Activation of the somatodendritic pool of the kinase, which appears to be homogeneous in stage III neurons, becomes regionalized in mature, stage V neurons. At this stage, it is possible to identify a proximal compartment of CaMKII activation, composed by the soma and proximal dendrites, and a distal compartment, localized in isolated spines. The absence of labeling in the distal dendritic shaft suggests partially independent generative mechanisms for distal and proximal CaMKII activation.
In the distal domain, where CaMKII activation can be analyzed without an interfering signal from the dendrite, KCl depolarization activates only 40% of the total number of spines. In contrast, the same stimulation appears to elicit activation of the kinase in >90% of the presynaptic terminals, as assayed by CaMKII-dependent synapsin I phosphorylation. The latter figure is in agreement with the percentage of presynaptic terminals in these cultures that respond to depolarization with Ca2+-dependent neurotransmitter release, as assayed by labeling with antibodies against the intravesicular lumen of the SV protein synaptotagmin, an indicator of SV recycling (Verderio et al., 1999b). The observation that the fraction of spines in which CaMKII is activated remains relatively constant during synaptogenesis, in contrast to the progressively increasing activation of the presynaptic pool, indicates that the maturation of the presynaptic and postsynaptic compartments is independently regulated and suggests that the postsynaptic receptive apparatus is tuned to a submaximal level, allowing synaptic efficiency to be effectively modulated in both directions by CaMKII activation.
Footnotes
This work was supported by Telethon Grants 1000 (F.V.), 1042 (M.M.), and 1131 (F.B.), by the Harvard-Armenise Foundation, by the Italian Ministry of Education (University Excellence Center on Physiopathology of Cell Differentiation and National Interest Research Program 2001), by European Community Grant QLGR3-CT-2000-01343 (M.M.), by Human Frontier Science Program Grant RGY0022/01 (M.M.), and by United States Public Health Service Grants MH39327 and AG15072 (P.G.).
Correspondence should be addressed to Flavia Valtorta, Unit of Experimental Neuropharmacology, San Raffaele Scientific Institute, Via Olgettina, 58, 20132 Milano, Italy. E-mail: valtorta.flavia@hsr.it.
REFERENCES
- 1.Banker G, Cowan W. Rat hippocampal neurons in dispersed cell culture. Science. 1977;209:809–811. doi: 10.1016/0006-8993(77)90594-7. [DOI] [PubMed] [Google Scholar]
- 2.Barria A, Muller D, Derkach V, Griffith LC, Soderling TR. Regulatory phosphorylation of AMPA-type glutamate receptors by CaM-KII during long-term potentiation. Science. 1997;276:2042–2045. doi: 10.1126/science.276.5321.2042. [DOI] [PubMed] [Google Scholar]
- 3.Benfenati F, Valtorta F, Rubenstein JL, Gorelick F, Greengard P, Czernik AJ. Synaptic vesicle-associated Ca2+/calmodulin-dependent protein kinase II is a binding protein for synapsin I. Nature. 1992;359:417–420. doi: 10.1038/359417a0. [DOI] [PubMed] [Google Scholar]
- 4.Benfenati F, Onofri F, Czernik AJ, Valtorta F. Biochemical and functional characterization of the synaptic vesicle-associated form of Ca2+/calmodulin-dependent protein kinase II. Brain Res Mol Brain Res. 1996;40:297–309. doi: 10.1016/0169-328x(96)00053-8. [DOI] [PubMed] [Google Scholar]
- 5.Booher J, Sensenbrenner M. Growth and cultivation of dissociated neurons and glial cells from embryonic chick, rat and human brain in flask cultures. Neurobiology. 1972;2:97–105. [PubMed] [Google Scholar]
- 6.Braun AP, Schulman H. The multifunctional calcium/calmodulin-dependent protein kinase: from form to function. Annu Rev Physiol. 1995;57:417–445. doi: 10.1146/annurev.ph.57.030195.002221. [DOI] [PubMed] [Google Scholar]
- 7.Czernik AJ, Girault JA, Nairn AC, Chen J, Snyder G, Kebabian J, Greengard P. Production of phosphorylation state-specific antibodies. Methods Enzymol. 1991;201:264–283. doi: 10.1016/0076-6879(91)01025-w. [DOI] [PubMed] [Google Scholar]
- 8.Dzhura I, Wu Y, Colbran RJ, Balser JR, Anderson ME. Calmodulin kinase determines calcium-dependent facilitation of L-type calcium channels. Nat Cell Biol. 2000;2:173–177. doi: 10.1038/35004052. [DOI] [PubMed] [Google Scholar]
- 9.Favre B, Turowski P, Hemmings BA. Differential inhibition and posttranslational modification of protein phosphatase 1 and 2A in MCF7 cells treated with caliculin-A, okadaic acid, and tautomycin. J Biol Chem. 1997;272:13856–13863. doi: 10.1074/jbc.272.21.13856. [DOI] [PubMed] [Google Scholar]
- 10.Gallo V, Suergiu R, Giovannini C, Levi G. Glutamate receptor subtypes in cultured cerebellar neurons: modulation of glutamate and gamma-aminobutyric acid release. J Neurochem. 1987;49:1801–1809. doi: 10.1111/j.1471-4159.1987.tb02439.x. [DOI] [PubMed] [Google Scholar]
- 11.Gardoni F, Schrama LH, Kamal A, Gispen WH, Cattabeni F, Di Luca M. Hippocampal synaptic plasticity involves competition between Ca2+/calmodulin-dependent protein kinase II and postsynaptic density 95 for binding to the NR2A subunit of the NMDA receptor. J Neurosci. 2001;21:1501–1509. doi: 10.1523/JNEUROSCI.21-05-01501.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science. 1993;259:780–785. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- 13.Hanson PI, Schulman H. Neuronal Ca2+/calmodulin-dependent protein kinases. Annu Rev Biochem. 1992;61:559–601. doi: 10.1146/annurev.bi.61.070192.003015. [DOI] [PubMed] [Google Scholar]
- 14.Harris MK. Structure, development, and plasticity of dendritic spines. Curr Opin Neurobiol. 1999;9:343–348. doi: 10.1016/s0959-4388(99)80050-6. [DOI] [PubMed] [Google Scholar]
- 15.Janssens V, Goris J. Protein phosphatase 2A: a highly regulated family of serine/threonine phosphatases implicated in cell growth and signaling. Biochem J. 2001;353:417–439. doi: 10.1042/0264-6021:3530417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kasahara J, Fukunaga K, Miyamoto E. Differential effects of a calcineurin inhibitor on glutamate-induced phosphorylation of Ca2+/calmodulin-dependent protein kinases in cultured rat hippocampal neurons. J Biol Chem. 1999;274:9061–9067. doi: 10.1074/jbc.274.13.9061. [DOI] [PubMed] [Google Scholar]
- 17.Kelly PT, McGuinness TL, Greengard P. Evidence that the major postsynaptic density protein is a component of a Ca2+/calmodulin-dependent protein kinase. Proc Natl Acad Sci USA. 1984;81:945–949. doi: 10.1073/pnas.81.3.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kennedy M. Signal-processing machines at the postsynaptic density. Science. 2000;290:750–754. doi: 10.1126/science.290.5492.750. [DOI] [PubMed] [Google Scholar]
- 19.Kennedy MB, Bennett MK, Erondu NE. Biochemical and immunochemical evidence that the “major postsynaptic density protein” is a subunit of a calmodulin-dependent protein kinase. Proc Natl Acad Sci USA. 1983;80:7357–7361. doi: 10.1073/pnas.80.23.7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 21.Lai Y, Nairn AC, Greengard P. Autophosphorylation reversibly regulates the Ca2+/calmodulin-dependence of Ca2+/calmodulin-dependent protein kinase II. Proc Natl Acad Sci USA. 1986;83:4253–4257. doi: 10.1073/pnas.83.12.4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lisman J. Tha CaM kinase II hypothesis for the storage of synaptic memory. Trends Neurosci. 1994;17:406–412. doi: 10.1016/0166-2236(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 23.Lisman J, Malenka RC, Nicoll RA, Malinow R. Learning mechanisms: the case for CaMKII. Science. 1997;276:2001–2002. doi: 10.1126/science.276.5321.2001. [DOI] [PubMed] [Google Scholar]
- 24.Maletic-Savatic M, Koothan T, Malinow R. Calcium-evoked dendritic exocytosis in cultured hippocampal neurons. Part II: mediation by calcium/calmodulin-dependent protein kinase II. J Neurosci. 1998;18:6814–6821. doi: 10.1523/JNEUROSCI.18-17-06814.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mayford MJ, Wang ER, Kandel TJ, O'Dell TJ. CaMKII regulates the frequency-response function of hippocampal synapses for the production of both LTD and LTP. Cell. 1995;81:891–904. doi: 10.1016/0092-8674(95)90009-8. [DOI] [PubMed] [Google Scholar]
- 26.McGuinness TL, Lai Y, Greengard P. Ca2+/calmodulin-dependent protein kinase II isozymic forms from rat forebrain and cerebellum. J Biol Chem. 1985;260:1696–1704. [PubMed] [Google Scholar]
- 27.Menegon A, Leoni C, Benfenati F, Valtorta F. Tat protein from HIV-1 activates MAP kinase in granule neurons and glial cells from the rat cerebellum. Biochem Biophys Res Commun. 1997;238:800–805. doi: 10.1006/bbrc.1997.7393. [DOI] [PubMed] [Google Scholar]
- 28.Menegon A, Dunlap DD, Castano F, Benfenati F, Czernik AJ, Greengard P, Valtorta F. Use of phosphosynapsin I-specific antibodies for image analysis of signal transduction in single nerve terminals. J Cell Sci. 2000;113:3573–3582. doi: 10.1242/jcs.113.20.3573. [DOI] [PubMed] [Google Scholar]
- 29.Miller SG, Kennedy MB. Regulation of brain type II Ca2+/calmodulin-dependent protein kinase by autophosphorylation: a Ca2+-triggered molecular switch. Cell. 1986;44:861–870. doi: 10.1016/0092-8674(86)90008-5. [DOI] [PubMed] [Google Scholar]
- 30.Pravettoni E, Bacci A, Coco S, Forbicini P, Matteoli M, Verderio C. Different localizations and functions of L-type and N-type calcium channels during development of hippocampal neurons. Dev Biol. 2000;227:581–594. doi: 10.1006/dbio.2000.9872. [DOI] [PubMed] [Google Scholar]
- 31.Rongo C, Kaplan JM. CaMKII regulates the density of central glutamatergic synapses in vivo. Nature. 1999;402:195–199. doi: 10.1038/46065. [DOI] [PubMed] [Google Scholar]
- 32.Scholz WK, Baitinger C, Schulman H, Kelly PT. Developmental changes in Ca2+/calmodulin-dependent protein kinase II in cultures of hippocampal pyramidal neurons and astrocytes. J Neurosci. 1988;8:1039–1051. doi: 10.1523/JNEUROSCI.08-03-01039.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen K, Meyer T. Dynamic control of CaMKII translocation and localization in hippocampal neurons by NMDA receptor stimulation. Science. 1999;284:162–166. doi: 10.1126/science.284.5411.162. [DOI] [PubMed] [Google Scholar]
- 34.Soderling TR. CaM-kinases: modulators of synaptic plasticity. Curr Opin Neurobiol. 2000;10:375–380. doi: 10.1016/s0959-4388(00)00090-8. [DOI] [PubMed] [Google Scholar]
- 35.Stevens CF, Tonegawa S, Wang Y. The role of calcium-calmodulin kinase II in three forms of synaptic plasticity. Curr Biol. 1994;4:687–693. doi: 10.1016/s0960-9822(00)00153-6. [DOI] [PubMed] [Google Scholar]
- 36.Strack S, Barban MA, Wadzinski BE, Colbran RJ. Differential inactivation of postsynaptic density-associated and soluble Ca2+/calmodulin-dependent protein kinase II by protein phosphatases 1 and 2A. J Neurochem. 1997;68:2119–2128. doi: 10.1046/j.1471-4159.1997.68052119.x. [DOI] [PubMed] [Google Scholar]
- 37.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4353. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vaccaro P, Dente L, Onofri F, Zucconi A, Martinelli S, Valtorta F, Greengard P, Cesareni G, Benfenati F. Epitope mapping on mammalian synapsins using phage display libraries. Brain Res Mol Brain Res. 1997;52:1–16. doi: 10.1016/s0169-328x(97)00219-2. [DOI] [PubMed] [Google Scholar]
- 39.Valtorta F, Iezzi N, Benfenati F, Lu B, Poo M-M, Greengard P. Accelerated structural maturation induced by synapsin I at developing neuromuscular synapses of Xenopus laevis. Eur J Neurosci. 1995;7:261–270. doi: 10.1111/j.1460-9568.1995.tb01062.x. [DOI] [PubMed] [Google Scholar]
- 40.Verderio C, Coco S, Pravettoni E, Bacci A, Matteoli M. Synaptogenesis in hippocampal cultures. Cell Mol Life Sci. 1999a;55:1448–1462. doi: 10.1007/s000180050384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verderio C, Coco S, Bacci A, Rossetto O, De Camilli P, Montecucco C, Matteoli M. Tetanus toxin blocks the exocytosis of synaptic vesicles clustered at synapses but not of synaptic vesicles in isolated axons. J Neurosci. 1999b;19:6723–6732. doi: 10.1523/JNEUROSCI.19-16-06723.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu G-Y, Cline HT. Stabilization of dendritic arbor structure in vivo by CaMKII. Science. 1998;279:222–226. doi: 10.1126/science.279.5348.222. [DOI] [PubMed] [Google Scholar]
- 43.Wu G-Y, Malinow R, Cline HT. Maturation of a central glutamatergic synapse. Science. 1996;274:972–976. doi: 10.1126/science.274.5289.972. [DOI] [PubMed] [Google Scholar]
- 44.Zou D-J, Cline HT. Expression of constitutively active CaMKII in target tissue modifies presynaptic axon arbor growth. Neuron. 1996;16:529–539. doi: 10.1016/s0896-6273(00)80072-0. [DOI] [PubMed] [Google Scholar]