

Abstract
Mice deficient in the neural cell adhesion molecule (NCAM) exhibit increased anxiety and anxiolytic sensitivity to serotonin 5-HT1A receptor agonists. Here, we investigate the relationship between NCAM and 5-HT1A receptor signaling pathways modulating G-protein-activated inwardly rectifying K+ (Kir3) channels. When studying this relationship in cultured hippocampal neurons, we observed that in cells from NCAM-deficient mice, inwardly rectifying K+ (Kir3) currents were increased compared with wild-type controls. Analysis of this modulatory mechanism in Xenopus oocytes and Chinese hamster ovary (CHO) cells revealed that the recombinantly expressed major transmembrane isoforms NCAM140 and NCAM180 specifically reduced inward currents generated by neuronal Kir3.1/3.2 and Kir3.1/3.3 but not by cardiac Kir3.1/3.4 channels. Using fluorescence measurements and surface biotinylation assays, we show that this effect was caused by a reduced surface localization of Kir3 channels. Furthermore, expression of flag-tagged Kir3 channels in cultured neurons of NCAM-deficient mice resulted in a higher transport of these channels into neurites and a higher cell-surface localization compared with wild-type neurons. Neuronal Kir3 channels and NCAM isoforms are associated with cholesterol-rich microdomains (lipid rafts) in CHO cells and in isolated brain membranes. Mutational and pharmacological disruption of the lipid raft association of NCAM140 normalizes surface delivery of channels. We conclude that the transmembrane isoforms of NCAM reduce the transport of Kir3 channels to the cell surface via lipid rafts. Thus, regulation of Kir3 channel activity by NCAM may represent a novel mechanism controlling long-term excitability of neurons.
Keywords: Kir3, GIRK, cell-surface localization, lipid rafts, palmitoylation, NCAM-deficient mice
The neural cell adhesion molecule (NCAM) is involved in neuronal migration, neurite outgrowth and fasciculation, synapse formation, synaptic plasticity, and emotional behavior (Schachner, 1997; Ronn et al., 1998; Murase and Schuman, 1999). NCAM-deficient mice show increased anxiety and aggression and an altered response of 5-HT1A receptors to their agonists 8-hydroxy-dipropylaminotetralin (8-OH-DPAT) and buspirone (Stork et al., 1997, 1999), suggesting a functional interdependence of the 5-HT1A receptor and NCAM. 5-HT1A receptor distribution and affinity or tissue concentrations of 5-HT or its metabolite 5-hydroxyindoacetic acid were not different between wild-type (NCAM+/+) and NCAM-deficient (NCAM−/−) mice, indicating that NCAM does not affect 5-HT turnover or 5-HT1A receptor function.
Therefore, we extended our studies on a possible influence of NCAM on the 5-HT1A receptor, which belongs to the seven membrane-spanning receptor family activating Gi/oproteins. In the hippocampus, the main effectors of the 5-HT1A receptor are G-protein-coupled inwardly rectifying K+ (Kir3) channels localized presynaptically and postsynaptically. These channels hyperpolarize the membrane potential and are thus involved in inhibitory activities (Lüscher et al., 1997). The Kir3 family consists of the Kir3.1, Kir3.2, Kir3.3, and Kir3.4 subunits, and the majority of functional Kir3 channels are believed to exist as heterotetramers containing the Kir3.1 subunit, although some studies report on functional Kir3.2 homomers and Kir3.2/3.3 combinations (Wischmeyer et al., 1997; Inanobe et al., 1999; Jelacic et al., 2000).
To investigate a possible influence of NCAM on Kir3 channel activity, we compared inwardly rectifying K+currents in hippocampal neurons from NCAM+/+ and NCAM−/− mice. After the observation that Kir3 channel activity was reduced in the presence of NCAM, we investigated this modulatory mechanism by recombinantly expressing combinations of the major glycosyl-phosphatidylinositol-linked NCAM120 and transmembrane NCAM140 and NCAM180 isoforms, Kir3 channels, and 5-HT1A receptor. Here, we show that the transmembrane isoforms of NCAM cause a reduction of Kir3 currents. This effect correlated with reduced cell-surface localization of Kir3 channels. We also show that neuronal Kir3 channels are associated with lipid rafts, and that mutational and pharmacological disruption of the lipid raft association of NCAM140 reverts the reduced surface delivery of Kir3 channels. We conclude that the transmembrane isoforms of NCAM reduce the cell-surface transport of Kir3 channels via a novel lipid raft-associated mechanism.
MATERIALS AND METHODS
DNA constructs. Rat NCAM140 and rat NCAM180/pcDNA3 were a gift from P. Maness (University of North Carolina, Chapel Hill, NC), and rat NCAM120 was a gift from E. Bock (University of Copenhagen, Copenhagen, Denmark). The plasmid coding for enhanced green fluorescent protein (GFP) was purchased from Clontech (Palo Alto, CA). Rat Kir2.1, rat Kir3.1, human Kir3.2, rat Kir3.3 and human Kir3.4, and the three NCAM isoforms were subcloned either into the pcDNA3 vector (Invitrogen, Leek, The Netherlands) for expression in Chinese hamster ovary (CHO) cells or into the psGEM vector, which provides the 5′ and 3′ untranslated regions of the Xenopus β-globin gene, for expression in Xenopus oocytes. The NCAM140Δ construct was generated as described previously (Little et al., 1998) with the exception that mutations were performed using anEcoRI/XhoI subfragment of NCAM140. The intracellular domain of NCAM140 (NCAM140id; base pairs 2185–2547) was amplified by PCR and cloned into the pDNA3 vector. The assembly of concatemeric Kir3.1/3.2 and Kir3.1/3.4 has been described previously (Wischmeyer et al., 1997). Flag-tagged Kir3.1 was a gift from D. Clapham (Children's Hospital, Boston, MA) and was fused in-frame to the Kir3.2 and Kir3.4 subunits. RNA was transcribed in vitrousing the Message Machine kit (Ambion, Austin, TX).
Kir3.1/3.2 and Kir3.1/3.4 chimeras and Kir3.2 mutants. For construction of the C-terminal and N-terminal chimeras of Kir3.2 and Kir3.4, standard PCR technique was used. Silent restriction sites were introduced for SalI and XhoI corresponding to amino acid positions 83 and 193 in Kir3.4 immediately before the first proposed membrane-spanning segment (TM1) and just downstream of the second membrane-spanning segment (TM2). Subfragments of the N- and C-terminal regions of Kir3.2 and Kir3.4 were subcloned into pBluescript II KS. Chimeras were produced by fusing the N or C termini of Kir3.2 or Kir3.4 with their SalI site in-frame on theXhoI site of the corresponding core channel. The sequence of all PCR-amplified products was verified by DNA sequence analysis. The following Kir3.2 mutants were constructed: (1) exchange of amino acids 1–35 in the N terminus, (2) exchange of amino acids 1–54 in the N terminus, (3) exchange of amino acids at positions 58, 61, and 72, and (4) exchange of amino acids at positions 58, 61, 72, 79, and 98 by the corresponding amino acids of Kir3.4. Amino acids 1–35 and 1–54 were exchanged between Kir3.2 and Kir3.4 using the PCR technique of splicing by overlap extension (Retzer et al., 1996). Mutation of the indicated amino acids in the N terminus of Kir3.2 was performed using the Quikchange Mutagenesis kit (Stratagene, Amsterdam, The Netherlands).
Kir3.1/3.2- and Kir3.1/3.4-GFP chimeras. Fusion proteins of the Kir channels with GFP chimeras (Kir3-GFP chimeras) were constructed by removing the stop codon and introducing an XhoI site at the 3′ end of the coding sequences of Kir3.2 and Kir3.4 using a standard PCR technique. SalI and XhoI sites were introduced by PCR into the 5′ and 3′ ends, respectively, of the GFP cDNA. The SalI site of GFP cDNA was fused in-frame to the 3′ end XhoI site of the Kir3.2 and Kir3.4 subunits, and the resulting construct was subcloned into pcDNA3.
Culturing and transfection of CHO cells. CHO cells were maintained in Glasgow modified Eagle's medium (GMEM) containing 10% fetal calf serum (FCS). CHO cells were seeded in six well plates and transfected at 90% confluence using the Lipofectamine Plus kit (Invitrogen, Gaithersburg, MD) according to the manufacturer's instructions. Cells were triple transfected with 0.4 μg of GFP cDNA, 0.6 μg of DNA of the indicated K+channel, and 1 μg of DNA of the specified NCAM isoform. Cell-surface localization of Kir3 and NCAM was studied 48 hr after transfection. For electrophysiological recordings, cells were detached with Versene solution (Invitrogen) 24 hr after transfection and seeded onto poly-l-lysine-coated glass coverslips. Recordings were performed 48 hr after transfection. For biochemical analysis and surface biotinylation assays, cells were detached with Versene solution 24 hr after transfection and reseeded in six well plates at a density of 40%. Where indicated, 10 μm lovastatin (Calbiochem, Bad Soden, Germany) together with 250 μm mevalonate (Sigma, Deisenhofen, Germany) (48 hr before analysis) were added to the medium. Lovastatin was used in the presence of mevalonate to allow synthesis of nonsterol products from mevalonate (Brown and Goldstein, 1980). Lovastatin was prepared in its open acid form before use as described previously (Fenton et al., 1992), and mevalonate was applied in its lactone form.
Calcium phosphate transfection of hippocampal neurons. Cells were transfected 24 hr after seeding using the calcium phosphate method (Ethell and Yamaguchi, 1999). Before transfection, cells were incubated for 1 hr at 3% CO2 and 35°C in serum-free culture medium. The DNA/calcium phosphate precipitate was prepared using a Mammalian Transfection Kit (Stratagene) according to the manufacturer's instructions. Fifty microliters of the DNA/calcium phosphate precipitate was mixed with 1 ml of serum-free medium and added to cultures for 3 hr at 3% CO2 and 35°C. The incubation was stopped by 5% glycerol in serum-free medium. Cells were then washed three times with medium and returned to 5% CO2 and 37°C for 11 d.
Surface biotinylation and determination of Kir3 channel internalization in transfected CHO cells. Internalization kinetics were measured after surface biotinylation as described previously (Schmidt et al., 1997). In brief, 48 hr after transfection, cells were washed twice with ice-cold PBS containing 2 mmMgCl2 and 0.2 mmCaCl2 (PBSCM). Surface proteins were biotinylated by incubating cells with 0.5 mg/ml sulfo-N-hydroxysuccinimide-disulfide-biotin (Pierce, Rockford, IL) in PBSCM for 10 min at 4°C. Biotinylation was terminated by incubation with 20 mm glycine in PBSCM at 4°C for 10 min followed by extensive washes with PBSCM. Biotinylated cells then either were returned to 37°C in GMEM, 10% FCS for 15 min, 30 min, or 1 hr or were lysed directly in radioimmunoprecipitation assay (RIPA) buffer containing 10 mm Tris, pH 7.5, 150 mmNaCl, 1 mm EDTA, 1% Triton X-100, and protease inhibitor mix (COMPLETE; Roche Diagnostics, Mannheim, Germany) and centrifuged for 15 min at 4°C. To determine the amount of protein that was internalized at 37°C after the indicated time periods, remaining surface-bound biotin was stripped off the surface proteins. Cells were washed twice with XXX (NT) buffer [20 mmN-tris(hydroxymethyl)methyl-3-aminopropane-sulfonic acid, pH 8.6, 150 mm NaCl, 0.5 mm CaCl2, 2 mm MgCl2, and 0.2% BSA] at 4°C and incubated twice for 10 min with NT buffer containing 10 mm sodium-2-mercaptoethanesulfonate at 4°C. The reaction was terminated by extensive washing with NT buffer. Finally, cells were lysed in RIPA buffer and centrifuged at 14,000 ×g at 4°C for 15 min. The supernatants were collected, and protein concentrations were determined using the bicinchoninic acid kit (Pierce). The amounts of surface-localized proteins and internalized proteins were determined by precipitating biotinylated proteins with streptavidin-coupled agarose beads (Pierce) at 4°C overnight. Agarose beads were pelleted by centrifugation and washed twice with RIPA buffer. Precipitated proteins were solubilized by the addition of 2× SDS sample buffer to the agarose beads. Proteins were separated by SDS-PAGE and quantified by immunoblot analysis using polyclonal Kir3.1 and NCAM (Simon et al., 1991) antibodies.
Isolation of detergent-resistant membrane fractions. Lipid rafts were isolated as described previously (Melkonian et al., 1999). In brief, monolayers of cotransfected CHO cells grown in 150 cm2 plates were detached using 4 ml of Versene solution and pelleted by centrifugation for 5 min at 500 × g and 4°C. Pellets were homogenized in TNE buffer (in mm: 25 Tris-HCl, pH 7.5, 150 NaCl, 5 EDTA) and COMPLETE protease inhibitor at 4°C using a Dounce homogenizer. Lysates were cleared by centrifugation at 500 × g and 4°C for 5 min to remove unlysed cells and debris. A small fraction of the supernatant was used to verify equal Kir3.1/3.2 and NCAM expression by immunoblot analysis. Membranes were collected by centrifugation at 100,000 × g for 1 hr at 4°C and resuspended in 500 μl of TNE buffer, pH 11, containing 1% Triton X-100. Membranes were incubated for 30 min on ice, adjusted to 40% sucrose, and placed in an SW 55Ti ultracentrifuge tube. The 40% fraction was overlaid with 1.5 ml of TNE 36% sucrose and 2 ml of 10% sucrose. After centrifugation (16 hr, 100,000 × g, 4°C), six 750 μl fractions were collected from the top, diluted with 3 ml of TNE buffer, pH 11, and centrifuged for 1.5 hr at 100,000 × g at 4°C. This procedure permits isolation of only the Triton X-100-insoluble proteins in the pelleted fractions, whereas Triton X-100-soluble proteins remain in the supernatant. Pellets were resuspended in 50 μl of 2× SDS sample buffer and subjected to SDS-PAGE. For preparation of lipid rafts from brain homogenates, 10 forebrains from 2-d-old C57BL/6J mice were homogenized in TNE buffer. Low-density fractions were prepared exactly as described for CHO cells.
Endoglycosidase H digestion. Cell lysates of transfected CHO cells were diluted with 2× citrate buffer (150 mm sodium-citrate, pH 5.5) and incubated overnight at 37°C with 0.1 U of endoglycosidase H (EndoH; Roche Diagnostics). Controls were treated identically without the addition of EndoH. Lysates were subjected to immunoblot analysis with Kir3.1 antibodies.
Electrophysiological recordings. Cultures of hippocampal neurons were prepared by a combination of enzymatic and mechanical dissociation (Dityatev et al., 2000) from 1- to 3-d-old, age-matched C57BL/6J or NCAM−/− mice (Cremer et al., 1994) that had been inbred for at least nine generations onto the C57BL/6J background. Cells were maintained in vitro for 12–20 d. K+ currents were recorded from pyramidal-like neurons in the whole-cell mode at room temperature. The extracellular solution used for recordings from hippocampal neurons contained (in mm): 140 NaCl, 4 KCl, 10 HEPES, 2 CaCl2, 1.5 MgCl2, 30 glucose, and 12 sucrose, pH 7.25. CHO cells were perfused with the solution at elevated concentrations of KCl (25 mm) and reduced concentrations of NaCl (119 mm). CHO cells with bright GFP signal were patched without knowledge of the identity of the plasmids used for transfection. Recordings of inward currents in neurons were performed in the presence of tetrodotoxin (1 μm; Calbiochem) and 0.1 mmCdCl2. Patch pipettes were filled with (in mm): 125 K-gluconate, 20 KCl, 10 HEPES, 0.2 EGTA, 2 Mg-ATP, 0.2 Na-GTP, and 10 glucose. The pH was adjusted with KOH to 7.2, and the osmolarity was 310–315 mOsm. G-protein-coupled inwardly rectifying K+ channels were activated in neurons either with 30 μm 8-OH-DPAT or 50 μm (RS)-baclofen (both from Tocris, Bristol, UK). Double whole-cell voltage-clamp recordings were performed using an EPC-9 amplifier and PULSE software (Heka Electronics, Lamprecht, Germany). During experiments, serial resistance as well as cell resistance and capacitance were measured routinely, and leak currents were digitally subtracted. There was no significant difference between neurons of different genotypes regarding these parameters.
For electrophysiological recordings from Xenopus oocytes, cDNAs of all NCAM and Kir3 isoforms were subcloned into the polyadenylating transcription plasmid psGEM, and capped run-off poly(A)+ cRNA transcripts (∼3 ng each) were injected into defolliculated oocytes (Wischmeyer et al., 1997). Oocytes were incubated at 20°C in ND96 solution containing (in mm): 96 NaCl, 2 KCl, 1 MgCl2, 1 CaCl2, and 5 HEPES, pH 7.4, supplemented with 100 μg/ml gentamicin and 2.5 mm sodium pyruvate. Seventy-two hours after injection, two-electrode voltage-clamp measurements were performed with a TURBO TEC-10 C amplifier (NPI Electronics, Tamm, Germany) in ND96 or high K+ solution containing (in mm): 96 KCl, 2 NaCl, 1 MgCl2, 1 CaCl2, and 5 HEPES, pH 7.4. Measurements were standardized in each experiment using cotransfected Kir3 channel/5-HT1A receptor oocytes as a positive control. Data were acquired and controlled by PULSE/PULSEFIT software (Heka Electronics).
Indirect immunocytochemistry. Before fixation, transfected neurons were stained with flag antibody (Sigma) and detected with Cy5-coupled secondary antibodies (Dianova, Hamburg, Germany). This procedure induces clustering of cell-surface-associated Kir3.1flag/3.2 channels. Cells were fixed with 4% paraformaldehyde in PBS for 15 min at room temperature and permeabilized with 0.25% Triton X-100 in PBS. Cells were then blocked with 3% BSA in PBS. Antibodies to Kir3.1 were applied in PBS containing 3% BSA for 1 hr at room temperature. Cultures were then washed with PBS and incubated with Cy3-coupled secondary antibodies (Dianova) for 1 hr at room temperature. Cultures were washed and postfixed with 2% paraformaldehyde in PBS. Finally, coverslips were embedded in Aqua Poly/Mount medium (Polysciences, Inc., Warrington, PA), and images were taken on a Zeiss (Jena, Germany) LSM510 confocal laser-scanning microscope.
Quantification of immunofluorescence. Cell-surface fluorescence in oocytes was measured with a Zeiss LSM410 argon–crypton laser-scanning microscope equipped with a 16 × oil-immersion objective lens. For quantification of fluorescence intensity, confocal images were taken under constant parameters from 10 average scans at different locations (n = 5 cells). Cell-surface fluorescence of transfected NCAM+/+ and NCAM−/− neurons was quantified using Scion Image software (Scion Corp., Frederick, MD).
RESULTS
Kir3 inward currents are increased in hippocampal neurons of NCAM−/− mice
To explore the interaction of NCAM and 5-HT1A receptor signaling, we investigated Kir3 channels in hippocampal neurons cultured from NCAM+/+ and NCAM−/− mice as a likely target for 5-HT1A receptor signaling (Lüscher et al., 1997). This analysis was also instigated by our previous findings that NCAM influences the activity of outward K+currents in cultured oligodendrocyte precursor cells (Sontheimer et al., 1990). Expression of Kir3 channels in pyramidal cell-like neurons could be demonstrated both immunocytochemically (data not shown) and by patch-clamp analysis. Whole-cell, voltage-clamp experiments revealed several features typical of Kir3 currents: slow current activation (Fig. 1A), current potentiation by the 5-HT1A receptor agonist 8-OH-DPAT (Fig. 1B) and the GABAB receptor agonist baclofen (Fig.1C), complete current block by Ba2+ (Fig. 1B,C), and current dependence on extracellular [K+] (data not shown). When compared in neurons from NCAM+/+ and NCAM−/− mice, it was found that the density of Kir3-like currents significantly increased to 215% in the absence of NCAM (Fig. 1E) (1.7 ± 1.7 pA/pS,n = 29, in NCAM+/+ neurons vs 3.6 ± 2.6 pA/pS, n = 20, in NCAM−/− neurons at a holding potential of −130 mV). In contrast, transient and sustained outward K+ currents remained unchanged (Fig.1D,E).
Fig. 1.

Increased inward currents in NCAM-deficient cultured hippocampal neurons. A, Slowly activating inward currents were evoked in wild-type (NCAM+/+) and NCAM-deficient (NCAM−/−) hippocampal neurons by voltage steps from −60 to −90, −110, and −130 mV. Inward currents activated in neurons by a voltage ramp from −130 to −40 mV were augmented by 30 μm 8-OH-DPAT (B) and by 50 μm (RS)-baclofen (C). These currents were blocked by the addition of 1 mm Ba2+ into the extracellular solution (B, C). D, Transient and sustained K+ outward currents were evoked in neurons by voltage steps from −60 to −30, −10, and +10 mV.E, Cumulative data show a significant difference in inward currents recorded in neurons from NCAM−/− and NCAM+/+ mice (∗p = 0.001; Student's ttest). The horizontal dotted line indicates current density in neurons from NCAM+/+ mice to which other current densities were normalized. Bars represent mean values of current density recorded in NCAM−/− relative to NCAM+/+ neurons; error bars represent SD. Absolute values corresponding to the bars are in pA/pS (number of cells): 3.6 ± 2.6 (n = 20), 1.7 ± 1.7 (n = 29), 63.1 ± 20.1 (n= 10), 55.5 ± 17 (n = 9), 30.5 ± 16.7 (n = 10), and 28.4 ± 10.9 (n = 9). Calibration: A, 100 pA, 50 msec; B, 100 pA, 200 msec; C, 50 pA, 200 msec; D, 1 nA, 50 msec.
NCAM140 and NCAM180 reduce neuronal Kir3 currents inXenopus oocytes and CHO cells
To determine the functional relationship between NCAM and Kir3 channels, we coinjected Xenopus oocytes with equimolar cRNA concentrations of NCAM isoforms and concatenated pairs of Kir3 subunits, which mimic the Kir3 subunit composition in native cells (Wischmeyer et al., 1997). 5-HT1A receptor cRNA was coinjected to assay the activity of receptor-activated Kir3 currents with ramp and voltage-step protocols. With 96 mm K+ and 10 μm 5-HT in the bath solution, a Kir3.1/3.2 combination (present in neurons) gave rise to robust inwardly rectifying K+ currents, which averaged −23.1 ± 7.1 μA (n = 100) at a holding potential of −100 mV. Independent of the presence of NCAM isoforms, both basal and ligand-activated macroscopic Kir3.1/3.2 currents exhibited biophysical properties, e.g., slow current activation (Fig.2A), K+ permeability, dependence of conductance on extracellular K+, or block by Ba2+ and Cs+, typical of native G-protein-activated Kir3 channels (data not shown). After coexpression with NCAM180 or NCAM140, total current amplitudes of Kir3.1/3.2 were strikingly reduced to 36 ± 14% (n = 10) and 25 ± 12% (n = 10), respectively (Fig. 2A,B). These values are of a magnitude similar to the reduction of Kir3-like currents in NCAM+/+ mice (42% reduction) compared with NCAM−/− mice. However, coexpression of NCAM120, which is devoid of an intracellular domain, or a receptor tyrosine kinase (e.g., the trkB receptor) used as additional control had no significant effect on Kir3.1/3.2 currents. When probed in CHO cells, cotransfected NCAM140 and NCAM180 isoforms suppressed Kir3.1/3.2 channel activity in a quantitatively similar manner (Fig.2C).
Fig. 2.

Suppression of neuronal Kir3 channels by NCAM180 and NCAM140 in Xenopus oocytes and CHO cells. Whole-cell currents of Xenopus oocytes injected with cRNAs encoding Kir3.1/3.2 channel subunits, 5-HT1A receptor, and one of the indicated NCAM isoforms are shown. A, Currents are responses to 2 sec voltage ramps between −150 and +60 mV and 500 msec voltage jumps to −80, −100, and −120 mV, respectively, in the presence of 10 μm 5-HT and 96 mm K+. Activation time constants at 0 mV were 20.3 msec in the absence and 19.7 msec in the presence of NCAM140. B, Bar graph showing the relative modulation of Kir3.1/3.2 currents by NCAM120 (1.1 ± 0.43; amplitude relative to control), NCAM140 (0.36 ± 0.14), NCAM180 (0.25 ± 0.12), and NCAM140id (expression vector encoding the intracellular domain of NCAM140) (0.9 ± 0.18) in oocytes. C, Bar graph showing the relative modulation of Kir3.1/3.2 currents by NCAM120 (1.24 ± 0.67, n = 10), NCAM140 (0.29 ± 0.33, n = 13), and NCAM180 (0.17 ± 0.09,n = 11) in transfected CHO cells. D, Macroscopic current responses to 2 sec voltage ramps between −150 and +60 mV of oocytes injected with NCAM180, 5-HT1Areceptor, and different Kir3 channels. E, Bar graph showing the relative inhibition by NCAM180 of Kir3.1/3.2 channels (0.25 ± 0.12 amplitude relative to control in the absence of NCAM), Kir3.1/3.3 (0.18 ± 0.12), Kir3.1/3.4 (0.92 ± 0.38), and Kir2.1 (0.86 ± 0.26). Error bars represent SD.Horizontal dotted lines in A and Dindicate baseline. Vertical dotted lines in A andD indicate 0 mV holding potential. Horizontal dotted lines in B, C, and E indicate current amplitudes to which other current densities were normalized. ∗Statistical significance (Student's t test;p < 0.01).
To investigate the specificity of this reduction in channel activity by NCAM isoforms, other Kir3 channel combinations were also tested. We found that the Kir3.1/3.3 channel expressed in neurons was also severely suppressed by NCAM180 to 18 ± 12% (n = 10). In contrast, both a Kir3.1/3.4 channel, constituting the cardiacKACh channel, as well as other neuronal, constitutively active Kir channels (e.g., Kir2.1), were not significantly affected (Fig. 2D,E).
NCAM140 reduces surface localization of GFP-tagged Kir3.2 channels in Xenopus oocytes
To visualize channel targeting to the cell surface ofXenopus oocytes, Kir3.2 and Kir3.4 channel subunits were tagged with enhanced GFP at their C termini. cRNAs were injected with or without NCAM120, NCAM140, and NCAM180, and the membrane GFP fluorescence of intact oocytes was inspected by confocal microscopy 48 hr after cRNA injection. Although uninjected oocytes showed no background fluorescence, injection of Kir3.1/3.2-GFP and Kir3.1/Kir3.4-GFP constructs resulted in strong fluorescence signals at the cell surface, which were quantified by line-scan luminometry using a photomultiplier (Fig. 3). Coexpression of NCAM140 caused a prominent decrease by ∼58% of Kir3.1/3.2-GFP fluorescence in the membrane (Figs. 3A,B), whereas the Kir3.1/3.4-GFP signal remained unaffected by coexpressed NCAM140 (Fig.3C,D). Similar results were obtained by coinjection of Kir3.1/Kir3.2-GFP with NCAM180 (data not shown). Coinjection of NCAM120, however, did not alter Kir3.1/3.2-GFP fluorescence levels. These results are in accordance with the measurements of ensemble and unitary Kir3 currents (see below) and indicate that NCAM140 and NCAM180 control membrane surface localization of neuronal Kir3 channels.
Fig. 3.

Cell-surface localization of Kir3 channels is reduced by NCAM. Representative confocal images of oocytes injected with cRNAs of GFP-tagged Kir3.1/3.2 (A, B) and Kir3.1/3.4 (C, D), respectively, in the absence (A, C) and presence (B, D) of NCAM140 are shown. Graphs show average fluorescence measurements of 10 line scannings perpendicular to the cell surface representative of five oocytes measured. Data are shown as mean ± SD of fluorescence signal.
The N terminus of Kir3.2 is the major structural determinant for NCAM sensitivity
From the differential sensitivity of Kir3 channels to NCAM inhibition, we sought to identify the structural determinants for the reduced Kir3 channel membrane localization. Because it was unlikely that Kir3.1 subunits, present in all tested combinations, conferred the sensitivity for NCAM, we analyzed hybrid channels in which the N or C termini of the Kir3.2 and Kir3.4 subunits were exchanged. When the C termini were exchanged, the hybrid channels maintained the NCAM sensitivity of the core channel (Fig.4A,B), whereas exchange of the N termini reverted the sensitivity of the core channel toward NCAM180 (Fig. 4C,D). Thus, Kir3.1/3.4 channels were suppressed by NCAM180 when the N terminus of Kir3.4 was substituted by the N terminus of Kir3.2. Although current suppression of hybrid channels was not as pronounced as in Kir3.1/3.2 wild-type channels, we conclude that the N terminus of the Kir3.1/3.2 channel harbors the major structural determinants for NCAM inhibition. To obtain additional insights into functionally important domains and amino acids, we exchanged subdomains and single amino acid residues that differ between the N termini of Kir3.2 and Kir3.4 (see Materials and Methods). The inhibitory effect of NCAM140 was not abolished in any of these mutants (data not shown), suggesting that multiple allosteric effects are involved in the NCAM-mediated Kir3 inhibition.
Fig. 4.

N termini of Kir3 subunits determine sensitivity to NCAM. A, C, Macroscopic current responses to 2 sec voltage ramps between −150 and +60 mV of oocytes injected with NCAM180, 5-HT1A receptor, and different combinations of hybrid Kir3.2/Kir3.4 subunits connected to Kir3.1 subunits. N and C termini of injected Kir3 subunits were exchanged as shown in the drawings with Kir3.2 components in gray and Kir3.4 inblack. B, Bar graph showing the relative inhibition by NCAM180 of Kir3.1/3.2 and Kir3.1/3.4 with the C terminus exchanged, Kir3.1/3.2 + C terminus 3.4 (0.6 ± 0.26), and Kir3.1/3.4 + C terminus 3.2 (0.82 ± 0.39). D, Relative inhibition by NCAM180 of Kir3.1/3.2 and Kir3.1/3.4 with the N terminus exchanged, Kir3.1/3.2 + N terminus 3.4 (0.84 ± 0.2), and Kir3.1/3.4 + N terminus 3.2 (0.56 ± 0.27; n = 10 each). Error bars represent SD. Horizontal dotted linesin A and C indicate baseline. Vertical dotted line in A and D indicates 0 mV holding potential. Horizontal dotted lines in Band D indicate current amplitudes to which the other currents were normalized. ∗Statistical significance (Student'st test; p < 0.01).
Because cell-surface localization of Kir3 channels is specifically reduced by the NCAM140 and NCAM180 isoforms that harbor an intracellular domain, we tested whether the intracellular domain of NCAM140 alone was sufficient to cause this effect. When a construct that encodes NCAM140id and that is membrane associated because of the presence of palmitoylated intracellular cysteines (Little et al., 1998) was coinjected with Kir3.1/3.2, it did not suppress Kir3 channel cell-surface localization or Kir3 inward currents (90 ± 18% of control) (Fig. 2B).
Stimulation of NCAM140- or NCAM180-associated signal transduction pathways do not alter surface localization of Kir3.1/3.2
We subsequently asked which mechanisms could account for the reduced surface expression of neuronal Kir3 channels in the presence of NCAM140 or NCAM180. First, we noted that acute or prolonged stimulation of NCAM by NCAM-specific antibodies known to induce NCAM-dependent signaling (Beggs et al., 1997) was without effect on the activity of Kir3.1/3.2 channels in Xenopus oocytes. Under our recording conditions, no significant differences in Kir3.1/3.2 current amplitudes were measured for NCAM140 or NCAM180 after application of polyclonal NCAM antibodies (200 μg/ml) for 30 min and 48 hr, respectively. Second, suppression was unaltered when assayed for known mediators of NCAM signaling [i.e., incubation of oocytes for 48 hr with 2′-amino-3′-methoxyflavone, a selective inhibitor of microtubule-associated protein kinase kinases (Kolkova et al., 2000), or 1-tert-butyl-3-[6-(3,5-dimethoxy-phenyl)-2-(4-diethylamino-butylamino)-pyrido [2,3-d]pyrimidin-7-yl]-urea (PD173074), a specific inhibitor (Mohammadi et al., 1998) of the FGF receptor known to be functionally linked to NCAM]. Third, single-channel measurements in the cell-attached configuration showed that both unitary conductance and open probability of Kir3.1/3.2 channels remained unchanged in the presence of NCAM180 (Fig.5A). These observations support the view that Kir3 current reduction by NCAM is attributable to a reduction in the number of functional channels at the cell surface, as revealed by immunofluorescence measurements. Finally, we were unable to demonstrate a direct physical interaction of Kir3.1/3.2 and NCAM140 by cocapping, coimmunoprecipitation, or yeast two-hybrid assays that tested the interaction of the NCAM140 intracellular domain with the N and C termini of Kir3.2 (data not shown).
Fig. 5.

Exclusion of NCAM140 from lipid rafts or depletion of cholesterol neutralizes the inhibitory effect of NCAM140 on Kir3 channel activity. A, Single channel cell-attached recordings at +60, −60, and −100 mV are shown from oocytes coinjected with Kir3.1/3.2 and NCAM180 (left panel). No significant difference in channel activity is observed in the presence or absence of NCAM180 (middle panel). The bar graph quantifies the product of open probability (Po) and unitary conductance (γ) for Kir3.1/3.2 channels in the presence and absence of NCAM. B, Schematic diagram of NCAM140 palmitoylation sites. Mutated cysteine residues are indicated in gray. Circular areas indicate immunoglobulin-like domains, and the fibronectin type III-like domains are shown as gray boxes. The surface membrane is indicated by the pair ofvertical lines. C, Immunoblot analysis of sucrose gradient fractions of Kir3.1/3.2 and NCAM140 (left) or NCAM140Δ (right)-cotransfected CHO cells. Kir3.1/3.2 (top panels) is present both in the low-density raft fractions (lanes 3 and 4) and in the high-density fraction (lane 6), possibly containing cytoskeleton-associated proteins. Analysis of the low-density sucrose gradient fractions shows that the amount of Kir3.1/3.2 in lipid rafts is identical in the presence of NCAM140 or NCAM140Δ. The blot was stripped and reprobed with polyclonal NCAM antibodies verifying that, in contrast to NCAM140, NCAM140Δ is hardly detectable in the lipid raft fractions (bottom panel,lanes 2–4). NCAM140 and the Kir3 channel are both present in the major lipid raft fraction (lane 3).D, In forebrain homogenates, the Kir3.1 subunit is present in the low-density fractions (lanes 3 and4) representing the lipid raft fraction and in the high-density, Triton X-100-insoluble fraction (lane 6, top panel). The blot was reprobed with an antibody for the raft-associated nonreceptor tyrosine kinase fyn (bottom panel) to confirm isolation of lipid rafts. E, Bar graph showing the relative inhibition of Kir3.1/3.2 currents in CHO cells mediated by NCAM140 (0.14 ± 0.04), NCAM140Δ (0.79 ± 0.19), and NCAM120 (1 ± 0.23). Lovastatin treatment rescued NCAM140-mediated inhibition (0.47 ± 0.07), leaving NCAM120-mediated inhibition unchanged (0.86 ± 0.14). Mean amplitudes relative to Kir3.1/3.2-NCAM120-cotransfected cells are shown. ∗Statistical significance from control.#Statistical significance between Kir3.1/3.2 + NCAM140 with and without lovastatin (Student's t test;p < 0.01). Error bars represent SEM.Horizontal dotted lines in A indicate the open and closed state of the channel. The horizontal dotted linein E indicates the current amplitude to which the other currents were normalized. The vertical dashed line inE separates the results without lovastatin from the results with lovastatin. IB, Immunoblot for the molecules indicated.
Impairment of NCAM140 raft association abrogates the inhibitory effect of NCAM140 on Kir3.1/3.2 cell-surface localization
We have reported recently that all NCAM isoforms are associated with lipid rafts, and that lipid raft association of NCAM140 is essential for its function as a neuritogenic receptor (Niethammer et al., 2002). Therefore, we asked whether lipid raft association of NCAM140 is essential to modulate Kir3 channel surface localization. These experiments were instigated by our observation that Kir3.1/3.2 is partly associated with lipid rafts in transfected CHO cells and brain homogenates using the well established criteria for identification of lipid raft-associated proteins, namely insolubility in cold nonionic detergents and flotation in defined sucrose density gradients (Hooper, 1997). As shown in Figure 5C,D, Kir3.1 immunoreactivity is present in the upper fractions (lanes 3 and4) of a sucrose density gradient of Triton X-100-insoluble protein fractions of transfected CHO cell lysates and mouse forebrain homogenates, indicating a partial association with the lipid raft fraction in CHO cells and mouse brains. Kir3.1 subunits were also present in the Triton X-100-insoluble bottom fraction of the gradient (Fig. 5C,D, lane 6), which is likely to contain cytoskeleton-associated Kir3.1 subunits (Montixi et al., 1998; Kennedy et al., 1999).
We then asked whether removal of NCAM140 from lipid rafts would affect Kir3 channel cell-surface localization. We have shown that raft association of an NCAM140 mutant (denoted NCAM140Δ) is reduced compared with NCAM140 when the palmitoylation sites, and thus a raft-targeting signal, are removed (Fig. 5B) (Niethammer et al., 2002). NCAM140 but not NCAM140Δ colocalizes with Kir3.1/3.2 in the lipid raft fraction of transfected CHO cells (Fig. 5C,left panel, lanes 3 and 4 vs Fig.5C, right panel, lanes 3and 4). Lipid raft localization of the Kir3 channel is not altered when NCAM140 is removed from the lipid rafts by ablation of palmitoylation sites (Fig. 5C, top panels). It needs to be emphasized in this context that NCAM140 and NCAM140Δ are present in equal amounts in total cell lysates and at the cell surface (Niethammer et al., 2002). The inhibitory effect of NCAM140 on Kir3 channel surface localization is abolished when NCAM140 is deprived of palmitoylation sites and therefore not present in lipid rafts (Fig.5E). Lipid rafts may thus function as a platform from which NCAM controls the surface localization of Kir3.1/3.2 channels.
To further document the involvement of lipid rafts in the NCAM-mediated surface localization of Kir3 channels, we disrupted lipid rafts in CHO cells by depletion of cholesterol using lovastatin, a blocker of the 3-hydroxy-3-methylglutaric acid CoA reductase and thus of cholesterol biosynthesis. This treatment not only disperses lipid rafts in the surface membrane but also affects assembly of lipid rafts in intracellular organelles, such as the trans-Golgi network. As determined electrophysiologically, disruption of lipid rafts by lovastatin partially rescued Kir3 channel surface localization in Kir3.1/3.2-NCAM140-cotransfected cells (53 ± 7% reduction by NCAM140 in the presence of lovastatin vs 86 + 4% reduction by NCAM140 in the absence of lovastatin) (Fig. 5E). Treatment with lovastatin per se did not impair the surface localization or channel properties of the Kir3.1/3.2, because currents were unaffected by lovastatin in Kir3.1/3.2-NCAM120-cotransfected cells (Fig.5E). One reason why NCAM140-mediated Kir3 channel inhibition was only partly rescued by lovastatin is most likely because of the incomplete block of cholesterol biosynthesis by this treatment (Keller and Simons, 1998).
Reduction of Kir3 channel cell-surface localization by NCAM140 altered delivery to or internalization from the surface membrane?
To further investigate the mechanisms of Kir3 channel cell-surface localization, we assayed the internalization of Kir3.1/3.2 and Kir3.1/3.4 biochemically by surface biotinylation in CHO cells after transfection with Kir3.1 that was tagged by a flag-epitope inserted into the first extracellular loop of the subunit (Kennedy et al., 1999). Single channel properties of Kir3.1flag/3.2 and Kir3.1flag/3.4 and current inhibition by NCAM140 were unaffected by the flag epitope (data not shown). Quantification of Kir3.1/3.2 and Kir3.1/3.4 cell-surface localization showed that NCAM140 but not NCAM140Δ reduced the amount of Kir3.1/3.2 protein (but not Kir3.1/3.4, data not shown) in the surface membrane by ∼50% (Fig.6A, lanes 8vs 7 and 9, B). Overall, protein expression of Kir3.1flag/3.2 was not significantly affected by NCAM140 (Fig. 6A, lanes 10–12, C). Lovastatin treatment also did not alter the overall protein expression of Kir3.1flag/3.2 (Fig. 6A, lanes 4–6 vs10–12).
Fig. 6.

NCAM140 reduces cell-surface localization of Kir3.1/3.2 in CHO cells but does not alter internalization rates of Kir3.1/3.2. A, Immunoblot analysis of Kir3.1/3.2 cell-surface expression in CHO cells. CHO cells were cotransfected with Kir3.1/3.2 and either NCAM120 (lanes 3, 6, 9, and 12), NCAM140 (lanes 2, 5, 8, and 11), or NCAM140Δ (lanes 1, 4, 7, and 10). Cell lysates (lanes 4–6 and 10–12) and proteins bound to streptavidin-agarose (lanes 1–3 and7–9) were separated by SDS-PAGE, and the amount of Kir3.1/3.2 or NCAM was quantified by immunoblot analysis using polyclonal Kir3.1 antibodies. NCAM140 reduces surface localization of Kir3.1/3.2 compared with NCAM120 and NCAM140Δ (lane 8vs lanes 7 and 9), whereas the overall intensity of Kir3.1/3.2-immunoreactive bands in the cell lysate is not altered by NCAM140 (lane 11 vs lanes 10and 12). Incubation with lovastatin does not influence Kir3.1/3.2 expression (lanes 4–6 vs10–12) but enhances surface localization of Kir3.1/3.2 (lane 2 vs lanes 1 and3). B, Normalized surface localization of Kir3.1/3.2 in the presence of NCAM140 (0.48 ± 0.03) and NCAM140Δ (0.84 ± 0.09) in comparison with NCAM120. Kir3.1-immunoreactive bands of streptavidin-agarose-precipitated proteins were quantified by densitometric analysis and expressed relative to NCAM120 (100%). ∗Values significantly different from that of NCAM120 (Student's t test;p < 0.01). Error bars represent SEM.C, Normalized protein expression of Kir3.1/3.2 in the presence of NCAM140 (0.86 ± 0.05) and NCAM140Δ (1.03 ± 0.04) in comparison with NCAM120. Kir3.1-immunoreactive bands in total cell lysates were quantified by densitometric analysis and expressed relative to NCAM120. Bars represent means ± SEM.D, Internalization kinetics of Kir3.1/3.2 in the presence of NCAM120 (lanes 1 and4), NCAM140 (lanes 2 and5), and NCAM140Δ (lanes 3 and6). Lanes 1–3, The initial amount of Kir3.1/3.2 channels present at the cell surface at 0 min.Lanes 4–6, The amount of Kir3.1/3.2 and the individual NCAM isoforms internalized after 60 min at 37°C. E, Internalization rates were calculated by dividing the relative intensities of Kir3.1-immunoreactive bands (normalized to cotransfection with NCAM120) after cells had been exposed to 60 min at 37°C (lanes 4–6 in D) by the band intensities at 0 min (lanes 1–3 in D). Kir3.1/3.2 channels are internalized with the same kinetics when cotransfected with NCAM140 (0.97 + 0.04) or NCAM140Δ (1 + 0.05) compared with cotransfection with NCAM120. Bar graphs show internalization relative to NCAM120. Bars show the mean ± SEM.F, EndoH digestion of CHO cell lysates transfected with Kir3.1/3.2 and NCAM140Δ (lanes 1 and2), Kir3.1/3.2 NCAM140 (lanes 3 and4), and the cell adhesion molecule L1 (lanes 5 and 6), in which the homophilic-binding site was deleted (Zhao et al., 1998). Cell lysates were either treated with EndoH (lanes 1, 3, and5) or treated without EndoH (lanes 2, 4, and 6). EndoH digestion does not result in a shift of the immunoreactive bands in their apparent molecular weights, indicating that Kir3.1/3.2 does not acquire an increased EndoH sensitivity in the presence of NCAM140. As a positive control, CHO cells were transfected with a protein known to be retained in the ER, namely the mutated L1 molecule (A. Rünker, personal communication). The L1 molecule is shifted to a lower apparent molecular weight by EndoH digestion (lane 5 vs6). The horizontal dotted lines inB, C, and E indicate the current amplitude to which the other currents were normalized.IB, Immunoblot for the molecules indicated.
Cell-surface biotinylation of the Kir3 channels allows the measurement of their internalization rates. Kir3 channel-transfected CHO cells showed that fewer Kir3 channels were internalized after 1 hr at 37°C in the presence of NCAM140 (Fig. 6D, lane 5) compared with NCAM120 and NCAM140Δ (Fig.6D, lanes 4 and 6). Because initially fewer Kir3 channels were surface localized in the presence of NCAM140 (Fig. 6D, lane 2 vslanes 1 and 3), the ratio between the initial amount of Kir3.1/3.2 present at the cell surface (lanes 1–3) and the amount internalized after 1 hr (lanes 4–6) was identical among the individual NCAM isoforms (Fig. 6E). These results were also obtained for shorter (15 and 30 min) internalization periods (data not shown). Our data thus show that NCAM140 does not enhance the internalization rates of Kir3 channels, suggesting that NCAM140 and NCAM180 reduce the delivery of Kir3.1/3.2 to the surface membrane.
To determine whether Kir3 channels remain in the endoplasmic reticulum (ER) in the presence of NCAM140, we measured the endoglycosidase H sensitivity of Kir3.1/3.2 in cells transfected with NCAM140 or NCAM140Δ. Sensitivity of the protein carbohydrate moieties toward EndoH is an indicator for the retention of proteins in the ER. We observed that Kir3 channels were not more sensitive to EndoH in NCAM140- compared with NCAM140Δ-transfected cells, indicating that the channels were not retained in the ER by NCAM140 (Fig.6F). Thus, control of the number of Kir3 channels at the cell surface by NCAM140 is likely to occur in compartments downstream of the ER en route to the surface membrane, presumably at the level of the Golgi network.
Kir3 channel transport in NCAM-deficient neurons
We also investigated whether increased Kir3 currents in neurons of NCAM-deficient (NCAM−/−) mice (Fig.1E) would correlate with altered surface targeting of Kir3.1/3.2. Cultured hippocampal neurons of wild-type (NCAM+/+) and NCAM−/− mice were cotransfected with Kir3.1flag/Kir3.2 channel and GFP. Transfected neurons were identified by the GFP signal, and live neurons were stained by an antibody recognizing the extracellular flag epitope of the Kir3 channel, allowing quantification of Kir3 channels at the cell surface. In live NCAM+/+ neurons, weak flag immunostaining was detectable at the neuronal cell surface (Fig.7A,c), showing that only small amounts of Kir3.1/3.2 are targeted to the cell surface. Immunostaining of permeabilized neurons, also monitoring intracellular localization of Kir3 channels, showed Kir3 channel accumulation primarily in the cytoplasm of the somata in vesicular structures around the nucleus. Only weak Kir3 staining was detected in the neurites of the permeabilized cells (Fig. 7A,e).
Fig. 7.
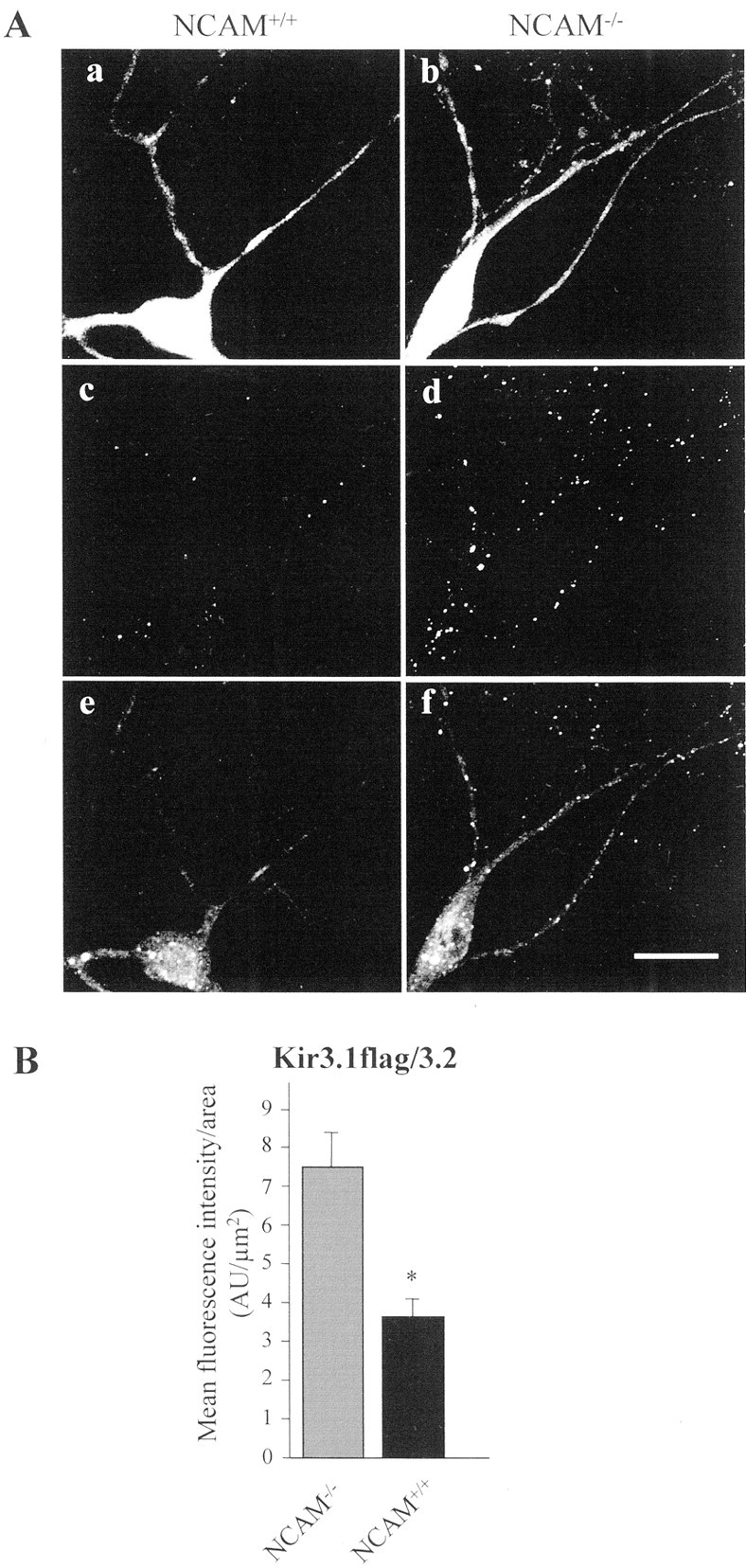
Kir3.1flag/3.2 channels are enriched at the cell surface and in the dendrites of NCAM−/− neurons.A, Confocal images of NCAM−/−(a, c, e) and NCAM+/+ (b, d, f) neurons cotransfected with Kir3.1flag/3.2 channels and GFP. Only cells expressing comparable amounts of GFP were selected for quantification (a, b). Neurons were stained with flag antibody before fixation (c, d). After permeabilization, cells were stained with Kir3.1 antibody to visualize intracellular Kir3.1flag/3.2 channels (e, f). In NCAM−/− neurons, more flag-immunoreactive clusters are detectable on the cell surface than in NCAM+/+neurons (d vs c), demonstrating that more Kir3.1flag/3.2 channels are present at the cell surface in NCAM−/− neurons. In NCAM+/+neurons, intracellular Kir3.1 immunoreactivity is prominent in the cell soma around the nucleus, whereas it is only weakly present in neurites (e). In contrast, NCAM−/−neurons show a more diffuse intracellular Kir3.1 immunoreactivity in neurites and cell soma (f).B, Bar graph showing in arbitrary units (AU) the mean fluorescence signal ± SEM of cell-surface flag immunostaining of Kir3.1flag/3.2-transfected NCAM−/− (7.45 ± 0.96 AU) and NCAM+/+ (3.7 ± 0.53 AU) neurons (n = 30 each), demonstrating that Kir3.1flag/3.2 surface localization is reduced by ∼50% in NCAM+/+ versus NCAM−/− neurons. Scale bar, 20 μm. ∗Statistically significant difference between genotypes (Student's t test; p < 0.01).
In contrast, live NCAM−/− neurons transfected with Kir3.1flag/3.2 showed prominent flag immunostaining at the cell surface, indicating considerable targeting of the Kir3 channel to the cell surface. Quantification of flag surface fluorescence showed that Kir3.1flag/3.2 surface localization was increased by 50% in NCAM−/− compared with NCAM+/+ neurons (3.7 ± 0.53 vs 7.45 ± 0.96; n = 30) (Fig. 7B). Moreover, immunostaining for the Kir3 channel in permeabilized NCAM−/− neurons showed that the Kir3 channel was not retained in the somata around the nucleus as in NCAM+/+ neurons but was transported into the neurites to be inserted into the surface membrane.
DISCUSSION
In a previous study on NCAM-deficient mice, 5-HT1A receptor signaling was identified to be hypersensitized to the receptor agonists 8-OH-DPAT and buspirone (Stork et al., 1999). The functions of the 5-HT1Areceptor itself, however, were unlikely to be altered, because receptor distribution and affinity or 5-HT metabolism in different brain regions of NCAM-deficient and wild-type mice proved to be independent of the genotype. Furthermore, coupling of the 5-HT1Areceptor to adenyl cyclase in transfected CHO cells was independent of the presence of any of the NCAM isoforms (M. Delling, unpublished observations), indicating that NCAM neither influenced the 5-HT1A receptor itself nor coupling of the receptor to G-proteins. Based on these results, we investigated the possibility that NCAM influences Kir3 channel activity as a downstream effector of the 5-HT1A receptor.
NCAM140 and NCAM180 reduce the cell-surface localization of Kir3 channels
In cultured hippocampal neurons of NCAM+/+ and NCAM−/− mice and in heterologous expression systems, Kir3 channel currents (Kir3.1/3.2 or Kir3.1/3.3) are reduced by ∼70% by the NCAM140 and NCAM180 isoforms. The reduction of Kir3 currents by NCAM140 and NCAM180 correlated with a decrease in the cell-surface localization of the Kir3 channel, whereas overall expression of the channel was not reduced. In addition, in NCAM+/+ hippocampal neurons, Kir3.1/3.2 surface localization was reduced by ∼50% compared with NCAM−/− neurons, and Kir3 channels were accumulated in intracellular organelles around the nucleus, suggesting that the Kir3 channel is retained by NCAM in Golgi apparatus-like structures.
Our results are in agreement with previous studies in which Kir3.1 subunits were not only detectable on dendritic spine membranes but also in the intracellular compartments of cell somata in the CA1 region of the hippocampus. These intracellular compartments were identified as Golgi network structures and as other, not fully characterized small vesicles (Drake et al., 1997). In primary thyrotroph cells, Kir3.1 subunits were found to be associated with intracellular dense core vesicles. After stimulation with thyrotropin-releasing hormone, they fuse with the surface membrane and thus enhanceKG currents (Morishige et al., 1999). Characterization of Kir3 channel internalization kinetics suggests that NCAM also regulates transport of Kir3 channels to the cell surface rather than internalization of the channel. Interestingly, activation of NCAM-dependent signaling cascades by antibody clustering and blockage of NCAM-linked kinase pathways did not influence Kir3 channel delivery to the cell surface, suggesting that NCAM regulates this delivery not via the known signaling pathways but via a novel mechanism.
Hence, our study extends the reports on the controlled transport of Kir3 channels to the membrane (Morishige et al., 1999; Ma et al., 2001) by the idea that the number of Kir3 channels in the membrane can also be controlled by a recognition molecule.
Lipid rafts are involved in the transport of Kir3.1/3.2 channels to the membrane
We have shown that both NCAM140 and the Kir3.1/3.2 channel are partially associated with lipid rafts, as demonstrated previously for a specific subgroup of K+ channels, the Kv1.5 and Shaker K+ channels (Martens et al., 2000a,b). Ablation of intracellular palmitoylation and thus raft targeting sites of NCAM140 recovered cell-surface localization of Kir3.1/3.2. In addition, disruption of lipid rafts by lovastatin treatment partly rescued the inhibition of NCAM140 on Kir3 channel delivery to the cell surface. Our observations underline the importance of lipid rafts in cellular protein trafficking, which has already been demonstrated in previous studies (for review, see Simons and Ikonen, 1997). It was also shown that lovastatin impairs surface delivery of the influenza virus hemagglutinin in transfected baby hamster kidney-21 and Madin–Darby canine kidney cells (Keller and Simons, 1998) and leads to a mis-sorting of the raft-associated aminopeptidase N in enterocytes (Hansen et al., 2000). However, in the present study, disruption of lipid rafts did not affect channel delivery per se, because lovastatin had no effect on current amplitudes in Kir3.1/3.2 and NCAM120 cotransfected cells. Instead, we observed an indirect effect of lipid raft disruption on Kir3 channel delivery, namely that removal of NCAM from lipid rafts enhances the transport of Kir3 channels to the plasma membrane. We cannot define whether NCAM-dependent modulation of Kir3 channels takes place in lipid rafts of the Golgi apparatus intracellularly or at the cell surface in the plasma membrane. Although treatment of CHO cells with methyl-cyclodextrin, an agent extracting cholesterol from the plasma membrane and thus dispersing plasma membrane lipid rafts, had no influence on NCAM140-mediated inhibition of Kir3 channel trafficking (our unpublished observations), suggesting that plasma membrane lipid rafts are not involved in the inhibitory effect, we cannot determine at which stage of intracellular processing NCAM blocks Kir3 channel delivery to the cell surface.
We thus hypothesize that NCAM140 modulates the retention of Kir3 channels via its presence in lipid rafts. The precise NCAM-mediated mechanism of channel retention operant within lipid rafts is presently unknown and deserves additional attention. Because we were unable to demonstrate a direct physical interaction of Kir3 channels with NCAM140, we suggest that intracellular retention of the Kir3 channel is controlled via an NCAM-linked mediator acting on the N terminus of the Kir3 channel (see Fig. 8 for a hypothetical model) that has been implicated in the cell-surface delivery of Kir3 channels (Stevens et al., 1997). Alternatively, yet unknown raft-associated second messenger systems under the control of NCAM140 and NCAM 180 might be involved in the retention of the Kir3 channel. However, raft localization of NCAM per se is not sufficient, because NCAM120, although present in lipid rafts (Niethammer et al., 2002), does not modulate Kir3 delivery, most likely because of the absence of an intracellular domain. Furthermore, the intracellular domain of NCAM140 per se is also not sufficient for inhibition of Kir3 transport, possibly because of incorrect configuration of this domain. Because raft localization of NCAM140 and NCAM180, and thus the degree of Kir3 channel inhibition, are determined by palmitoylation (Fig. 8), the regulatory mechanisms controlling palmitoylation deserve major attention (Resh, 1999). An important role for palmitoylation has been demonstrated by previous studies showing that a reduction in palmitoylation of growth cone proteins is sufficient to stop neurite extension (Hess et al., 1993; Patterson and Skene, 1994). There is growing evidence that different subtypes of lipid rafts exist in the plasma membrane (Pierini and Maxfield, 2001). Further analysis of the exact lipid raft subtype containing NCAM and the Kir3 channels may yield additional insights into the mechanisms by which NCAM inhibits Kir3 channel delivery to the cell surface.
Fig. 8.

Hypothetical scheme of NCAM-mediated inhibition of Kir3 channel transport to the cell surface. Kir3 channels are transported along the secretory pathway to the cell surface (pathway A) or to yet-unidentified intracellular compartments (pathway B) (Drake et al., 1997). Palmitoylation of the intracellular domain of NCAM140 or NCAM180 and thus targeting to lipid rafts favors the transport of Kir3 channels into intracellular compartments rather than to the cell surface. Both NCAM140 and Kir3 channels are present in lipid rafts. Disruption of lipid rafts by lovastatin or mutation of NCAM140 palmitoylation sites directs the Kir3 channels into pathway A. Because no direct molecular association of the transmembrane isoforms of NCAM and the Kir3 channel could be demonstrated, a raft-associated functional linker (arrow) might be involved. Both the N terminus of the Kir3.2 subunit and the intracellular domain of NCAM140 in its complete transmembrane configuration favor pathway B.
Physiological relevance of recognition molecule-dependent regulation of Kir3 channel activity
The combined observations tempt us to speculate on the involvement of NCAM in the 5-HT1A receptor-mediated regulation of Kir3 channels. We have shown previously that the number of NCAM180-positive spine synapses in the hippocampus is increased after induction of long-term potentiation (Schuster et al., 1998). This increase has been suggested to enhance synaptic strength (Schachner, 1997). As suggested by the present study, another consequence of NCAM180 upregulation would be a downregulation of the inhibitory Kir3 subgroup of potassium channels in the cell membrane and thus a reduction in the amplitudes of slow inhibitory postsynaptic potentials triggered by several neurotransmitters. These neurotransmitter receptors would be coupled to the G-protein-activated Kir3 channels and thereby control the degree of postsynaptic hyperpolarization. As a consequence of reduced Kir3 channel activity during periods of NCAM protein upregulation correlating with increase in synaptic efficacy, postsynaptic cell impedance increases, and neurons become more sensitive to synaptic inputs. These effects would also be important during neuritogenesis and synapse formation. In NCAM−/− mice, increased activity of 5-HT1A receptor-stimulated Kir3 channels may be causal for a lower excitability of target neurons for serotonergic fibers in the limbic system, and thus altered levels of aggression and anxiety (Stork et al., 1999, 2000). Although the behavioral consequences of NCAM-dependent Kir3 channel regulation remain to be elucidated at the cellular level, our study provides the first insights into some of the molecular determinants regulating neuronal long-term excitability by controlling the number of active K+ channels in the surface membrane.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grants Scha 185/17-1 (M.S.), Ka 1175/1-3 (A.K.), and Ve 187/1-2 (R.W.V.). M.D. is a scholar of the Studienstiftung des Deutschen Volkes. We thank Dr. Harold Cremer for his gift of NCAM-deficient mice, Galina Dityateva for hippocampal cultures, Drs. Patricia Maness and Elisabeth Bock for the gift of NCAM cDNAs, Dr. D. Clapham for the gift of the flag-tagged Kir3.1 channel, Dr. Olaf Pongs for his comments on this manuscript, and Dr. Günter Gercken for support. We thank M. Honemann and A. Sporning for oocyte preparation and P. Niethammer for help with the sucrose gradients. The FGF inhibitor PD173074 was a gift from Parke-Davis (Ann Arbor, MI).
Correspondence should be addressed to Melitta Schachner, Zentrum für Molekulare Neurobiologie, Universität Hamburg, 20246 Hamburg, Germany. E-mail: melitta.schachner@zmnh.uni-hamburg.de.
REFERENCES
- 1.Beggs HE, Baragona SC, Hemperly JJ, Maness PF. NCAM140 interacts with the focal adhesion kinase p125(fak) and the SRC-related tyrosine kinase p59 (fyn). J Biol Chem. 1997;272:8310–8319. doi: 10.1074/jbc.272.13.8310. [DOI] [PubMed] [Google Scholar]
- 2.Brown MS, Goldstein JL. Multivalent feedback regulation of HMG CoA reductase, a control mechanism coordinating isoprenoid synthesis and cell growth. J Lipid Res. 1980;21:505–517. [PubMed] [Google Scholar]
- 3.Cremer H, Lange R, Christoph A, Plomann M, Vopper G, Roes J, Brown R, Baldwin S, Kraemer P, Scheff S. Inactivation of the N-CAM gene in mice results in size reduction of the olfactory bulb and deficits in spatial learning. Nature. 1994;367:455–459. doi: 10.1038/367455a0. [DOI] [PubMed] [Google Scholar]
- 4.Dityatev A, Dityateva G, Schachner M. Synaptic strength as a function of post- versus presynaptic expression of the neural cell adhesion molecule NCAM. Neuron. 2000;26:207–217. doi: 10.1016/s0896-6273(00)81151-4. [DOI] [PubMed] [Google Scholar]
- 5.Drake CT, Bausch SB, Milner TA, Chavkin C. GIRK1 immunoreactivity is present predominantly in dendrites, dendritic spines, and somata in the CA1 region of the hippocampus. Proc Natl Acad Sci USA. 1997;94:1007–1012. doi: 10.1073/pnas.94.3.1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ethell IM, Yamaguchi Y. Cell surface heparan sulfate proteoglycan syndecan-2 induces the maturation of dendritic spines in rat hippocampal neurons. J Cell Biol. 1999;144:575–586. doi: 10.1083/jcb.144.3.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fenton RG, Kung HF, Longo DL, Smith MR. Regulation of intracellular actin polymerization by prenylated cellular proteins. J Cell Biol. 1992;117:347–356. doi: 10.1083/jcb.117.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hansen GH, Niels-Christiansen LL, Thorsen E, Immerdal L, Danielsen EM. Cholesterol depletion of enterocytes. Effect on the Golgi complex and apical membrane trafficking. J Biol Chem. 2000;275:5136–5142. doi: 10.1074/jbc.275.7.5136. [DOI] [PubMed] [Google Scholar]
- 9.Hess DT, Patterson SI, Smith DS, Skene JH. Neuronal growth cone collapse and inhibition of protein fatty acylation by nitric oxide. Nature. 1993;366:562–565. doi: 10.1038/366562a0. [DOI] [PubMed] [Google Scholar]
- 10.Hooper NM. Glycosyl-phosphatidylinositol anchored membrane enzymes. Clin Chim Acta. 1997;266:3–12. doi: 10.1016/s0009-8981(97)00161-7. [DOI] [PubMed] [Google Scholar]
- 11.Inanobe A, Yoshimoto Y, Horio Y, Morishige KI, Hibino H, Matsumoto S, Tokunaga Y, Maeda T, Hata Y, Takai Y, Kurachi Y. Characterization of G-protein-gated K+ channels composed of Kir3.2 subunits in dopaminergic neurons of the substantia nigra. J Neurosci. 1999;19:1006–1017. doi: 10.1523/JNEUROSCI.19-03-01006.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jelacic TM, Kennedy ME, Wickman K, Clapham DE. Functional and biochemical evidence for G-protein-gated inwardly rectifying K+ (GIRK) channels composed of GIRK2 and GIRK3. J Biol Chem. 2000;275:36211–36216. doi: 10.1074/jbc.M007087200. [DOI] [PubMed] [Google Scholar]
- 13.Keller P, Simons K. Cholesterol is required for surface transport of influenza virus hemagglutinin. J Cell Biol. 1998;140:1357–1367. doi: 10.1083/jcb.140.6.1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kennedy ME, Nemec J, Corey S, Wickman K, Clapham DE. GIRK4 confers appropriate processing and cell surface localization to G-protein-gated potassium channels. J Biol Chem. 1999;274:2571–2582. doi: 10.1074/jbc.274.4.2571. [DOI] [PubMed] [Google Scholar]
- 15.Kolkova K, Novitskaya V, Pedersen N, Berezin V, Bock E. Neural cell adhesion molecule-stimulated neurite outgrowth depends on activation of protein kinase C and the Ras-mitogen-activated protein kinase pathway. J Neurosci. 2000;20:2238–2246. doi: 10.1523/JNEUROSCI.20-06-02238.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Little EB, Edelman GM, Cunningham BA. Palmitoylation of the cytoplasmic domain of the neural cell adhesion molecule N-CAM serves as an anchor to cellular membranes. Cell Adhes Commun. 1998;6:415–430. doi: 10.3109/15419069809109150. [DOI] [PubMed] [Google Scholar]
- 17.Lüscher C, Jan LY, Stoffel M, Malenka RC, Nicoll RA. G protein-coupled inwardly rectifying K+ channels (GIRKs) mediate postsynaptic but not presynaptic transmitter actions in hippocampal neurons. Neuron. 1997;19:687–695. doi: 10.1016/s0896-6273(00)80381-5. [DOI] [PubMed] [Google Scholar]
- 18.Ma D, Zerangue N, Lin YF, Collins A, Yu M, Jan YN, Jan LY. Role of ER export signals in controlling surface potassium channel numbers. Science. 2001;291:316–319. doi: 10.1126/science.291.5502.316. [DOI] [PubMed] [Google Scholar]
- 19.Martens JR, Navarro-Polanco R, Coppock EA, Nishiyama A, Parshley L, Grobaski TD, Tamkun MM. Differential targeting of Shaker-like potassium channels to lipid rafts. J Biol Chem. 2000a;275:7443–7446. doi: 10.1074/jbc.275.11.7443. [DOI] [PubMed] [Google Scholar]
- 20.Martens JR, Sakamoto N, Sullivan SA, Grobaski TD, Tamkun MM. Isoform-specific localization of voltage-gated K+ channels to distinct lipid raft populations: targeting of Kv1.5 to caveolae. J Biol Chem. 2000b;276:8409–8414. doi: 10.1074/jbc.M009948200. [DOI] [PubMed] [Google Scholar]
- 21.Melkonian KA, Ostermeyer AG, Chen JZ, Roth MG, Brown DA. Role of lipid modifications in targeting proteins to detergent-resistant membrane rafts. Many raft proteins are acylated, while few are prenylated. J Biol Chem. 1999;274:3910–3917. doi: 10.1074/jbc.274.6.3910. [DOI] [PubMed] [Google Scholar]
- 22.Mohammadi M, Froum S, Hamby JM, Schroeder MC, Panek RL, Lu GH, Eliseenkova AV, Green D, Schlessinger J, Hubbard SR. Crystal structure of an angiogenesis inhibitor bound to the FGF receptor tyrosine kinase domain. EMBO J. 1998;17:5896–5904. doi: 10.1093/emboj/17.20.5896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montixi C, Langlet C, Bernard AM, Thimonier J, Dubois C, Wurbel MA, Chauvin JP, Pierres M, He HT. Engagement of T cell receptor triggers its recruitment to low-density detergent-insoluble membrane domains. EMBO J. 1998;17:5334–5348. doi: 10.1093/emboj/17.18.5334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morishige K, Inanobe A, Yoshimoto Y, Kurachi H, Murata Y, Tokunaga Y, Maeda T, Maruyama Y, Kurachi Y. Secretagogue-induced exocytosis recruits G protein-gated K+ channels to plasma membrane in endocrine cells. J Biol Chem. 1999;274:7969–7974. doi: 10.1074/jbc.274.12.7969. [DOI] [PubMed] [Google Scholar]
- 25.Murase S, Schuman EM. The role of cell adhesion molecules in synaptic plasticity and memory. Curr Opin Cell Biol. 1999;11:549–553. doi: 10.1016/s0955-0674(99)00019-8. [DOI] [PubMed] [Google Scholar]
- 26.Niethammer P, Delling M, Sytnyk V, Dityatev A, Fukami K, Schachner M. Cosignaling of NCAM via lipid rafts and the FGF receptor is required for neuritogenesis. J Cell Biol. 2002;157:521–532. doi: 10.1083/jcb.200109059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patterson SI, Skene JH. Novel inhibitory action of tunicamycin homologues suggests a role for dynamic protein fatty acylation in growth cone-mediated neurite extension. J Cell Biol. 1994;124:521–536. doi: 10.1083/jcb.124.4.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pierini LM, Maxfield FR. Flotillas of lipid rafts fore and aft. Proc Natl Acad Sci USA. 2001;98:9471–9473. doi: 10.1073/pnas.181353098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Resh MD. Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim Biophys Acta. 1999;1451:1–16. doi: 10.1016/s0167-4889(99)00075-0. [DOI] [PubMed] [Google Scholar]
- 30.Retzer MD, Kabani A, Button LL, Yu RH, Schryvers AB. Production and characterization of chimeric transferrins for the determination of the binding domains for bacterial transferrin receptors. J Biol Chem. 1996;271:1166–1173. doi: 10.1074/jbc.271.2.1166. [DOI] [PubMed] [Google Scholar]
- 31.Ronn LC, Hartz BP, Bock E. The neural cell adhesion molecule (NCAM) in development and plasticity of the nervous system. Exp Gerontol. 1998;33:853–864. doi: 10.1016/s0531-5565(98)00040-0. [DOI] [PubMed] [Google Scholar]
- 32.Schachner M. Neural recognition molecules and synaptic plasticity. Curr Opin Cell Biol. 1997;9:627–634. doi: 10.1016/s0955-0674(97)80115-9. [DOI] [PubMed] [Google Scholar]
- 33.Schmidt A, Hannah MJ, Huttner WB. Synaptic-like microvesicles of neuroendocrine cells originate from a novel compartment that is continuous with the plasma membrane and devoid of transferrin receptor. J Cell Biol. 1997;137:445–458. doi: 10.1083/jcb.137.2.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schuster T, Krug M, Hassan H, Schachner M. Increase in proportion of hippocampal spine synapses expressing neural cell adhesion molecule NCAM180 following long-term potentiation. J Neurobiol. 1998;37:359–372. doi: 10.1002/(sici)1097-4695(19981115)37:3<359::aid-neu2>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- 35.Simon H, Klinz S, Fahrig T, Schachner M. Molecular association of the neural cell adhesion molecules L1 and N-CAM in the surface membrane of neuroblastoma cells is shown by chemical cross-linking. Eur J Neurosci. 1991;3:634–640. doi: 10.1111/j.1460-9568.1991.tb00849.x. [DOI] [PubMed] [Google Scholar]
- 36.Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. doi: 10.1038/42408. [DOI] [PubMed] [Google Scholar]
- 37.Sontheimer H, Kettenmann H, Schachner M, Trotter J. The neural cell adhesion molecule (N-CAM) modulates K+ channels in cultured glial precursor cells. Eur J Neurosci. 1990;3:230–236. doi: 10.1111/j.1460-9568.1991.tb00084.x. [DOI] [PubMed] [Google Scholar]
- 38.Stevens EB, Woodward R, Ho IH, Murrell-Lagnado R. Identification of regions that regulate the expression and activity of G protein-gated inward rectifier K+ channels in Xenopus oocytes. J Physiol (Lond) 1997;503:547–562. doi: 10.1111/j.1469-7793.1997.547bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stork O, Welzl H, Cremer H, Schachner M. Increased intermale aggression and neuroendocrine response in mice deficient for the neural cell adhesion molecule (NCAM). Eur J Neurosci. 1997;9:1117–1125. doi: 10.1111/j.1460-9568.1997.tb01464.x. [DOI] [PubMed] [Google Scholar]
- 40.Stork O, Welzl H, Wotjak CT, Hoyer D, Delling M, Cremer H, Schachner M. Anxiety and increased 5-HT1A receptor response in NCAM null mutant mice. J Neurobiol. 1999;40:343–355. doi: 10.1002/(sici)1097-4695(19990905)40:3<343::aid-neu6>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 41.Stork O, Welzl H, Wolfer D, Schuster T, Mantei N, Stork S, Hoyer D, Lipp H, Obata K, Schachner M. Recovery of emotional behaviour in neural cell adhesion molecule (NCAM) null mutant mice through transgenic expression of NCAM180. Eur J Neurosci. 2000;12:3291–3306. doi: 10.1046/j.1460-9568.2000.00197.x. [DOI] [PubMed] [Google Scholar]
- 42.Wischmeyer E, Doring F, Wischmeyer E, Spauschus A, Thomzig A, Veh R, Karschin A. Subunit interactions in the assembly of neuronal Kir3.0 inwardly rectifying K+ channels. Mol Cell Neurosci. 1997;9:194–206. doi: 10.1006/mcne.1997.0614. [DOI] [PubMed] [Google Scholar]
- 43.Zhao X, Yip PM, Siu CH. Identification of a homophilic binding site in immunoglobulin-like domain 2 of the cell adhesion molecule L1. J Neurochem. 1998;71:960–971. doi: 10.1046/j.1471-4159.1998.71030960.x. [DOI] [PubMed] [Google Scholar]