

Abstract
Ifenprodil is a noncompetitive antagonist of NMDA receptors highly selective for the NMDA receptor 2B (NR2B) subunit. It is widely used as a pharmacological tool to discriminate subpopulations of NMDA receptors, and derivatives are currently being developed as candidate neuroprotectants. Despite numerous studies on the mechanism of action of ifenprodil on NMDA receptors, the structural determinants responsible for the subunit selectivity have not been identified. By combining functional studies on recombinant NMDA receptors and biochemical studies on isolated domains, we now show that ifenprodil binds to the N-terminal leucine/isoleucine/valine-binding protein (LIVBP)-like domain of NR2B. In this domain, several residues, both hydrophilic and hydrophobic, were found to control ifenprodil inhibition. Their location in a modeled three-dimensional structure suggests that ifenprodil binds in the cleft of the LIVBP-like domain of NR2B by a mechanism (Venus-flytrap) resembling that of the binding of Zn on the LIVBP-like domain of NR2A. These results reinforce the proposal that the LIVBP-like domains of NMDA receptors, and possibly of other ionotropic glutamate receptors, bind modulatory ligands. Moreover, they identify the LIVBP-like domain of the NR2B subunit as a promising therapeutic target and provide a framework for designing structurally novel NR2B-selective antagonists.
Keywords: glutamate receptors, NMDA, ifenprodil, phenylethanolamine, LIVBP, neuroprotection
Ionotropic glutamate receptors (iGluRs) are made of subunits sharing a common membrane topology: a large N-terminal extracellular region, three transmembrane segments (TM1, TM3, and TM4), a P loop region (initially called TM2) that forms the pore selectivity filter, and a cytoplasmic C-terminal region (Dingledine et al., 1999). The agonist binding domain, made of ∼150 amino acids preceding TM1 together with the extracellular loop between TM3 and TM4, is distantly related to the bacterial periplasmic-binding protein (PBP) glutamine binding protein (GlnBP) (Stern-Bach et al., 1994). It has been crystallized in the case of the AMPA subunit GluR2 and the prokaryotic glutamate receptor subunit GluR0, showing a bilobed structure with the agonist bound in a central cleft (Armstrong et al., 1998; Mayer et al., 2001).
Eukaryotic iGluR subunits possess an additional extracellular N-terminal domain made of the first ∼380 amino acids that is weakly related to leucine/isoleucine/valine-binding protein (LIVBP), another PBP (O'Hara et al., 1993). In AMPA and kainate receptors, this domain participates in subunit oligomerization (Kuusinen et al., 1999;Leuschner and Hoch, 1999; Ayalon and Stern-Bach, 2001). In NMDA receptors (NRs; heteromers made of NR1 and NR2A–NR2D subunits), we have proposed recently that the LIVBP-like domains of the NR2 subunits also have a bilobed structure, and we have shown that in NR2A, this domain forms a high-affinity Zn binding site (Paoletti et al., 2000) (also see Choi and Lipton, 1999; Fayyazuddin et al., 2000; Low et al., 2000).
Ifenprodil is representative of a class of NMDA receptor antagonists (phenylethanolamines) with high selectivity for NR2B-containing receptors (Williams, 1993; Chenard and Menniti, 1999). Several phenylethanolamines are neuroprotective both in vitro and inin vivo models of a variety of neurological disorders and lack many of the side effects associated with non-subunit-selective NMDA receptor antagonists (references in Kew and Kemp, 1998); they also produce antinociceptive effects (Chizh et al., 2001). Ifenprodil acts as a noncompetitive, partial, and voltage-independent antagonist (Carter et al., 1988; Legendre and Westbrook, 1991; Williams, 1993). Its potency strongly depends on the extracellular pH and is only weakly affected by the insertion of the NR1 exon 5 (Pahk and Williams, 1997;Mott et al., 1998). Finally, ifenprodil displays use dependence such that binding of glutamate increases binding of ifenprodil and vice versa (Kew et al., 1996; Zheng et al., 2001). On the basis of binding experiments on chimeric NR2 subunits, Gallagher et al. (1996) have proposed that determinants of ifenprodil inhibition locate to the N terminus of NR2B. However, using a mutagenesis approach, Masuko et al. (1999) concluded in favor of a binding site located in the N terminus of NR1. Thus, despite the detailed functional characterization of the mechanism of ifenprodil inhibition, the precise location of the ifenprodil binding site has remained for the most part elusive.
All the functional properties of the ifenprodil inhibition of NR2B-containing receptors listed above also apply to the high-affinity Zn inhibition of NR2A-containing receptors (Westbrook and Mayer, 1987;Christine and Choi, 1990; Paoletti et al., 1997; Traynelis et al., 1998; Choi and Lipton, 1999; Low et al., 2000; Zheng et al., 2001). This striking similarity between both antagonisms suggests that Zn and ifenprodil share a common mechanism of modulation at the structural level. We now show that, similarly to the Zn binding site on the NR2A LIVBP-like domain, the LIVBP-like domain of NR2B forms in its central cleft a high-affinity binding site for ifenprodil.
MATERIALS AND METHODS
Molecular biology. The expression plasmids, mutagenesis strategy, RNA synthesis, and NR2A/NR2B chimera constructions have been described previously by Paoletti et al. (1997,2000). Each mutation was verified by sequencing across the mutated region (∼400–600 bp; Genome Express, Montreuil, France). For each mutation strongly affecting ifenprodil inhibition, two independent clones were isolated, sequenced, and functionally characterized (except for NR2B-K234A, for which only one clone has been isolated). Point mutations in isolated LIVBP-like domains were made by using mismatch PCR (QuikChange; Stratagene Europe, Amsterdam, The Netherlands) and verified by sequencing the entire domain.
Electrophysiology and data analysis. Xenopus laevis oocytes were prepared, kept, injected with cRNAs, voltage-clamped, and superfused as described by Paoletti et al. (1995,1997). Oocytes were injected with 30–40 nl of a mixture of NR1 and NR2 cRNAs (ratio, 1:2) at a final concentration of 100 ng/μl and recorded in the following 1–4 d. The control solution superfusing the oocytes contained (in mm): 100 NaCl, 5 HEPES, 0.3 BaCl2, and 10 Tricine (used to chelate traces amount of contaminating Zn; Paoletti et al., 1997). The pH was adjusted to 7.3 with KOH. Both l-glutamate and glycine were prepared as 250 μl aliquots (in bidistilled water) at 100 mm and stored at −20°C. NMDA currents were induced by application of the agonist solution containing a saturating concentration of both l-glutamate and glycine (100 μm each). Ifenprodil (a gift from B. Scatton, Sanofi-Synthélabo, Bagneux, France) was prepared as 50 μl aliquots (in bidistilled water) at 10 mm and stored at −20°C. Ifenprodil (0.03–30 μm) was extemporaneously diluted in agonist solution, protected from light, and used within 4 hr. All experiments were performed at room temperature (18–24°C) with oocytes that exhibited agonist-induced currents within the 150–1500 nA range at −60 mV (except for NR2B-I150A, for which currents were never >200 nA).
The kinetics of ifenprodil inhibition are particularly slow (see Fig.2A; for 1 μm ifenprodil, they are much slower than the estimated rate of complete exchange of the solution in our recording chamber, ∼2 sec; Paoletti et al., 1997) and therefore could be estimated directly from the current relaxations at the onset and offset of ifenprodil applications. The kinetic parameters shown in Figure 1B were estimated with the fitting procedure of Clampfit 6.0.5 (Axon Instruments, Foster City, CA). Off relaxations could be well fitted with a single exponential (yielding τoff), whereas on relaxations had to be fitted with either two exponentials or one exponential and a sloping baseline; however, during the first 30 sec of ifenprodil application (leading to 90–95% of the total inhibition at equilibrium), on relaxations could be satisfactorily fitted with a single exponential (yielding τon).
Fig. 2.

Parameters of ifenprodil inhibition of the chimeric and wild-type NMDA receptors. A, Concentration–response curves at equilibrium and at −60 mV. Datapoints were fitted with Hill equations (see Materials and Methods). Each point is the mean value of 3–15 oocytes. The estimated values of IC50,nH, and maximal inhibition are, respectively, 155 nm, 0.98, and 96% for NR2B wt and 215 nm, 1.00, and 94% for NR2A-(LIVBP NR2B). For NR2A wt and NR2B-(LIVBP NR2A), the estimated IC50 values are 28 and 75 μm, respectively. B, The low-affinity ifenprodil inhibition of NR2A- and NR2B-(LIVBP NR2A)-containing receptors is voltage-dependent. Leak-subtracted agonist-induced NMDA currents were recorded in the absence (cont) and presence of 30 μm ifenprodil (ifen) during 2 sec voltage ramps from −70 to +50 mV. Insets, Mean relative currents (percentage) measured at −60 and +40 mV.
Fig. 1.
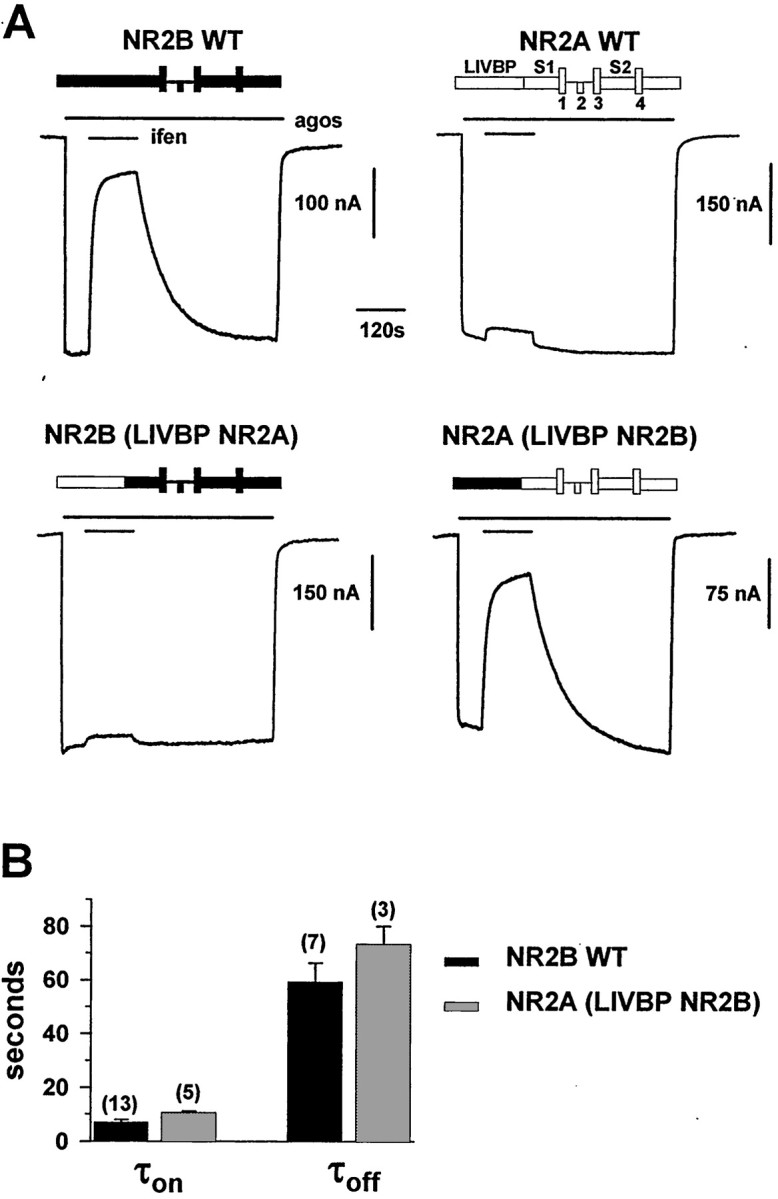
The LIVBP-like domain of NR2B controls the high-affinity ifenprodil inhibition of NMDA receptors.A, Swapping the LIVBP-like domain between NR2B and NR2A subunits transfers the NR2B-specific high-affinity ifenprodil (ifen) inhibition from NR2B to NR2A. Eachtrace shows the inhibition of the current response to agonists (agos; glutamate and glycine, 100 μm each) by 1 μm ifenprodil inXenopus oocytes coexpressing either NR2B wt, NR2A wt, or chimeric NR2B/NR2A subunits with the NR1 wt subunit. Note that the slow kinetics of ifenprodil inhibition of NR1/NR2B wt (most particularly the recovery rate) are also observed in NR1/NR2A-(LIVBP NR2B) receptors. The recordings were made at −60 mV. The bars above the current traces indicate the duration of agonists and ifenprodil applications. A schematic diagram of the NR2 construct is shown on top of each trace:LIVBP, LIVBP-like domain; S1 S2, agonist-binding GlnBP-like domain; 1, 3, 4, transmembrane segments; 2, reentrant pore loop.B, NR1/NR2B wt and NR1/NR2A-(LIVBP NR2B) receptors display similar kinetic parameters of ifenprodil inhibition. The time constants of onset (τon) and offset (τoff) of the inhibition of agonist-induced currents by 1 μm ifenprodil were estimated by single-exponential fits to traces such as those shown inA (see Materials and Methods for the fitting procedures). The mean τon and τoff values are as follows: NR2B wt, 7.1 ± 1 sec (n = 13) and 59 ± 7 (n = 7), respectively; NR2A-(LIVBP NR2B), 10.6 ± 0.6 sec (n = 5) and 73 ± 7 sec (n = 3), respectively.
Because of the slow on rate of ifenprodil inhibition, ifenprodil solutions had to be applied for 30 sec (30 μm), 60 sec (3–10 μm), 60–90 sec (0.3–1 μm), or 120 sec (30–100 nm) to reach equilibrium. Regarding the very slow dissociation rate constant of ifenprodil, recovery to control agonist-induced current was usually not attempted (except in those experiments aimed at evaluating τoff). Thus, the typical protocol used in this study was control agonist solution applied for 40 sec (to verify the stability of the control current), immediately followed by two increasing ifenprodil concentrations successively applied (90–150 sec total).
Experimental points were fitted with the following Hill equation: (100 ×Iifen/Icontrol) = 100 − a/((1 + (IC50/[ifen])nH), where 100 ×Iifen/Icontrolis the mean relative current (percentage), [ifen] is the ifenprodil concentration, nH is the Hill coefficient, IC50 is the concentration of ifenprodil producing 50% of the maximal inhibition, and ais the maximal inhibition at the saturating ifenprodil concentration. IC50, a andnH were set as free parameters. For NR1/NR2B mutated receptors (see Fig. 6), ifenprodil concentration–response curves at −60 mV were fitted without any attempt to correct for the ifenprodil voltage-dependent block (correction would have been ∼10% at 10 μmifenprodil and negligible for lower ifenprodil concentrations). For mutants displaying mean relative currents >60% even at the highest ifenprodil concentration tested (10 μm), no Hill fit was attempted, but rather, experimental points were graphically linked by a constrained sigmoidal curve, and IC50 was arbitrarily reported to be >10 μm.
Fig. 6.

Ifenprodil concentration–response curves of receptors mutated at critical residues. Each graphcorresponds to a region in the LIVBP-like domain of NR2B in which one or more critical residues controlling ifenprodil inhibition were identified. The dotted curves are the fits of the ifenprodil concentration–response curves of the NR1/NR2B wt receptors (left dotted curve) and the chimeric NR1/NR2B-(LIVBP NR2A) receptors (right dotted curve) obtained in Figure2B. The estimated values of IC50 of the different mutated receptors are listed in Table 2. Estimated values of Hill coefficients (0.92–1.10) and maximal inhibitions (88–100%) are in the same range as those obtained with NR2B wt receptors (Fig.2A). Each data point is the mean value obtained from 3 to 22 oocytes. The recordings were made at −60 mV.
To study the voltage-dependent block in NR2A wild-type (wt)- and NR2B (LIVBP NR2A)-containing receptors at high (10 and 30 μm) ifenprodil concentrations, 2 sec −70/+50 mV voltage ramps were used (capacitive and leakage currents were recorded before agonist application and subtracted from the agonist-induced currents).
Error bars represent the SD of the mean relative currents.
Production of isolated LIVBP-like domains in Escherichia coli and proteolysis experiments. LIVBP-like domains of the NR2A and of the NR2B subunits were produced as thrombin-cleavable glutathione S-transferase (GST) fusion proteins in E. coli. LIVBP-like domains of the rat NR2A (Glu28-Val375) and of the mouse NR2B (ε2; Ser28-Val376) were subcloned in the pGEX-2T vector (Amersham Biosciences, Buckinghamshire, UK). After transformation, BL21(DE3) cells were grown in 1 liter of Luria–Bertani medium supplemented with ampicillin (100 μg/ml) at 37°C until OD600 reached 0.7–0.8. Protein production was induced by 1 mmisopropyl-β-d-thiogalactopyranoside (Roche Biochemicals, Meylan, France) for 2.5 hr at 37°C. The following steps, adapted from those of Chen and Gouaux (1997), were performed either on ice or at 4°C. Cells were harvested and resuspended in buffer 1 (in mm: 200 NaCl, 20 Tris, and 1 EDTA, pH 7.5). The cells were sonicated for 2 min. Inclusion bodies were collected by centrifugation (15,000 rpm, 20 min), resuspended in buffer 1 (20 ml), and purified by a first incubation (30 min) in the presence of DNase1 (1 mg), deoxycholic acid (120 mg), and lysozyme (100 mg), followed by a second incubation (30 min) with 0.5% Triton X-100. Purified inclusion bodies were then solubilized overnight in buffer 2 (6 m GuHCl, 50 mm Tris, and 10 mm DTT, pH 8.0). Proteins (∼1 mg/ml) were refolded by 16 hr of dialysis against a 20-fold higher volume of buffer 3 (in mm: 500 NaCl, 50 Tris, and 1 DTT, pH 8.0), using a membrane tube with a molecular mass cutoff of 15,000 Da (Fisher, Illkirch, France). Buffer 3 was changed after 8 hr of incubation. Refolded proteins were separated from the precipitate by centrifugation (1 hr, 40 000 rpm) and purified using glutathione-Sepharose beads (Amersham Biosciences). LIVBP-like domains of NR2A and NR2B were cleaved from the GST by a 2 hr digestion with human thrombin (Roche Biochemicals), which was stopped by addition of 2 mm PMSF. Protein concentration was measured at OD280 using extinction coefficients of 57,160 and 52,180 m−1cm−1 for NR2A and NR2B, respectively. With 1 liter of bacterial culture, we usually obtain ∼10 mg of purified soluble protein.
Trypsin proteolysis experiments were performed using a protein solution at 0.2 or 0.5 mg/ml (NR2A or NR2B LIVBP-like domain), and the trypsin concentration was adjusted to have a final protease/protein ratio of 1:500. Reactions were performed at room temperature. Preincubations with ifenprodil or Zn were done for 5 min before adding trypsin. Proteolysis was stopped at varied times by mixing aliquots of the reaction solution to the SDS-containing 2× sample buffer. Samples were analyzed on 12% SDS-PAGE gels (Bio-Rad, Hercules, CA) and stained with SyPro Orange (Molecular Probes, Leiden, The Netherlands), and SyPro Orange fluorescence was revealed using a fluorophosphoimager (FLA-3000; Fujifilm, St-Quentin-en-Yvelines, France) at λ = 475 nm.
Alignments and three-dimensional modeling. The alignments of Figure 4 were primarily adapted from the alignments presented byPaoletti et al. (2000). E. coli LIVBP [GenBank accession number, 230609; Protein Data Bank (PDB) coordinates, 2LIV;Sack et al., 1989], rat metabotropic GluR1 (mGluR1; GenBank accession number, P23385; PDB coordinates, 1EWT; Kunishima et al., 2000), and human atrial natriuretic peptide clearance receptor type C (ANP-C; GenBank accession number, P17342; PDB coordinates, 1JDN; He et al., 2001) were aligned by structure superimposition deduced from visual inspection of the secondary structure elements. For NR1, conserved patterns of clusters of hydrophobic residues between the LIVBP-like domain of NR1 and LIVBP were identified using hydrophobic cluster analysis (Callebaut et al., 1997). This conservation, which indicates a conserved fold, was used to constrain the alignment of the LIVBP-like domain of NR1 with LIVBP (Paoletti et al., 2000). The 3D structure of the NR2B LIVBP-like domain was modeled by homology to the structure of the unliganded form of LIVBP (PDB coordinates, 2LIV; Sack et al., 1989) on the basis of the sequence alignment shown in Figure 4. The model was produced by Modeler 4 (Sali and Blundell, 1993).
Fig. 4.
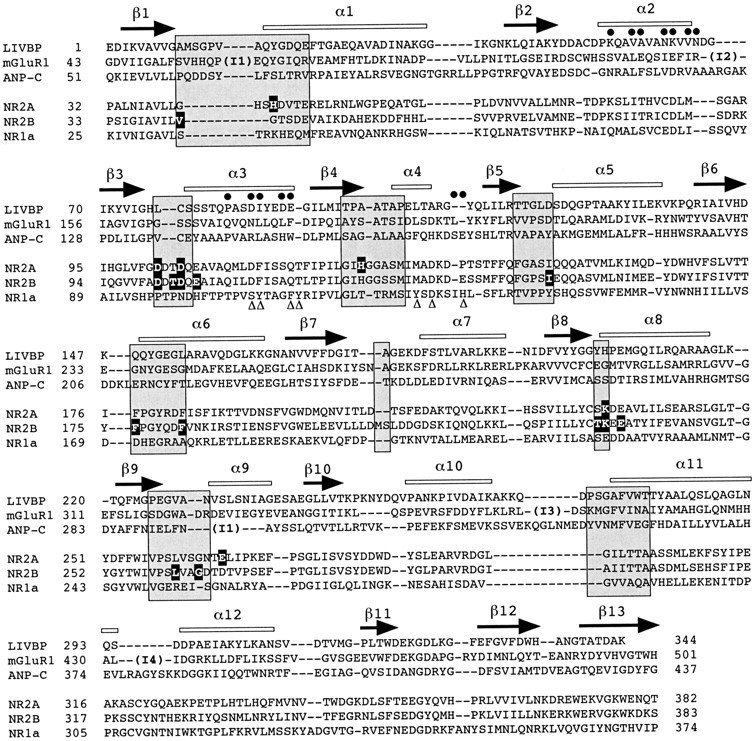
Ligand-contacting regions in proteins containing LIVBP-like domains. Amino acid sequence alignment of the LIVBP-like domains of the NMDA receptor subunits NR2A, NR2B, and NR1 and the LIVBP-like domains of known structures from LIVBP (Sack et al., 1989), mGluR1 (Kunishima et al., 2000), and ANP-C (He et al., 2001). For LIVBP (E. coli), mGluR1, and ANP-C, the alignments were obtained from structure superimposition; for NR2A and NR2B, the alignments were adapted from those of Paoletti et al. (2000); for NR1, the alignment was based on the conserved pattern of a cluster of hydrophobic residues within LIVBP-like domains (see Materials and Methods). The β strands (arrows) and α helices (open bars) identified in LIVBP, mGluR1, and ANP-C crystal structures are indicated on top of the alignment. The insertions found in mGluR1 with respect to LIVBP are indicated by I1 (14 residues), I2 (31 residues), I3 (47 residues), and I4 (13 residues) and in ANP-C by I1 (21 residues).Shaded boxes indicate regions (mostly loops) known to contact the ligand molecules in LIVBP-like domains (adapted fromPaoletti et al., 2000). Residues of NR2A controlling high-affinity Zn inhibition (see Paoletti et al., 2000) and residues of NR2B identified as controlling ifenprodil inhibition (present study) are highlighted. Residues of NR1 mutated by Masuko et al. (1999) and affecting ifenprodil inhibition are indicated by triangles. Residues of mGluR1 and ANP-C participating in dimerization of the LIVBP-like domains (Kunishima et al., 2000; He et al., 2001) are indicated by closed circles.
RESULTS
The LIVBP-like domain of the NR2B subunit confers high sensitivity to ifenprodil
To test our prediction that the target of ifenprodil is the LIVBP-like domain of NR2B, we first assessed the ifenprodil sensitivity of chimeric NMDA receptors in which the LIVBP-like domains were swapped from one NR2 subunit to the other. Chimeric NR2 subunits NR2A-(LIVBP NR2B) and the complementary subunit NR2B-(LIVBP NR2A) were obtained by substituting the entire LIVBP-like domain (i.e., the first ∼390 amino acids; Paoletti et al., 2000; Fig.1A) of NR2A (a subunit with poor ifenprodil sensitivity; Williams, 1993) by that of NR2B. NMDA receptors were expressed in Xenopus oocytes, and NMDA currents were induced by saturating concentrations ofl-glutamate (100 μm) and glycine (100 μm). As shown in Figure1A, replacing the LIVBP-like domain of NR2A by that of NR2B transfers the NR2B-specific high-sensitivity ifenprodil inhibition from NR2B to the chimera. In contrast, the converse chimera shows very little ifenprodil sensitivity. Indeed, 1 μm ifenprodil strongly inhibited NR2B- and NR2A-(LIVBP NR2B)-containing receptors [mean residual relative currents, 16 ± 2% (n = 12) and 23 ± 4% (n = 5), respectively], whereas the same concentration had little effect on either NR2A- or NR2B-(LIVBP NR2A)-containing receptors [mean residual relative currents, of 94 ± 1% (n = 4) and 97 ± 1% (n = 5), respectively].
One striking feature of the high-affinity ifenprodil antagonism of NMDA receptors is the slowness of both the onset of blockade on drug application and the offset after drug removal. The full recovery of inhibition by ifenprodil of wt NR1/NR2B receptors requires >5 min (Williams, 1993; Kew et al., 1996; Fig. 1A, top left). Similar slow kinetics were observed with the NR2A-(LIVBP NR2B)-containing receptor using protocols with long applications of both agonists and ifenprodil (Fig. 1A,B).
We also obtained ifenprodil concentration–response curves at equilibrium (Fig. 2A). For wt NR1/NR2B receptors, as already shown on recombinant (Williams, 1993; Mott et al., 1998; Masuko et al., 1999) and native (Kew et al., 1996) NMDA receptors, ifenprodil inhibition was partial (maximal inhibition of ∼96% at saturating ifenprodil concentrations), had an IC50 in the hundreds of nanomolar range (156 nm), and had annH value very close to 1 (0.99). Similar values were obtained with the NR1/NR2A-(LIVBP NR2B) receptors: maximal inhibition of 94%, IC50 of 215 nm, and nH of 1.0. NR1/NR2A and NR1/NR2B-(LIVBP NR2A) receptors were only slightly inhibited by ifenprodil. At the highest concentration of ifenprodil tested (30 μm), the mean residual current was 48 ± 6% (n = 8) for NR2A wt receptors and 67 ± 3% (n = 7) for NR2B-(LIVBP NR2A), thus corresponding to a decrease of at least 200-fold in ifenprodil sensitivity.
Williams (1993) showed that the high- and low-affinity ifenprodil inhibitions differ markedly in their voltage dependency. The high-affinity NR2B-specific ifenprodil inhibition is voltage-independent, whereas the inhibitory effects of high concentrations of ifenprodil at NR2A-containing receptors are mostly voltage-dependent. This difference suggests that ifenprodil binds on two distinct sites, one extracellular (accounting for the high-affinity inhibition) and the other within the pore (accounting for the low-affinity inhibition). The low-affinity ifenprodil pore blockade of wt NR2A-containing receptors is illustrated in Figure2B1, which compares agonist-induced currents obtained during voltage ramps (−70/+50 mV) before and after application of a high ifenprodil concentration (30 μm). The current is very weakly inhibited by ifenprodil at +40 mV compared with −60 mV (see inset). As shown in Figure2B2, a qualitatively similar voltage-dependent ifenprodil block is observed with NR2B-(LIVBP NR2A)-containing receptors. [Note that in wt NR2B-containing receptors, high ifenprodil concentrations (≥30 μm) also produce an outward rectification of the residual currents (data not shown).] Therefore, as for wt NR2A-containing receptors, the blocking effect of ifenprodil at NR2B-(LIVBP NR2A)-containing receptors most probably involves the binding of ifenprodil to the pore site rather than to the “extracellular” site. In consequence, the apparent 200-fold shift in ifenprodil sensitivity deduced from the concentration–response curves constructed at negative potentials underestimates the selectivity of the ifenprodil binding for the extracellular site associated with the LIVBP-like domain of NR2B versus that of NR2A.
Ifenprodil protects the isolated LIVBP-like domain of NR2B against digestion by trypsin
The experiments presented above using chimeric NMDA receptors suggest that the LIVBP-like domain of NR2B contains molecular determinants that are required for ifenprodil to produce its high-affinity inhibitory effects. Two hypotheses could account for an involvement of the NR2B LIVBP-like domain in the ifenprodil modulation: (1) the LIVBP-like domain of NR2B contains (at least part of) the ifenprodil binding site; and (2) the LIVBP-like domain of NR2B does not bind ifenprodil but is required for the transduction mechanism that links binding of ifenprodil to receptor inhibition. We sought to discriminate between the two hypotheses by testing whether ifenprodil could bind to the isolated LIVBP-like domain of NR2B using a proteolysis protection assay. The rationale underlying such experiments is that ligand binding, by stabilizing the protein in its folded conformation, may decrease the accessibility of proteolytic sites (Hubbard, 1998).
Isolated LIVBP-like domains of NR2A and NR2B were produced in E. coli as thrombin-cleavable GST fusion proteins (see Materials and Methods). On SDS-PAGE gels, both domains run as a major band of the expected molecular mass (∼40 kDa) (Fig.3). Minor bands of lower molecular mass, mostly seen at ∼30 kDa in the case of NR2B, partly result from thrombin overdigestion (see below). We performed trypsin digestion of the isolated NR2B LIVBP-like domain in the presence and absence of 10 μm ifenprodil (a saturating concentration for the high-affinity inhibition) (Fig. 2A), using a trypsin/protein ratio of 1:500. In the absence of ifenprodil, digestion of the NR2B LIVBP-like domain occurs quickly, as shown by the complete disappearance of the 40 kDa band after 5 min. In contrast, in the presence of ifenprodil, the rate of digestion is greatly slowed, with a substantial amount of undigested protein still present after 5 min (Fig. 3A). Similar results were obtained in nine different experiments with ifenprodil concentrations of 10 μm (n = 6), 30 μm (n = 2), and 100 μm (n = 1). Thus, the presence of ifenprodil markedly slows the trypsin digestion of the LIVBP-like domain of NR2B.
Fig. 3.
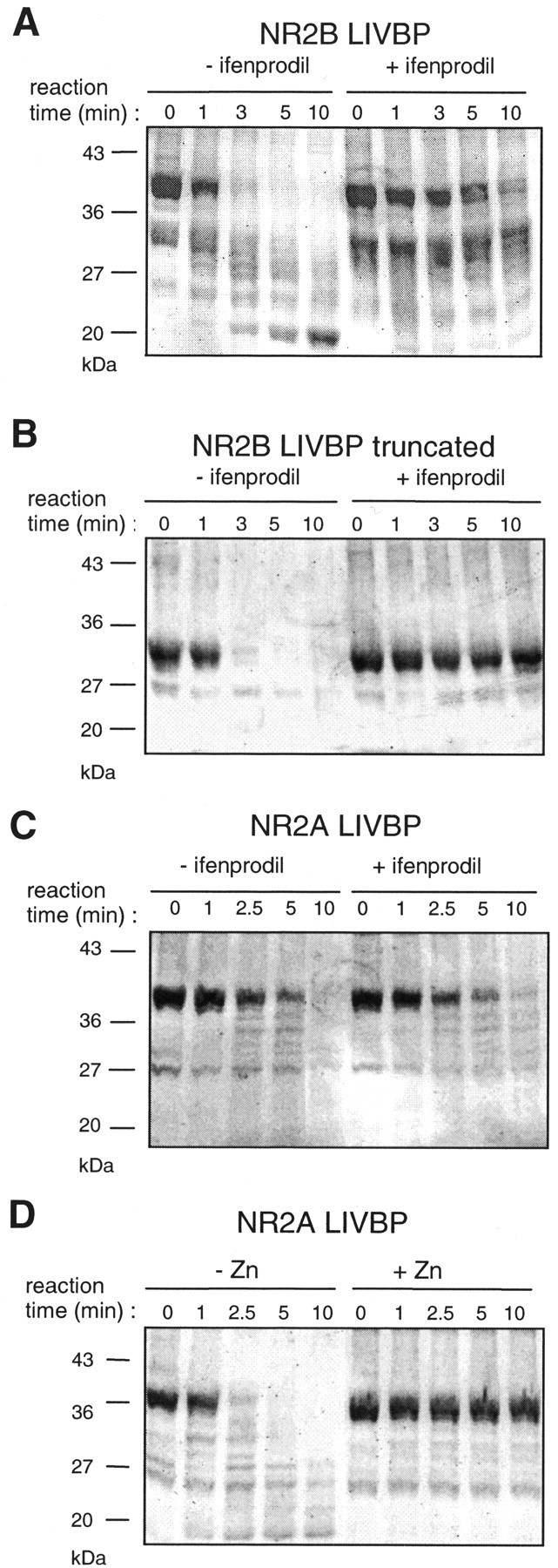
Ifenprodil protects the isolated LIVBP-like domain of NR2B but not that of NR2A against hydrolysis by trypsin. Isolated LIVBP-like domains of NR2A and NR2B were produced in E. coli and subjected to trypsinization with or without ifenprodil or Zn for various amounts of time (up to 10 min). Fifty micrograms of the purified LIVBP-like domains were mixed with 0.1 μg of trypsin. The samples were analyzed on 12% SDS-PAGE gels. Lane 0corresponds to the protein solution just before trypsin addition.A, Ifenprodil (10 μm) protects the NR2B LIVBP-like domain (main band at ∼40 kDa) against trypsin digestion.B, Ifenprodil (10 μm) also protects a C-terminal truncated NR2B LIVBP-like domain (main band at ∼30 kDa) against trypsin digestion. C, Ifenprodil (100 μm) does not protect the NR2A LIVBP-like domain (main band at ∼40 kDa) against trypsin digestion. D, Zn (10 μm) protects the NR2A LIVBP-like domain against trypsin digestion.
Interestingly, we consistently observed an ifenprodil-induced protection of the minor band migrating at 30 kDa (Fig. 3A). We have obtained experimental evidence that this band is not a bacterial contaminant but most probably represents a thrombin-truncated product of the LIVBP-like domain (thrombin is used to cleave off the GST). Indeed, we have expressed, refolded, and purified a shortened version of the NR2B LIVBP-like domain [in which Val293, the residue after a putative thrombin cleavage site (Arg292), has been replaced by a stop codon] and have obtained a highly purified band, migrating at the expected molecular mass (∼30 kDa) and very efficiently protected against proteolysis by ifenprodil (10 μm; n = 3) (Fig.3B).
To eliminate the possibility that ifenprodil inhibits trypsin, we repeated the experiments on the isolated LIVBP-like domain of NR2A. As shown in Figure 3C, the rate of trypsin digestion of the NR2A domain is not affected by 100 μmifenprodil, indicating that ifenprodil does not inhibit trypsin. Similar results were obtained eight times [10 μm (n = 4) or 100 μm (n = 4) ifenprodil]. In contrast, Zn (10 μm), a known ligand of the NR2A LIVBP-like domain (Paoletti et al., 2000), is very efficient at protecting the isolated domain of NR2A against trypsin digestion (n = 3) (Fig. 3D).
In conclusion, ifenprodil specifically protects the isolated LIVBP-like domain of NR2B against proteolysis. This implies that the NR2B LIVBP-like domain contains the ifenprodil binding site.
Identification of residues of the LIVBP-like domain of NR2B controlling the high-affinity ifenprodil inhibition
We searched for residues of the LIVBP-like domain of NR2B putatively involved in ifenprodil binding using site-directed mutagenesis. We had shown in our previous work on high-affinity Zn inhibition that the residues known (or proposed) to contact the ligand in various LIVBP-like domains cluster in a few discrete homologous regions scattered throughout the sequence (Paoletti et al., 2000) (Fig.4, shaded boxes). In LIVBP and LIVBP-like domains of known 3D structure, these regions are mostly made of loops between secondary structure elements (usually one β-strand followed by one α-helix) and line a central cleft separating two globular subdomains (Quiocho and Ledvina, 1996; Kunishima et al., 2000). We used the sequence alignments between the bacterial protein LIVBP and the LIVBP-like domains of the NR2 subunit that we had proposed previously (based on the conservation of patterns of hydrophobic clusters; Paoletti et al., 2000) to delineate putative ligand-binding regions in the LIVBP-like domain of NR2B (Fig. 4). We then performed point mutagenesis targeted to these regions and assessed ifenprodil sensitivity of the NR2B mutated receptors. Given that ifenprodil binding to NMDA receptors may involve a variety of interactions (electrostatic, hydrophobic, and hydrogen bonds; see Discussion), we did not restrict our mutagenesis to specific residues chosen for their chemical properties but rather mutated into alanine each residue of the studied regions (alanine scan).
Mutants were screened by measuring the inhibition of agonist-induced current at two concentrations of ifenprodil: 300 nm, a concentration approximately twice the IC50, and 3 μm, a nearly saturating concentration for NR1/NR2B wt receptors. We arbitrarily considered as “significant” (or “critical”) those mutants that had a current of >70% of the control at 300 nm ifenprodil (vs 37% for wt NR2B receptors; Table 1). Of the 41 mutations tested (all of which gave functional receptors), we detected 13 mutations resulting in a significant decrease in ifenprodil sensitivity (Table 1). Typical current traces are shown in Figure5 for three different mutated receptors and wt NR1/NR2B receptors. NR2B-V42A exemplify mutant receptors with an ifenprodil inhibition clearly decreased at 300 nm but still potent at 3 μm, whereas NR2B-D101A and NR2B-F176A exemplify mutants with the strongest effects. Such mutants receptor are almost insensitive to 300 nm ifenprodil and only weakly affected by 3 μm ifenprodil.
Table 1.
Screening of point mutations in the LIVBP-like domain of NR2B for effects on ifenprodil sensitivity
| NR2B mutant | Mean relative current (%) | n | |
|---|---|---|---|
| 300 nm Ifenprodil | 3 μm Ifenprodil | ||
| wt | 37 ± 5 | 8 ± 2 | 55 |
| Loop β1/α1 | |||
| V39A | 45 ± 4 | 16 ± 2 | 7 |
| I40A | 52 ± 1 | 19 ± 1 | 4 |
| L41A | 50 ± 2 | 12 ± 1 | 4 |
| V42A1-a | 84 ± 4 | 35 ± 4 | 6 |
| G43A | 32 ± 3 | 9 ± 2 | 4 |
| T44A | 33 ± 4 | 9 ± 2 | 6 |
| S45A | 40 ± 1 | 13 ± 1 | 4 |
| D46A | 37 ± 1 | 12 ± 2 | 5 |
| E47A | 39 ± 3 | 9 ± 1 | 6 |
| V48A | 44 ± 2 | 11 ± 2 | 4 |
| Loop β3/α3 | |||
| D101A1-a | 99 ± 1 | 91 ± 2 | 9 |
| D102A | 52 ± 4 | 16 ± 1 | 3 |
| T103A1-a | 84 ± 1 | 39 ± 1 | 3 |
| D104A1-a | 94 ± 1 | 60 ± 4 | 5 |
| Q105A | 59 ± 3 | 16 ± 1 | 4 |
| E106A1-a | 86 ± 3 | 45 ± 4 | 6 |
| Loop β4/α4 | |||
| I126A | 47 ± 4 | 23 ± 1 | 8 |
| H127A | 32 ± 3 | 7 ± 2 | 6 |
| S130A | 39 ± 2 | 9 ± 1 | 4 |
| S130A–S131A | 36 ± 2 | 8 ± 1 | 4 |
| Linker β5/α5 | |||
| S149A | 43 ± 7 | 12 ± 3 | 5 |
| I150A1-a | 98 ± 2 | 83 ± 5 | 7 |
| E151A | 44 ± 1 | 11 ± 1 | 4 |
| Loop β6/α6 | |||
| Y175A | 64 ± 1 | 8 ± 2 | 4 |
| F176A1-a | 95 ± 4 | 82 ± 5 | 12 |
| Y179A | 58 ± 3 | 15 ± 1 | 6 |
| Q180A | 45 ± 9 | 9 ± 3 | 4 |
| D181A | 50 ± 3 | 10 ± 1 | 4 |
| F182A1-a | 97 ± 2 | 86 ± 4 | 10 |
| Loop β8/α8 | |||
| T233A1-a | 93 ± 2 | 57 ± 4 | 6 |
| K234A1-a | 86 ± 2 | 41 ± 6 | 10 |
| E235A | 57 ± 5 | 25 ± 3 | 5 |
| E236A1-a | 74 ± 4 | 23 ± 3 | 4 |
| Loop β9/α9 | |||
| S260A | 43 ± 2 | 12 ± 1 | 7 |
| L261A1-a | 76 ± 5 | 9 ± 2 | 15 |
| V262A | 50 ± 4 | 11 ± 1 | 8 |
| A263S | 40 ± 4 | 18 ± 4 | 3 |
| G264A1-a | 74 ± 5 | 24 ± 6 | 3 |
| D265A | 39 ± 5 | 10 ± 1 | 5 |
| T266A | 45 ± 4 | 12 ± 4 | 2 |
| D267A | 30 ± 4 | 8 ± 3 | 3 |
Mutations resulting in a significant decrease in ifenprodil sensitivity.
Fig. 5.

Identification in the LIVBP-like domain of NR2B critical residues controlling high-affinity ifenprodil inhibition. A comparison of the current traces obtained from oocytes coexpressing NR1 with either wt or mutated NR2B subunits is shown. Ifenprodil was applied at two increasing concentrations (300 nm and 3 μm) during an application of agonists. Thebars above the current traces indicate the duration of agonists (agos) and ifenprodil (ifen) applications. The holding potential was −60 mV. These current traces are typical of NR2B mutants having an intermediate (V42A) or strong (D101A, F176A) effect on ifenprodil sensitivity.
Concentration–response curves of ifenprodil antagonism were constructed for each critical mutant (Fig.6). Ifenprodil concentrations were maintained at <10 μm to minimize the contribution of the low-affinity voltage-dependent block (Fig. 2B). Critical mutants can be subdivided into two groups according to the degree of reduction of ifenprodil sensitivity. One group includes the mutants V42A, T103A, D104A, E106A, T233A, K234A, E236A, L261A, and G264A, for which IC50 values for ifenprodil are in the low micromolar range (4- to 25-fold higher than wt NR2B-containing receptors; Table 2). The second group includes D101A, I150A, F176A, and F182A, the four mutants having the most pronounced effects on ifenprodil sensitivity. For these mutants, IC50 values were estimated to be >10 μm (>60-fold higher than for wt NR2B receptors; Table 2). In fact, substituting Asp101, Ile150, Phe176, and Phe182 into Ala is almost as effective in shifting the apparent ifenprodil sensitivity as replacing the entire LIVBP-like domain of NR2B by that of NR2A. In that latter receptors, we have shown that the ifenprodil inhibition results mostly from ifenprodil binding into the pore (Fig. 2B2). A similar mechanism may account for part of the residual ifenprodil inhibition observed with receptors mutated at “critical” positions. Thus, with these mutants, the decrease in affinity of the NR2B LIVBP-like domain for ifenprodil could be significantly higher than the apparent 60-fold deduced from the concentration–response curves.
Table 2.
Parameters of the ifenprodil inhibition of NMDA receptors mutated at critical NR2B residues
| NR2B mutant | IC50(μm) | nH | IC50(mutant)/IC50 (wt) |
|---|---|---|---|
| wt | 0.156 | 0.99 | 1 |
| Charged residues | |||
| D101A | >10 | ND | >60 |
| D104A | 3.5 | 1.11 | 22 |
| E106A | 1.8 | 0.93 | 11 |
| K234A | 1.9 | 1.02 | 12 |
| E236A | 0.7 | 1.07 | 4.5 |
| Polar residues | |||
| T103A | 1.7 | 0.92 | 11 |
| T233A | 3.8 | 0.98 | 25 |
| Aliphatic residues | |||
| V42A | 1.3 | 1.10 | 8 |
| I150A | >10 | ND | >60 |
| L261A | 0.7 | 1.10 | 4.5 |
| G264A | 0.6 | 1.14 | 4 |
| Aromatic residues | |||
| F176A | >10 | ND | >60 |
| F182A | >10 | ND | >60 |
Critical residues are listed according to the chemical nature of their side chain. ND, Not determined.
Mutations at critical positions suppress ifenprodil-induced protection of the isolated NR2B LIVBP-like domains against trypsin proteolysis
The results obtained above suggest that the critical residues identified in the NR2B LIVBP-like domain are closely associated with the ifenprodil binding site. We obtained an additional and independent evidence showing that these residues are required for high-affinity ifenprodil binding using the biochemical assay on isolated LIVBP-like domains. As illustrated in Figure 7, mutating the isolated LIVBP-like domain at the critical position Asp101 (Fig. 7A;n = 4) or Phe176 (Fig.7B; n = 4) abolishes the ifenprodil-induced protection against proteolysis (compare with Fig. 3A).
Fig. 7.
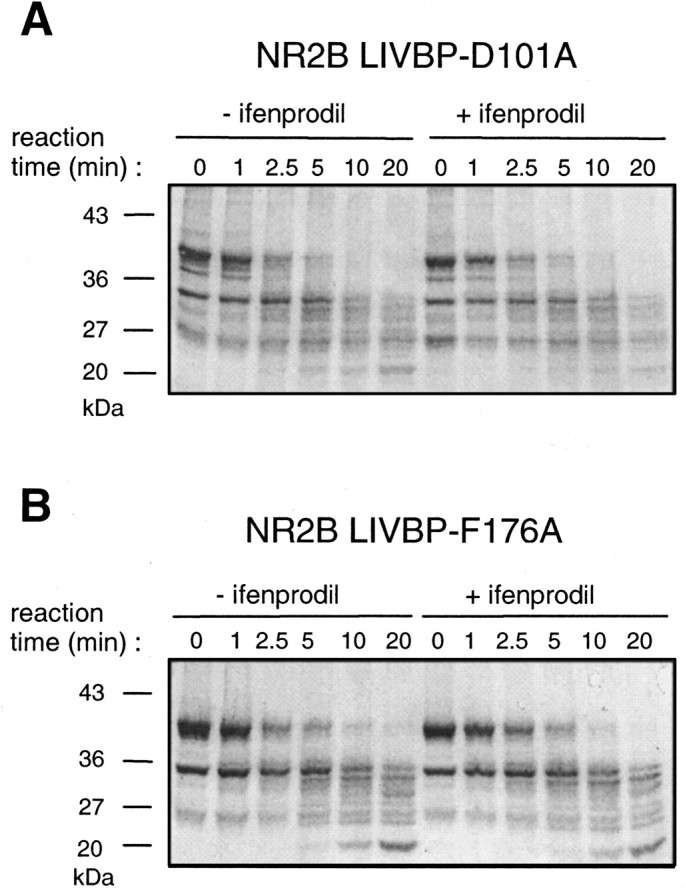
Lack of ifenprodil-induced protection against trypsin digestion of NR2B LIVBP-like domains mutated at critical positions. Point mutations (D101A and F176A) were introduced in the plasmid pGEX-2T-LIVBP NR2B and the mutated domains produced inE. coli. Protection by ifenprodil (10 μm) against trypsin digestion was tested using the protocol described in Figure 3. A, Mutation D101A. B, Mutation F176A.
DISCUSSION
By combining a functional and biochemical approach on wild-type and mutated NMDA receptors, we show in the present work that the high-affinity binding site of ifenprodil is situated in the N-terminal LIVBP-like domain of the NR2B subunit. This domain, isolated from the rest of the receptor, is sufficient to form an ifenprodil binding site, and we have identified a number of residues in this domain that are closely associated with ifenprodil binding.
An alanine mutagenesis scan in the NR2B LIVBP-like domain targeted to regions known to contain ligand-contacting residues in other proteins with the LIVBP-like domain allowed the identification of 13 residues (Table 1) likely to participate in the formation of the high-affinity ifenprodil binding site. These critical residues are highly diverse in their chemical nature. As shown in Table 2, their side chain can be either charged (Asp101, Asp104, Glu106, Lys234, and Glu236) or uncharged but polar (Thr103 and Thr233), aliphatic (Val42, Ile150, Leu261, and Gly264), or aromatic (Phe176 and Phe182). Such a diversity is expected given the complex chemical nature of ifenprodil and the variety of interactions it may have with its receptor site. In a study investigating the structure–activity relationships of a series of bis(phenylalkyl)amines (including ifenprodil) assayed for their potency as NR2B-selective antagonists, Tamiz et al. (1998) proposed that the binding site for NR2B-selective, ifenprodil-like antagonists is made of (at least) three major subsites or “pockets” (Chenard and Menniti, 1999): one hydrophobic, accommodating the phenyl ring; one interacting electrostatically with the central basic nitrogen atom; and one being both hydrophobic and a hydrogen bond acceptor and accommodating the phenol group. The optimum intramolecular distances between these subsites have been evaluated to ∼8Å from the nitrogen atom to the phenolic hydroxylic group and ∼10Å from the nitrogen binding site to the hydrophobic pocket interacting with the phenyl ring.
To evaluate whether the spatial distribution of the critical residues could match the proposed pharmacophore model, we constructed a 3D model of the NR2B LIVBP-like domain. The 3D structure of the LIVBP-like domain of NR2B was modeled by homology to the known structure of the unliganded form of LIVBP (Sack et al., 1989) on the basis of the sequence alignment shown in Figure 4 (see Materials and Methods). The modeled structure of the LIVBP-like domain of NR2B consists of two globular lobes, each lobe being made of alternation of β strands and α helices, interconnected by a hinge made of three short linkers delineating a deep central cleft (Fig.8A). The critical residues all belong to regions lining the central cleft, and most of them have their side chain projecting from both lobes into the cleft. This suggests that, as in the case of Zn binding to the LIVBP-like domain of NR2A (Paoletti et al., 2000), ifenprodil binds in the cleft of the LIVBP-like domain of NR2B and could promote its closure (Venus-flytrap mechanism). Thus, on ifenprodil binding, the critical residues may group according to the pharmacophore model presented above, with a central highly polar and negatively charged cluster (made of residues from β3/α3 and β8/α8 loops and interacting with the positively charged protonated nitrogen) surrounded by two hydrophobic clusters, one comprising the two aromatic Phe176 and Phe182 together with Ile150 (and interacting with the phenyl ring) and the other, less well defined, made of Val42, Gly264, and Leu261 (Fig. 8A). The estimated width of the modeled cleft (∼25Å) is sufficiently large to accommodate one molecule of ifenprodil.
Fig. 8.

LIVBP-like domain of NR2 subunits as binding domains for extracellular modulators of NMDA receptor activity.A, 3D model of the LIVBP-like domain of NR2B and the putative ifenprodil binding site. This model was produced by homology modeling using the sequence alignment shown in Figure 4 and the Protein Data Bank coordinates of LIVBP (2LIV), the unliganded form of LIVBP (Sack et al., 1989). The residues identified as critical for high-affinity ifenprodil inhibition (numbered 1–13 for clarity) are displayed in the space fill representation and according to the following color code: Corey–Pauling–Koltun (CPK) for polar and charged residues (lobe I: Asp101(2), Thr103(3), Asp104(4), Glu106(5); lobe II: Thr233(9), Lys234(10), Glu236(11)), greenfor aliphatic residues (lobe I: Val42(1); lobe II: Leu261(12), Gly264(13); hinge: Ile150(6)), andyellow for aromatic residues (lobe II: Phe176(7), Phe182(8)). On theright of the model is a space fill representation of the ifenprodil molecule (CPK color code). B, Functional organization of the extracellular regions of the NR2 subunits. See Discussion for more details.
In this putative ifenprodil binding site, some residues could contact ifenprodil through ligand–protein backbone interactions. This could be true for Gly264 and Leu261, which in our model has its side chain buried in lobe 2. Some critical residues may not contact ifenprodil directly but rather act “downstream” of ifenprodil binding. This could be the case for Ile150, a residue localized in the hinge (linker β5/α5) between the two lobes. The mutation of this residue produces receptors with very small currents (50–150 nA at −60 mV) and with a marked desensitization (data not shown). Thus, Ile150 could participate in the mechanism of flytrap closure of the domain rather than directly contact the ligand. Finally, one cannot exclude that there are other additional critical residues for ifenprodil inhibition.
Despite the lack of direct proof that the critical residues do contact ifenprodil, both our functional and our biochemical results support the proposed model of ifenprodil binding within the cleft of the LIVBP-like domain of NR2B. In particular, the three residues Asp101, Phe176, and Phe182, the substitutions of which produce the largest effects on ifenprodil sensitivity (Fig. 6, Table 2) and suppress the ifenprodil-induced protection of the isolated NR2B LIVBP-like domain against proteolysis (Fig. 7; not tested for Phe182), seem to be mandatory for high-affinity ifenprodil binding. The fact that these residues are present on both lobes (Asp101 on lobe 1 and Phe176 and Phe182 on lobe 2) is an additional argument in favor of a Venus-flytrap type of mechanism.
Although our results are fully consistent with the initial observation of Gallagher et al. (1996), that an N-terminal region of NR2B controls the high-affinity ifenprodil inhibition, they are in clear discrepancy with that of Masuko et al. (1999), who proposed that the ifenprodil binding site resides in the LIVBP-like domain of NR1. These authors based their conclusion on the fact that some mutations in the LIVBP-like domain of NR1 strongly affect ifenprodil sensitivity of NR1/NR2B responses. However, the analysis of sequence alignments between NR1 and other LIVBP-like domains favors another interpretation. X-ray structures of homodimers of LIVBP-like domains have been obtained recently for two receptors, rat mGluR1 (Kunishima et al., 2000) and ANP-C (He et al., 2001), and in both receptors, the LIVBP-like domain (which forms the agonist-binding domain) dimerizes through a central interface made of highly hydrophobic residues (Fig. 4, filled circles). Strikingly, the residues of NR1 identified by Masuko et al. (1999) are also mostly hydrophobic (including three tyrosines and one phenylalanine) and align to positions homologous to those shown to participate in the dimer interface in mGluR1 and ANP-C (Fig. 4,triangles). Therefore, even if we do not discount the possibility that a large molecule such as ifenprodil might contact residues from both NR1 and NR2B, we propose that the critical residues identified on NR1, rather than being directly involved in contacting ifenprodil, form an interface between two LIVBP-like domains (either NR1/NR1 or NR1/NR2B). As demonstrated for the activation mechanism of mGluR1 (Kunishima et al., 2000), a rotation of this intersubunit dimer interface might be fundamental in transferring to the gating machinery the ifenprodil-induced conformational change in the NR2B LIVBP-like domain.
The present results reinforce our proposal that LIVBP-like domains of NMDA receptors, and possibly of other eukaryotic iGluRs, bind modulatory ligands (Paoletti et al., 2000). Moreover, the results also strengthen the hypothesis of a modular architecture of iGluR subunits, with an extracellular region made of a tandem of functionally specialized Venus-flytrap domains, one (GlnBP-like) binding the agonist (glutamate for all iGluR subunits except NR1, which binds glycine) and the other (LIVBP-like) binding a modulatory ligand (Zn for NR2A and ifenprodil for NR2B) (Fig. 8B). This latter proposal is particularly well supported by the finding that domains separated from the rest of the receptor retain their ligand-binding properties (for the GlnBP-like domain; see Kuusinen et al., 1995; present study for the LIVBP-like domains). In NMDA receptors, it has been shown recently that during gating, both domains, the LIVBP- and GlnBP-like, functionally interact to produce one form of receptor desensitization (Krupp et al., 1998; Villarroel et al., 1998; Zheng et al., 2001). This interaction might be a common mechanistic feature of iGluRs, stressing the need for additional studies aimed at identifying the interdomain contacts as well as their connection to the channel gate.
Finally, our results identify the LIVBP-like domain of the NR2B subunit as a new target for neuroprotective and analgesic agents. This target has a particularly attractive neuropharmacological profile. First, it is selective of a subpopulation of NMDA receptors (those containing NR2B subunits), thus reducing the unacceptable side effects usually associated with broad-spectrum NMDA antagonists. Second, the blockers will be most efficient at high levels of glutamate (because of the use dependency; Kew et al., 1996) and at low pH (because of the strong pH dependency; Mott et al., 1998), and they will be still potent at depolarized potentials (because of the voltage independence; Williams, 1993), all conditions often encountered in pathological situations such as stroke, seizures, and pain states. It has been shown on the GlnBP-like domain that the efficacy of different agonists is directly related to the degree of cleft closure that they induce: the more the domain closes on agonist binding, the more efficient the agonist (Armstrong and Gouaux, 2000). Similarly, if ifenprodil binding were to promote the closure of the NR2B LIVBP-like domain (which remains to be proved), inhibition of channel activity could be directly related to the degree of lobe separation in this domain. One could then imagine NR2B-selective inhibitors with different potency according to the level of domain closure they produce. In that respect, a partial antagonist (inducing an intermediate level of domain closure) would be an attractive drug, because it would both maintain a persistent level of NMDA receptor activity even at saturating concentrations and allow chronic treatment with reduced side effects. Our results provide a structural framework for designing such new neuroprotective agents.
Footnotes
This work was supported by Assistance Publique des Hôpitaux de Paris (F.P.D.), Ministère de la Recherche (J.R.), and Institut National de la Santé et de la Recherche Médicale (P.P.). We are most grateful to Roderick MacKinnon and members of his laboratory who helped much in starting the biochemical approach. We thank Philippe Ascher and Roderick MacKinnon for comments on this manuscript. We also thank Sanofi-Synthélabo for the gift of ifenprodil.
Correspondence should be addressed to Dr. Pierre Paoletti, Laboratoire de Neurobiologie, Ecole Normale Supérieure, 46 rue d'Ulm, 75005 Paris, France. E-mail: paoletti@biologie.ens.fr.
REFERENCES
- 1.Armstrong N, Sun Y, Chen GQ, Gouaux E. Structure of a glutamate-receptor ligand-binding core in complex with kainate. Nature. 1998;395:913–917. doi: 10.1038/27692. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong N, Gouaux E. Mechanisms for activation and antagonism of an AMPA-sensitive glutamate receptor: crystal structures of the GluR2 ligand binding core. Neuron. 2000;28:165–181. doi: 10.1016/s0896-6273(00)00094-5. [DOI] [PubMed] [Google Scholar]
- 3.Ayalon G, Stern-Bach Y. Functional assembly of AMPA and kainate receptors is mediated by several discrete protein-protein interactions. Neuron. 2001;31:103–113. doi: 10.1016/s0896-6273(01)00333-6. [DOI] [PubMed] [Google Scholar]
- 4.Callebaut I, Labesse G, Durand P, Poupon A, Canard L, Chomilier J, Henrissat B, Mornon JP. Deciphering protein sequence information through hydrophobic cluster analysis (HCA): current status and perspectives. Cell Mol Life Sci. 1997;53:621–645. doi: 10.1007/s000180050082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carter C, Benavides J, Legendre P, Vincent JD, Noel F, Thuret F, Lloyd KG, Arbilla S, Zivkovic B, MacKenzie ET, Scatton B, Langer SZ. Ifenprodil and SL 82.0715 as cerebral anti-ischemic agents: II. Evidence for N-methyl-d-aspartate receptor antagonist properties. J Pharmacol Exp Ther. 1988;247:1222–1232. [PubMed] [Google Scholar]
- 6.Chen GQ, Gouaux E. Overexpression of a glutamate receptor (GluR2) ligand binding domain in Escherichia coli: application of a novel protein folding screen. Proc Natl Acad Sci USA. 1997;94:13431–13436. doi: 10.1073/pnas.94.25.13431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chenard BL, Menniti FS. Antagonists selective for NMDA receptors containing the NR2B subunit. Curr Pharm Des. 1999;5:381–404. [PubMed] [Google Scholar]
- 8.Chizh BA, Headley PM, Tzschentke TM. NMDA receptor antagonists as analgesics: focus on the NR2B subtype. Trends Pharmacol Sci. 2001;22:636–642. doi: 10.1016/s0165-6147(00)01863-0. [DOI] [PubMed] [Google Scholar]
- 9.Choi YB, Lipton SA. Identification and mechanism of action of two histidine residues underlying high-affinity Zn2+ inhibition of the NMDA receptor. Neuron. 1999;23:171–180. doi: 10.1016/s0896-6273(00)80763-1. [DOI] [PubMed] [Google Scholar]
- 10.Christine CW, Choi DW. Effect of zinc on NMDA receptor-mediated channel currents in cortical neurons. J Neurosci. 1990;10:108–116. doi: 10.1523/JNEUROSCI.10-01-00108.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dingledine R, Borges K, Bowie D, Traynelis S. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 12.Fayyazuddin A, Villarroel A, Le Goff A, Lerma J, Neyton J. Four residues of the extracellular N-terminal domain of the NR2A subunit control high-affinity Zn2+ binding to NMDA receptors. Neuron. 2000;25:683–694. doi: 10.1016/s0896-6273(00)81070-3. [DOI] [PubMed] [Google Scholar]
- 13.Gallagher MJ, Huang H, Pritchett DB, Lynch DR. Interactions between ifenprodil and the NR2B subunit of the N-methyl-d-aspartate receptor. J Biol Chem. 1996;271:9603–9611. doi: 10.1074/jbc.271.16.9603. [DOI] [PubMed] [Google Scholar]
- 14.He X, Chow D, Martick MM, Garcia KC. Allosteric activation of a spring-loaded natriuretic peptide receptor dimer by hormone. Science. 2001;293:1657–1662. doi: 10.1126/science.1062246. [DOI] [PubMed] [Google Scholar]
- 15.Hubbard SJ. The structural aspects of limited proteolysis of native proteins. Biochim Biophys Acta. 1998;1382:191–206. doi: 10.1016/s0167-4838(97)00175-1. [DOI] [PubMed] [Google Scholar]
- 16.Kew JN, Kemp JA. An allosteric interaction between the NMDA receptor polyamine and ifenprodil sites in rat cultured cortical neurones. J Physiol (Lond) 1998;512:17–28. doi: 10.1111/j.1469-7793.1998.017bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kew JN, Trube G, Kemp JA. A novel mechanism of activity-dependent NMDA receptor antagonism describes the effect of ifenprodil in rat cultured cortical neurones. J Physiol (Lond) 1996;497:761–772. doi: 10.1113/jphysiol.1996.sp021807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Krupp JJ, Vissel B, Heinemann SF, Westbrook GL. N-terminal domains in the NR2 subunit control desensitization of NMDA receptors. Neuron. 1998;20:317–327. doi: 10.1016/s0896-6273(00)80459-6. [DOI] [PubMed] [Google Scholar]
- 19.Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamamoto M, Kumasaka T, Nakanishi S, Jingami H, Morikawa K. Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature. 2000;407:971–977. doi: 10.1038/35039564. [DOI] [PubMed] [Google Scholar]
- 20.Kuusinen A, Arvola M, Keinanen K. Molecular dissection of the agonist binding site of an AMPA receptor. EMBO J. 1995;14:6327–6332. doi: 10.1002/j.1460-2075.1995.tb00323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuusinen A, Abele R, Madden DR, Keinanen K. Oligomerization and ligand-binding properties of the ectodomain of the alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor subunit GluRD. J Biol Chem. 1999;274:28937–28943. doi: 10.1074/jbc.274.41.28937. [DOI] [PubMed] [Google Scholar]
- 22.Legendre P, Westbrook GL. Ifenprodil blocks N-methyl-d-aspartate receptors by a two-component mechanism. Mol Pharmacol. 1991;40:289–298. [PubMed] [Google Scholar]
- 23.Leuschner WD, Hoch W. Subtype-specific assembly of α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor subunits is mediated by their N-terminal domains. J Biol Chem. 1999;274:16907–16916. doi: 10.1074/jbc.274.24.16907. [DOI] [PubMed] [Google Scholar]
- 24.Low CM, Zheng F, Lyuboslavsky P, Traynelis SF. Molecular determinants of coordinated proton and zinc inhibition of N-methyl-d-aspartate NR1/NR2A receptors. Proc Natl Acad Sci USA. 2000;97:11062–11067. doi: 10.1073/pnas.180307497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Masuko T, Kashiwagi K, Kuno T, Nguyen ND, Pahk AJ, Fukuchi J, Igarashi K, Williams K. A regulatory domain (R1–R2) in the amino terminus of the N-methyl-d-aspartate receptor: effects of spermine, protons, and ifenprodil, and structural similarity to bacterial leucine/isoleucine/valine binding protein. Mol Pharmacol. 1999;55:957–969. doi: 10.1124/mol.55.6.957. [DOI] [PubMed] [Google Scholar]
- 26.Mayer ML, Olson R, Gouaux E. Mechanisms for ligand binding to GluR0 ion channels: crystal structures of the glutamate and serine complexes and a closed apo state. J Mol Biol. 2001;311:815–836. doi: 10.1006/jmbi.2001.4884. [DOI] [PubMed] [Google Scholar]
- 27.Mott DD, Doherty JJ, Zhang S, Washburn MS, Fendley MJ, Lyuboslavsky P, Traynelis SF, Dingledine R. Phenylethanolamines inhibit NMDA receptors by enhancing proton inhibition. Nat Neurosci. 1998;1:659–667. doi: 10.1038/3661. [DOI] [PubMed] [Google Scholar]
- 28.O'Hara PJ, Sheppard PO, Thogersen H, Venezia D, Haldeman BA, McGrane V, Houamed KM, Thomsen C, Gilbert TL, Mulvihill ER. The ligand-binding domain in metabotropic glutamate receptors is related to bacterial periplasmic binding proteins. Neuron. 1993;11:41–52. doi: 10.1016/0896-6273(93)90269-w. [DOI] [PubMed] [Google Scholar]
- 29.Pahk AJ, Williams K. Influence of extracellular pH on inhibition by ifenprodil at N-methyl-d-aspartate receptors in Xenopus oocytes. Neurosci Lett. 1997;225:29–32. doi: 10.1016/s0304-3940(97)00176-6. [DOI] [PubMed] [Google Scholar]
- 30.Paoletti P, Neyton J, Ascher P. Glycine-independent and subunit-specific potentiation of NMDA responses by extracellular Mg2+. Neuron. 1995;15:1109–1120. doi: 10.1016/0896-6273(95)90099-3. [DOI] [PubMed] [Google Scholar]
- 31.Paoletti P, Ascher P, Neyton J. High-affinity zinc inhibition of NMDA NR1–NR2A receptors. J Neurosci. 1997;17:5711–5725. doi: 10.1523/JNEUROSCI.17-15-05711.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paoletti P, Perin-Dureau F, Fayyazuddin A, Le Goff A, Callebaut I, Neyton J. Molecular organization of a zinc binding N-terminal modulatory domain in a NMDA receptor subunit. Neuron. 2000;28:911–925. doi: 10.1016/s0896-6273(00)00163-x. [DOI] [PubMed] [Google Scholar]
- 33.Quiocho FA, Ledvina PS. Atomic structure and specificity of bacterial periplasmic receptors for active transport and chemotaxis: variation of common themes. Mol Microbiol. 1996;20:17–25. doi: 10.1111/j.1365-2958.1996.tb02484.x. [DOI] [PubMed] [Google Scholar]
- 34.Sack JS, Saper MA, Quiocho FA. Periplasmic binding protein structure and function. Refined X-ray structures of the leucine/isoleucine/valine-binding protein and its complex with leucine. J Mol Biol. 1989;206:171–191. doi: 10.1016/0022-2836(89)90531-7. [DOI] [PubMed] [Google Scholar]
- 35.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 36.Stern-Bach Y, Bettler B, Hartley M, Sheppard PO, O'Hara PJ, Heinemann SF. Agonist selectivity of glutamate receptors is specified by two domains structurally related to bacterial amino acid-binding proteins. Neuron. 1994;13:1345–1357. doi: 10.1016/0896-6273(94)90420-0. [DOI] [PubMed] [Google Scholar]
- 37.Tamiz AP, Whittemore ER, Zhou ZL, Huang JC, Drewe JA, Chen JC, Cai SX, Weber E, Woodward RM, Keana JF. Structure-activity relationships for a series of bis(phenylalkyl)amines: potent subtype-selective inhibitors of N-methyl-d-aspartate receptors. J Med Chem. 1998;41:3499–3506. doi: 10.1021/jm980235+. [DOI] [PubMed] [Google Scholar]
- 38.Traynelis SF, Burgess MF, Zheng F, Lyuboslavsky P, Powers JL. Control of voltage-independent zinc inhibition of NMDA receptors by the NR1 subunit. J Neurosci. 1998;18:6163–6175. doi: 10.1523/JNEUROSCI.18-16-06163.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Villarroel A, Regalado MP, Lerma J. Glycine-independent NMDA receptor desensitization: localization of structural determinants. Neuron. 1998;20:329–339. doi: 10.1016/s0896-6273(00)80460-2. [DOI] [PubMed] [Google Scholar]
- 40.Westbrook GL, Mayer ML. Micromolar concentrations of Zn2+ antagonize NMDA and GABA responses of hippocampal neurons. Nature. 1987;328:640–643. doi: 10.1038/328640a0. [DOI] [PubMed] [Google Scholar]
- 41.Williams K. Ifenprodil discriminates subtypes of the N-methyl-d-aspartate receptor: selectivity and mechanisms at recombinant heteromeric receptors. Mol Pharmacol. 1993;44:851–859. [PubMed] [Google Scholar]
- 42.Zheng F, Erreger K, Low CM, Banke T, Lee CJ, Conn PJ, Traynelis SF. Allosteric interaction between the amino terminal domain and the ligand binding domain of NR2A. Nat Neurosci. 2001;4:894–901. doi: 10.1038/nn0901-894. [DOI] [PubMed] [Google Scholar]