

Abstract
The reelin signaling pathway plays a crucial role during the development of laminated structures in the mammalian brain. Reelin, which is synthesized and secreted by Cajal–Retzius cells in the marginal zone of the neocortex and hippocampus, is proposed to act as a stop signal for migrating neurons. Here we show that a decreased expression of reelin mRNA by hippocampal Cajal–Retzius cells correlates with the extent of migration defects in the dentate gyrus of patients with temporal lobe epilepsy. These results suggest that reelin is required for normal neuronal lamination in humans, and that deficient reelin expression may be involved in migration defects associated with temporal lobe epilepsy.
Keywords: human hippocampus, extracellular matrix, neuronal migration disorder, Cajal–Retzius cells, dentate gyrus, Ammon's horn sclerosis
Newborn forebrain neurons migrate from their site of origin to their definitive positions in the cortical plate. Defects in neuronal migration are often associated with epileptic disorders (Palmini et al., 1991). Temporal lobe epilepsy (TLE), one of the most common neurological disorders in humans (Margerison and Corsellis, 1966), is accompanied by Ammon's horn sclerosis (AHS) and granule cell dispersion (GCD), a defect in granule cell migration (Houser, 1990; Armstrong, 1993). A similar altered granule cell migration is observed in a mouse mutant, thereeler mouse (Rakic and Caviness, 1995).
Recently, the gene deleted in this mouse mutant was cloned and the protein it encodes was named reelin (D'Arcangelo et al., 1995;Hirotsune et al., 1995). Reelin is synthesized and secreted by Cajal–Retzius (CR) cells in the marginal zones of the neocortex and hippocampus (D'Arcangelo et al., 1995, 1997; Hirotsune et al., 1995). Mice lacking reelin and its lipoprotein receptors apolipoprotein E receptor 2 (ApoER2) and very low density lipoprotein receptor (VLDLR) as well as mice deficient in the adaptor protein disabled 1 (dab1) show severe migration defects in the neocortex, hippocampus, and cerebellum (Rakic and Caviness, 1995; Howell et al., 1997; Sheldon et al., 1997;D'Arcangelo et al., 1999; Trommsdorff et al., 1999). Reelin deficiency in humans is associated with autosomal recessive lissencephaly that is characterized by malformations of the cerebellum, hippocampus, and brainstem (Hong et al., 2000). Recently, increased numbers of CR cells were found both in reeler mice (Coulin et al., 2001) and in tissue samples from TLE patients (Blümcke et al., 1999), raising the possibility that alterations in CR cells and in the reelin pathway underlie neuronal migration defects in reeler mutants and in humans with TLE.
To this end, we have studied the expression of reelin, VLDLR, ApoER2, and dab1 in tissue samples of hippocampus removed from TLE patients for therapeutic reasons. We correlated reelin expression to the extent of GCD and compared these data with control tissue from tumor patients and with tissue from autopsy cases with no identified neurological abnormalities.
MATERIALS AND METHODS
Patient selection. A total of 22 patients (mean age, 36.9 ± 9.6 years) undergoing selective amygdalohippocampectomy or two-thirds temporal lobectomy with amygdalohippocampectomy for medically intractable TLE were included in this study (Table 1). In all patients, the removal of the hippocampus was clinically warranted to achieve seizure control. Presurgical assessment consisted of the documentation of a detailed history and neurological examination. All patients experienced pharmacoresistant complex partial seizures (CPS). The epileptogenic focus was localized to the temporal lobe in all patients. For comparison, the hippocampi from seven subjects (mean age, 28.3 ± 9.2 years) with no history of neurological or psychiatric disorder were collected at autopsy within 48 hr of death. Informed consent was obtained from patients and families of controls according to the declaration of Helsinki. The Ethics Committee at the University Clinic Freiburg (Freiburg, Germany) approved the selection process and procedures.
Table 1.
Clinical data of TLE patients with AHS and autopsy cases used in this study
| Patient | Age | Sex | Onset | Duration | Frequency | Seizure | |
|---|---|---|---|---|---|---|---|
| TLE group 1 (for ISH) | I1 | 17 | M | 14 | 3 | Not known | SPS, CPS |
| I2 | 16 | M | 4.5 | 11.5 | 3–4/month | SPS, CPS | |
| I3 | 32 | F | 2 | 30 | 2–3/month | SPS, CPS | |
| I4 | 53 | M | 38 | 15 | Not known | SPS, CPS | |
| I5 | 31 | M | 2 | 29 | Not known | CPS, sGS | |
| I6 | 39 | F | 1.5 | 37.5 | 2–8/month | SPS, CPS, sGS | |
| I7 | 43 | M | 35 | 8 | 3–4/month | SPS, CPS | |
| I8 | 57 | F | 7 | 50 | Not known | SPS, CPS | |
| I9 | 33 | F | 20 | 13 | 5/month | SPS, CPS, sGS | |
| I10 | 44 | F | 6 | 36 | 3/month | CPS | |
| I11 | 18 | M | 2 | 16 | 2–3/month | SPS, CPS, sGS | |
| I12 | 44 | M | 6 | 38 | 2–3/month | SPS, CPS, sGS | |
| I13 | 40 | M | 30 | 10 | 6–10/year | SPS, CPS, sGS | |
| I14 | 34 | M | 4 | 30 | 2–3/month | SPS, CPS, sGS | |
| I15 | 52 | F | 8 | 44 | 3–4/month | SPS, CPS, sGS | |
| TLE group 2 (for PCR) | P1 | 27 | F | 13 | 14 | 1–3/month | SPS, CPS, sGS |
| P2 | 29 | M | 1.5 | 27 | Not known | SPS, CPS, sGS | |
| P3 | 41 | F | 18 | 23 | 6–7/month | SPS, CPS, sGS | |
| P4 | 32 | M | 8 | 24 | 2/week | SPS, CPS, sGS | |
| P5 | 22 | F | 11 | 11 | 2/week | SPS, CPS | |
| P6 | 38 | M | 8 | 30 | Not known | SPS, CPS | |
| P7 | 37 | F | Not known | Not known | Not known | Not known | |
| Autopsy group 3 | A1 | 24 | M | — | — | — | — |
| A2 | 33 | F | — | — | — | — | |
| A3 | 34 | M | — | — | — | — | |
| A4 | 48 | M | — | — | — | — | |
| A5 | 19 | F | — | — | — | — | |
| A6 | 5 | M | — | — | — | — | |
| A7 | 35 | M | — | — | — | — |
Groups indicate the patients and cases used for ISH and PCR experiments. Age represents the age of the patients at surgery (TLE) or death (autopsy). Onset refers to the age at which a first epileptic seizure was documented. Duration indicates the duration of the epileptic disorder in years. Frequency indicates the frequency of seizure attacks, and seizure indicates the type of seizure (SPS, single partial seizure; sGS, secondary generalized seizure). M, Male; F, female.
Tissue preparation. Hippocampi were collected in isotonic saline, and 2 mm coronal sections at the midlevel of the hippocampus were cut. Slices for PCR and Western blot analysis were immediately frozen and kept at −80°C. Tissue for morphological analysis was immersion-fixed in buffered 4% paraformaldehyde followed by cryoprotection.
In situ hybridization histochemistry. Serial sections (coronal plane, 40 μm) were cut on a cryostat and alternately processed for cresyl violet staining or for in situhybridization (ISH). Reelin, synapsin I, and dab1 mRNAs were detected with digoxigenin-labeled riboprobes (Haas et al., 2000).
Measurement of GCD. The average width of the granule cell layer (GCL) of the dentate gyrus was determined in cresyl violet-stained sections of epileptic (n = 22) and autopsy (n = 7) hippocampi. Twenty consecutive measurements were taken at 50 μm intervals covering a region of 1000 μm within the lower (infrapyramidal), relatively straight limb of the dentate gyrus (Houser, 1990). The distances from the inner (hilar) border of the GCL to the outer border of the most distal granule cell somata were determined using an image analysis system (StereoInvestigator; MicroBrightField Inc., Colchester, VT). The mean and SD of 20 measurements in five sections were calculated for each case.
Cell counting. Reelin mRNA-positive cells were counted along the hippocampal fissure in five consecutive sections of epilepsy (n = 15) and autopsy (n = 7) hippocampal specimens at 200× magnification using a counting grid, defining an area of interest to a width of 500 μm along the hippocampal fissure.
Microdissection of the dentate gyrus for PCR analysis.Cryosections (50 μm) of the hippocampus were collected on RNase-free slides and fixed in −20°C ethanol (75%; 20 min). Three sections were used for measurements of GCD, and three sections were stained with toluidin blue (1% in 75% ethanol; −20°C) for microdissection. For this, the dentate gyrus was excised using a scalpel blade and collected in Trizol (Invitrogen, Carlsbad, CA). The remainder of the section was kept for measurements of the microdissected volume, which was determined with an image analysis system. The averaged volume from three sections per patient was used for normalization.
RNA extraction and reverse transcription. Total RNA was isolated according to the manufacturer's instructions along with 0.5 ng of Drosophila poly(A+) RNA (Clontech, Palo Alto, CA) added as external standard. RNA was treated with DNase (Invitrogen), and reverse transcription (RT) was performed in 30 μl reactions containing ≤2 μg of RNA, 1 μg of oligo(dT), 500 μm deoxyNTPs, 30 U of RNase inhibitor, and 200 U of Moloney murine leukemia virus-reverse transcriptase (all from Promega, Madison, WI) for 60 min at 40°C following a standard protocol.
Real time quantitative RT-PCR. Abundance of transcripts was determined by real-time quantitative PCR on a GeneAmp 5700-System with SYBR Green (Applied Biosystems, Foster City, CA). Primers [human reelin: forward 5′-GCACCAGCCAAAGGACTTCA-3′, reverse 5′-GTTGCCACCAGCGCAGTAA-3′; human VLDLR: forward 5′-CCAG-TGGCCTAACGGAATTACA-3′, reverse 5′-CCTACGATCTTGGCC-ATTCAAG-3′; human ApoER2: forward 5′-GCTGAGCCAGCGCT- TGTACT-3′, reverse 5′-GGAGGAGATCAGCGTCTTTCTG-3′; human dab1: forward 5′-ATCGCAGTGAAGCCACTTTGATA-3′, reverse 5′-AGCTGCGGAAACTTCATCAATC-3′; Drosophilaglucose-6-phosphate dehydrogenase: forward 5′-ACACCGCCCTGGATCTCATA-3′, reverse 5′-ACGAACGTGTGCGGAATCTT-3′] were used at 250 nm. Cycling conditions were as follows: 10 min at 95°C followed by 40 cycles of 1 min at 95°C and 1 min at 60°C. Monitoring the fluorescence signal, which is proportional to the amount of double-stranded product, yielded complete amplification profiles. Melting curves of the amplified products were used to control for specificity of the amplification reaction.
From the amplification curves obtained, a threshold cycle number (Ct) was calculated, corresponding to the cycle number at which a user-defined fluorescence signal is reached. Differences in Ct values were used to calculate relative amounts of PCR product. Details of this relative quantification can be found at:http://www.appliedbiosystems.com/support/(Relative quantitation of gene expression; User Bulletin #2). Because of heterogeneity of the tissue samples with respect to disease-related cell loss, an internal standard was found to be unreliable for normalization. Therefore, we used an external standard and the dissected tissue volume to normalize gene expression levels from different patients.
Western blot analysis. Protein extracts were prepared from isolated human dentate gyrus, size-fractionated by SDS-PAGE, and transferred to nylon membrane by electroblotting. Immunodetection of dab1 was performed by treatment with an anti-dab1 antibody (1:5000;Chemicon International, Temecula, CA) followed by incubation with an alkaline phosphatase-coupled secondary antibody and enhanced chemiluminescence for detection.
RESULTS
In hippocampal sections of control tissue (Fig.1a–c), many reelin mRNA-expressing neurons were found by ISH. Reelin mRNA-positive cells showed a bipolar morphology resembling CR cells and were predominantly located at the hippocampal fissure (Fig. 1c). To test for RNA integrity in the autoptic hippocampi, ISH for synapsin I mRNA was performed in parallel with the tissue samples probed for reelin. Synapsin I mRNA, encoding for a neuron-specific protein, could be detected in virtually every neuron of the dentate gyrus, confirming the RNA integrity of the autoptic tissue (data not shown).
Fig. 1.
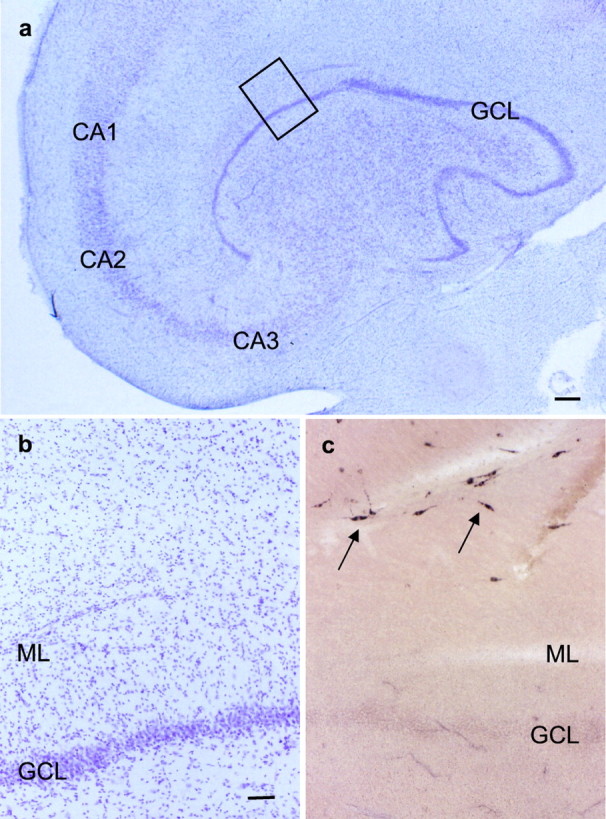
Reelin mRNA expression in a representative sample of a human control hippocampus. a, Hippocampal areas in cresyl violet stain. The inset is shown inb. Scale bar, 400 μm. b, Portion of the dentate gyrus framed in a. The GCL is densely packed. Scale bar, 80 μm. c, ISH for reelin mRNA. Many reelin mRNA-positive cells with bipolar morphology resembling CR cells are observed at the hippocampal fissure (arrows). Same magnification as in b. CA1, CA2, CA3, Hippocampal subfields; ML, Dentate molecular layer.
Next, we investigated the expression of reelin mRNA in hippocampal specimens from patients with TLE. In specimens with mild GCD (visualized by cresyl violet staining), many reelin mRNA-positive cells were observed, located predominantly at the hippocampal fissure (Fig.2a,b). In contrast, in TLE cases with severe GCD, we found only very few reelin mRNA-positive cells (Fig. 2c,d). In a quantitative analysis of 15 TLE patients and seven autopsy cases (see also Table 1), the number of reelin mRNA-expressing neurons was counted. In parallel, the width of the GCL, used as an indicator of GCD, was measured in cresyl violet-stained serial sections of the same hippocampi. This analysis revealed a striking correlation: In epileptic hippocampi with little GCD, many reelin mRNA-positive cells were counted, similar to values of nonepileptic controls. In contrast, in hippocampal sections from patients with severe GCD, we found only low numbers of reelin mRNA-expressing neurons, indicating an inverse correlation between the extent of GCD and reelin mRNA expression in individual cases of epileptic disorder (Fig. 2e,f).
Fig. 2.

Reelin mRNA expression in the dentate gyrus of control and epileptic cases. a–d, Reelin mRNA expression in representative samples of epileptic hippocampi with mild (a, b) and severe (c, d) GCD. Consecutive sections are shown in cresyl violet stain and after ISH for reelin mRNA. Arrows point to reelin mRNA-positive cells at the hippocampal fissure. Scale bars: a, b, 100 μm;c, d, 50 μm. e, f, Correlation of the GCD (width of the GCL) and number of reelin mRNA-expressing CR cells in sections of seven control hippocampi (white bars) and 15 epileptic hippocampi with AHS (gray bars). Data are represented as mean ± SD. ML, Dentate molecular layer.
To substantiate this semiquantitative result obtained by cell counting, we applied a quantitative RT-PCR approach. Because the epileptic material varied considerably with respect to cell death and gliosis, we developed a quantification procedure that included three additional steps: (1) microdissection of the dentate gyrus, (2) morphometric measurement of the microdissected volume, and (3) addition of an external, unrelated standard RNA [Drosophilapoly(A+)] before reverse transcription and PCR analysis. This procedure made it possible to normalize gene expression levels from different patients.
This quantitative RT-PCR approach was used to measure reelin mRNA expression in sections of seven epileptic hippocampi. The PCR results were compared with the extent of GCD measured in cresyl violet-stained sections of the same cases. The relative abundance of reelin mRNA varied considerably between the individual epileptic hippocampi (Fig.3a); however, when compared with the width of the GCL, a clear correlation was observed. In cases with small GCD (Fig. 3a,c, case 1), high levels of reelin mRNA were found. In contrast, in cases with severe GCD (Fig.3a,c, case 7), only small amounts of reelin mRNA were measured. In conclusion, a similar inverse correlation between reelin mRNA expression and GCD as observed by cell counting was found by quantitative RT-PCR (Fig. 3e).
Fig. 3.

Expression profiles of reelin signal-transduction components in epileptic human hippocampi.a, Histogram showing the amounts of reelin mRNA in the dentate gyrus of individual epileptic human hippocampi (n = 7). Reelin mRNA levels were determined by quantitative RT-PCR in extracts from microdissected dentate gyri. Two measurements for each individual case were performed. Quantification included the addition of an external standard and volume measurement of the microdissected region. b, d, f, Histograms showing the expression levels of VLDLR (b), ApoER2 (d), and dab1 (f) mRNA in the dentate gyrus of individual epileptic human hippocampi. The epileptic cases (n = 7) were identical to the ones shown in a. c, Histogram showing the width of the GCL in the dentate gyrus of individual epileptic hippocampi. The width of the GCL was measured in cresyl violet-stained sections of the same cases (n = 7) as shown ina. e, Correlation of reelin mRNA levels and the width of the GCL. The reelin mRNA levels shown ina were plotted against the width of the GCL (c). g, Expression of dab1 mRNA in granule cells of a normal human dentate gyrus visualized by ISH.h, Expression of dab1 mRNA in the dentate gyrus of a TLE patient. Note dab1 mRNA in dispersed granule cells. Scale bars, 80 μm (g, h). ML, Dentate molecular layer.
Reelin, acting as a stop signal for migrating neurons, requires the presence of reelin receptors on the respective target cells. Therefore, we investigated simultaneously the expression of VLDLR (Fig.3b), ApoER2 (Fig. 3d), and dab1 (Fig.3f) mRNAs in the same epileptic cases by quantitative RT-PCR. All three mRNA species are expressed in human hippocampi with AHS. The relative dab1 mRNA levels were high in contrast to the low expression levels of VLDLR and ApoER2 mRNA. No correlation to GCD was observed for these three mRNA species. However, dab1 mRNA was localized to dentate granule neurons in the human dentate gyrus by ISH (Fig.3g,h), and dab1 protein was detected by immunoblotting in extracts from human hippocampus (data not shown), suggesting that reelin signaling takes place in granule cells.
DISCUSSION
Our results show an inverse correlation between reelin expression and GCD in human epileptic hippocampal tissue. Lack of reelin during development prevents the formation of a densely packed GCL inreeler mice. We observed very few reelin mRNA-synthesizing CR cells and low reelin mRNA levels in tissue samples from TLE patients with pronounced GCD, indicating that reelin is also essential in humans for the formation of a densely packed GCL. It should be pointed out, however, that reelin expression is reduced, not absent, in the present tissue samples from TLE patients. A defect in the humanreelin gene resulting in a lack of reelin expression is associated with autosomal recessive lissencephaly (Hong et al., 2000). We do not know whether reelin expression is similarly reduced in other brain regions of the present patients with TLE. Alternatively, focal lesions (e.g., focal ischemic insults) may have caused CR cell degeneration or dysfunction in the affected regions, eventually leading to local deficits in reelin expression and, as a result, to local migration defects. Focal ischemia during development is associated with local migration defects (Gressens et al., 1996), which in turn are known to underlie the development of certain forms of epileptic disorders (Gleeson and Walsh, 2000). Moreover, ischemia leads to increased release of the excitatory neurotransmitter glutamate (Nishizawa, 2001), and glutamate agonists have been shown to cause CR cell degeneration (Del Rio et al., 1997). Morphological analysis of our samples from TLE patients revealed no similar dispersion of nearby hippocampal pyramidal cells, thus being in favor of locally restricted processes underlying GCD.
A unique feature of the granule cells is their enduring postnatal generation (Altman and Das, 1965), which is even stimulated by epileptic activity in the dentate gyrus (Parent et al., 1997). Thus, local GCD could represent abnormal migration of newly generated neurons. One is tempted to speculate that seizures in epileptic patients might have caused CR cell degeneration (and hence reduced reelin expression) followed by an altered migration of recently generated granule cells. This interpretation is supported by the finding that in slice culture experiments CR cells were selectively caused to degenerate after application of excitotoxic drugs (Del Rio et al., 1997). Moreover, in a mouse model of TLE, GCD can be provoked by kainic acid injection (Bouillert et al., 2000). If seizures caused CR cell death, one would expect to see fewer CR cells and lower reelin mRNA levels in early onset epilepsies and in patients with frequent seizures than in cases with rare seizures. No such correlation was found in the present material. However, besides seizure frequency, the intensity and duration of seizures may vary considerably among patients. Along this line, the lack of immunostaining in our tissue samples for caspase-3, a marker for neuronal degeneration (C. A. Haas, unpublished observation), does not rule out seizure-induced degeneration of CR cells followed by an altered migration of recently generated granule cells.
The observed decrease in reelin expression is not contradictory to previous studies that reported an increased number of CR cells in TLE patients (Blümcke et al., 1999) and reeler mice (Coulin et al., 2001). Unlike in the present study, CR cells were immunostained for calretinin in these studies, and we have to be aware of different CR cell types (Meyer and Goffinet, 1998; Chen et al., 2001) and different types of calretinin-positive neurons (Maglóczky et al., 2000). Double labeling of the present human tissue for reelin mRNA and calretinin indicated that there is only a partial overlap of reelin mRNA-expressing CR cells and the calretinin-positive CR cell population in the dentate gyrus (Haas, unpublished observation).
In conclusion, the reelin pathway seems to be important for granule cell lamination in the human dentate gyrus, because decreased reelin expression in tissue samples from epileptic patients was found to be directly correlated to the extent of GCD. Moreover, we demonstrated here that the various molecules of the reelin signaling cascade, reelin receptors and dab1, are expressed in the human dentate gyrus. Although these data provide evidence for a role of reelin deficiency in the development of GCD in AHS, they do not explain why this migration defect is accompanied by epileptic disorder. Additional studies will have to analyze whether or not a loss of dense granule cell packing is accompanied by an increased synaptic coupling of these neurons.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (Transregio Sonderforschungsbereich TR-3). We are grateful to Dr. Patrick Carroll for the gift of a dab1 plasmid. We thank H. Banse and S. Huber for excellent technical assistance.
Correspondence should be addressed to Dr. Carola A. Haas, Institute of Anatomy I, University of Freiburg, Albertstrasse 17, D-79104 Freiburg, Germany. E-mail: Carola.Haas@anat.uni-freiburg.de.
REFERENCES
- 1.Altman J, Das GD. Autoradiographic and histological evidence of postnatal hippocampal neurogenesis in rats. J Comp Neurol. 1965;124:319–335. doi: 10.1002/cne.901240303. [DOI] [PubMed] [Google Scholar]
- 2.Armstrong DD. The neuropathology of temporal lobe epilepsy. J Neuropath Exp Neurol. 1993;52:433–443. doi: 10.1097/00005072-199309000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Blümcke I, Beck H, Suter B, Hoffmann D, Fodisch HJ, Wolf HK, Schramm J, Elger CE, Wiestler OD. An increase of hippocampal calretinin-immunoreactive neurons correlates with early febrile seizures in temporal lobe epilepsy. Acta Neuropathol. 1999;97:31–39. doi: 10.1007/s004010050952. [DOI] [PubMed] [Google Scholar]
- 4.Bouillert V, Loup F, Kiener T, Marescaux C, Fritschy JM. Early loss of interneurons and delayed subunit-specific changes in GABA(A)-receptor expression in a mouse model of mesial temporal lobe epilepsy. Hippocampus. 2000;10:305–324. doi: 10.1002/1098-1063(2000)10:3<305::AID-HIPO11>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 5.Chen Y, Bender RA, Frotscher M, Baram TZ. Novel and transient populations of corticotropin-releasing hormone-expressing neurons in developing hippocampus suggest unique functional roles: a quantitative spatiotemporal analysis. J Neurosci. 2001;21:7171–7181. doi: 10.1523/JNEUROSCI.21-18-07171.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coulin C, Drakew A, Frotscher M, Deller T. Stereological estimates of total neuron numbers in the hippocampus of adult reeler mutant mice: evidence for an increased survival of Cajal-Retzius cells. J Comp Neurol. 2001;439:19–31. doi: 10.1002/cne.1332. [DOI] [PubMed] [Google Scholar]
- 7.D‘Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- 8.D‘Arcangelo G, Nakajima K, Miyata T, Ogawa M, Mikoshiba K, Curran T. Reelin is a secreted glycoprotein recognized by the CR-50 monoclonal antibody. J Neurosci. 1997;17:23–31. doi: 10.1523/JNEUROSCI.17-01-00023.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.D‘Arcangelo G, Homayouni R, Keshvara L, Rice DS, Sheldon M, Curran T. Reelin is a ligand for lipoprotein receptors. Neuron. 1999;24:471–479. doi: 10.1016/s0896-6273(00)80860-0. [DOI] [PubMed] [Google Scholar]
- 10.Del Rio JA, Heimrich B, Borrell V, Förster E, Drakew A, Alcantara S, Nakajima K, Miyata T, Ogawa M, Mikoshiba K, Derer P, Frotscher M, Soriano E. A role for Cajal-Retzius cells and reelin in the development of hippocampal connections. Nature. 1997;385:70–74. doi: 10.1038/385070a0. [DOI] [PubMed] [Google Scholar]
- 11.Gleeson JG, Walsh CA. Neuronal migration disorders: from genetic diseases to developmental mechanisms. Trends Neurosci. 2000;23:352–359. doi: 10.1016/s0166-2236(00)01607-6. [DOI] [PubMed] [Google Scholar]
- 12.Gressens P, Marret S, Evrard P. Developmental spectrum of the excitotoxic cascade induced by ibotenate: a model of hypoxic insults in fetuses and neonates. Neuropath Appl Neurobiol. 1996;22:498–502. doi: 10.1111/j.1365-2990.1996.tb01123.x. [DOI] [PubMed] [Google Scholar]
- 13.Haas CA, Deller T, Krsnik Z, Tielsch A, Woods A, Frotscher M. Entorhinal cortex lesion does not alter reelin mRNA expression in the dentate gyrus of young and adult rats. Neuroscience. 2000;97:25–31. doi: 10.1016/s0306-4522(00)00007-5. [DOI] [PubMed] [Google Scholar]
- 14.Hirotsune S, Takahare T, Sasaki N, Hirosè K, Yoshiki A, Ohashi T, Kusakabe M, Murakami Y, Muramatsu M, Watanabe S, Nakao K, Katsuki M, Hayashizaki Y. The reeler gene encodes a protein with an EGF-like motif expressed by pioneer neurons. Nat Genet. 1995;10:77–83. doi: 10.1038/ng0595-77. [DOI] [PubMed] [Google Scholar]
- 15.Hong SE, Shugart YY, Hung DT, Al Shahwan S, Garnt PE, Hourihane JOB, Martin NDT, Walsh C. Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nat Genet. 2000;26:93–96. doi: 10.1038/79246. [DOI] [PubMed] [Google Scholar]
- 16.Houser CR. Granule cell dispersion in the dentate gyrus of humans with temporal lobe epilepsy. Brain Res. 1990;535:195–204. doi: 10.1016/0006-8993(90)91601-c. [DOI] [PubMed] [Google Scholar]
- 17.Howell BW, Hawkes R, Soriano P, Cooper JA. Neuronal positioning in the developing brain is regulated by mouse disabled-1. Nature. 1997;389:733–737. doi: 10.1038/39607. [DOI] [PubMed] [Google Scholar]
- 18.Maglóczky ZS, Wittner L, Borheghyi ZS, Halász P, Vajda J, Czirják S, Freund TF. Changes in the distribution and connectivity of interneurons in the epileptic human dentate gyrus. Neuroscience. 2000;96:7–25. doi: 10.1016/s0306-4522(99)00474-1. [DOI] [PubMed] [Google Scholar]
- 19.Margerison JH, Corsellis JAN. Epilepsy and the temporal lobe. Brain. 1966;89:499–530. doi: 10.1093/brain/89.3.499. [DOI] [PubMed] [Google Scholar]
- 20.Meyer G, Goffinet AM. Prenatal development of reelin-immunoreactive neurons in the human neocortex. J Comp Neurol. 1998;397:29–40. [PubMed] [Google Scholar]
- 21.Nishizawa Y. Glutamate and neuronal damage in ischemia. Life Sci. 2001;69:369–381. doi: 10.1016/s0024-3205(01)01142-0. [DOI] [PubMed] [Google Scholar]
- 22.Palmini A, Andermann F, Olivier A, Tampieri D, Robitaille Y, Andermann E, Wright G. Focal neuronal migration disorders and intractable partial epilepsy: a study of 30 patients. Ann Neurol. 1991;30:741–749. doi: 10.1002/ana.410300602. [DOI] [PubMed] [Google Scholar]
- 23.Parent JM, Yu TW, Leibowitz RT, Geschwind DH, Sloviter RS, Lowenstein DH. Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci. 1997;17:3727–3738. doi: 10.1523/JNEUROSCI.17-10-03727.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rakic P, Caviness VS., Jr Cortical development: view from neurological mutants two decades later. Neuron. 1995;14:1101–1104. doi: 10.1016/0896-6273(95)90258-9. [DOI] [PubMed] [Google Scholar]
- 25.Sheldon M, Rice DS, D’Arcangelo G, Yoneshima H, Nakajima M, Mikoshiba K, Howell BW, Cooper JA, Goldowitz D, Curran T. Scrambler and yotari disrupt the disabled gene and produce a reeler-like phenotype in mice. Nature. 1997;389:730–733. doi: 10.1038/39601. [DOI] [PubMed] [Google Scholar]
- 26.Trommsdorff M, Gotthardt T, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J. Reeler/disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell. 1999;97:689–701. doi: 10.1016/s0092-8674(00)80782-5. [DOI] [PubMed] [Google Scholar]