

Abstract
This study addresses the hypothesis that the previously described capacity of D1 dopamine receptors (D1Rs) to regulate dendritic growth in developing cortical neurons may involve alterations in the phosphorylation state of microtubule-associated protein-2 (MAP2). The changes in phosphorylation of this protein are known to affect its ability to stabilize the dendritic cytoskeleton. The study involved two systems: primary cultures of mouse cortical neurons grown in the presence of the D1R agonists, SKF82958 or A77636, and the cortex of neonatal transgenic mice overexpressing the D1A subtype of D1R. In both models, a decrease in dendritic extension corresponded with an elevation in MAP2 phosphorylation. This phosphorylation occurred on all three amino acid residues examined in this study: serine, threonine, and tyrosine. In cultured cortical neurons, D1R stimulation-induced increase in MAP2 phosphorylation was blocked by the protein kinase A (PKA) inhibitor, H-89, and mimicked by the PKA activator, Sp-cAMPS. This indicates that D1Rs modulate MAP2 phosphorylation through PKA-associated intracellular signaling pathways. We also observed that the elevations in MAP2 phosphorylation in neuronal cultures in the presence of D1R agonists (or Sp-cAMPS) were maintained for a prolonged time (up to at least 96 hr). Moreover, MAP2 phosphorylation underwent a substantial increase between 24 and 72 hr of exposure to these drugs. Our findings are consistent with the idea that D1Rs can modulate growth and maintenance of dendrites in developing cortical cells by regulating the phosphorylation of MAP2. In addition, our observations suggest that MAP2 phosphorylation by long-term activation of D1Rs (and PKA) can be divided into two phases: the initial ∼24-hr-long phase of a relatively weak elevation in phosphorylation and the delayed phase of a much more robust phosphorylation increase taking place during the next ∼48 hr.
Keywords: D1 dopamine receptor, cerebral cortex, development, dendrites, MAP2, protein phosphorylation
Multiple investigations have demonstrated the ability of D1 dopamine receptors (D1Rs) to regulate the extension of dendritic processes in cerebral cortical and other neurons (Lankford et al., 1987; Rodrigues and Dowling, 1990; Reinoso et al., 1996; Schmidt et al., 1996; Jones et al., 2000; Zachor et al., 2000). In a great majority of these studies, D1R activation resulted in reduced dendritic length. The intracellular mechanisms underlying such actions of D1Rs, however, have not been examined.
The signal transduction pathways coupled to D1Rs have been characterized in various cellular systems (for review, see Huff, 1997). In most of these systems, stimulation of D1Rs increases the intracellular levels of cAMP, leading to activation of cAMP-dependent protein kinase A (PKA) (Kebabian, 1997). D1Rs may also interact with other intracellular pathways (Undie and Friedman, 1990; Wang et al., 1995) and activate additional protein kinases, such as protein kinase C (PKC) (Rodrigues and Dowling, 1990). Furthermore, D1R-associated signaling pathways may be involved in regulating certain serine–threonine and tyrosine phosphatases (Snyder et al., 1998; Paul et al., 2000). It is likely that D1R exert its regulatory activity by modulating the phosphorylation states of specific cellular proteins through these kinases and phosphatases.
Cytoskeletal proteins have long been recognized to play an important role in neuronal growth and maturation (Yamada et al., 1970; Daniels et al., 1975; Keith, 1990; Johnson and Jope, 1992; Shea and Beermann, 1994; Kobayashi and Mundel, 1998). Among these proteins, microtubule-associated protein-2 (MAP2) is a major agent responsible for promoting assembly and preservation of dendritic microtubules (for review, see Maccioni and Cambiazo, 1995; Sanchez et al., 2000), which are the principal cytoskeletal constituents involved in the growth and maintenance of dendrites (Yamada et al., 1970; Daniels, 1975; Keith, 1990; Shea and Beermann, 1994; Kobayashi and Mundel, 1998). This activity of MAP2 is regulated predominantly via phosphorylation at multiple sites by PKA, PKC, and other protein kinases (Goldenring et al., 1985; Kadowaki et al., 1985; Walaas and Nairn, 1989; Hiraga et al., 1993; Krueger et al., 1997) and via dephosphorylation by various serine–threonine and tyrosine phosphatases (Yamamoto et al., 1988;Hiraga et al., 1993; Gong et al., 2000). Changes in the phosphorylation state of MAP2 have been shown to affect the stability of the dendritic microtubular cytoskeleton (Wiche et al., 1991; Maccioni and Cambiazo, 1995; Sanchez et al., 2000) and, consequently, the number, length, and branching of dendrites (Diez-Guerra and Avila, 1993, 1995; Audesirk et al., 1997).
We hypothesize that at least one of the mechanisms responsible for the D1R-associated control of dendritic length in cortical neurons involves regulation of the phosphorylation state of MAP2. The present paper describes results of our investigations in primary cortical neuronal cultures and in D1R-overexpressing mice that are consistent with this hypothesis. We also show that D1Rs are capable of long-term regulation of MAP2 phosphorylation through PKA-associated pathways. Moreover, these pathways were able to affect MAP2 phosphorylation not only on serine residues, as can be predicted on the basis of studies with purified PKA and MAP2 (Goldenring et al., 1985; Walaas and Nairn, 1989), but also on threonine and tyrosine residues. These findings reveal an additional unanticipated level of complexity in PKA–MAP2 interactions in live neurons. Furthermore, our observations suggest that MAP2 phosphorylation resulting from long-term activation of D1R involves at least two phases: an initial relatively weak phosphorylation of serine and threonine residues with no change in tyrosine phosphorylation, and a delayed more robust increase in the phosphorylation of all three residues.
MATERIALS AND METHODS
Primary cultures of cortical neurons. Primary cortical neuronal cultures were prepared as described previously byVaccarino et al. (1995) and Reinoso et al. (1996) with some modifications. Briefly, cerebral cortex of the frontal lobe, selected as a cortical region rich in D1 dopamine receptors (Schambra et al., 1994; Gaspar et al., 1995; Lidow, 1995, 1999; Reinoso et al., 1996), was dissected from CD1 mouse fetuses at embryonic day 15 (E15) in ice-cold sterile Ca2+- and Mg2+-free HBSS (Biofluids). The dissected tissue was triturated with a fire-polished Pasteur pipette to obtain a suspension of individual cells. The dispersed cells were plated on 100-mm-diameter Petri dishes (Nalge Nunc International) precoated with poly-l-ornithine and laminin (Sigma, St. Louis, MO) at a density of 4 × 106 cells per dish in DMEM/F12 medium supplemented with 10% (v/v) fetal bovine serum, 25 mm HEPES, 2.5 mm glutamine, 50 IU/ml penicillin, and 50 mg/ml streptomycin (Biofluids). Petri dishes were placed in a water-jacketed incubator (Forma Scientific) in 5% CO2/95% air at 37°C. Three hours later, the cultures were washed with HBSS, and culturing was continued in serum-free glia-conditioned media (GCM). The GCM used was produced from cultures of the human astrocytoma cell line U-251 MG. Initial culturing of the latter cells was performed in Porter flasks (Nalge Nunc International) under conditions recommended by the cell supplier (American Type Cell Culture). When cells approached confluence, the flasks were rinsed with HBSS, and the original medium was replaced with serum-free DMEM/F12 medium, with the addition of 25 mm HEPES, 2.5 mml-glutamine, (Biofluids), 1 mm sodium pyruvate, 100 mg/ml transferrin, 5 mg/ml insulin, 20 nm progesterone, 30 nm sodium selenite, 60 mmputrescine, 50 nm hydrocortisone, 0.5 mg/ml prostaglandin F2a, 10 ng/ml epidermal growth factor, 50 IU/ml penicillin, and 50 mg/ml streptomycin (Sigma). Four days later, GCM was collected for use in cortical cultures.
Drug exposure. Cultures of cortical cells were allowed to grow for 3 d before addition of specific drugs (with the exception of one experiment when drugs were added on the sixth day of culturing). Three days of culturing before the initiation of experimental manipulations was needed to allow accumulation of detectable amounts of high molecular weight neuron-specific MAP2. It also provided for a relatively stable basal level of phosphorylation of this protein (Diez-Guerra and Avila, 1995). After these first 3 d, media was replaced once a day in all cultures. For analysis of the dose–response effects, 10−7-5 × 10−5m of the D1R agonists, SKF82958 or A77636 (RBI), were added for 96 hr. For analysis of the time course of the effects, 5 × 10−5m of the same agonists were added for periods ranging from 5 min to 96 hr. The D1R specificity of the agonist actions was tested by the addition of 10−5m of the D1R antagonist, SCH23390 (RBI). The controls consisted of cortical cells cultured for comparable periods of time either without any dopaminergic drugs or with 10−5m SCH23390. To examine whether the observed effects of D1R stimulation involved PKA-associated intracellular pathways, 3-d-old cultures were coincubated for 2 hr with 5 × 10−5m of SKF82958 or A77636 and 10−6m of the PKA inhibitor, H-89 (Calbiochem). Incubation with H-89 for longer periods of time was not used because prolonged exposure to this agent markedly reduced cell viability (see Results). Insight into the prolonged effects of PKA, however, was gained by examining cell cultures that were incubated for 2 or 72 hr with 10−6m of the PKA activator, Sp-cAMPS (RBI).
Analysis of cell viability in cultures. First, we stained live cultures with propidium iodine (PI) (Sigma; 3 nm for 5 min), which is known to selectively visualize apoptotic and necrotic cells (Lizard et al., 1995; Ertel et al., 1998; Leite et al., 1999; Foglieni et al., 2001). The bright red PI-stained nuclei were counted under a Leitz Diavert microscope equipped for detection of fluorescence. For each experimental condition examined, seven separately generated cultures were evaluated, and for each culture the counting was performed in 15 randomly selected 0.1 mm2 fields. Second, live cultures were stained with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT; Sigma), which visualizes only living cells the active mitochondria of which are capable of cleaving the tetrazolium ring in MTT (Mosmann, 1983; Reinoso et al., 1996). For this purpose, 10 nm MTT and 300 μl of 0.04m HCl in isopropanol were added to the cultures for 4 hr, after which the resultant fluorescence at the test wave length of 570 nm and the reference wave length of 360 nm was measured with a C&L Fluorometer mounted on a Leitz Diavert microscope. As with PI staining, the measurements were performed in 15 randomly selected 0.1 mm2 fields in each of seven separately generated cultures for every experimental condition examined. For both staining methods, controls consisted of 7-d-old cultures grown in glia-unconditioned medium (non-GCM) for 48 hr before examination.
Immunocytochemistry. For immunocytochemical detection of MAP2 or neuron-specific enolase (NSE), cell cultures were fixed for 5 min in 4% formaldehyde in PBS, pH. 7.6, and cleared in 0.1% Triton X-100 in PBS for 30 min. The cultures were then preincubated for 1 hr in 5% normal goat serum (Sigma). The primary antibodies used were rabbit polyclonal anti-NSE (1:500 dilution; Biodesign International) and mouse monoclonal anti-MAP2, which label only the neuron-specific high molecular weight a and b isoforms of MAP2 (1:1000 dilution; Sigma). Cultures were incubated with the primary antibodies for 18–24 hr at 4°C and then rinsed in PBS. For NSE detection, the secondary antibodies were Cy3-conjugated goat anti-rabbit IgGs (Jackson Laboratory). The cultures were incubated with these antibodies (1:1000 dilution) for 1 hr at room temperature and then counterstained for 3 min with 300 nmbis-benzimidazole (BZM; Molecular Probes). For detection of anti-MAP2 primary antibodies, the secondary antibodies were biotinylated goat anti-mouse IgGs (1:1000 dilution; Jackson Laboratories). The incubation with these antibodies was also conducted for 1 hr at room temperature. This was followed by visualization of the labeling with Vectastain Elite ABC kit (Vector Laboratories) and fast DAB tablets (Sigma). Controls included omission of the primary antibodies. In the absence of the primary antibodies, virtually no immunostaining was observed (see Fig. 3F).
Fig. 3.
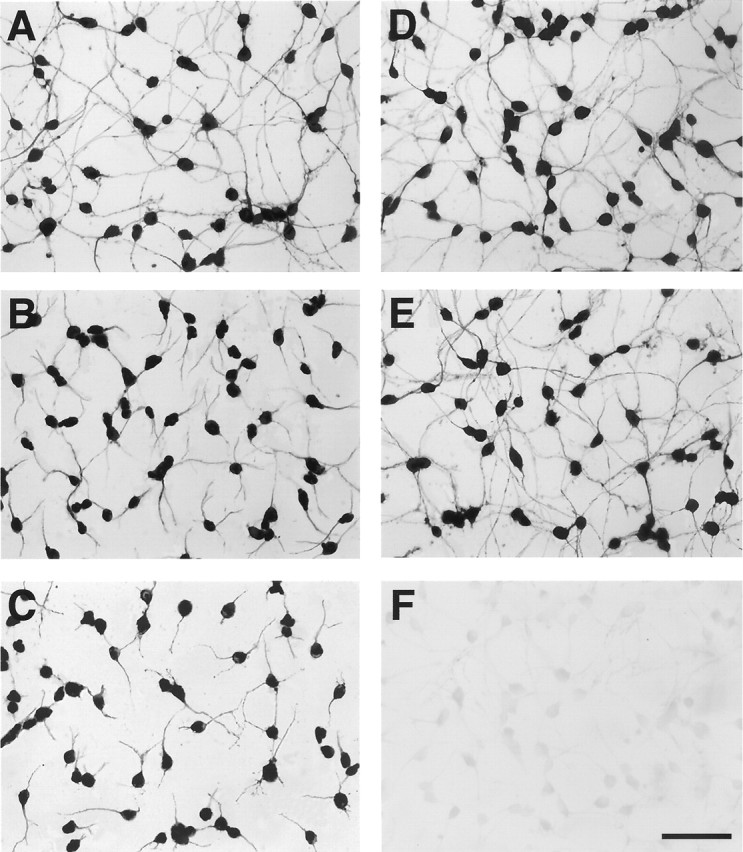
Micrographs depicting typical effects of D1R stimulation on neuritic extension in primary cultures of cortical neurons from the frontal lobe of fetal mouse exemplified by the actions of the D1R antagonist, A77636. Primary cultures of cortical cells obtained from the frontal lobe of E15 mice were maintained for 3 d in drug-free medium and then grown for an additional 96 hr in the presence of culture medium alone (A), 10−5mA77636(B), 5 × 10−5mA77636 (C), 5 × 10−5mA77636 in combination with 10−5m SCH23390 (D), and 10−5mSCH23390 (E). The soma and neurites of neuronal cells were visualized by the immunolabeling of the heavy molecular weight neuron-specific MAP2. F, Control culture in which immunolabeling was performed in the absence of the primary antibodies. Scale bar, 60 μm. Note the A77636-induced concentration-dependent reductions in the lengths of MAP2-positive neurites. Although SCH23390 does not show any effects on its own, this D1R antagonist prevents the reductions in neurite extension induced by A77636.
Analysis of the proportion of neurons in cultured cells. This analysis was performed by comparing the number of neuronal cell bodies identified by NSE immunolabeling (Schmechel et al., 1980) with the total number of cell nuclei in the same cultures visualized by BZM counterstaining (Wang and Lidow, 1997). Images of the stained cultures were captured and digitized with a SensiCam high-resolution digital video camera (Cooke) mounted on a Leitz Diavert microscope. Captured images of 15 randomly selected 0.1 mm2fields from each of seven separately generated 3-d-old cultures were used to count the number of green-fluorescent NSE-immunolabeled cell bodies and blue-fluorescent BZM-stained nuclei. Cell counting was performed using IPLab software (Scanalytics).
Morphometric analysis of MAP2-labeled neurites. This analysis was performed on the basis of the methodology described inJones et al. (2000) with modifications by Miguel-Hidalgo et al. (2000). For every experimental condition examined, images of MAP2 immunolabeling in 15 randomly selected 0.1 mm2 fields from each of seven separately generated cultures were digitized with a SensiCam digital video camera (Cooke) mounted on a Leitz Diavert microscope. For each digitized image, the percentage of the area occupied by MAP2 staining was determined using IPLab software (Scanalytics). To verify that the alterations in the area of MAP2 staining in drug-treated cultures were related to the treatment-induced changes in neurites, the number and average area of MAP2-expressing soma were also calculated for each field. This method of analysis was selected, rather than measuring the length of individual neurites, because outgrowth in the cultures was usually too extensive to allow consistent tracing of individual processes. Although plating at lower densities may permit direct measurement of neurite length, we chose not to use low density plating because such conditions may subject cultured neurons to extensive stress and alter their responses to pharmacologic treatments (Jones et al., 2000).
Animals. Heterozygous male and female transgenic Line-19 mice overexpressing the D1A subtype of D1R (Dracheva et al., 1999;Dracheva and Haroutunian, 2001) were bred, and their offspring were genotyped at birth. The genotyping was performed by quantitative PCR as described earlier [(Dracheva et al., 1999); this paper also gives levels of D1R in different cortical regions of transgenic animals]. The sex of the pups was determined by examining the anogenital distance. The pups were killed at postnatal day 6, and their brains were collected for analysis. Six-day-old pups were chosen for this study because by that age all cortical neurons in this species should reach their adult position and begin to grow their dendritic trees (Rakic et al., 2000). Procedures for handling of the animals were approved by the University of Maryland Animal Care and Use Committee.
Morphological analysis of cortical tissue. In this part of the study, we used transgenic mice from five litters, with each litter containing two wild-type, two D1R-overexpressing heterozygous, and two D1R-overexpressing homozygous males. The mice were anesthetized with 100 mg/kg Nembutal (Abbott Laboratories) and perfused intracardially with 2% paraformaldehyde and 2% glutaraldehyde in 0.2m phosphate buffer, pH 7.4. The brains were dissected out, blocked, and postfixed in the same fixative at 4°C for 7 d. The blocks from the right hemisphere were then sectioned into 20-μm-thick sections on a cryostat. The sections were stained with cresyl violet and used for the microscopic examination (under a ZeissAxioscope microscope) of the general cortical morphology in the dorsal part of the anterior cingulate region and in the primary visual cortex [defined according to Franklin and Paxinos (1997)]. The thickness of these cortical areas was measured as described by Selemon et al. (1999). For both areas, we examined at least three sections from every animal, with the measurement of the cortical thickness being performed at three random positions along a given area per section. The tissue blocks from the left hemisphere were washed in distilled water and placed in 2.5% potassium dichromate (Sigma) in the dark at 4°C for 21 d. After that, they were washed again and immersed in 0.75% silver nitrate (Polyscience) in the dark at room temperature for 48 hr. The impregnated blocks were embedded in 12% celloidin (Fisher Scientific) and sectioned at 250 μm on a sliding microtome (Warner Lambert Technologies). These sections were used for measuring the length of the apical dendrites of the layer V pyramidal neurons in the same two cortical areas that were subjected to a general morphological examination on cresyl violet-stained material. For each cortical area, the measurement was performed in 10 randomly selected pyramidal neurons per animal, providing that their overall appearance was similar to that of the general population of such cells. In addition, the apical dendrites selected for the analysis had to be fully silver impregnated and running parallel to the section cut [as recommended by Uylings et al. (1986)]. Glass slides with Golgi-stained sections were placed on an XYZ motor stage (Ludl Electronics) fitted on a Zeiss Axioscope microscope equipped with a SensiCam digital video camera (Cooke). The analysis was performed with Oncor Imaging Software, which drove the motor stage and automatically collected a series of images focused through the thickness of the section. Collected images were used to automatically reconstruct a three-dimensional (3D) image of the digitized portion of the tissue section. All visible branches of the apical dendrites were then traced manually. On the basis of these 3D tracings, we estimated the length of the apical dendrite from the cell body to its most distal point (Jones et al., 2000) as well as the combined length of all traced branches of the apical dendrite (Uylings et al., 1986). We also counted the number of cuts through the side-branches produced by the microtome knife when the section containing the main body of the dendrite was made. Because all samples were prepared in a similar manner, no corrections for tissue shrinkage were introduced. Morphometric analysis was performed by investigators who were unaware whether the samples were from control or experimental animals.
Analysis of MAP2 phosphorylation. The phosphorylation of MAP2 on serine, threonine, and tyrosine residues was examined using a nonradio-isotopic method as described in Lidow et al. (2001). Culture medium was aspirated from Petri dishes containing primary neuronal cultures, and 1 ml of ice-cold lysis buffer (20 mm Tris, pH 7.5, 50 mmNaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 5 mm EDTA, 50 mm sodium fluoride, 40 mm β-glycerophosphate, 0.5 mm sodium orthovanadate, 1 mm 4-(2-aminoethyl)-benzenesulfonyl fluoride, 20 μg/ml aprotinin, 2 μg/ml leupeptin, 2 μg/ml pepstatin A; all from Sigma) was added to each dish for 2 min. The lysate buffer with dissociated cells was collected and sonicated for 30 sec using a Sonic Dismembrator 60 (Fisher Scientific). The dissected frontal and occipital cortices from 6-d-old mice were homogenized for 3 min in 1 ml of ice-cold lysis buffer using a Cole-Palmer TriRS63C Homogenizer. The lysates were clarified by centrifugation at 20,000 × gfor 30 min at 4°C (DuPont RC-S Superspeed Refrigerated Centrifuge), and the supernatants were processed for MAP2 purification with an IMMUNOcatcher protein immunoprecipitation kit (CytoSignal). The immunoprecipitating agent (monoclonal MAP2 antibody specific for the a and b isoforms of MAP2; Sigma) was used at a concentration of 2.5 μg/500 μg of the total sample protein (determined using Bradford Protein Assay Kit; Sigma). The resultant immunoprecipitates were mixed 1:5 with the loading buffer, containing 0.1 mTris-HCl, pH 6.8, 20% SDS, 50% glycerol, 0.2% Bromophenol blue, and 4.9% DTT (Sigma), and boiled for 5 min. The proteins in the boiled mixtures (30 μl per well) were resolved by SDS-PAGE for 1.5 hr at 100 V using Ready-Made 4–15% Gradient SDS Gel in a Ready Gel Cell containing Tris/Glycin/SDS Running Buffer (Bio-Rad) The resolved proteins were transferred onto Hybond ECL nitrocellulose membranes (Amersham) for 2 hr at 100 V using a the same Bio-Rad Gel Cell containing Bio-Rad Tris/Glycine Buffer with the addition of 0.1% SDS (w/v; Sigma) to ensure a complete transfer of high molecular weight MAP2 (Wang et al., 1989). The completeness of transfer was verified by post-staining of gels with Coomassie blue (Bio-Rad). The membranes were preincubated for 2 hr with a Membrane Blocking Solution (Zymed Laboratories). To reveal the phosphorylation states of serine, threonine, and tyrosine residues, blots were incubated with one of the three antibodies from Zymed: rabbit anti-phosphoserine (1:250 dilution), anti-phosphothreonine (1:250 dilution), or anti-phosphotyrosine (1:1000 dilution) at 4°C overnight. Membranes were rinsed five times in TBS containing 0.1% (v/v) Tween 20 (Sigma) and then incubated for 1 hr with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgGs (Jackson Laboratories; 1:6500). After five washes in TBS, bands were visualized using SuperSignal Chemiluminescence kit (Pierce) and X-Omat AR Film (Kodak). To detect any cross-reactivity between antibodies to different phospho-amino acids, some of the blots were incubated with antibodies that had been preincubated either with 20 ng of the phospho-amino acid to which the antibodies were supposed to bind or with a mixture of the other two phospho-amino acids (20 ng each) as described in Glenney et al. (1988),Levine et al. (1989), and Heffeth et al. (1991). We found that preabsorption of all three antibodies with their specific phospho-amino acids completely abolished the blot immunolabeling (see Fig. 5). In contrast, preincubation with the mixture of the other two phospho-amino acids did not affect the immunolabeling of blots by any of the three antibodies (Fig. 5).
Fig. 5.

Typical Western blots of MAP2 immunoprecipitates from the primary cultures of E15 mouse cerebral cortical neurons, which after 3 d of culturing were maintained for an additional 96 hr in the presence of 0–5 × 10−5m of the D1 dopamine receptor agonist, A77636. Blots of immunoprecipitates from cultures maintained in the presence of a combination of A77636 and 10−5m of the D1R antagonist, SCH23390, are also shown. The blots were initially immunolabeled for phosphoserine, phosphothreonine, or phosphotyrosine. Control blots were preincubated either with phospho-amino acids (20 ng) specific for a given antibody (SPAA) or a mixture of phospho-amino acids nonspecific for these antibodies (NSPAA) (20 ng each). After visualization of the phospho-amino acids, the labeling was stripped, and the same blots were immunolabeled again for heavy molecular weight neuron-specific MAP2. Note an increase in the levels of phosphorylation on all three residues with an increase in the concentration of A77636. This effect is blocked by SCH23390. Also note the lack of cross-reactivity between antibodies to specific phospho-amino acids.
To detect MAP2 in the same blots, the anti-phospho-amino acid antibodies were stripped from the membranes by washing in a buffer containing 62.5 mm Tris-HCl, pH 6.7, 2% SDS, and 100 mm 2-mercaptoethanol at 65°C for 30 min. Membranes were rinsed in PBS, blocked in Zymed blocking solution for 1 hr, and incubated for 2 hr at room temperature with monoclonal anti-MAP2 antibodies (1:2000 dilution; the same clone used for immunocytochemistry and immunoprecipitation). This was followed by 1 hr incubation at room temperature with HRP-conjugated goat anti-mouse IgGs (Jackson Laboratories; 1:4000 dilution) and visualization by chemiluminescence as described above. The resultant films were digitized using a Umax UC1260 flat bed scanner, and the densitometric analysis of the bands was performed with Universal Software (Advanced American Biotechnology).
Samples from all the experimental cell cultures were always processed together with their controls, and each experiment was repeated seven times. For each cortical region examined, the samples from wild-type, heterozygous, and homozygous D1R-overexpressing littermates (two males of each genotype per litter) were also processed together for all five litters used in this part of the study. In addition, on all the films used for comparison of the optical densities of bands generated by experimental and control samples, these optical densities were within the linear range of the amount of antigen-generated optical density curve. Such curves for MAP2, phosphoserine, phosphothreonine, and phosphotyrosine were obtained using dot blots of serial dilutions of adult mouse cerebral cortex followed by immunochemistry with appropriate antibodies as described in Lidow et al. (2001). For every sample, the phosphorylation of MAP2 on each residue was expressed as a ratio of the optical density of the film images generated by the immunolabeling of MAP2 bands with the appropriate phospho-amino acid-specific antibodies and the optical density of the film images generated by the immunolabeling of the same bands with MAP2-specific antibodies. For the tissue cultures, the ratios of bands generated by the experimental samples were normalized to the ratios generated on the same film by their controls, with the latter ratios being designated as equal to 1. For the litters of D1R-overexpressing mice, the optical density ratios generated by all samples were normalized to the mean ratio for the samples from the wild-type littermates, with the latter ratio being designated as equal to 1.
Statistical analysis. The statistical analyses of the effects of different doses of D1-specific drugs and the effects of PKA-acting substances in cell cultures were performed with one-way ANOVAs followed by Tukey's post hoc comparisons between individual groups. The same statistical methodology was also used for the evaluation of the data collected from transgenic animals. The statistical analyses of the time course of drug effects and the data on cell survival in the cultures were conducted by two-way ANOVAs with treatment type and duration as variables. These were followed by Tukey's post hoc comparisons between individual groups. The differences were considered significant at p < 0.05.
RESULTS
Characterization of the in vitro and in vivo systems used in this study
Cell cultures
Given that the focus of the present investigation was on cerebral cortical neurons, it was reasonable to determine the proportion of neurons in the cortical cell cultures used in this study. The analysis was performed by comparing the number of neuronal cell bodies, identified by NSE immunolabeling, and the total number of cell nuclei, identified by BZM counterstaining, in 3-d-old cultures (Fig.1A,B). The analysis was conducted after 3 d of culturing because this was the time-point at which most drug treatments were initiated. We found that NSE-immunolabeled cell bodies constituted ∼97% of the total number of all cell nuclei present in the cultures (Fig. 1C). This demonstrates that the cortical cell cultures used in this study were essentially pure neuronal cultures.
Fig. 1.

Analysis of the proportion of the neuronal phenotype in cultured cerebral cortical cells. A, Fluorescent immunolabeling of the neuronal marker, NSE, in paraformaldehyde-fixed cortical cells obtained from the frontal lobe of E15 mouse fetuses and cultured for 3 d. B, The same cells stained with BZM, which is a fluorescent stain for cell nuclei. Scale bar, 30 μm. C, Histograms showing the densities of NSE-immunolabeled neuronal cell bodies and BZM-stained nuclei in cortical cell cultures. Each bar represents the mean ± SEM of seven separately generated cultures. Note that ∼97% of all nuclei in these cultures belong to NSE-expressing cell bodies.
Decreased neurite density and altered protein phosphorylation might reflect a general decline in cell viability that is unrelated to physiologic regulation. To exclude this possibility, we examined whether the D1R-selective agonists used in the present study altered viability of cultured cortical cells. Here, two complementary techniques were applied in live cell cultures: PI labeling, which accesses cell death, and MTT labeling, which accesses cell survival and metabolic activity. The counts of PI-stained cells showed that even the highest used concentration (5 × 10−5m) of the D1R agonists, SKF82958 and A77636, did not increase the occurrence of apoptotic–necrotic cells in cultures exposed to these drugs for up to 96 hr (Fig.2A,B,D). In both treated and untreated cultures, the density of PI-stained cells remained at three to five cells per 0.1 mm2 (Fig. 2D). Quantitative analysis of the fluorescence resulting from MTT transformation in mitochondria of living cells also revealed no statistically significant differences between drug-free cultures and cultures grown in the presence of 5 × 10−5m of SKF82958 or A77636 for 24–96 hr (Fig. 2E). These observations were not associated with poor staining techniques. The latter was demonstrated by positive controls, in which 7-d-old cultures were kept for the last 48 hr in non-GCM. These controls displayed >80 PI-stained cells per 0.1 mm2 as well as a nearly 75% decrease in MTT-associated fluorescence (Fig.2C–E).
Fig. 2.

Analysis of cell viability in cortical cultures.A–C, PI staining of apoptotic–necrotic nuclei in live cultures of cortical cells obtained from the frontal lobe of E15 mouse fetuses and cultured for 7 d. A, Cultures grown in the absence of D1R antagonists; B, cultures that for the last 96 hr of culturing were exposed to 5 × 10−5m of the D1R agonist, SKF82958.C, Cultures in which cell death was induced by 48 hr exposure to glia-unconditioned medium (Non-GCM). Scale bar, 25 μm. D, Histogram showing the density of PI-stained apoptotic–necrotic nuclei in cultures of cortical cells obtained from the frontal lobe of E15 mouse fetuses, which after 3 d of drug-free culturing were grown for 96 hr either in the absence of D1R-specific ligands or in the presence of 5 × 10−5m of the D1R agonists, SKF82958 and A77636. E, Histogram showing fluorescence resulting from MTT transformation in mitochondria of live cells in cultures of cortical cells obtained from the frontal lobe of E15 mouse fetuses, which after 3 d of drug-free culturing were grown for an additional 24–96 hr either in the absence of D1R-specific ligands or in the presence of 5 × 10−5m of SKF82958 or A77636. Histograms D and Ealso include control cultures in which cell death was induced by 48 hr exposure to non-GCM. In both histograms, each columnrepresents the mean value from seven separately generated cultures ± SEM. Note that D1R agonist exposure does not induce a significant decrease in the viability of cultured cortical cells.
PI and MTT labeling techniques were also used to determine viability of cell cultures exposed to the other three drugs used in this study, SCH23390, Sp-cAMPS, and H-98. We found that neither SCH23390 nor Sp-cAMPS reduced the viability of cells that were exposed to these drugs after 3 d of culturing at the doses (10−5 and 10−6m, respectively) and time periods used in the main body of our research (72 and 96 hr, respectively; data not shown). However, the concentrations of H-89, which we found to be effective in our study (10−6m), significantly reduced the viability of cultured cells if applied for >2 hr (for example, after 2 hr exposure to H-89, the MTT-induced fluorescence of cortical cell cultures decreased by only 3.1 ± 1.4%; 4 hr exposure, however, reduced this fluorescence by 23.0 ± 3.7%, and 8 hr exposure produced a 49.8 ± 4.6% reduction in the fluorescence).
The major goal of this study was to evaluate the hypothesis that the capacity of D1Rs to regulate growth and maintenance of dendrites may be related to their ability to affect long-term phosphorylation of MAP2. Therefore, it is important for us to demonstrate the appropriateness of the selected in vitro models for this task by demonstrating that stimulation of D1R in our cell cultures resulted in detectable alterations in dendritic trees. This was addressed by analyzing the effects of D1R-specific ligands on the percentage of the area occupied by MAP2-specific immunostaining (which visualized neurites and bodies of cultured neurons) within an average 0.1 mm2 fields of cultured cells. Such an approach was possible because none of the ligand exposure conditions used in the study significantly affected either soma density or average soma area of cultured cells. Neither were there any discernable dose- or exposure time-related trends in these two parameters (Table1). Hence, any changes in the proportion of MAP2 immunolabeling in our cultures should reflect changes in the area occupied by dendritic trees.
Table 1.
Effects of the exposure to D1R- and PKA-acting drugs on the percentage of the area occupied by MAP2 immunolabeling as well as density and average area of the soma of MAP2-expressing cells in cultures obtained from the cortex of the frontal lobe of E15 mouse fetuses
| Total days in culture | Hours of treatment | Drugs | % of Culture area occupied by MAP2 immunostaining | Number of MAP2-immunolabeled cells per 0.1 mm2 | Average soma area of MAP2-immunolabeled cells (μm2) |
|---|---|---|---|---|---|
| 3 | 0 | None | 15.8 ± 1.4 | 87 ± 11 | 66.9 ± 5.7 |
| 3 | 2 | SKF82958 (5 × 10−5m) | 12.1 ± 1.3* | 90 ± 13 | 71.4 ± 6.5 |
| 3 | 2 | SKF82958 (5 × 10−5m) H-89 (10−6m) | 16.0 ± 1.7 | 85 ± 14 | 65.5 ± 5.6 |
| 3 | 2 | Sp-cAMPS (10−6m) | 11.8 ± 1.5* | 85 ± 15 | 67.8 ± 5.1 |
| 4 | 0 | None | 17.2 ± 1.5 | 91 ± 8 | 68.3 ± 4.3 |
| 4 | 24 | SKF82958 (5 × 10−5m) | 12.4 ± 1.6* | 86 ± 12 | 70.0 ± 5.3 |
| 5 | 0 | None | 18.6 ± 2.5 | 88 ± 13 | 66.5 ± 6.8 |
| 5 | 48 | SKF82958 (5 × 10−5m) | 9.9 ± 1.7** | 85 ± 13 | 73.1 ± 6.6 |
| 6 | 0 | None | 19.9 ± 2.6 | 83 ± 11 | 63.4 ± 5.4 |
| 6 | 72 | SKF82958 (5 × 10−5m) | 7.6 ± 1.4** | 89 ± 14 | 72.6 ± 6.3 |
| 6 | 72 | Sp-cAMPS (10−6m) | 7.9 ± 1.5** | 84 ± 13 | 67.3 ± 4.9 |
| 7 | 0 | None | 21.8 ± 3.6 | 84 ± 11 | 69.9 ± 4.7 |
| 7 | 96 | SKF82958 (10−7m) | 18.3 ± 2.8 | 95 ± 17 | 64.7 ± 5.2 |
| 7 | 96 | SKF82958 (10−6m) | 15.6 ± 2.3 | 87 ± 12 | 74.1 ± 7.3 |
| 7 | 96 | SKF82958 (10−5m) | 12.0 ± 2.4* | 93 ± 14 | 64.8 ± 6.0 |
| 7 | 96 | SKF82958 (5 × 10−5m) | 7.7 ± 1.1** | 89 ± 14 | 67.5 ± 5.9 |
| 7 | 96 | SKF82958 (5 × 10−5m) SCH23390 (10−5m) | 21.5 ± 2.6 | 82 ± 13 | 68.9 ± 6.0 |
| 7 | 96 | SCH23390 (10−5m) | 22.9 ± 3.5 | 87 ± 12 | 66.7 ± 5.8 |
The cultures were allowed to grow in drug-free media for 3 d and then some of them were exposed to drugs. The time course of the effects of D1R agonists was examined by comparing cultures exposed to 5 × 10−5m of these ligands for 2, 24, 48, 72, and 96 hr with the age-matched drug-free cultures (exemplified here by the data produced by SKF82958). The dose dependence of the effects of D1R agonists was examined in cultures exposed for 96 hr to 0–5 × 10−5m of these ligands (also exemplified here by the data produced by SKF82958). The D1 receptor specificity of the effects of the agonists was demonstrated by the addition of 10−5m of D1R antagonist, SCH23390. The ability of the PKA stimulator, Sp-cAMPS, to replicate the effects of D1R agonists was examined in cultures exposed to 10−6m of this drug for 2 and 72 hr, and the ability of 10−6m of the PKA inhibitor, H-89, to block the effects D1R agonists was examined in cultures exposed to a combination of these drugs for only 2 hr (longer exposure to H-89 was not used because of significant decline in cell viability). Each data point represents a mean of seven separately performed experiments ± SEM. The statistically significant differences between drug-exposed cultures and the age-matched drug-free controls are marked by asterisks (*p < 0.05; **p < 0.01; Tukey's post-test). Note D1R agonist-induced dose- and time-dependent reductions in the percentage of the area occupied by MAP2-immunolabeled cells without any significant changes either in the density or in the area of the soma of these cells. The addition of a D1R antagonist blocked this reduction, whereas the antagonist alone had no effect. Furthermore, PKA stimulator replicated the effects of D1R agonists, whereas PKA inhibitor counteracted the effects of these agonists.
The dose dependence of the effects of the D1R agonists, SKF82958 andA77636, were examined in cultures, which after 3 d of drug-free culturing were grown for 4 additional days in the presence of increasing concentrations of these agonists. Age-match cultures grown in the absence of these ligands served as controls. We found that all drug-exposed cultures displayed a reduced percentage of the area occupied by MAP2-specific immunostaining (Fig.3A–C; Table 1). The magnitude of this reduction was concentration dependent, with statistically significant alterations being detectable beginning at 10−5m for both agonists (Fig. 3A–C; Table 1). At all agonist concentrations examined, the reduction was blocked by the D1R antagonist, SCH23390 (10−5m) (Fig. 3D; Table 1), whereas the antagonist alone did not produce any significant effects (Fig.3E; Table 1).
The time course of the D1R agonist effects was examined in 3-d-old cultures exposed for an additional 2–96 hr to 5 × 10−5m of either SKF82958 orA77636. Compared with age-matched cultures grown in the absence of these agents, there was a significant reduction in the proportion of MAP2 staining in cultures exposed to either agonist at all time periods examined (Table 1). Interestingly, the percentage of the area occupied by MAP2 immunostaining in cultures exposed to the D1R agonists for 2 hr was very similar to that in cultures exposed to these agonists for 24 hr (Table 1). However, 48 hr exposure to the D1 agonists produced a further decrease in the percentage of the area occupied by MAP2 immunostaining (Table 1). This decline was even more severe in cultures exposed to the agonists for 72 hr (Table 1). Extension of the agonist exposure from 72 to 96 hr did not result in any further decrease in the area occupied by MAP2-expressing cells (Table 1). The latter was not because this area could no longer be significantly reduced by a further decrease in neuritic extension, because the area occupied by MAP2-positive soma was still small enough for additional reductions in the proportion of MAP2 staining to occur.
This study used PKA modulating agents to test whether D1R affects MAP2 phosphorylation through the PKA-associated cascade. Therefore, it was of interest to examine the ability of these drugs to regulate neuritic extension. We found that 2 and 72 hr applications of 10−6m of the PKA activator, Sp-cAMPS, caused reductions in the area of the culture occupied by MAP2 immunostaining (without reducing either soma density or average soma area of cultured cells) that were comparable to those induced by the D1R agonists (Table 1). As noted above, our cell cultures could not be exposed for >2 hr to the effective concentrations of the PKA inhibitor, H-89, without significantly reducing their viability. Nevertheless, during the 2 hr period, 10−6m H-89 fully prevented a decrease in the area of the culture occupied by MAP2 immunostaining induced by 5 × 10−5mof the two D1R agonists used in this study (Table 1).
D1R-overexpressing mice
Cortical cell cultures are invaluable for establishing the ability of D1R to regulate specific biochemical and morphological aspects of these cells and for investigating the molecular mechanisms of such regulatory activities. However, the in vitro preparations can reveal only the potential of D1R to be involved in mediating similar effects in vivo. To demonstrate that this potential is realized in the brain, we complemented the in vitroassays with studies in 6-d-old transgenic D1R-overexpressing mice. In preparation for the analysis of MAP2 phosphorylation in cortical tissues of the above-mentioned transgenic mice, we examined the overall morphology and determined the lengths of the apical dendrites of the layer V pyramidal neurons in the dorsal part of the anterior cingulate and primary visual cortices of these animals. The anterior cingulate cortex was selected to represent dopamine D1R-rich cortical areas of the frontal lobe where one would expect to clearly see the effects of D1R activation (Levitt et al., 1997; Jones et al., 2000). The primary visual cortex was selected as a control cortical tissue that is poor in dopaminergic receptors even in D1R-overexpressing mice (Dracheva et al., 1999) and where one would expect little manifestation of D1R activity (Levitt et al., 1997; Jones et al., 2000). The layer V pyramidal neurons were chosen for analysis as cortical cells most heavily expressing D1R (Bergson et al., 1995).
Microscopic examinations of cresyl violet-stained sections through either of the two cortical regions showed that the appearance of cortical lamination and the positioning of pyramidal and nonpyramidal cells within individual laminas were not different between homozygous and heterozygous D1R-overexpressing mice and their wild-type littermates (Fig.4A,B), nor were there any significant differences between these animal groups in the thickness of the cerebral cortex (Fig. 4E). These observations suggest that the alterations in the length of the apical dendrites of cortical pyramidal cells described in the next paragraph are not a reflection of some general disorganization of cortical morphology in D1R-overexpressing animals.
Fig. 4.
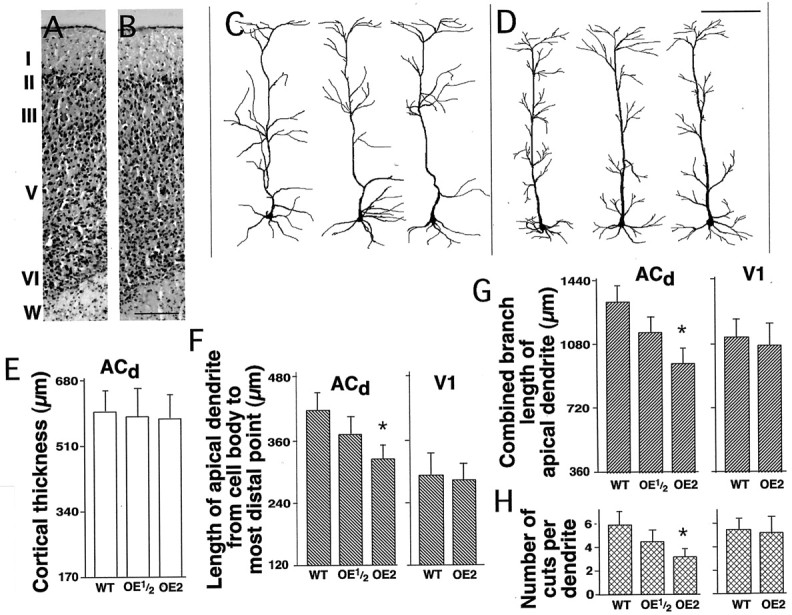
Effects of D1R overexpression on dendritic development in the cortex of 6-d-old transgenic mice. Typical images of cresyl violet-stained sections through the dorsal part of the anterior cingulate cortex in wild-type mice (A) and their homozygous D1R-overexpressing littermates (B).I–VI, Cortical layers; W, white matter. Scale bar, 500 μm. Examples of the tracing of Golgi silver-impregnated pyramidal layer V neurons from the dorsal part of the anterior cingulate cortex of the wild-type mice (C) and their homozygous D1R-overexpressing littermates (D). Scale bar, 100 μm.E, Histogram showing the thickness of the dorsal part of the anterior cingulate cortex in wild-type mice (WT) and their heterozygous (OE1/2) or homozygous (OE2) D1R-overexpressing littermates.F, Histograms showing the lengths of the apical dendrites measured from the cell body to the most distal point in layer V pyramidal neurons of the dorsal part of the anterior cingulate (ACd) and primary visual (V1) cortical regions in wild-type mice and their D1R-overexpressing littermates. G, Histograms showing the combined length of all traceable branches of the same apical dendrites. E, Histograms showing the number of cuts of the side branches of these apical dendrites that resulted from sectioning of the tissue. In all histograms, each columnrepresents the mean ± SEM of the data pooled from five litters of transgenic animals (n = 5), each containing two male representatives of each genotype. Statistically significant differences between wild-type and D1R-overexpressing littermates are indicated by asterisks (*p < 0.05 compared with wild-type by Tukey's post hoc test). Note that the layer V pyramidal neurons of the dorsal part of the anterior cingulate cortex display a significant decrease in the length of their apical dendritic trees (accompanied by a decrease in the proportion of the cuts of dendritic branches produced by tissue sectioning) in homozygous D1R-overexpressing mice as compared with their wild-type littermates. This takes place in the absence of any noticeable changes either in cortical cytoarchitecture or in the cortical thickness. In contrast, no corresponding significant changes in dendritic length are present in the primary visual cortex.
Measurements of the length of the apical dendrites (from the cell body to their most distal point) of the layer V pyramidal neurons in the dorsal part of the anterior cingulate cortex showed them to be significantly shorter in homozygous D1R-overexpressing mice than in their wild-type littermates (Fig. 4C–F). Homozygous D1R-overexpressing animals also displayed a significant decrease in the combined length of the traceable branches of the apical dendrites in the same pyramidal neurons (Fig.4C,D,G). Furthermore, the apical dendrites from the latter animals had fewer cuts produced by a microtome knife during generation of the tissue sections (Fig.4G). This indicates that the decrease in the overall dendritic length observed in homozygous D1R-overexpressing animals does not result from the extension of dendritic branches outside of the tissue sections. Moreover, the decrease in the overall dendritic length in homozygous D1R-overexpressing animals is most likely underestimated, because wild-type mice had more cuts through their dendritic branches. Dendrites of heterozygous animals were of intermediate length. These same results were obtained both when separate comparisons were conducted between members of each of the five litters examined and when the data for each genotype were pooled together before analysis.
The comparison of the apical dendrites of the layer V pyramidal neurons from the primary visual cortex of wild-type mice and their D1R-overexpressing littermates revealed no significant differences either in their length from the cell body to the most distal end or in the combined length of the traceable branches, or in the number of branches cut in the preparation of the tissue sections (Fig.4F–H).
D1R regulation of MAP2 phosphorylation on serine, threonine, and tyrosine residues in in vitro and in vivo models
Blots of neuron-specific high molecular weight MAP2 immunoprecipitated from either cultured cortical neurons or cortical tissue homogenates revealed a single band of ∼280 kDa (Fig.5). We believe this to be theb isoform of this protein, which is present in developing neurons (Binder et al., 1984; Burgoyne and Cumming, 1984). This MAP2 always displayed phosphorylation of serine, threonine, and tyrosine residues at all time-points of culturing that were examined in the present study (Fig. 5).
Studies in in vitro model
We found that 96 hr exposure of cerebral cortical cell cultures to 10−7-5 × 10−5m of the D1R agonists, SKF82958 and A77636 (after these cultures were grown for 3 d in the absence of dopaminergic drugs), resulted in a significant increase in MAP2 phosphorylation on all three residues examined in this study. For all residues, this increase was agonist concentration dependent, with statistically significant effects being observed at concentrations 10−5m and above (Figs. 5,6). The agonist effects were prevented by the presence of 10−5m of the D1R agonist, SCH23390 (Figs. 5, 6), whereas this antagonist alone was unable to affect MAP2 phosphorylation (Fig. 6).
Fig. 6.
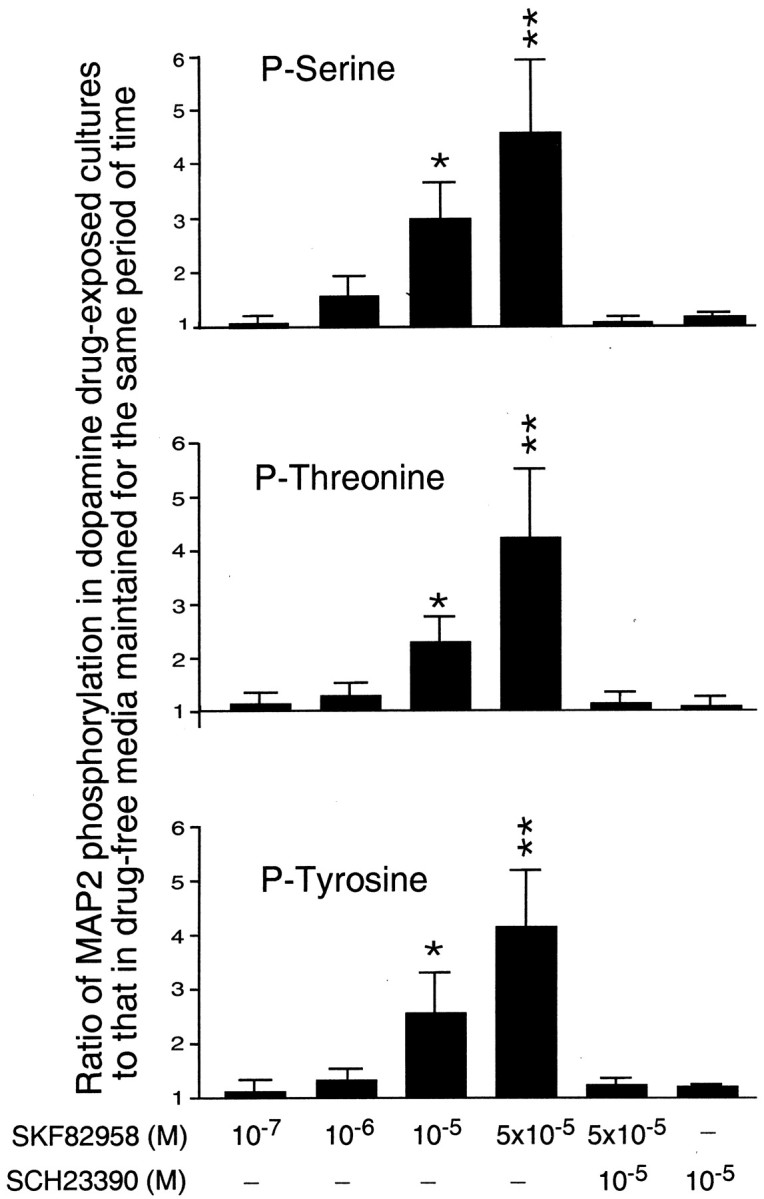
Histograms showing D1R agonist concentration dependence of MAP2 phosphorylation on serine, threonine, and tyrosine residues. The data are presented as the ratios between MAP2 phosphorylation on specific residues in cultures of E15 mouse cortical neurons, which after 3 d of drug-free culturing were incubated for an additional 96 hr with 10−7– 5 × 10−5m of the D1R agonist, SKF82958, and MAP2 phosphorylation on the same residues in cultures maintained for the comparable periods of time without the addition of D1R ligands. In addition, the histograms show the ratios between MAP2 phosphorylation in cultures incubated with either a combination of SKF82958 and 10−5m of the D1R antagonist, SCH23390, or with 10−5m of this antagonist alone and MAP2 phosphorylation in corresponding drug-free cultures. All of these ratios were obtained using seven drug-exposed and seven drug-naive cultures maintained for the same period of time. Error bars are SEMs. The ratios representing statistically significant differences between experimental and corresponding control cultures are marked by asterisks(*p < 0.05; **p < 0.01; Tukey's post-test). Note that increasing concentrations of the D1R agonist induce corresponding increases in the levels of MAP2 phosphorylation on all three residues. These effects are blocked by the D1R antagonist.
The time course of MAP2 phosphorylation in response to D1R stimulation was examined in cortical cells that after 3 d of drug-free culturing were incubated with 5 × 10−5m of SKF82958 or A77636for periods from 5 min to 96 hr. Age-matched drug-naive cultures served as controls. Five minutes of incubation with either of the D1R agonists resulted in a statistically significant increase in the phosphorylation of MAP2 on serine and threonine residues (Fig.7). Approximately the same levels of elevation in phosphorylation on these residues were also obtained after 30 min, 2 hr, and 24 hr of agonist exposure (Fig. 7). After 48 hr of incubation with the D1R agonists, the increase in MAP2 phosphorylation on both serine and threonine residues not only failed to dissipate, but became considerably more prominent than during the first 24 hr of agonist exposure (Fig. 7). The increase in phosphorylation on these two residues was still more robust after 72 hr of agonist exposure and remained at these levels through 96 hr of the exposure (Fig. 7). MAP2 phosphorylation at tyrosine residues was unchanged from basal levels within the first 24 hr of incubation with the D1R agonists. Phosphorylation on this residue, however, increased significantly between 48 and 72 hr of the agonist exposure and then remained steady through 96 hr of the exposure (Fig. 7). One possible explanation for the increase in MAP2 phosphorylation seen after 72 hr of agonist exposure is that it resulted from an altered efficacy of D1R caused by maturation of cultured cells, which by that time were in culture for 6 d. Such a possibility was investigated in 6-d-old cultures subjected to 5 min applications of SKF82958 or A77636. This produced only a moderate elevation in MAP2 phosphorylation very similar to that seen in 3-d-old cultures exposed to the agonists for the same length of time. This elevation in no way resembled the robust increase in phosphorylation of MAP2 observed after 72 hr of the agonist exposure (Fig. 7).
Fig. 7.
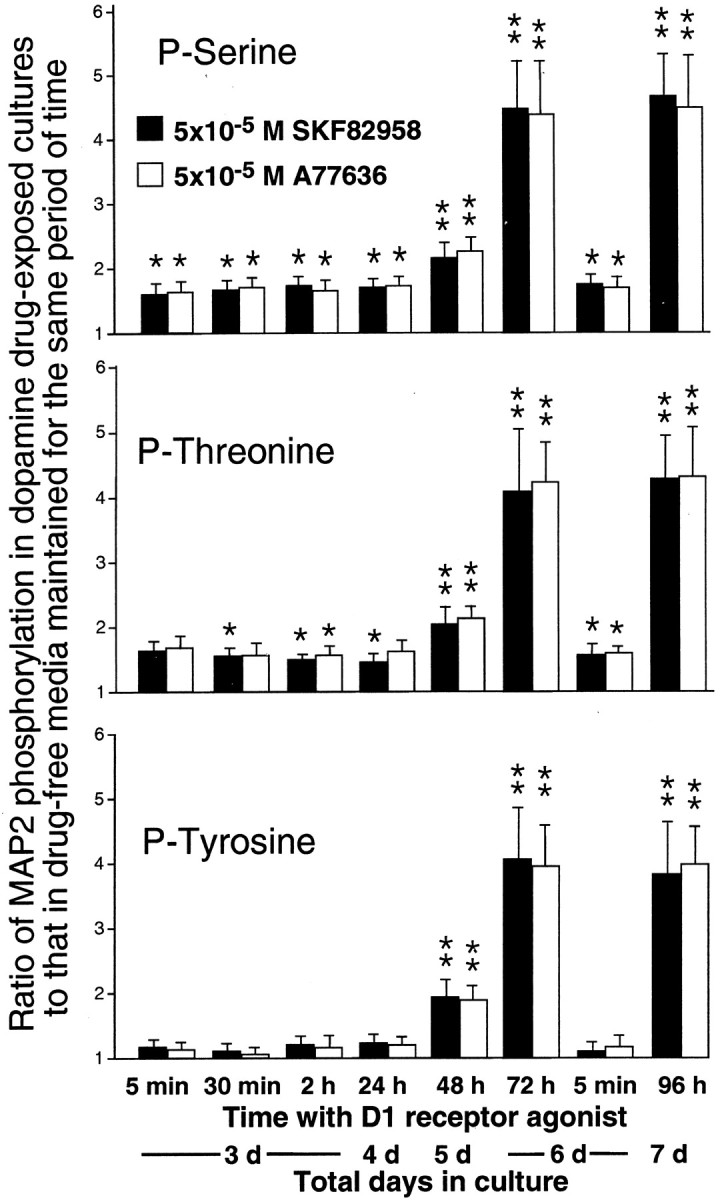
Histograms showing the time course of MAP2 phosphorylation on serine, threonine, and tyrosine residues in cultures of E15 mouse cortical neurons that after 3 d of culturing were exposed from 5 min to 96 hr to 5 × 10−5m of the D1R agonists, SKF82958 and A77636. MAP2 phosphorylation in cultures that on the sixth day were exposed to SKF82958 for 5 min are also presented. The MAP2 phosphorylation in all of these cultures was expressed as a ratio to that in cultures maintained for the same period of time without the addition of D1R ligands. All of the ratios were obtained using seven drug-exposed and seven drug-naive cultures maintained for the same period of time. Error bars are SEMs. The ratios representing statistically significant differences between experimental and corresponding control cultures are marked by asterisks (*p < 0.05; **p < 0.01; Tukey's post-test). Note that 5 min to 24 hr of exposure to D1R agonists results in a moderate increase in MAP2 phosphorylation on serine and threonine residues. Also, note further enhancements in MAP2 phosphorylation on serine and threonine residues as well as significant increases in phosphorylation on tyrosine residues after 48 hr of agonist exposure. Even larger increases in phosphorylation on all three residues are detectable after 72 and 96 hr of the exposure. Finally, note the similarity in the magnitudes of increase in MAP2 phosphorylation produced by 5 min exposure to D1R agonists in 3- and 6-d-old cultures.
To examine the role of PKA in the D1R agonist induction of MAP2 phosphorylation, 3-d-old cultures of cerebral cortical cells were exposed for 2 hr to 5 × 10−5m of either SKF82958 or A77636 in the presence or absence of 10−6m of the PKA inhibitor, H-89. The application of PKA inhibitor abolished the increase in MAP2 phosphorylation on serine and threonine residues, which was observed in the presence of SKF82958 or A77636 alone (Fig.8). H-89 had no effect on MAP2 phosphorylation at tyrosine residues (Fig. 8), which, as indicated above, was also unaltered by 2 hr exposure to the D1R agonists used in this study (Figs. 7). The PKA activator, Sp-cAMPS, significantly elevated MAP2 phosphorylation on serine and threonine residues after 2 hr of exposure and on serine, threonine, and tyrosine residues after 72 hr of exposure (Fig. 8). Furthermore, the increases in serine or threonine phosphorylation were much greater after 72 hr than after 2 hr of exposure to Sp-cAMPS (Fig. 8). Therefore, the actions of Sp-cAMPS closely mimicked those of D1R agonists.
Fig. 8.

Histograms providing the comparison of the levels of MAP2 phosphorylation on serine, threonine, and tyrosine residues between cultures of E15 mouse cortical neurons, which after 3 d of culturing were exposed for an additional 2 or 72 hr to 5 × 10−5m of the D1R agonist, A77636, and cultures exposed for the same durations either to 10−6m of the PKA activator, Sp-cAMPS, or to 5 × 10−5mA77636 combined with 10−6m of the PKA inhibitor, H-89. The latter drug combination was applied only for 2 hr, because longer exposure to H-89 led to a significant decline in cell viability. MAP2 phosphorylation in all of these cultures was expressed as a ratio to that in control cultures maintained for the same period of time without any drugs being added to the culture medium. All of the ratios were obtained using seven drug-exposed and seven drug-naive cultures maintained for the same period of time. Error bars are SEMs. The ratios representing statistically significant differences between experimental and corresponding control cultures are marked by asterisks(*p < 0.05; **p < 0.01; Tukey's post-test). Note the similarity in the effects of the D1R agonist and the PKA activator. Also note that the effects of the D1R agonist are blocked by the PKA inhibitor.
Studies in the in vivo model
Analysis of MAP2 immunoprecipitates obtained from cortical preparations of the frontal lobe revealed significant increases in phosphorylation of all three amino acid residues in homozygous D1R-overexpressing mice as compared with their wild-type littermates (Fig. 9A). MAP2 phosphorylation in heterozygous animals had intermediate values (Fig. 9A). Similar conclusions were reached when separate comparisons were conducted between members of each of the five litters examined and when the data for each genotype were pooled together before analysis. In contrast, MAP2 immunoprecipitates from the control dopamine receptor-poor occipital cortex showed no statistically significant differences in phosphorylation between wild-type animals and their D1R-overexpressing littermates (Fig. 9B).
Fig. 9.

Histograms showing the effects of D1 dopamine receptor overexpression in the cortex of 6-d-old transgenic mice on the levels of MAP2 phosphorylation on serine, threonine, and tyrosine residues. A, Cortex of the frontal lobe;B, cortex of the occipital lobe. WT, Wild-type animals;OE1/2, their heterozygous D1R-overexpressing littermates; OE2, their homozygous D1R-overexpressing littermates. In both histograms, each column represents the mean value of the data pulled from five litters of transgenic animals (n = 5), each containing two male representatives of each genotype. The error bars are SEMs. The statistically significant differences between wild-type and D1R-overexpressing littermates are marked byasterisks (p < 0.05; Tukey's post-test). Note a significant increase in MAP2 phosphorylation on all three residues in the cortex of the frontal lobe of D1R-overexpressing homozygous mice. No such significant increase in MAP2 phosphorylation is observed in the occipital cortex of same animals.
DISCUSSION
The central finding of this study is that D1Rs are capable of regulating the phosphorylation of MAP2 on all three phosphorylatable residues examined: serine, threonine, and tyrosine. Moreover, in cultures of both mouse cortical neurons and transgenic D1R-overexpressing neonatal mice, decreases in the extension of dendritic processes corresponded with increases in the levels of MAP2 phosphorylation. In particular, alterations in MAP2 phosphorylation and in neurite development in cultured cortical neurons displayed matching dose-dependence for the two D1R-specific agonists used in our investigations, with both effects reaching statistically significant levels in cultures exposed to the agonists at concentrations of 10−5m or greater. Furthermore, both the decreases in neurite extension and the increases in MAP2 phosphorylation induced by D1R agonists were inhibited by the addition of the D1R antagonist, SCH23390. There was also a remarkable similarity between the time courses of changes in MAP2 phosphorylation and in neurite extension in cultures grown in the presence of D1R agonists. For both parameters, the initial, rather modest changes were maintained for the first 24 hr. These changes, however, become more pronounced after 48 hr of agonist exposure and increased even further during the subsequent 72 hr of exposure. Longer durations of agonist exposure (up to 96 hr), however, produced no additional augmentations either in MAP2 phosphorylation or neurite extension. In addition, the application of a drug that prevented the D1R agonist-induced increase in MAP2 phosphorylation also abolished the reduction in neuritic extension caused by these agonists, whereas the application of a drug that mirrored D1R agonists in increasing MAP2 phosphorylation also resulted in a decrease in neuritic extension. Finally, the presence of both reduced dendritic length and increased MAP2 phosphorylation was seen in the cortex of the frontal lobe of D1R-overexpressing mice, which is the region where the number of D1Rs should be sufficiently high to allow them to influence neuronal development (these alterations were observed not only in 6-d-old mice described in this study, but also in adult 50-d-old animals; M. S. Lidow, unpublished observations). In contrast, no significant alterations either in dendritic length or in MAP2 phosphorylation were detected in the occipital cortex of the same animals, which is the region where the number of D1Rs should be too low to have a developmental impact. The latter observation, along with the apparent absence of any cytoarchitectonic abnormalities in the cortex of the transgenic mice, indicates that the alterations in dendritic length and MAP2 phosphorylation seen in the cortex of the frontal lobe of D1R-overexpressing animals were specific consequences of an increase in the levels of D1R in this cortical region. Taken together, our in vitro and in vivo findings support the idea that stimulation of D1R downregulates dendritic extension in developing cerebral cortical neurons, at least in part, by increasing the phosphorylation of MAP2 and thus suppressing the ability of this protein to promote the assembly and stability of the dendritic microtubular cytoskeleton (for review, see Maccioni and Cambiazo, 1995;Sanchez et al., 2000).
The present study suggests that the ability of D1Rs to increase phosphorylation of MAP2 most likely involves activation of PKA-associated intracellular signaling pathways because a PKA activator was able to replicate and a PKA inhibitor was able to block these effects of D1R agonists. Previous analyses of phosphorylation in purified MAP2 preparations suggested that PKA directly phosphorylates this protein only on serine residues (Goldenring et al., 1985; Walaas and Nairn, 1989). In our study, however, the increases in MAP2 phosphorylation were detected not only on serine but also on threonine and tyrosine residues of this protein (Tsuyama et al., 1987). We speculate that such an effect is possible because in intact cortical neurons PKA may be involved in indirect regulation of MAP2 phosphorylation on the latter two residues. For example, it has been shown that stimulation of neuronal D1R leads to a PKA-mediated activation of Mr 32 kDa phosphoprotein, DARPP-32, which acts as an inhibitor of protein phosphatase-1 (PP1) (Snyder et al., 1998). The latter phosphatase is involved in dephosphorylation of MAP2 on serine and threonine residues (Yamamoto et al., 1988; Sanchez et al., 1996). Therefore, deactivation of PP1 should lead to a net increase in MAP2 phosphorylation on these residues. PKA may also be involved in deactivation of tyrosine phosphatases (Ito et al., 1997;Paul et al., 2000), which would result in an increased level of tyrosine phosphorylation on their respective substrate proteins, including MAP2 (Hiraga et al., 1993). In addition, PKA is known to activate intracellular cascades that are associated with other kinases (Kim et al., 1997; Vossler et al., 1997; Blanco-Aparicio et al., 1999;Ambrosini et al., 2000) capable of phosphorylating MAP2 on multiple residues (for review, see Sanchez et al., 2000).
An important discovery of this study is that the increase in MAP2 phosphorylation was detectable as early as 5 min after D1R agonists were added and was maintained for as long as these agonists were present in the culture medium (up to 4 d). In addition, an increase in MAP2 phosphorylation was observed in the cortex of neonatal mice genetically manipulated to permanently overexpress the D1A subtype of D1R. These observations suggest that the changes in MAP2 phosphorylation are involved not only in short-term modulation but also in long-term control of the activity of this protein by D1Rs. Chronic alterations in MAP2 phosphorylation have previously been described in the cerebral cortex after prolonged administration of aluminum, antidepressant, and antipsychotic medications (Johnson and Jope, 1988;Miyamoto et al., 1997; Lidow et al., 2001). Thus, it is possible that long-term regulation of MAP2 phosphorylation is a common neuroregulatory mechanism used by diverse endogenous, therapeutic, and toxic agents.
The present data further show that persistent D1R stimulation increases MAP2 phosphorylation in two phases. The first phase, taking place during the first 24 hr of the stimulation, consists of a moderate increase in phosphorylation of this protein on serine and threonine residues, with no statistically significant change in tyrosine phosphorylation. The second phase consists of an additional robust increase in MAP2 phosphorylation that is evident on all three residues. We detected this delayed elevation in MAP2 phosphorylation after 48 hr of exposure to the D1R agonists used in this study and found it to be particularly notable in cultures maintained in the presence of these agonists for 72 hr or longer. The second phase of the augmentation in MAP2 phosphorylation is likely induced by the prolonged D1R stimulation rather than being related to maturation-associated changes in cell physiology. This is demonstrated by the observation that a 5 min exposure of cultured cells to D1R agonists produced elevations in MAP2 phosphorylation that were similar in magnitude in both 3- and 6-d-old cultures. Furthermore, the delayed increase in MAP2 phosphorylation (as well as the accompanying decrease in the extension of neurites) could not be easily explained by a general toxicity of our D1R-specific ligands. Such toxicity should have lead to a significant decline in cell viability, yet it remained unchanged in our cell cultures even at the highest D1R ligand concentrations and the longest exposures used in the present studies. In addition, there were no ligand exposure-associated changes in the size or density of neuronal soma in our cultures. Finally, that the delayed increase in MAP2 phosphorylation is a normal regulatory response to prolonged stimulation of D1R is suggested by the fact that the alterations in phosphorylation on tyrosine residues, which are a part of this delayed increase, are also seen in D1R-overexpressing mice.
The reasons for the existence of these two phases in the induction of MAP2 phosphorylation by D1R stimulation are presently unknown. We speculate that although the first phase likely results directly from increased activation of cAMP–PKA-associated intracellular cascades, the second phase may reflect possible recruitment of some additional mechanisms that ultimately produce an enhancement of protein phosphorylation. Such additional mechanisms could include upregulation of PKA expression and translocation of PKA from the cytosolic compartments to the cytoskeletal or membrane compartments. Both of these effects have been observed after prolonged treatment with antidepressants (Perez et al., 1991; Racagni et al., 1992; Mori et al., 1998a,b). Long-term changes in the expression and/or location of other protein kinases and phosphatases also cannot be excluded. We are presently evaluating these possibilities.
Footnotes
This study was supported by National Institutes of Health Grants MH44866 and DA08057 (M.S.L.) and a National Alliance for Research on Schizophrenia and Depression Young Investigator Award (Z.-M.S.).
Correspondence should be addressed to Dr. Michael S. Lidow, Department of Oral and Craniofacial Biological Sciences, University of Maryland, Baltimore, 5-A-12, HHH, Baltimore, MD 21201. E-mail:mlidow@umaryland.edu.
REFERENCES
- 1.Ambrosini A, Tininini S, Barassi A, Racagni G, Sturani E, Zippel R. cAMP cascade leads to Ras activation in cortical neurons. Mol Brain Res. 2000;75:54–60. doi: 10.1016/s0169-328x(99)00294-6. [DOI] [PubMed] [Google Scholar]
- 2.Audesirk G, Cabell L, Kern M. Modulation of neurite branching by protein phosphorylation in cultured rat hippocampal neurons. Dev Brain Res. 1997;102:247–260. doi: 10.1016/s0165-3806(97)00100-4. [DOI] [PubMed] [Google Scholar]
- 3.Bergson C, Mrzljak L, Smiley JF, Pappy M, Levenson R, Goldman-Rakic PS. Regional, cellular, and subcellular variations in the distribution of D1 and D5 dopamine receptors in primate brain. J Neurosci. 1995;15:7821–7836. doi: 10.1523/JNEUROSCI.15-12-07821.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Binder LI, Frankfurter A, Kim H, Cacares A, Payne MR, Rebhun LI. Heterogeneity of microtubular-associated protein 2 during rat brain development. Proc Natl Acad Sci USA. 1984;81:5613–5617. doi: 10.1073/pnas.81.17.5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blanco-Aparicio C, Torres J, Pulido R. A novel regulatory mechanism of MAP kinase activation and nuclear translocation mediated by PKA and PTP-SL tyrosine phosphatase. J Cell Biol. 1999;147:1129–1136. doi: 10.1083/jcb.147.6.1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Burgoyne RD, Cumming R. Ontogeny of microtubule-associated protein 2 in rat cerebellum: differential expression of the doublet polypeptides. Neuroscience. 1984;11:156–167. doi: 10.1016/0306-4522(84)90220-3. [DOI] [PubMed] [Google Scholar]
- 7.Daniels M. The role of microtubules in the growth and stabilization of nerve fibers. Ann NY Acad Sci. 1975;253:535–544. doi: 10.1111/j.1749-6632.1975.tb19227.x. [DOI] [PubMed] [Google Scholar]
- 8.Diez-Guerra FJ, Avila J. MAP2 phosphorylation parallels dendrite arborization in hippocampal neurons in culture. NeuroReport. 1993;4:419–422. doi: 10.1097/00001756-199304000-00020. [DOI] [PubMed] [Google Scholar]
- 9.Diez-Guerra FJ, Avila J. An increase in phosphorylation of microtubule-associated protein 2 accompanies dendrite extension during the differentiation of cultured hippocampal neurons. Eur J Biochem. 1995;227:68–77. doi: 10.1111/j.1432-1033.1995.tb20360.x. [DOI] [PubMed] [Google Scholar]
- 10.Dracheva S, Haroutunian V. Locomotor behavior of dopamine D1 receptor transgenic/D2 receptor deficient hybrid mice. Brain Res. 2001;905:142–151. doi: 10.1016/s0006-8993(01)02522-7. [DOI] [PubMed] [Google Scholar]
- 11.Dracheva S, Xu M, Kelley KA, Haroutunian V, Holstein GR, Haun S, Silverstein JH, Sealfon SC. Paradoxical locomotor behavior of dopamine D1 receptor transgenic mice. Exp Neurol. 1999;157:169–179. doi: 10.1006/exnr.1999.7037. [DOI] [PubMed] [Google Scholar]
- 12.Ertel W, Keel M, Infanger M, Ungethum U, Steckolzer U, Trenz O. Circulating mediators in serum of injured patients with septic complications inhibit neutrophil apoptosis through up-regulation of protein tyrosine phosphorylation. J Trauma. 1998;44:767–775. doi: 10.1097/00005373-199805000-00005. [DOI] [PubMed] [Google Scholar]
- 13.Foglieni C, Meoni C, Davalli M. Fluorescent dyes for cell viability: an application on prefixed conditions. Histochem Cell Biol. 2001;115:223–229. doi: 10.1007/s004180100249. [DOI] [PubMed] [Google Scholar]
- 14.Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Academic; New York: 1997. [Google Scholar]
- 15.Gaspar P, Bloch B, Le Moine C. D1 and D2 receptor gene expression in the rat frontal cortex: cellular localization in different classes of efferent neurons. Eur J Neurosci. 1995;7:1050–1063. doi: 10.1111/j.1460-9568.1995.tb01092.x. [DOI] [PubMed] [Google Scholar]
- 16.Glenney JR, Zokas L, Kamps MP. Monoclonal antibodies to phosphotyrosine. J Immunol Methods. 1988;109:277–285. doi: 10.1016/0022-1759(88)90253-0. [DOI] [PubMed] [Google Scholar]
- 17.Goldenring JR, Vallano ML, DeLorenzo RJ. Phosphorylation of microtubule-associated protein 2 at distinct sites by calmodulin-dependent and cyclic-AMP-dependent kinases. J Neurochem. 1985;45:900–905. doi: 10.1111/j.1471-4159.1985.tb04078.x. [DOI] [PubMed] [Google Scholar]
- 18.Gong C-X, Wegiel J, Lidsky T, Zuck L, Avila J, Wisnewski HM, Grundke-Iqhbal A, Iqhbal K. Regulation of phosphorylation of neuronal microtubule-associated proteins MAP1b and MAP2 by protein phosphatase-2A and –2B in rat brain. Brain Res. 2000;853:299–309. doi: 10.1016/s0006-8993(99)02294-5. [DOI] [PubMed] [Google Scholar]
- 19.Heffeth D, Fridkin M, Zick Y. Generation and use of antibodies to phosphothreonine. Methods Enzymol. 1991;201:44–53. doi: 10.1016/0076-6879(91)01007-o. [DOI] [PubMed] [Google Scholar]
- 20.Hiraga A, Hata K, Suzuki Y, Tsuiki S. Identification of a rat liver protein-tyrosine phosphatase similar to human placental PTPase 1B using quantitatively phosphorylated protein substrates. J Biochem (Tokyo) 1993;113:180–188. doi: 10.1093/oxfordjournals.jbchem.a124023. [DOI] [PubMed] [Google Scholar]
- 21.Huff RM. Signaling pathways modulated by dopamine receptors. In: Neve KA, Neve RL, editors. The dopamine receptors. Humana; Totowa, NJ: 1997. pp. 167–185. [Google Scholar]
- 22.Ito Y, Uchijma Y, Ariga M, Seki T, Takenaka A, Hakuno F, Takahashi S-I, Ariga T, Noguchi T. Interaction between cAMP-dependent and insulin-dependent signal pathways in tyrosine phosphorylation in primary cultures of rat hepatocytes. Biochem J. 1997;324:379–388. doi: 10.1042/bj3240379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnson GV, Jope RS. Phosphorylation of rat brain cytoskeletal proteins is increased after orally administered aluminium. Brain Res. 1988;456:95–103. doi: 10.1016/0006-8993(88)90350-2. [DOI] [PubMed] [Google Scholar]
- 24.Johnson GV, Jope RS. The role of microtubule-associated protein 2 (MAP-2) in neuronal growth, plasticity, and degeneration. J Neurosci Res. 1992;33:505–512. doi: 10.1002/jnr.490330402. [DOI] [PubMed] [Google Scholar]
- 25.Jones LS, Stanwood GD, Reinoso BS, Washington RA, Wang HY, Friedman E, Levitt P. In utero cocaine-induced dysfunction of dopamine D1 receptor signaling and abnormal differentiation of cerebral cortical neurons. J Neurosci. 2000;20:4606–4614. doi: 10.1523/JNEUROSCI.20-12-04606.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kadowaki T, Fujita-Yamaguchi Y, Nishida E, Takaku F, Akiyama T, Kathuria A, Akanuma Y, Kasuga M. Phosphorylation of tubulin and microtubule-associated proteins by the purified insulin receptor kinase. J Biol Chem. 1985;260:4016–4020. [PubMed] [Google Scholar]
- 27.Kebabian JW. A phosphorylation cascade in the basal ganglia of the mammalian brain: regulation by the D1 dopamine receptor. A mathematical model of known biochemical reactions. J Neural Trans [Suppl] 1997;49:145–153. doi: 10.1007/978-3-7091-6844-8_16. [DOI] [PubMed] [Google Scholar]
- 28.Keith CH. Neurite elongation is blocked if microtubule polymerization is inhibited in PC12 cells. Cell Motil Cytoskeleton. 1990;17:95–105. doi: 10.1002/cm.970170205. [DOI] [PubMed] [Google Scholar]
- 29.Kim HA, DeClue, Ratner N. cAMP-dependent protein kinase A is required for Schwann cell growth: interaction between the cAMP and neuregulin/tyrosine kinase pathways. J Neurosci Res. 1997;49:236–247. [PubMed] [Google Scholar]
- 30.Kobayashi N, Mundel P. A role of microtubules during the formation of cell processes in neuronal and non-neuronal cells. Cell Tissue Res. 1998;291:163–174. doi: 10.1007/s004410050988. [DOI] [PubMed] [Google Scholar]
- 31.Krueger KA, Bhatt H, Landt M, Easom RA. Calcium-stimulated phosphorylation of MAP-1 in pancreatic βTC3-cells is mediated by Ca2+/calmodulin-dependent kinase II. J Biol Chem. 1997;272:27464–27469. doi: 10.1074/jbc.272.43.27464. [DOI] [PubMed] [Google Scholar]
- 32.Lankford K, De Mello FG, Klein WL. A transient embryonic dopamine receptor inhibits growth cone motility and neurite outgrowth in a subset of avian retina neurons. Neurosci Lett. 1987;75:169–174. doi: 10.1016/0304-3940(87)90292-8. [DOI] [PubMed] [Google Scholar]
- 33.Leite M, Quinta-Costa M, Leite PS, Guimaraes JE. Critical evaluation of techniques to detect and measure cell death-study in a model of UV radiation on the leucaemic cell line HL60. Anal Cell Pathol. 1999;19:139–151. doi: 10.1155/1999/176515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levine L, Gjika HB, Van Vunkis H. Antibodies and radioimmunoassays for phosphoserine, phosphothreonine and phosphotyrosine. J Immunol Methods. 1989;124:239–249. doi: 10.1016/0022-1759(89)90360-8. [DOI] [PubMed] [Google Scholar]
- 35.Levitt P, Harvey JA, Friedman E, Simansky K, Murphy EH. New evidence for neurotransmitter influences on brain development. Trend Neurosci. 1997;20:269–274. doi: 10.1016/s0166-2236(96)01028-4. [DOI] [PubMed] [Google Scholar]
- 36.Lidow MS. D1 and D2 dopaminergic receptors in the developing cerebral cortex of macaque monkey: a film autoradiographic study. Neuroscience. 1995;65:439–452. doi: 10.1016/0306-4522(94)00475-k. [DOI] [PubMed] [Google Scholar]
- 37.Lidow MS, Trakht T, Howard RL. Cocaine-induced alterations in the density of monoaminergic receptors in the embryonic guinea pig cerebral wall. Synapse. 1999;32:225–237. doi: 10.1002/(SICI)1098-2396(19990601)32:3<225::AID-SYN8>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 38.Lidow MS, Song Z-M, Castner SA, Allen PB, Greengard P, Goldman-Rakic PS. Antipsychotic treatment induces alterations in dendrite- and spine-associated proteins in dopamine-rich areas of the primate cerebral cortex. Biol Psychiatry. 2001;49:1–12. doi: 10.1016/s0006-3223(00)01058-1. [DOI] [PubMed] [Google Scholar]
- 39.Lizard G, Fournel S, Genestier L, Dhedin N, Chaput C, Flacher M, Mutin M, Panaye G, Revillard JP. Kinetics of plasma membrane and mitochondrial alterations in cells undergoing apoptosis. Cytometry. 1995;45:675–683. doi: 10.1002/cyto.990210308. [DOI] [PubMed] [Google Scholar]
- 40.Maccioni RB, Cambiazo V. Role of microtubule-associated proteins in the control of microtubule assembly. Physiol Rev. 1995;75:835–864. doi: 10.1152/physrev.1995.75.4.835. [DOI] [PubMed] [Google Scholar]
- 41.Miguel-Hidalgo JJ, Baucom C, Dilley G, Overholser JC, Meltzer HY, Stockmeier CA, Rajkowska G. GFAP-immunoreactivity in the prefrontal cortex distinguishes young from old adults in major depressive disorder. Biol Psychiatry. 2000;48:860–872. doi: 10.1016/s0006-3223(00)00999-9. [DOI] [PubMed] [Google Scholar]
- 42.Miyamoto S, Asakura M, Sasuga Y, Osada K, Bodaija N, Imafuku J, Aoba A. Effects of long-term treatment with desipramine on microtubule proteins in rat cerebral cortex. Eur J Pharmacol. 1997;333:279–287. doi: 10.1016/s0014-2999(97)01140-0. [DOI] [PubMed] [Google Scholar]
- 43.Mori S, Garbini S, Caivano M, Pertez J, Racagli G. Time-course changes in rat cerebral cortex subcellular distribution of the cyclic-AMP binding after treatment with selective serotonin reuptake inhibitors. Int J Neuropsychopharmacol. 1998a;1:3–10. doi: 10.1017/S1461145798001084. [DOI] [PubMed] [Google Scholar]
- 44.Mori S, Zanardi R, Pipoli M, Garbili S, Smeraldi E, Racagni G, Perez J. cAMP-dependent phosphorylation system after short and long-term administration of moclobemide. J Psychiatr Res. 1998b;32:111–115. doi: 10.1016/S0022-3956(98)00003-X. [DOI] [PubMed] [Google Scholar]
- 45.Mosmann T. Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 46.Paul S, Snyder GL, Yakakura H, Picciotto MR, Narin AC, Lombroso PJ. The dopamine/D1 receptor mediates the phosphorylation and inactivation of the protein tyrosine phosphatase STEP via a PKA-dependent pathway. J Neurosci. 2000;20:5630–5638. doi: 10.1523/JNEUROSCI.20-15-05630.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perez J, Tinelli D, Bianchi E, Brunello N, Racagni G. cAMP binding proteins in the rat cerebral cortex after administration of selective 5-HT and NE reuptake blockers with antidepressant activity. Neuropsychopharmacology. 1991;4:57–64. [PubMed] [Google Scholar]
- 48.Racagni G, Brunello N, Tinelli D, Perez J. New biochemical hypothesis on the mechanism of action of antidepressant drug: cAMP-dependent phosphorylation system. Pharmacopsychiatry. 1992;25:51–55. doi: 10.1055/s-2007-1014388. [DOI] [PubMed] [Google Scholar]
- 49.Rakic P, Scheer U, Nover L, Hennig W. Mouse brain development. Springer; New York: 2000. [Google Scholar]
- 50.Reinoso BS, Undie AS, Levitt P. Dopamine receptors mediate differential morphological effects on cerebral cortical neurons in vitro. J Neurosci Res. 1996;43:439–453. doi: 10.1002/(SICI)1097-4547(19960215)43:4<439::AID-JNR5>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 51.Rodrigues PDS, Dowling JE. Dopamine induces neurite retraction in retinal horizontal cells via diacylglycerol and protein kinase C. Proc Natl Acad Sci USA. 1990;87:9693–9697. doi: 10.1073/pnas.87.24.9693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sanchez C, Tompa P, Szucs K, Friedrich P, Avila J. Phosphorylation and dephosphorylation in the proline-rich C-terminal domain of microtubule-associated protein 2. Eur J Biochem. 1996;241:765–771. doi: 10.1111/j.1432-1033.1996.00765.x. [DOI] [PubMed] [Google Scholar]
- 53.Sanchez C, Diaz-Nido J, Avila J. Phosphorylation of microtubule-associated protein 2 (MAP2) and its relevance for the regulation of the neuronal cytoskeleton function. Prog Neurobiol. 2000;61:133–168. doi: 10.1016/s0301-0082(99)00046-5. [DOI] [PubMed] [Google Scholar]
- 54.Schambra UB, Duncan GE, Breese GR, Fornaretto MG, Caron MG, Friemeau RT. Ontogeny of D1A and D2 dopamine receptor subtypes in rat brain using in situ hybridization and receptor binding. Neuroscience. 1994;62:65–85. doi: 10.1016/0306-4522(94)90315-8. [DOI] [PubMed] [Google Scholar]
- 55.Schmechel DE, Brighman MW, Marangos PJ. Neurons switch from non-neuronal enolase to neuron-specific enolase during differentiation. Brain Res. 1980;190:195–214. doi: 10.1016/0006-8993(80)91169-5. [DOI] [PubMed] [Google Scholar]
- 56.Schmidt U, Beyer C, Oestereicher AB, Reisert I, Schilling K, Pilgrim C. Activation of dopamine D1 receptors promotes morphogenesis of developing striatal neurons. Neuroscience. 1996;74:453–460. doi: 10.1016/0306-4522(96)00201-1. [DOI] [PubMed] [Google Scholar]
- 57.Selemon LD, Lidow MS, Goldman-Rakic PS. Increased volume and glial density in primate prefrontal cortex associated with chronic antipsychotic drug exposure. Biol Psychiatry. 1999;46:161–172. doi: 10.1016/s0006-3223(99)00113-4. [DOI] [PubMed] [Google Scholar]
- 58.Shea TB, Beermann ML. Respective roles of neurofilaments, microtubules, MAP1B, and tau in neurite outgrowth and stabilization. Mol Cell Biol. 1994;5:863–875. doi: 10.1091/mbc.5.8.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Snyder GL, Fienberg AA, Huganir RL, Greengard P. A dopamine/D1 receptor/protein kinase A/dopamine- and cAMP-regulated phosphoprotein (Mr 32 kDa)/protein phosphatase-1 pathway regulates dephosphorylation of the NMDA receptor. J Neurosci. 1998;18:10297–10303. doi: 10.1523/JNEUROSCI.18-24-10297.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tsuyama S, Terayama Y, Mastsuyama S. Numerous phosphates of microtubule-associated protein 2 in living rat brain. J Biol Chem. 1987;262:10886–10892. [PubMed] [Google Scholar]
- 61.Undie AS, Friedman E. Stimulation of a dopamine D1 receptor enhances inositol phosphate formation in rat brain. J Pharmacol Exp Ther. 1990;253:987–992. [PubMed] [Google Scholar]
- 62.Uylings HBM, Ruiz-Marcos A, van Pelt J. The metric analysis of three-dimensional dendritic tree patterns: a methodological review. J Neurosci Methods. 1986;18:127–151. doi: 10.1016/0165-0270(86)90116-0. [DOI] [PubMed] [Google Scholar]
- 63.Vaccarino FM, Schwartz ML, Hartigan D, Leckman JF. Basic fibroblast growth factor increases the number of excitatory neurons containing glutamate in the cerebral cortex. Cereb Cortex. 1995;5:64–78. doi: 10.1093/cercor/5.1.64. [DOI] [PubMed] [Google Scholar]
- 64.Vossler MR, Yao H, York RD, Pan MG, Rim CS, Stork PJ. cAMP activates MAP kinase and Elk-1 through B-Raf- and Rap1-dependent pathway. Cell. 1997;89:73–82. doi: 10.1016/s0092-8674(00)80184-1. [DOI] [PubMed] [Google Scholar]
- 65.Walaas SI, Nairn AC (1989) Multisite phosphorylation of microtubule-associated protein 2 (MAP2) in rat brain: peptide mapping distinguishes between cyclic AMP-, calcium/calmodulin-, and calcium/phospholipid-regulated phosphorylation mechanisms. J Mol Neurosci 1117–1127. [DOI] [PubMed]
- 66.Wang F, Lidow MS. Alpha 2A-adrenergic receptors are expressed by diverse cell types in the fetal primate cerebral wall. J Comp Neurol. 1997;378:493–507. [PubMed] [Google Scholar]
- 67.Wang H-Y, Undie AS, Friedman E. Evidence for the coupling of Gq protein to D1-like dopamine sites in rat striatum: possible role in dopamine-mediated inositol phosphate formation. Mol Pharmacol. 1995;48:988–994. [PubMed] [Google Scholar]
- 68.Wang K, Fanger BO, Guyer CA, Staros JV. Electrophoretic transfer of high-molecular-weight proteins for immunostaining. Methods Enzymol. 1989;172:687–696. doi: 10.1016/s0076-6879(89)72038-3. [DOI] [PubMed] [Google Scholar]
- 69.Wiche G, Oberkanins C, Himmler A. Molecular structure and function of microtubule-associated proteins. Int Rev Cytol. 1991;124:217–237. doi: 10.1016/s0074-7696(08)61528-4. [DOI] [PubMed] [Google Scholar]
- 70.Yamada KM, Spooner BS, Wessells NK. Axon growth: roles of microfilaments and microtubules. Proc Natl Acad Sci USA. 1970;66:1206–1212. doi: 10.1073/pnas.66.4.1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yamamoto H, Saitoh Y, Fukunaga K, Nishimura H, Miyamoto E. Dephosphorylation of microtubule proteins by brain protein phosphatases 1 and 2A, and its effect on microtubule assembly. J Neurochem. 1988;50:1614–1623. doi: 10.1111/j.1471-4159.1988.tb03051.x. [DOI] [PubMed] [Google Scholar]
- 72.Zachor DA, Moore JF, Bezausek C, Theibert A, Percy AK. Cocaine inhibits NGF-induced PC12 cells differentiation through D(1)-type dopamine receptors. Brain Res. 2000;869:85–97. doi: 10.1016/s0006-8993(00)02355-6. [DOI] [PubMed] [Google Scholar]