

Abstract
The GABAB receptor plays important roles in the tuning of many synapses. Although pharmacological differences have been observed between various GABAB-mediated effects, a single GABAB receptor composed of two subunits (GB1 and GB2) has been identified. Although GB1 binds GABA, GB2 plays a critical role in G-protein activation. Moreover, GB2 is required for the high agonist affinity of GB1. Like any other family 3 G-protein-coupled receptors, GB1 and GB2 are composed of a Venus Flytrap module (VFTM) that usually contains the agonist-binding site and a heptahelical domain. So far, there has been no direct demonstration that GB2 binds GABA or another endogenous ligand. Here, we have further refined the GABA-binding site of GB1 and characterized the putative-binding site in the VFTM of GB2. None of the residues important for GABA binding in GB1 appeared to be conserved in GB2. Moreover, mutation of 10 different residues, alone or in combination, within the possible binding pocket of GB2 affects neither GABA activation of the receptor nor the ability of GB2 to increase agonist affinity on GB1. These data indicate that ligand binding in the GB2 VFTM is not required for activation. Finally, although in either GB1 or the related metabotropic glutamate receptors most residues of the binding pocket are conserved fromCaenorhabditis elegans to human, no such conservation is observed in GB2. This suggests that the GB2 VFTM does not constitute a binding site for a natural ligand.
Keywords: ligand recognition, evolution, three-dimensional modeling, dimerization, GPCR, allostery, baclofen
In addition to the ligand-gated channels, GABA also activates a G-protein-coupled receptor (GPCR), the GABAB receptor (Bettler et al., 1998; Couve et al., 2000). This receptor is found on either presynaptic or postsynaptic elements in various types of synapses. The GABAB receptor is the target of the antispastic drug baclofen and may also be involved in various types of epilepsy, as well as in nociception and drug addiction (Couve et al., 2000).
In contrast to many other GPCRs, the GABABreceptor requires two distinct subunits, GB1 and GB2, to activate G-proteins (Marshall et al., 1999). GB1 has been shown to bind all known GABAB ligands but with a 100-fold lower affinity for agonists compared with the native receptor (Kaupmann et al., 1997). Moreover, GB1 does not reach the cell surface alone, because of an intracellular retention signal (IRS) in its C-terminal tail (Margeta-Mitrovic et al., 2000; Calver et al., 2001; Pagano et al., 2001). Even when the IRS is mutated such that GB1 reaches the cell surface, GB1 is still unable to activate G-proteins. Several roles of GB2 have been identified. First, GB2 masks the IRS of GB1, such that the heteromer GB1+GB2 reaches the cell surface. Second, GB2 increases the agonist affinity on GB1 (Kaupmann et al., 1998; Galvez et al., 2001). Third, GB2 contains all the determinants required for G-protein coupling and plays a pivotal role in G-protein activation by the heteromer (Galvez et al., 2001; Margeta-Mitrovic et al., 2001b; Robbins et al., 2001; Duthey et al., 2002; Havlickova et al., 2002). However, although GABAB ligands have been demonstrated to bind on GB1, their possible interaction on GB2 remains elusive (Kaupmann et al., 1998; Martin et al., 1999; Galvez et al., 2000a). Indeed, binding of GABA or another endogenous compound on GB2 may be required for the increased GABA affinity on GB1 observed in the heteromer. Moreover, both GB1 and GB2 share sequence similarity with the other family 3 GPCRs: both subunits are composed of a heptahelical domain (HD) and large extracellular domain [the so-called Venus Flytrap module (VFTM)], which is responsible for agonist binding in both the metabotropic glutamate (mGlu) receptors (Kunishima et al., 2000; Tsuchiya et al., 2002) and GB1 (Galvez et al., 1999, 2000a).
The GB1+GB2 heteromer appears to be the only GABAB receptor in the brain, because the knock-out of the GB1 subunit is enough to suppress all studied GABAB-mediated effects (Prosser et al., 2001;Schuler et al., 2001). However, pharmacological studies suggest the existence of different GABAB receptor subtypes (Bonanno and Raiteri, 1993; Kerr and Ong, 1995), observations that still cannot be explained by the presence of various GB1 and GB2 splice variants (Billinton et al., 2001). Ligand binding in GB2 may possibly affect the pharmacological profile of the GABABreceptor.
In the present study, we refined the analysis of GABA binding on GB1 and used this information to examine whether or not ligand binding in the GB2 VFTM would be required for GABAB receptor activation or modulation.
MATERIALS AND METHODS
Materials. GABA, baclofen, 3-aminopropylphosphinic acid (APPA), 3-{1-(R)-[2-(S)-hydroxy-3-(hydroxy-{5-[3-(4-hydroxy-3-iodo-phenyl)-propionylamino]-pentyl}-phosphinoyl)-propylamino]-ethyl}-benzoic acid (CGP64213), and [125I]CGP64213 were gifts from Drs. W. Froestl and K. Kaupman (Novartis Pharma, Basel, Switzerland). Fetal bovine serum, culture media, and other solutions used for cell culture were from Invitrogen (Cergy Pontoise, France).myo-[3H]inositol (23.4 Ci/mol) was purchased from PerkinElmer Life Sciences (NEN) (Paris, France). All other reagents used were of molecular or analytical grade where appropriate.
Sequence comparison and molecular modeling. Protein sequence database searches were performed with the Position-Specific Iterated–Basic Local Alignment Search Tool program, version 2.0.5 (Altschul et al., 1997) with default parameters. Alignment refinement was subsequently performed using the Tool for Incremental Threading Optimization (TITO) program (Labesse and Mornon, 1998) using various templates: pdb codes: 2liv, 1ewk, 1dp4, 1jdn, or 1pea. The validity of the refined alignment was assessed through pseudo-energy and visual inspection. The secondary structures (α-helix and β-strand) of GB1 and GB2 VFTMs were predicted using Jpred2 (Cuff et al., 1998) and were also deduced by similarity during TITO processing. These secondary structure predictions were merged by consensus and used as additional restraints in the following modeling steps. Three-dimensional models were built using the 1ewk:A as a template in Modeler 6.0 α (Sali and Blundell, 1993) and assessed using PROSA (Sippl, 1993), ERRAT (Colovos and Yeates, 1993), and Verify3D (Eisenberg et al., 1997). These three-dimensional structures were visualized on a UNIX workstation using XmMol (Tuffery, 1995). Figures were prepared using the SwissPdbViewer program (version 3.7) (Guex and Peitsch, 1997).
Plasmids and site-directed mutagenesis. The plasmids encoding the wild-type GB1a and GB2 subunits epitope tagged with the hemagglutinin (HA) at their N-terminal ends [pRK-GB1a-XXX (HA) and pRK-GB2-HA], under the control of a cytomegalovirus promoter, were described previously (Galvez et al., 2001; Pagano et al., 2001).
Mutant subunits, carrying single or multiple mutations, were obtained using the Quick-Change strategy (Stratagene, La Jolla, CA). Briefly, cDNA fragments encoding part of wild-type GB1a (EcoRI–XbaI) and GB2 (EcoRI–BamHI) were cloned into the pBluescript SK(−) vector. For each mutagenesis, two complementary 27 mer primers (Genaxis Biotechnologie, Nîmes, France) were designed to contain the desired mutation. To allow a rapid screening of mutated clones, primers carried an additional silent mutation introducing (or removing) a restriction site. The presence of the desired mutations and the absence of additional ones were confirmed by DNA sequencing. For multiple mutants, several Quick-Change reactions were performed successively. Finally, short fragments surrounding the mutations were subcloned in the place of corresponding wild-type fragments of pRK-GB1a-HA (PshAI–BamHI) or pRK-GB2-HA (EcoRI–BamHI).
Cell culture and expression in human embryonic kidney 293.Human embryonic kidney (HEK) 293 cells were cultured and transfected by electroporation as described previously using 2 μg of plasmid DNA containing wild-type or mutated receptor for 10 × 106 cells, unless otherwise indicated. In the case of the mutants GB2-F208A and D256W and those bearing the D256Y mutation, which were expressed at a low level, 4 μg of plasmid DNA were used. For all functional studies, both GB1 and GB2 constructs were transfected together with the chimeric Gαqi9 G-protein. The latter G-protein allows the coupling of the recombinant heteromeric GABAB receptor to phospholipase C.
Western blotting. After transfection (48 hr), cells were washed with ice-cold PBS and scraped in lysis buffer (Tris–EDTA). Membranes were then pelleted and solubilized in buffer containing 0.1% SDS and 2% Triton X-100 at a final concentration of 10 μg/μl. Membrane (10 μg) was loaded on a 7.5% tricine–SDS–polyacrylamide gel and transferred on a nitrocellulose membrane. After overnight incubation in Tris-buffered saline-Tween 20 (TBST: 10 mm Tris, 150 mm NaCl, and 0.1% Tween 20)–milk 5%, the membranes were incubated for 2 hr at room temperature with monoclonal anti-HA antibody (1/3000 in TBST–milk 2%). After washing, the membranes were incubated for 1 hr at room temperature with the anti-mouse HRP antibody (1/5000). The signal was revealed using an enhanced chemiluminescence assay.
Immunohistochemistry. Twenty-four hours after transfection, HEK 293 cells were plated onto glass coverslips, washed twice with PBS, and incubated for 90 min at 37°C with monoclonal mouse 12CA5 at 1.3 μg/ml in PBS/gelatin (0.2%), as described previously (Ango et al., 1999). For detection, Cy3 secondary antibody (Jackson ImmunoResearch, West Grove, PA) was used at 1:1000. Coverslips were mounted and observed on an upright Axiophot 2 microscope (Zeiss, Thornwood, NY).
Ligand-binding assay. A ligand-binding assay on intact HEK 293 cells was performed as described previously using 0.1 nm [125I]CGP64213 (Galvez et al., 2001). Displacement curves were performed with at least seven different concentrations of the displacer, and the curves were fitted according to the following equation: y = (ymax −ymin)/[1 + (x/IC50)nH] + ymin, where the IC50 is the concentration of the compound that inhibits 50% of bound radioligand and nH is the Hill coefficient.Ki values were calculated according to the equation IC50 =Ki[1 + (RL)/Kd], where RL andKd are the concentration and dissociation constant of the radioligand.Kd was determined assumingKi =Kd in the case of CGP64213.
Determination of inositol phosphate accumulation.Determination of inositol phosphate (IP) accumulation in transfected cells was performed after labeling the cells overnight withmyo-[3H]inositol (23.4 Ci/mol) as described previously (Brabet et al., 1998; Blahos et al., 2001). Curves were fitted with Kaleidagraph software using the equationy = (ymax −ymin)/[1 + (x/EC50)nH] + ymin, where the EC50 is the concentration of the compound necessary to obtain 50% of the maximal effect and nH is the Hill coefficient.
Construction of the evolutionary trees. The sequences of the VFTMs of GB1 from human (Swissprot accession number Q9UBS5), rat (Swissprot accession number Q9Z0U4), mouse (Swissprot accession number Q9WV18), Drosophila melanogaster (GenBank accession numberAAK13420), and Caenorhabditis elegans (from the cosmid Y41G9A) and those of the GB2 VFTMs from human (Swissprot accession number O75899), rat (Swissprot accession number O88871), D. melanogaster (GenBank accession number AAK13421), and C. elegans (from the cosmid ZK180) were aligned using Clustal W (version 1.6) (Thompson et al., 1994) with the default parameters (Gap open penalty, 10; Gap extension penalty, 0.1; protein weight matrix Blosum30). A multiple alignment of all mGlu receptor VFTMs was also generated using the same procedure. These two multiple alignments were then aligned according to the alignment presented in Figure1. The phylogenetic tree was then constructed using the neighbor-joining method (Saitou and Nei, 1987) with the command interface of the Clustal W program. The positions with gaps were excluded, and only the GB1, GB2, and mGlu receptor group-II sequences were taken into account. The tree was then visualized using TreeView (version 1.6.2) (Page, 1996). For the analysis of the binding pocket, only positions of residues aligning with those that are at a distance inferior to 7 Å of the bound ligand, glutamate, in the closed form of the mGlu1 VFTM (pdb code: 1ewk:A) were taken into account in the alignment used to calculate the tree.
Fig. 1.

Alignment of the GB1 and GB2 VFTMs with those of mGlu1 (1EWK), LIVBP (2LIV), AmiC (1PEA), NPRA (1DP4), and NPRC (1JDN). Highlighted in black andgray are the α helices and β strand secondary elements, respectively, as observed in the crystal structures (1EWK, 2LIV, 1PEA,1DP4, and 1JDN) or as predicted according to Jpred2 (GB1 andGB2). Highlighted in red are residues that directly contact the ligand in mGlu1, LIVBP, and AmiC, as well as those we propose to contact GABA in GB1. Highlighted inyellow are the cysteines involved (or proposed to be for the GB subunits) in intramolecular disulfide bonds. Inblue are residues involved in a network of interaction within lobe-I.
RESULTS
Functional studies suggested that GB2 may be activated by GABA and baclofen (Kaupmann et al., 1998; Martin et al., 1999). However, binding studies with radiolabeled GABAB ligands revealed no significant binding on the GB2 subunit expressed alone (Jones et al., 1998; Kaupmann et al., 1998; White et al., 1998). This does not exclude a possible interaction of GABAB ligands on GB2. Indeed, this may simply result from a low affinity of these radioligands on GB2, as observed with GB1, on which no [3H]GABA or [3H]APPA binding could be detected, although GABA and APPA displaced bound [125I]CGP64213 (Kaupmann et al., 1997). A comparison of the putative-binding site within the cleft that separates both lobes of the GB2 VFTM with that of GB1 may help unravel this important issue.
Several studies have examined in detail the agonist-binding domain of GB1. Although all three-dimensional models described a bilobate VFTM-like structure with GABA interacting primarily with lobe-I, they differ in the region contacting the amino group of GABA. Indeed, some authors proposed that Asp471 plays such a role (Galvez et al., 2000a;Costantino et al., 2001), whereas others involved Glu465 (Couve et al., 2000; Bernard et al., 2001). These models were all built using the structure of the leucine/isoleucine/valine-binding protein (LIVBP) and the negative regulator of amidase operon (AmiC) as the only templates. Since then, the structures of several other proteins with a similar threefold dimension have been solved by x-ray crystallography. These include the VFTM of the mGlu1 receptor with and without bound glutamate (Kunishima et al., 2000; Tsuchiya et al., 2002) and those of natriuretic peptide receptor A (NPRA) (van den Akker et al., 2000) and NPRC (He et al., 2001). We then first aimed at refining the GABA-binding site of GB1 using these new data.
Three-dimensional modeling and identification of the GABA-binding site of GB1
A structural alignment of the VFTM of LIVBP (pdb code: 2liv), AmiC (pdb code: 1pea), mGlu1 (pdb code: 1ewk), NPRA (pdb code: 1dp4), and NPRC (pdb code: 1jdn) was deduced from the superposition of their structure. The sequence of the GB1 VFTM was then aligned on top of this alignment as described in Materials and Methods (Fig. 1), and three-dimensional models were generated using Modeler (see Materials and Methods). The best model was selected according to the energy of constraint violation in Modeler, the pseudo-energy computed by PROSA II (Sippl, 1993), as well as the score ERRAT (Colovos and Yeates, 1993) and Verify3D (Eisenberg et al., 1997). The main error concentrates in a large insertion/deletion (between the βI and βJ strands) (Fig. 1) outside the ligand-binding site, whereas the remains of the structure have pseudo-energy below zero according to PROSA II. The mean pseudo-energy was −0.7, whereas the ERRAT score reached 62% for a monomeric GB1. Although confirming the conservation of the overall fold, as well as the carboxylic function of GABA making H-bonds with the hydroxyl of both Ser246 (lobe-I) and Tyr366 (lobe-II) (Galvez et al., 1999, 2000a), the current model refines some conclusions drawn on the previous models.
In this new model, the acidic moiety of Asp471 of GB1 is not part of the GABA-binding site but rather is involved in a network of H-bonds stabilizing lobe-I (Fig. 2a). This Asp side-chain points toward a β-hairpin also observed in the NPRs, mGlu1, and bacterial VFTMs LIVBP and AmiC. The β-hairpin is primarily stabilized by hydrogen bonding to the Asp side-chain (or Asn in mGlu1) in the receptor structures. Analysis of the neighboring residues in the known three-dimensional structures revealed a hydrogen network leading from this Asp to a buried Arg (Arg284 in GB1) lying below a ligand-binding loop (Fig. 2a) via another Arg (532 in GB1), a Thr, and a Ser (285 and 531 in GB1, respectively). The side-chain of Arg284 interacts with two backbone carbonyls, stabilizing the conformation of the loop-bearing residues (Gly267, Ser269, and Ser270) that interact with the ligand via their backbone groups (Fig.2). This Arg, as well as the other residues involved in this network, are well conserved in the previous structures (see the blue residues in Fig. 1). This globally conserved interaction network might be essential for the correct folding of the VFTM, explaining the absence of ligand binding and G-protein coupling of a GB1-D471A+GB2 heteromeric receptor (Galvez et al., 2000a).
Fig. 2.
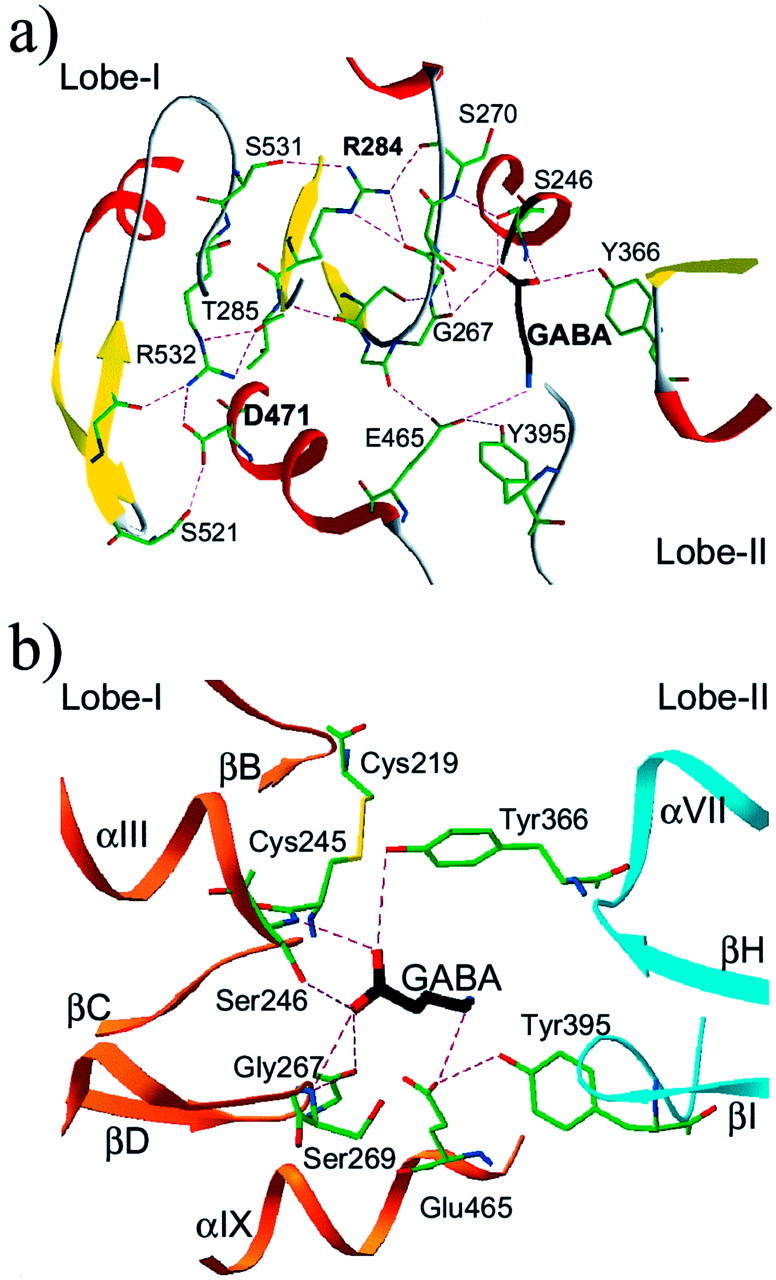
Details of the three-dimensional model of the GB1 VFTM. A, View of the hydrogen network leading from the common Asp (Asp471 in GB1) to a buried Arg (Arg284 in GB1) lying below the ligand-binding loop βD–βE. As depicted in Figure 1, most residues that are part of this network are conserved in many VFTMs and play a similar role. The β strands are in yellow, and the α helices are in red. H bonds are indicated bydashed magenta lines. B, Detailed view of the GB1-binding pocket with bound GABA. The orange andcyan ribbons correspond to lobe-I and lobe-II. The nomenclature used for the secondary structural elements is as shown in Figure 1.
Other residues (Tyr395, Asp397, Asn398, and Glu465) were predicted to be part of the ligand-binding site, and some side-chain conformations of both Tyr395 and Glu465 allow close contacts with the ligand (primarily hydrogen bonding). The N group of GABA can be modeled at contact distance of the side-chains of Glu465 from lobe-I and Tyr395 from lobe-II (Fig. 2b). To validate these possibilities, new GB1 mutants were generated, in which Tyr395, Asp397, and Asn398 were mutated into Ala, and Glu465 was replaced by either Ala, Ser, or Asp. The binding and functional properties of these mutants were then analyzed and compared with those of the wild-type and S246A mutant GB1 coexpressed with GB2.
Functional analysis of GB1 mutants
As shown in Table 1, and in agreement with our modeling studies, [125I]CGP64213 binding on intact cells was not affected by the D397A and N398A mutations, andKi values for GABA were similar to those measured on the wild-type receptor. These results are in agreement with the modeling of these two residues at the edge of the ligand-binding cleft with their side-chains pointing toward the solvent. In contrast, the mutants S246A, Y395A, E465A, E465D, and E465S no longer bind [125I]CGP64213, although they are correctly expressed as shown by Western blotting (Fig.3a) and targeted to the cell surface in the presence of GB2, as shown by immunolabeling of intact cells (Fig. 3b and data not shown). This is consistent with these three residues playing an important role in CGP64213 binding.
Table 1.
Effect of mutations of various residues in the GB1 VFTM on the binding and functional properties of the heteromeric GABAB receptor
| [125I]CGP64213 binding % | CGP64213 Ki nm | GABAKi μm | GABA EC50μm | |
|---|---|---|---|---|
| Wild type | 100 | 2.8 ± 0.6 | 3.7 ± 0.4 | 0.36 ± 0.05 |
| S246A | N.B.1-a | 261 ± 47 | ||
| Y395A | N.B. | 2.60 ± 0.43 | ||
| D397A | 74 ± 11 | 0.49 ± 0.15 | 2.9 ± 0.7 | 0.73 ± 0.34 |
| N398A | 71 ± 10 | 0.88 ± 0.39 | 4.0 ± 0.3 | 0.72 ± 0.04 |
| E465A | N.B. | 66 ± 18 | ||
| E465D | N.B. | 0.19 ± 0.04 | ||
| E465S | N.B. | 49 ± 3 |
Specific [125I]CGP64213 binding was measured on intact cells expressing the indicated GB1 mutant cotransfected with the wild-type GB2 and is expressed as a percentage of that measured on cells expressing the wild-type subunits. Kivalues for GABA and CGP64213 and EC50 values for GABA were determined as described in Materials and Methods. Values are means ± SEM of at least three independent experiments performed in triplicate.
N.B., No significant specific binding.
Fig. 3.

Expression and plasma membrane insertion of the wild-type and mutated GB1 and GB2 subunits. a, Immunoblot obtained with membrane proteins prepared from cells expressing the wild-type GB1 (WT) or the E465A, E465D, E465S, Y395A, D397A, or N398A GB1 mutants and labeled with the HA antibody. b, Immunolabeling with the HA antibody of intact cells [or cells permeabilized with 0.05% Triton X-100 (perm)] expressing the indicated subunits. Because the HA epitope is fused at the N-terminal extracellular end of the subunits, labeling of intact cells is indicative of a plasma membrane insertion of the subunit. Note that GB1-HA can be detected in permeabilized cells only when expressed alone and in intact cells when coexpressed with GB2.
The possible activation of these mutant receptors by GABA was also analyzed by measuring IP formation after coexpression with the wild-type GB2 and the chimeric G-protein Gαqi9. As shown in Table 1 and Figure 4, the GB1-D397A, N398A, and E465D behave like the wild-type receptor. However, an increase in the EC50 value for GABA by a factor of 10, 100, and 1000 was observed with the Y395A, E465A, E465S, and S246A mutants, respectively. It is interesting to note that the E465D mutant led to a functional receptor that can be activated by GABA, although it did not bind [125I]CGP64213, suggesting that this Glu is important for the proper binding of this antagonist but not for GABA. Together, these data are consistent with the proposed model for GABA binding in GB1 (Fig. 2b). The better characterization of the ligand-binding site of GB1 was in turn used to analyze the equivalent region in GB2.
Fig. 4.

S246 and E465 of GB1 are critical for agonist potency at the heteromeric GABAB receptor. The effect of increasing concentrations of GABA on IP formation in cells coexpressing the wild-type (WT; ●), S246A (○), E465A (■), E465D (▵), or E465S (▴) GB1 subunit with the wild-type GB2 and Gαqi9 is shown. Data are expressed as the IP production over the radioactivity remaining in the membrane and are means ± SEM of triplicate determinations from a typical experiment.
Characterization and functional importance of the putative ligand-binding site of GB2
Based on the alignment of GB2 and GB1 VFTM sequences and of the above described structural alignment (Fig. 1), a three-dimensional model for the GB2 VFTM was generated (Fig.5). This model satisfies both statistical (Verify 3D) and energetic (PROSA II) criteria for a correctly folded protein. Interestingly, among the three residues identified in GB1 that likely interact with GABA (Ser246, Tyr366, and Glu465), none are conserved in GB2 (the homologous residues being Pro136, Asp256, and Phe354, respectively). Moreover, the residue Ser269 of GB1 that has been shown to be responsible for the increased GABA affinity in the presence of Ca2+ (Galvez et al., 2000b;Costantino et al., 2001) is replaced by a Thr (Thr159) in GB2.
Fig. 5.
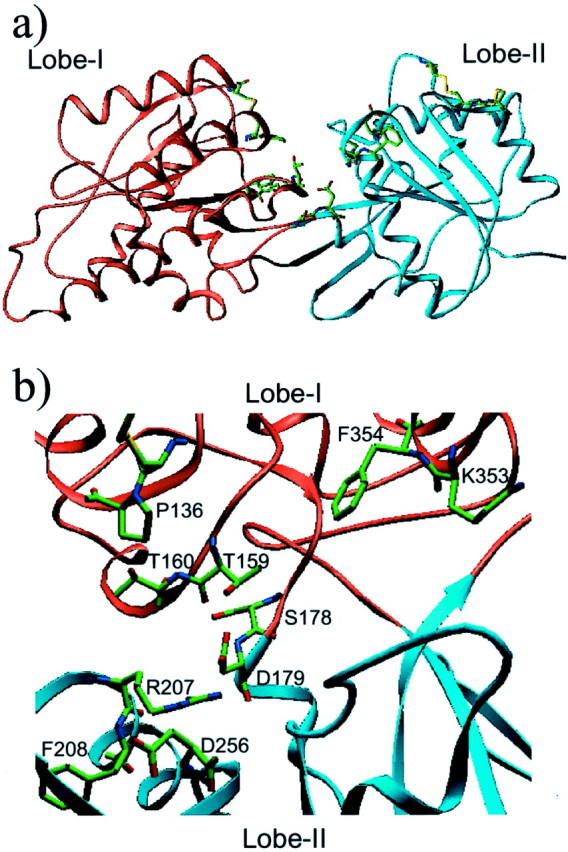
Three-dimensional model of the GB2 VFTM. Lobe-I is in orange, and lobe-II is in cyan. Indicated are residues that have been mutated in the present study and the Cys residues involved in putative intramolecular disulfide bonds.
To examine the possible importance of this putative-binding site of GB2, the residues homologous to those responsible for GABA binding in GB1 were mutated into residues with very different properties (Table 2). For example, the acidic residue Asp256 was replaced either by the neutral residue Ala or the aromatic residues Tyr or Trp, and Phe354 was replaced by a Glu. Additional residues that are part of the binding pocket and that may possibly form an H-bond or ionic interaction with a ligand were also mutated into Ala (Thr160, Ser178, Asp179, Arg207, Phe208, and Lys353). GB2 receptor mutants simultaneously carrying two to five of these mutations were also generated, in such a way that the putative GB2-binding site is highly modified.
Table 2.
Effect of mutations of various residues in the GB2 VFTM on the binding and functional properties of the heteromeric GABAB receptor
| [125I]CGP64213 binding % | CGP64213Ki nm | GABAKi μm | GABA EC50μm | |
|---|---|---|---|---|
| WT | 100 | 2.39 ± 0.55 | 3.64 ± 0.35 | 0.35 ± 0.05 |
| 1ASA | 102 ± 8 | 2.70 ± 0.67 | 17.20 ± 3.48 | N.E. |
| Binding site residues | ||||
| P136S | 90 ± 17 | 0.8 ± 0.3 | 2.3 ± 0.8 | 0.45 ± 0.10 |
| T159A | 63 ± 17 | 3.0 ± 0.4 | 3.1 ± 0.8 | 0.29 ± 0.21 |
| T160A | 85 ± 7 | 0.8 ± 0.1 | 3.1 ± 0.9 | 0.28 ± 0.12 |
| D256A | 71 ± 8 | 2.5 ± 0.3 | 3.3 ± 0.7 | 0.51 ± 0.04 |
| D256E | 85 ± 16 | 1.7 ± 0.5 | 4.2 ± 0.1 | 0.63 ± 0.13 |
| D256W | 12 ± 3 | 1.8 ± 0.3 | 2.1 ± 0.3 | 11.70 ± 2.35 |
| D256Y | 23 ± 7 | 3.2 ± 1.0 | 3.4 ± 0.2 | 4.54 ± 0.04 |
| K353A | 57 ± 5 | 2.6 ± 1.3 | 1.9 ± 0.1 | 0.57 ± 0.10 |
| F354E | 69 ± 7 | 1.6 ± 0.2 | 3.2 ± 1.1 | 0.48 ± 0.10 |
| Multiple mutants | ||||
| P136S-D256Y | 13 ± 2 | 3.9 ± 1.1 | 2.0 ± 0.6 | 6.68 ± 0.62 |
| T159S-T160S | 75 ± 5 | 1.9 ± 0.1 | 2.0 ± 0.1 | 0.26 ± 0.00 |
| P136S-T159S-T160S-D256Y | 13 ± 1 | 2.6 ± 0.6 | 2.1 ± 0.9 | 4.90 ± 1.92 |
| P136S-T159S-T160S-D256Y-F354E | 9 ± 1 | 1.9 ± 1.1 | 3.7 ± 0.5 | 26.7 ± 3.5 |
| Conserved residues | ||||
| S178A | 73 ± 3 | 2.2 ± 0.4 | 4.6 ± 2.0 | 0.60 ± 0.21 |
| D179A | 92 ± 12 | 2.9 ± 0.6 | 4.2 ± 0.1 | 0.32 ± 0.12 |
| R207A | 79 ± 9 | 2.7 ± 0.3 | 3.8 ± 0.8 | 0.63 ± 0.18 |
| F208A | 22 ± 4 | 2.7 ± 0.4 | 4.9 ± 0.3 | 1.74 ± 0.31 |
| GB2 equivalent of D471 | ||||
| D360A | N.B. | N.E. |
Specific [125I]CGP64213 binding was measured on intact cells expressing the indicated GB2 mutant cotransfected with the wild-type GB1 and is expressed as a percentage of that measured on cells expressing the wild-type subunits. Kivalues for GABA and CGP64213 and EC50 values for GABA were determined as described in Materials and Methods. Values are means ± SEM of at least three independent experiments performed in triplicate. N.B., No significant specific binding; N.E., no significant effect.
When coexpressed with GB1, all of these mutant GB2 subunits were able to target GB1 to the cell surface, as demonstrated by the significant [125I]CGP64213 binding measured on intact cells (Table 2). Moreover, all GB2 mutants were still able to increase GABA affinity like the wild-type GB2, indicating that none of these mutations affect the allosteric effect of GB2 on GB1. Finally, all GB2 mutants formed a functional GABABreceptor when coexpressed with GB1. However, a higher EC50 value for GABA (Table 2), as well as for the other agonists, baclofen or APPA (data not shown), was observed with the GB2-F208A, D256Y, D256W, and multiple mutant receptors bearing the D256Y mutation. Indeed, the higher the agonist EC50 value, the lower the [125I]CGP64213 binding measured on intact cells (Table 2). Was this decrease in agonist potency caused by the lower level of expression only, or does it result from a lower efficacy of GABA to activate the GB2 HD? To answer this question, GABA potency on the wild-type receptor was measured on cells expressing various levels of the GB1+GB2 heteromer. As shown in Figure6, a 10-fold decrease in CGP64213 binding results in a threefold to fourfold increase in the GABA EC50 value. Interestingly, although most GB2 mutants fit on the curve obtained with the wild-type heteromer, those bearing the D256Y or D256W mutation have a much larger decrease in GABA potency than would be expected if this was attributable to their low level of expression only. Accordingly, these data show that the mutation of Asp256 into the large residues Tyr or Trp decreases the efficacy of the bound GB1 VFTM to activate the G-protein coupling domain of the receptor.
Fig. 6.

Influence of receptor expression level on GABA potency (EC50). HEK 293 cells were transfected with 2 μg of pRKGB1 and various concentrations of pRKGB2 (from 0.05 to 2 μg), and the specific [125I]CGP64213 binding (expressed as the percentage of that measured on cells transfected with 2 μg of both pRKGB1 and pRKGB2) and GABA EC50 values were determined. Values obtained are indicated by open circles in this plot. Closed circles correspond to the data obtained with the various GB2 mutants coexpressed with the wild-type GB1 and indicated in Table 2.Numbers indicate the data obtained with the GB2-P136S, T159S, T160S, D256Y, F354E (1); GB2-D256W (2); GB2-P136S, D256Y (3); GB2-P136S, T159S, T160S, D256Y (4); GB2-D256Y (5); and GB2-F208A (6).
We also analyzed the possible role of Asp360, the GB2 homologous residue of Asp471 of GB1. According to our model, this Asp residue is also involved in the same interaction network as that observed in GB1 and may therefore play a role in the correct folding of the lobe-I of GB2 VFTM. In agreement with this proposal, the mutation of Asp360 into Ala is sufficient to prevent GB2 to target GB1 to the cell surface, as shown by the absence of [125I]CGP64213 binding on intact cells expressing both subunits (Table 2), although this GB2 mutant is expressed and reached the cell surface alone, as shown by immunohistochemistry (Fig. 3b).
Evolution analysis of GB1 and GB2 VFTMs
Regions of proteins involved in ligand recognition (the ligand being either another protein or a small molecule) are subjected to a high pressure during evolution and, as such, are more conserved than the other area of the protein. This has been the basis for the generation of the “evolutionary trace” method to identify possible ligand-interacting sites of proteins (Lichtarge et al., 1996a,b). Therefore, we examined whether a higher selective pressure could be observed on the binding pocket of various family 3 GPCRs, including the GB2 subunit, compared with the rest of the protein.
The sequences of the group-II mGlu receptors, the GB1 and GB2 subunits from C. elegans, D. melanogaster, and various mammalian species, were retrieved from the data bank, and their VFTM sequences were aligned. The deduced phylogenetic tree revealed a similar distance between the C. elegans and mammalian proteins regardless of whether group-II mGlu receptors or GB1 or GB2 subunits were considered (Fig.7a). The same analysis was then performed taking into account the residues that constitute the binding pocket only. As shown on Figure 7b, the phylogenetic distance between the C. elegans and mammalian binding pockets was much shorter for the GB1 and group-II mGlu receptors than that observed with the entire VFTMs of these proteins. This clearly indicates a high pressure during evolution on these binding pockets. In contrast, the phylogenetic distances between the C. elegans,D. melanogaster, and mammalian GB2 proteins remain the same regardless of which residues constituting the putative-binding pocket, or the entire sequence of the VFTM, are considered (Fig.7b). This clearly shows that there has been no higher selective pressure on the putative GB2-binding pocket than on the rest of the protein.
Fig. 7.

Absence of selective evolutionary pressure of the putative GB2-binding site. a, Evolutionary trees constructed with the sequences of the VFTM of C. elegans(Cel), D. melanogaster(Dro), or various mammalian [human (Hum), rat, or mouse (Mus)] GB1 or GB2 subunits or group-II mGlu receptors. b, Tree obtained with the residues lining the putative-binding pocket (see Materials and Methods). Note that the phylogenetic distances (expressed as the percentage of divergence divided by 100) between the GB2 subunits remain similar regardless of whether the entire VFTM is considered or only the residues that lined the putative-binding pocket.c, Residues close to the putative-binding pocket of the GB1 and GB2 subunits. These residues correspond to the position of those residues located at <8 Å of glutamate in the mGlu1 VFTM. Residues highlighted in black are those that are not conserved during evolution. The boxed residues are conserved in all GB subunits and are likely to be structurally important. Positions in GB2 indicated with an asteriskare those that have been subjected to mutagenesis in the rat GB2 (present study).
Comparison of the residues that constitute the binding pocket of GB1 revealed that only 14 of 46 are not conserved during evolution (Fig.7c). In contrast, 34 of 46 residues are not conserved in the GB2 pocket (Fig. 7c). As shown above, even when residues conserved in both D. melanogaster and mammalian GB2 sequences and able to form H-bonds with a ligand are mutated (Ser178, Asp179, Arg207), the heteromeric GABAB receptor retains its functional properties (Table 2).
DISCUSSION
The present study shows that mutations of as many as 10 different residues within the putative-binding pocket of GB2 do not prevent GABA from activating the heteromeric GABAB receptor, nor do they inhibit the positive allosteric effect of the GB2 subunit on the agonist affinity of GB1. Moreover, our data revealed that this putative-binding pocket of GB2 has not been subjected to high pressure during evolution, in contrast to the equivalent binding site of GB1 or mGlu receptors.
Thanks to the recent resolution of the structure of various VFTMs, we have been able to further refine our three-dimensional model for the GB1-binding site. In agreement with previous studies, the carboxylic moiety of GABA interacts with Ser246 as well as with backbone atoms of the loop βD–βE, like the α-carboxylic function of glutamate in the mGlu1 VFTM (Kunishima et al., 2000) or of Leu in LIVBP (Sack et al., 1989), and with Tyr366 from lobe-II. Our analysis also revealed a network of hydrogen bonds (in which Asp471 is involved) that is likely to be important for the correct folding of lobe-I and is conserved in many of these VFTMs. Finally, our data are consistent with Glu465 interacting with the N group of GABA. This residue aligns with Lys409 of mGlu1, which forms an ionic interaction with the γ-carboxylic group of glutamate.
The GB2 VFTM has been reported previously to be required to obtain a functional GABAB receptor (Jones et al., 2000;Galvez et al., 2001; Margeta-Mitrovic et al., 2001a). Moreover, the GB2 VFTM appears to be crucial to increase the agonist affinity on GB1 (Galvez et al., 2001; Duthey et al., 2002). Finally, the extracellular domain of the GB2 subunit is structurally related to that of the other family 3 GPCRs, which usually contain the agonist binding site (O'Hara et al., 1993; Okamoto et al., 1998; Galvez et al., 1999, 2000a;Hammerland et al., 1999; Hampson et al., 1999; Malitschek et al., 1999;Bessis et al., 2000; Kunishima et al., 2000). These observations suggest that a ligand interacts with the GB2 VFTM, such an interaction being possibly required either for GABAB receptor activation or for the allosteric modulation of agonist affinity on GB1. A rapid comparison of the GB1 GABA-binding site and the GB2 putative-binding pocket revealed that the three residues of GB1 that likely contact GABAB agonists are not conserved in GB2. However, GABAB agonists could still interact in this pocket but in a manner different from that in GB1. Our present data exclude this possibility, because none of the mutations introduced in the GB2 putative site prevent GABA activation of the receptor or the positive allosteric effect of GB2 on GB1. This conclusion is reinforced when one takes into account the change in the properties of the putative-binding pocket in some of the mutants analyzed as well as the number of mutations introduced (less than or equal to five). These data also exclude the possibility that an endogenous compound that may be produced by HEK 293 cells acts at this putative GB2 site.
One may also consider that the GB2 putative-binding site could be occupied by a specific ligand that may be present in the brain under certain circumstances. This may for instance influence the functioning of the GB1 subunit and may possibly explain the different pharmacological properties of GABAB receptors reported in the literature (Bonanno and Raiteri, 1993; Kerr and Ong, 1995). Because the GB2 subunit is conserved from C. elegansto mammals and insects, one may expect that such a ligand is also conserved in these various species and that, accordingly, its binding site is also conserved. Indeed, surface area of special physiological importance for the protein function, such as regions of interaction with a ligand or with another protein, must conserve their properties during evolution. Accordingly, such areas are found to be better conserved than the rest of the protein (Lichtarge et al., 1996a,b). In agreement with this hypothesis, a higher degree of conservation of the binding pocket compared with the rest of the protein was found for GB1 as well as for mGlu receptors. This is clearly not the case for GB2. Accordingly, the cleft that separates both lobes of the GB2 VFTM does not appear to constitute a ligand-binding site. However, a second GABAB receptor ligand could interact at another site in GB2.
If the GB2 VFTM does not bind a ligand, why is it necessary for GABAB receptor activation? Indeed, a GABAB receptor heteromer in which the GB2 VFTM is deleted (Jones et al., 2000) or in which both VFTMs are from GB1 (the GB1+GB1/2 combination) (Galvez et al., 2001; Margeta-Mitrovic et al., 2001a; Robbins et al., 2001) is not activated by GABA. However, the latter combination is able to activate a G-protein, as indicated by its high basal activity (Galvez et al., 2001; Margeta-Mitrovic et al., 2001a). This indicates that the GB2 VFTM is required (1) to maintain the receptor in its inactive state and (2) to allow GABA binding in GB1 to activate the receptor. The recent resolution of the crystal structure of the dimeric mGlu1 VFTM with and without bound glutamate sheds light on the possible role of the GB2 VFTM (Kunishima et al., 2000). In the mGlu1 receptor, the VFTM forms homodimers by interacting at the level of its lobe-I. In the absence of ligand, both VFTMs are in an open conformation and in a relative orientation in such a way that the lobes-II are far apart (the C-terminal ends of the VFTM being part of lobe-II and being at a 87 Å distance) (Fig.8). In the presence of glutamate, both VFTMs are occupied by glutamate, but one is found in the closed state, whereas the other remains in the open state (Fig. 8, step 1). Moreover, the closure of one VFTM appears sufficient to induce a change in the relative orientation of the VFTMs so that the lobes-II, and therefore the C-terminal ends, become closer (63 Å) (Fig. 8, step 2). This state is supposed to be the active state that, by bringing together the HDs within the dimer, would lead to G-protein activation. Although the C-terminal tails of the GABAB receptor subunits play an important role in the dimerization process (White et al., 1998;Kuner et al., 1999), this is not the only part involved (Pagano et al., 2001). Indeed, the HDs also likely dimerize, as well as the GB1 and GB2 VFTMs (Schwarz et al., 2000). Accordingly, one may propose that GB1 and GB2 VFTMs may interact with each other in such a way that they prevent the dimer of HD from reaching its active state (Fig. 8). Binding of GABA in GB1 would stabilize the closed state (Galvez et al., 1999,2000a) (Fig. 8, step 1) and change the relative orientation of the two VFTMs so that the two HDs become closer (Fig. 8, step 2). As such, the GB2 VFTM would not be required to recognize a ligand but rather to allow the GB1 VFTM to activate the dimer of HDs with agonist binding.
Fig. 8.
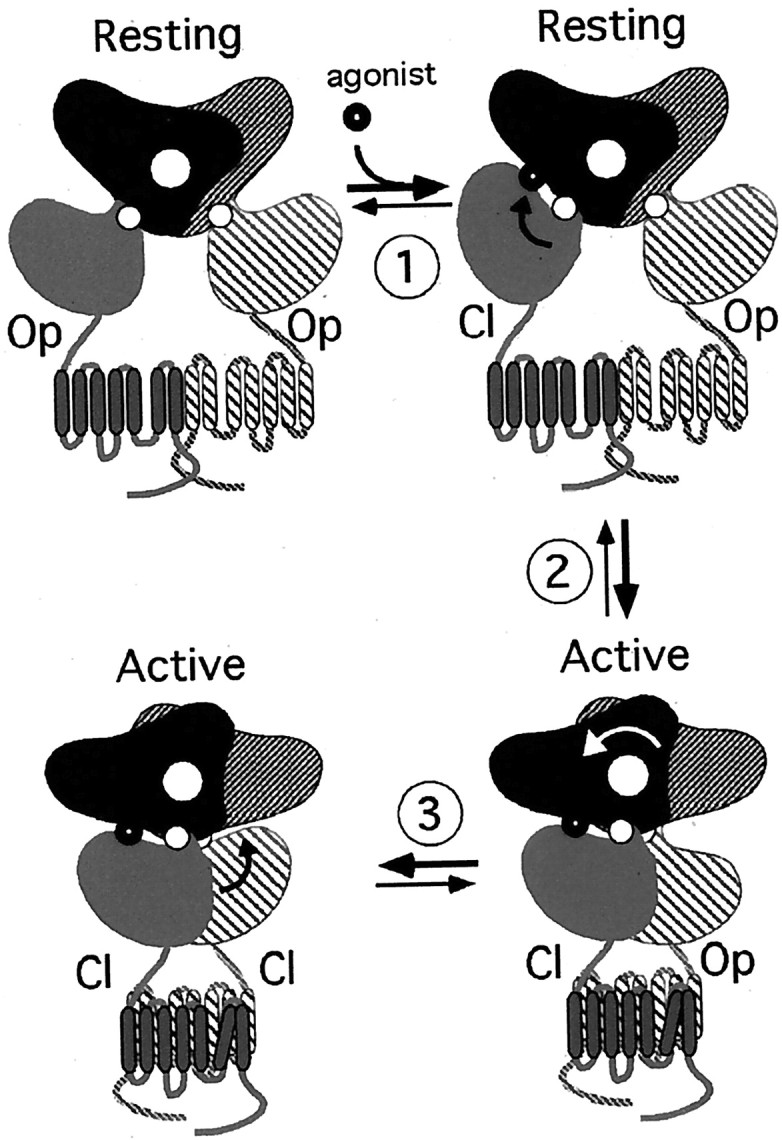
Schematic representation of the putative activation mechanism of family 3 GPCRs. Top left, Schematic representation of the inactive receptor with both VFTMs in the open state (Op) and in the resting orientation [according to the structure of the free dimeric mGlu1 VFTM (pdb code: 1ewt)] (Kunishima et al., 2000). Step 1: Binding of an agonist in at least one VFTM induces its closing (Cl). Step 2: One closed, liganded VFTM induces a change in the relative orientation of the two VFTMs to reach the active orientation [according to the structure of the agonist-bound form of the dimeric mGlu1 VFTM (pdb code: 1ewk)] (Kunishima et al., 2000). Step 3: Addition of Gd3+ allows the dimeric VFTM of mGlu1, with both sites occupied by glutamate, to reach a novel active conformation in which both VFTMs are in the closed state [according to the structure of the agonist and Gd3+-bound form of the dimeric mGlu1 VFTM (pdb code: 1 isr)] (Tsuchiya et al., 2002). In the case of the heteromeric GABAB receptor, binding of GABA in the GB1 VFTM would allow step 1 and step 2 to occur, although no ligand is bound in the GB2 VFTM. The possible closure without ligand of the GB2 VFTM would correspond to step 3. The small white circlesrepresent the axis allowing the opening or closing of each VFTM. Thelarge white circle represents the axis for the change in the relative orientation of the two VFTMs.
For most VFTMs, both closed and open conformations have been observed, even in the absence of ligand (Quiocho, 1990; Walmsley et al., 1992;Wolf et al., 1994). Would such an opening and closing of the GB2 VFTM in the absence of ligand play a role in GABABreceptor function? In the case of the mGlu1 receptor, Gd3+ stabilizes a conformation of the dimer of VFTMs in which both are in the closed state (Tsuchiya et al., 2002) (Fig. 8, step 3), possibly leading to an increased efficacy of the receptor to activate G-proteins (Kubo et al., 1998; Saunders et al., 1998; Hammerland et al., 1999). Accordingly, the possible spontaneous closure of the GB2 VFTM may stabilize the active orientation of the dimer of VFTMs (Fig. 8, step 3). In agreement with this proposal, we found that the mutation of Asp256 into either Tyr or Trp decreases the G-protein coupling efficacy of the heteromeric receptor, as indicated by the high EC50 values for agonists, despite their wild-type agonist affinity. Because the side-chain of Asp256 points toward the cleft that separates both lobes, it is possible that the large side-chains of Tyr and Trp prevent the closure of the GB2 VFTM, therefore preventing step 3, as depicted in Figure 8.
In conclusion, our data are consistent with a single GABA molecule being sufficient to activate the heteromeric GABAB receptor and reveal that the GB2 subunit is unlikely to bind a ligand at the level of its VFTM. The recent determination of the structure of the dimer of mGlu1 VFTMs sheds light on the possible role of the GB2 VFTM in the GABABreceptor activation process.
Footnotes
This work was supported by the “Action Incitative Physique et Chimie du Vivant” (PCV00-134) from the Centre National de la Recherche Scientifique (CNRS) (J.-P.P.), the program “Molécules et Cibles Thérapeutiques” from Institut National de la Santé et de la Recherche Médicale and CNRS (J.-P.P.), and Novartis Pharma (Basel, Switzerland). We thank Drs. F. Acher, A.-S. Bessis, B. Bettler, L. Prézeau, and P. Rondard for constructive discussions and constant support throughout this work and I. Brabet for her help in some experiments. We also thank Drs. W. Froestl and K. Kaupmann (Novartis) for the supply of GABAB ligands and A. Turner-Madeuf for help with our English.
Correspondence should be addressed to Dr. Jean-Philippe Pin, Centre National de la Recherche Scientifique–Unité Propre de Recherche 9023, Mécanismes Moléculaires des Communications Cellulaires, 141 rue de la Cardonille, 34094 Montepellier Cedex 5, France. E-mail: pin@montp.inserm.fr.
T. Galvez's present address: Department of Molecular Pharmacology, Stanford University Medical Center, Stanford, CA 94304.
REFERENCES
- 1.Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ango F, Albani-Torregrossa S, Joly C, Robbe D, Michel J-M, Pin J-P, Bockaert J, Fagni L. A simple method to transfer plasmid DNA into neuronal primary cultures: functional expression of mGluR5 in cerebellar granule cells. Neuropharmacology. 1999;38:793–803. doi: 10.1016/s0028-3908(99)00005-2. [DOI] [PubMed] [Google Scholar]
- 3.Bernard P, Guedin D, Hibert M. Molecular modeling of the GABA/GABAB receptor complex. J Med Chem. 2001;44:27–35. doi: 10.1021/jm000915o. [DOI] [PubMed] [Google Scholar]
- 4.Bessis A-S, Bertrand H-O, Galvez T, De Colle C, Pin J-P, Acher F. Three-dimensional model of the extracellular domain of the type 4a metabotropic glutamate receptor: new insights into the activation process. Protein Sci. 2000;9:2200–2209. doi: 10.1110/ps.9.11.2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bettler B, Kaupmann K, Bowery N. GABAB receptors: drugs meet clones. Curr Opin Neurobiol. 1998;8:345–350. doi: 10.1016/s0959-4388(98)80059-7. [DOI] [PubMed] [Google Scholar]
- 6.Billinton A, Ige AO, Bolam JP, White JH, Marshall FH, Emson PC. Advances in the molecular understanding of GABAB receptors. Trends Neurosci. 2001;24:277–282. doi: 10.1016/s0166-2236(00)01815-4. [DOI] [PubMed] [Google Scholar]
- 7.Blahos J, Fischer T, Brabet I, Stauffer D, Rovelli G, Bockaert J, Pin J-P. A novel site on the Gα-protein that recognizes heptahelical receptors. J Biol Chem. 2001;276:3262–3269. doi: 10.1074/jbc.M004880200. [DOI] [PubMed] [Google Scholar]
- 8.Bonanno G, Raiteri M. Multiple GABAB receptors. Trends Pharmacol Sci. 1993;14:259–261. doi: 10.1016/0165-6147(93)90124-3. [DOI] [PubMed] [Google Scholar]
- 9.Brabet I, Parmentier M-L, De Colle C, Bockaert J, Acher F, Pin J-P. Comparative effect of L-CCG-I, DCG-IV and γ-carboxy–l-glutamate on all cloned metabotropic glutamate receptor subtypes. Neuropharmacology. 1998;37:1043–1051. doi: 10.1016/s0028-3908(98)00091-4. [DOI] [PubMed] [Google Scholar]
- 10.Calver AR, Robbins MJ, Cosio C, Rice SQ, Babbs AJ, Hirst WD, Boyfield I, Wood MD, Russell RB, Price GW, Couve A, Moss SJ, Pangalos MN. The C-terminal domains of the GABAB receptor subunits mediate intracellular trafficking but are not required for receptor signaling. J Neurosci. 2001;21:1203–1210. doi: 10.1523/JNEUROSCI.21-04-01203.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Colovos C, Yeates TO. Verification of protein structures: patterns of nonbonded atomic interactions. Protein Sci. 1993;2:1511–1519. doi: 10.1002/pro.5560020916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Costantino G, Macchiarulo A, Entrena Guadix A, Pellicciari R. QSAR and molecular modeling studies of baclofen analogues as GABAB agonists. Insights into the role of the aromatic moiety in GABAB binding and activation. J Med Chem. 2001;44:1827–1832. doi: 10.1021/jm0100133. [DOI] [PubMed] [Google Scholar]
- 13.Couve A, Moss SJ, Pangalos MN. GABAB receptors: a new paradigm in G protein signaling. Mol Cell Neurosci. 2000;16:296–312. doi: 10.1006/mcne.2000.0908. [DOI] [PubMed] [Google Scholar]
- 14.Cuff JA, Clamp ME, Siddiqui AS, Finlay M, Barton GJ. Jpred: a consensus secondary structure prediction server. Bioinformatics. 1998;14:892–893. doi: 10.1093/bioinformatics/14.10.892. [DOI] [PubMed] [Google Scholar]
- 15.Duthey B, Caudron S, Perroy J, Bettler B, Fagni L, Pin J-P, Prézeau L. A single subunit (GB2) is required for G-protein activation by the heterodimeric GABAB receptor. J Biol Chem. 2002;277:3236–3241. doi: 10.1074/jbc.M108900200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Eisenberg D, Luthy R, Bowie JU. VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol. 1997;277:396–404. doi: 10.1016/s0076-6879(97)77022-8. [DOI] [PubMed] [Google Scholar]
- 17.Galvez T, Parmentier M-L, Joly C, Malitschek B, Kaupmann K, Kuhn R, Bittiger H, Froestl W, Bettler B, Pin J-P. Mutagenesis and modeling of the GABAB receptor extracellular domain support a Venus flytrap mechanism for ligand binding. J Biol Chem. 1999;274:13362–13369. doi: 10.1074/jbc.274.19.13362. [DOI] [PubMed] [Google Scholar]
- 18.Galvez T, Prézeau L, Milioti G, Franek M, Joly C, Froestl W, Bettler B, Bertrand H-O, Blahos J, Pin J-P. Mapping the agonist binding site of GABAB type 1 subunit sheds light on the activation process of GABAB receptors. J Biol Chem. 2000a;275:41166–41174. doi: 10.1074/jbc.M007848200. [DOI] [PubMed] [Google Scholar]
- 19.Galvez T, Urwyler S, Prézeau L, Mosbacher J, Joly C, Malitschek B, Heid J, Brabet I, Froestl W, Bettler B, Kaupmann K, Pin J-P. Ca2+-requirement for high affinity γ-aminobutyric acid (GABA) binding at GABAB receptors: involvement of serine 269 of the GABABR1 subunit. Mol Pharmacol. 2000b;57:419–426. doi: 10.1124/mol.57.3.419. [DOI] [PubMed] [Google Scholar]
- 20.Galvez T, Duthey B, Kniazeff J, Blahos J, Rovelli G, Bettler B, Prézeau L, Pin J-P. Allosteric interactions between GB1 and GB2 subunits are required for optimal GABAB receptor function. EMBO J. 2001;20:2152–2159. doi: 10.1093/emboj/20.9.2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 22.Hammerland LG, Krapcho KJ, Garrett JE, Alasti N, Hung BC, Simin RT, Levinthal C, Nemeth EF, Fuller FH. Domains determining ligand specificity for Ca2+ receptors. Mol Pharmacol. 1999;55:642–648. [PubMed] [Google Scholar]
- 23.Hampson DR, Huang XP, Pekhletski R, Peltekova V, Hornby G, Thomsen C, Thogersen H. Probing the ligand-binding domain of the mGluR4 subtype of metabotropic glutamate receptor. J Biol Chem. 1999;274:33488–33495. doi: 10.1074/jbc.274.47.33488. [DOI] [PubMed] [Google Scholar]
- 24.Havlickova M, Prezeau L, Duthey B, Bettler B, Pin J-P, Blahos J. The intracellular loops of the GB2 subunit are crucial for G-protein coupling of the heteromeric GABAB receptor. Mol Pharmacol. 2002;62:343–350. doi: 10.1124/mol.62.2.343. [DOI] [PubMed] [Google Scholar]
- 25.He X-L, Chow D-C, Martick MM, Garcia KC. Allosteric activation of a spring-loaded natriuretic peptide receptor dimer by hormone. Science. 2001;293:1657–1662. doi: 10.1126/science.1062246. [DOI] [PubMed] [Google Scholar]
- 26.Jones KA, Borowsky B, Tamm JA, Craig DA, Durkin MM, Dai M, Yao W-J, Johnson M, Gunwaldsen C, Huang L-Y, Tang C, Shen Q, Salon JA, Morse K, Laz T, Smith KE, Nagarathnam D, Noble SA, Branchek TA, Gerald C. GABAB receptors function as a heteromeric assembly of the subunits GABABR1 and GABABR2. Nature. 1998;396:674–679. doi: 10.1038/25348. [DOI] [PubMed] [Google Scholar]
- 27.Jones KA, Tamm JA, Craig DA, Yao W-J, Panico R. Signal transduction by GABAB receptor heterodimers. Neuropsychopharmacology. 2000;23:S41–S49. doi: 10.1016/S0893-133X(00)00145-7. [DOI] [PubMed] [Google Scholar]
- 28.Kaupmann K, Huggel K, Heid J, Flor PJ, Bischoff S, Mickel SJ, McMaster G, Angst C, Bittiger H, Froestl W, Bettler B. Expression cloning of GABAB receptors uncovers similarity to metabotropic glutamate receptors. Nature. 1997;386:239–246. doi: 10.1038/386239a0. [DOI] [PubMed] [Google Scholar]
- 29.Kaupmann K, Malitschek B, Schuler V, Heid J, Froestl W, Beck P, Mosbacher J, Bischoff S, Kulik A, Shigemoto R, Karschin A, Bettler B. GABAB-receptor subtypes assemble into functional heteromeric complexes. Nature. 1998;396:683–687. doi: 10.1038/25360. [DOI] [PubMed] [Google Scholar]
- 30.Kerr DIB, Ong J. GABAB receptors. Pharmacol Ther. 1995;67:187–246. doi: 10.1016/0163-7258(95)00016-a. [DOI] [PubMed] [Google Scholar]
- 31.Kubo Y, Miyashita T, Murata Y. Structural basis for a Ca2+-sensing function of the metabotropic glutamate receptors. Science. 1998;279:1722–1725. doi: 10.1126/science.279.5357.1722. [DOI] [PubMed] [Google Scholar]
- 32.Kuner R, Kohr G, Grunewald S, Eisenhardt G, Bach A, Kornau HC. Role of heteromer formation in GABAB receptor function. Science. 1999;283:74–77. doi: 10.1126/science.283.5398.74. [DOI] [PubMed] [Google Scholar]
- 33.Kunishima N, Shimada Y, Tsuji Y, Sato T, Yamamoto M, Kumasaka T, Nakanishi S, Jingami H, Morikawa K. Structural basis of glutamate recognition by a dimeric metabotropic glutamate receptor. Nature. 2000;407:971–977. doi: 10.1038/35039564. [DOI] [PubMed] [Google Scholar]
- 34.Labesse G, Mornon J-P. Incremental threading optimization (TITO) to help alignment and modeling of remote homologues. Bioinformatics. 1998;14:206–211. doi: 10.1093/bioinformatics/14.2.206. [DOI] [PubMed] [Google Scholar]
- 35.Lichtarge O, Bourne HR, Cohen FE. Evolutionarily conserved G(alpha beta gamma) binding surfaces support a model of the G protein-receptor complex. Proc Natl Acad Sci USA. 1996a;93:7507–7511. doi: 10.1073/pnas.93.15.7507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lichtarge O, Bourne HR, Cohen FE. An evolutionary trace method defines binding surfaces common to protein families. J Mol Biol. 1996b;257:342–358. doi: 10.1006/jmbi.1996.0167. [DOI] [PubMed] [Google Scholar]
- 37.Malitschek B, Schweizer C, Keir M, Heid J, Froestl W, Mosbacher J, Kuhn R, Henley J, Joly C, Pin J-P, Kaupmann K, Bettler B. The N-terminal domain of γ-aminobutyric acidB receptors is sufficient to specify agonist and antagonist binding. Mol Pharmacol. 1999;56:448–454. doi: 10.1124/mol.56.2.448. [DOI] [PubMed] [Google Scholar]
- 38.Margeta-Mitrovic M, Jan YN, Jan LY. A trafficking checkpoint controls GABAB receptor heterodimerization. Neuron. 2000;27:97–106. doi: 10.1016/s0896-6273(00)00012-x. [DOI] [PubMed] [Google Scholar]
- 39.Margeta-Mitrovic M, Jan YN, Jan LY. Ligand-induced signal transduction within heterodimeric GABAB receptor. Proc Natl Acad Sci USA. 2001a;98:14643–14648. doi: 10.1073/pnas.251554798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Margeta-Mitrovic M, Jan YN, Jan LY. Function of GB1 and GB2 subunits in G protein coupling of GABAB receptors. Proc Natl Acad Sci USA. 2001b;98:14649–14654. doi: 10.1073/pnas.251554498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marshall FH, Jones KA, Kaupmann K, Bettler B. GABAB receptors–the first 7TM heterodimers. Trends Pharmacol Sci. 1999;20:396–399. doi: 10.1016/s0165-6147(99)01383-8. [DOI] [PubMed] [Google Scholar]
- 42.Martin SC, Russek SJ, Farb DH. Molecular identification of the human GABABR2: cell surface expression and coupling to adenylyl cyclase in the absence of GABABR1. Mol Cell Neurosci. 1999;13:180–191. doi: 10.1006/mcne.1999.0741. [DOI] [PubMed] [Google Scholar]
- 43.O'Hara PJ, Sheppard PO, Thøgersen H, Venezia D, Haldeman BA, McGrane V, Houamed KM, Thomsen C, Gilbert TL, Mulvihill ER. The ligand-binding domain in metabotropic glutamate receptors is related to bacterial periplasmic binding proteins. Neuron. 1993;11:41–52. doi: 10.1016/0896-6273(93)90269-w. [DOI] [PubMed] [Google Scholar]
- 44.Okamoto T, Sekiyama N, Otsu M, Shimada Y, Sato A, Nakanishi S, Jingami H. Expression and purification of the extracellular ligand binding region of metabotropic glutamate receptor subtype 1. J Biol Chem. 1998;273:13089–13096. doi: 10.1074/jbc.273.21.13089. [DOI] [PubMed] [Google Scholar]
- 45.Pagano A, Rovelli G, Mosbacher J, Lohmann T, Duthey B, Stauffer D, Ristig D, Schuler V, Heid J, Meigel I, Lampert C, Stein T, Prézeau L, Pin J-P, Froestl W, Kuhn R, Kaupmann K, Bettler B. C-terminal interaction is essential for surface trafficking but not for heteromeric assembly of GABAB receptors. J Neurosci. 2001;21:1189–1202. doi: 10.1523/JNEUROSCI.21-04-01189.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Page RDM. TreeView: an application to display phylogenetic trees on personal computers. Comput Appl Biosci. 1996;12:357–358. doi: 10.1093/bioinformatics/12.4.357. [DOI] [PubMed] [Google Scholar]
- 47.Prosser HM, Gill CH, Hirst WD, Grau E, Robbins M, Calver A, Soffin EM, Farmer CE, Lanneau C, Gray J, Schenck E, Warmerdam BS, Clapham C, Reavill C, Rogers DC, Stean T, Upton N, Humphreys K, Randall A, Geppert M. Epileptogenesis and enhanced prepulse inhibition in GABAb1-deficient mice. Mol Cell Neurosci. 2001;17:1059–1070. doi: 10.1006/mcne.2001.0995. [DOI] [PubMed] [Google Scholar]
- 48.Quiocho FA. Atomic structures of periplasmic binding proteins and the high-affinity active transport systems in bacteria. Philos Trans R Soc Lond B Biol Sci. 1990;326:341–351. doi: 10.1098/rstb.1990.0016. [DOI] [PubMed] [Google Scholar]
- 49.Robbins MJ, Calver AR, Filippov AK, Hirst WD, Russell RB, Wood MD, Nasir S, Couve A, Brown DA, Moss SJ, Pangalos MN. GABAB2 is essential for G-protein coupling of the GABAB receptor heterodimer. J Neurosci. 2001;21:8043–8052. doi: 10.1523/JNEUROSCI.21-20-08043.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sack JS, Saper MA, Quiocho FA. Periplasmic binding protein structure and function. Refined X-ray structures of the leucine/isoleucine/valine-binding protein and its complex with leucine. J Mol Biol. 1989;206:171–191. doi: 10.1016/0022-2836(89)90531-7. [DOI] [PubMed] [Google Scholar]
- 51.Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987;4:406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- 52.Sali A, Blundell TL. Comparative protein modeling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 53.Saunders R, Nahorski SR, Challiss RAJ. A modulatory effect of extracellular Ca2+ on type 1α metabotropic glutamate receptor-mediated signalling. Neuropharmacology. 1998;37:273–276. doi: 10.1016/s0028-3908(98)00027-6. [DOI] [PubMed] [Google Scholar]
- 54.Schuler V, Luscher C, Blanchet C, Klix N, Sansig G, Klebs K, Schmutz M, Heid J, Gentry C, Urban L, Fox A, Spooren W, Jaton A, Vigouret J, Pozza M, Kelly PH, Mosbacher J, Froestl W, Kaslin E, Korn R. Epilepsy, hyperalgesia, impaired memory, and loss of pre- and postsynaptic GABAB responses in mice lacking GABAB1. Neuron. 2001;31:47–58. doi: 10.1016/s0896-6273(01)00345-2. [DOI] [PubMed] [Google Scholar]
- 55.Schwarz DA, Barry G, Eliasof SD, Petroski RE, Conlon PJ, Maki RA. Characterization of gamma-aminobutyric acid receptor GABAB(1e), a GABAB(1) splice variant encoding a truncated receptor. J Biol Chem. 2000;275:32174–32181. doi: 10.1074/jbc.M005333200. [DOI] [PubMed] [Google Scholar]
- 56.Sippl MJ. Recognition of errors in three-dimensional structures of proteins. Proteins. 1993;17:355–362. doi: 10.1002/prot.340170404. [DOI] [PubMed] [Google Scholar]
- 57.Thompson JD, Higgins DG, Gibson TJ. Clustal W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tsuchiya D, Kunishima N, Kamiya N, Jingami H, Morikawa K. Structural views of the ligand-binding cores of a metabotropic glutamate receptor complexed with an antagonist and both glutamate and Gd3+. Proc Natl Acad Sci USA. 2002;99:2660–2665. doi: 10.1073/pnas.052708599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tuffery P. XmMol: an X11 and motif program for macromolecular visualization and modeling. J Mol Graph. 1995;13:67–72. doi: 10.1016/0263-7855(94)00011-g. [DOI] [PubMed] [Google Scholar]
- 60.van den Akker F, Zhang X, Miyagi M, Huo X, Misono KS, Yee VC. Structure of the dimerized hormone-binding domain of a guanylyl-cyclase-coupled receptor. Nature. 2000;406:101–104. doi: 10.1038/35017602. [DOI] [PubMed] [Google Scholar]
- 61.Walmsley AR, Shaw JG, Kelly DJ. Perturbation of the equilibrium between open and closed conformations of the periplasmic C4-dicarboxylate binding protein from Rhodobacter capsulatus. Biochemistry. 1992;31:11175–11181. doi: 10.1021/bi00160a031. [DOI] [PubMed] [Google Scholar]
- 62.White JH, Wise A, Main MJ, Green A, Fraser NJ, Disney GH, Barnes AA, Emson P, Foord SM, Marshall FH. Heterodimerization is required for the formation of a functional GABAB receptor. Nature. 1998;396:679–682. doi: 10.1038/25354. [DOI] [PubMed] [Google Scholar]
- 63.Wolf A, Shaw EW, Nikaido K, Ames GF-L. The histidine-binding protein undergoes conformational changes in the absence of ligand as analyzed with conformation-specific monoclonal antibodies. J Biol Chem. 1994;269:23051–23058. [PubMed] [Google Scholar]