

Abstract
Visinin-like protein-1 (VILIP-1) belongs to the family of neuronal calcium sensor (NCS) proteins, a neuronal subfamily of EI-hand calcium-binding proteins that are myristoylated at their N termini. NCS proteins are discussed to play roles in calcium-dependent signal transduction of physiological and pathological processes in the CNS. The calcium-dependent membrane association, the so-called calcium–myristoyl switch, localizes NCS proteins to a distinct cellular signaling compartment and thus may be a critical mechanism for the coordinated regulation of signaling cascades. To study whether the biochemically defined calcium–myristoyl switch of NCS proteins can occur in living neuronal cells, the reversible and stimulus-dependent translocation of green fluorescent protein (GFP)-tagged VILIP-1 to subcellular targets was examined by fluorescence microscopy in transfected cell lines and hippocampal primary neurons. In transiently transfected NG108–15 and COS-7 cells, a translocation of diffusely distributed VILIP-1–GFP but not of myristoylation-deficient VILIP-1–GFP to the plasma membrane and to intracellular targets, such as Golgi membranes, occurred after raising the intracellular calcium concentration with a calcium ionophore. The observed calcium-dependent localization was completely reversed after depletion of intracellular calcium by EGTA. Interestingly, a fast and reversible translocation of VILIP-1–GFP and translocation of endogenous VILIP-1 to specialized membrane structures was also observed after a depolarizing stimulus or activation of glutamate receptors in hippocampal neurons. These results show for the first time the reversibility and stimulus-dependent occurrence of the calcium–myristoyl switch in living neurons, suggesting a physiological role as a signaling mechanism of NCS proteins, enabling them to activate specific targets localized in distinct membrane compartments.
Keywords: activity-dependent, calcium–myristoyl switch, hippocampal neurons, GFP, Golgi, membrane compartments, NCS protein, signaling, VILIP-1
To explain the regulation and organization of the large number of existing signaling proteins in space and time, a dynamic model of signal transduction based on the mechanism of translocation and reversible colocalization of signaling proteins has been postulated (Teruel and Meyer, 2000). Thus, the reversible localization of signaling proteins to distinct signaling compartments, such as adaptor complexes, cytoskeletal structures, the plasma and intracellular membranes, or signal effector molecules themselves, is a critical event for the selective activation of downstream signaling cascades. The underlying molecular mechanisms comprise, for example, different domains within proteins that can function as modules for selective targeting to both proteins and lipids but also lipid modifications of proteins, such as myristoylation, palmitoylation, isoprenylation, or a glycosyl-phosphatidylinositol anchor, which enable a specific targeting and membrane association (for review, see Casey, 1995).
One of the most common forms of protein lipid modification is the cotranslational attachment of myristate, a 14-carbon saturated fatty acid, to a specific consensus site at the N-terminal part of a protein via the myristoyl–CoA:proteinN-myristoyltransferase (Towler et al., 1988; Resh, 1999). In some proteins, this type of acylation is critical for structure and enzymatic activity (Kennedy et al., 1996); it can mediate the interaction between two proteins (Takasaki et al., 1999) and facilitates the binding to membranes (Zozulya and Stryer, 1992). Moreover, for many myristoylated proteins, including Src tyrosine kinases, G-proteins, ADP-ribosylation factors (Randazzo et al., 1995), myristoylated alanine-rich C kinase substrate proteins (McLaughlin and Aderem, 1995), or neuronal calcium sensor (NCS) proteins (Braunewell et al., 1997), myristoylation has been shown to be critical for their function in signal transduction processes (for review, see Resh, 1999).
The regulation of membrane association of myristoylated proteins can occur via different switch mechanisms i.e., electrostatic, pH- or ligand-dependent switch mechanism, which involve a conformational change within the myristoylated protein. One example for myristoyl switch proteins in the nervous system is the family of NCS proteins. These proteins constitute a subfamily of EI-hand calcium binding proteins that have been shown to be involved in calcium-dependent signal transduction processes, such as cyclic nucleotide metabolism, neurotransmitter release, modulation of ion channel function, and regulation of gene expression (Braunewell and Gundelfinger, 1999;Burgoyne and Weiss, 2001). In the calcium–myristoyl switch model (Zozulya and Stryer, 1992), the myristoyl moiety is sequestered in a hydrophobic pocket in the calcium-free conformation, whereas in the alternative calcium-bound conformation, the myristate is extruded and becomes available for membrane binding. Using crystallography and nuclear magnetic resonance (NMR) spectroscopy, the detailed molecular mechanism of the calcium–myristoyl switch has been studied for some NCS proteins (Ames et al., 1995, 1999, 2000a,b), but the subcellular localization in response to a calcium signal in living neurons has not been investigated yet.
Therefore, we explored whether the calcium–myristoyl switch can serve as a cellular signaling mechanism in living cells. In this study, the translocation of myristoylated and non-myristoylated visinin-like protein-1 (VILIP-1)–green fluorescent protein (GFP) to subcellular membrane structures was examined in transfected cell lines and primary hippocampal neurons. The stimuli-induced and reversible translocation of the GFP fusion protein was monitored by fluorescence and time-lapse microscopy.
MATERIALS AND METHODS
Materials. Specific primers and transfection reagent were obtained from Eurogentec (Brussels, Belgium). Materials for cell culture were purchased from Invitrogen (San Diego, CA). Ionomycin was obtained from Calbiochem (San Diego, CA). Unless otherwise specified, all other reagents were from Sigma (St. Louis, MO) or Roth (Karlsruhe, Germany).
Antibodies. Rabbit polyclonal antibodies were raised against recombinant His-tagged VILIP-1 fusion proteins (Braunewell et al., 1997) and were affinity purified as described previously (Braunewell et al., 2001a). For immunocytochemistry, the anti-VILIP-1 antibody was preincubated with a 100-fold excess of recombinant glutathioneS-transferase (GST)-tagged VILIP-2 fusion protein, which was purified from Escherichia coli lysates as described previously (Spilker et al., 2000). Monoclonal antibodies used in this study included anti-GFP (Clontech, Palo Alto, CA) and anti-syntaxin-6 (Transduction Laboratories, Lexington, KY). Monoclonal anti-microtubule-associated protein-2 (MAP-2) antibody and phalloidin-tetramethylrhodamine isothiocyanate (TRITC) was purchased from Sigma. Cy3- and Alexa Fluor 488-labeled secondary antibodies were purchased from Dianova (Hamburg, Germany) and Molecular Probes (Eugene, OR).
Cloning of VILIP-1–GFP fusion constructs. To generate VILIP-1–GFP fusion constructs, wild-type VILIP-1 cDNA (myrVILIP-1) and a non-myristoylable mutant cDNA (G2AVILIP-1; glycine 2 of the myristoylation consensus site was changed into alanine) were amplified by PCR with appropriate oligonucleotide primers and cloned into the pEGFP-N1 vector (Clontech) via HindIII/SacII restriction sites. The accuracy of the fusion constructs with GFP attached to the 3′ end of VILIP-1 to leave the N-terminal myristoylation site accessible was confirmed by sequencing. Endotoxin-free plasmid DNA was prepared from E. coli lysates using the EndoFree plasmid kit (Qiagen, Hilden, Germany).
Cell culture and transfection of cell lines. NG108–15 and COS-7 cells were cultured essentially as described previously (Spilker et al., 2000). Briefly, cells were maintained at 37°C in DMEM supplemented with 10% fetal calf serum, 2 mml-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin in a humidified atmosphere containing 5% CO2. Medium for NG108–15 cells additionally contained 0.1 mm sodium hypoxanthine, 1 μm aminopterin, and 16 μm thymidine to obtain the hybridoma status. For transfection, cells were grown on poly-d-lysine-coated glass coverslips to 50–70% confluency. Transient transfection was performed with lipid-mediated gene transfer using DAC-30 (Eurogentec) according to the protocol of the manufacturer. Fluorescence became detectable 12–14 hr after transfection. All experiments with cell lines were performed 2 d after transfection.
Culture of hippocampal primary neurons and transient transfection. Neuronal cultures were prepared from hippocampi of 18-d-old, fetal Wistar rats essentially following the protocol ofGoslin and Banker (1998). Briefly, hippocampi were dissociated by enzyme digestion with 0.1% trypsin at 37°C for 20 min, followed by trituration through two different-sized syringes. Cells were plated onto poly-d-lysine-coated glass coverslips (12 mm in diameter) at a density of 60 × 103 cells for transient transfection experiments and 10–20 × 103 cells for immunocytochemistry in DMEM containing 10% fetal calf serum, 2 mml-glutamine, and antibiotics. Twenty-four hours after plating, culture medium was exchanged for Neurobasal medium (Invitrogen) supplemented with 2% B27, 0.5 mml-glutamine, and antibiotics, and cells were maintained in a humidified 37°C atmosphere containing 5% CO2. Hippocampal neurons were transfected after 3–6 d in culture using the DNA–calcium phosphate precipitation method essentially as described byKöhrmann et al. (1999). In brief, 1 hr before transfection, the culture medium was removed from the neurons and kept for later use. Prewarmed Opti-MEM (500 μl; Invitrogen) was added on each coverslip, and the cells were placed into a 37°C, 5% CO2incubator for 30 min. To prepare the DNA–calcium phosphate precipitate, 15 μl of 2× BBS (50 mmN,N-bis[2-hydroxyethyl]-2-aminoethanesulfonic acid, pH 7.1, 280 mm NaCl, and 1.5 mmNa2HPO4) was drop-wisely added to 1 μg of DNA dissolved in 15 μl of 250 mm CaCl2 (volumes for one coverslip). After 30 min incubation at room temperature, the precipitate was added to the neurons, and the cells were incubated for ∼1 hr at 37°C and 5% CO2. During the incubation time, the formation of the DNA–calcium phosphate precipitate was controlled several-fold to prevent cell death caused by too large precipitates. Subsequently, the neurons were washed twice with Opti-MEM, the original medium was added, and cells were incubated for additional 3–6 d before performing translocation experiments.
Visualization of VILIP-1–GFP translocation and image analysis. Time-lapse microscopy of transiently transfected living COS-7 and NG108–15 cells or hippocampal neurons was performed using a Leica (Wetzlar, Germany) DMR upright fluorescence microscope with a 63× water immersion Plan apochromate objective, a 50 W mercury arc bulb (Osram, Berlin, Germany), a Leica L5 480/40 bandpass filter, and a Spot RT cooled CCD camera (Visitron Systems, Puchheim, Germany) controlled by the Visitron MetaMorph software. Cells grown on glass coverslips were transferred into 2 ml of physiological buffer (in mm: 20 HEPES, pH 7.4, 135 NaCl, 5 KCl, 30 glucose, 1.5 MgCl2, and 1.5 CaCl2) in a 3.5 cm culture dish. During stimulation, images were taken every 30 sec (COS-7 and NG108–15 cells) or every 10 sec (hippocampal neurons). Translocation of VILIP-1–GFP in COS-7 and NG108–15 cells was monitored after raising the intracellular calcium concentration by addition of 2 mmionomycin (Calbiochem) to the surrounding buffer. Hippocampal neurons were stimulated with either 50 mm KCl or 100 μm glutamate/10 μmglycine. Reversal of the translocation of VILIP-1–GFP fusion protein was achieved by addition of 5 mm EGTA to the buffer to deplete calcium.
Immunocytochemistry. Hippocampal neurons were fixed with 4% paraformaldehyde in PBS, pH 7.4, for 20 min at room temperature. Before permeabilization, cells were washed twice with 25 mm glycine in PBS to quench background staining attributable to free reactive groups of paraformaldehyde. Subsequently, the cells were permeabilized and blocked in 0.2% Triton X-100, 3% bovine serum albumin, and 10% horse serum in PBS (blocking solution) for 30 min. Cells were incubated with primary antibodies diluted in blocking solution at 4°C overnight. After washing three times with PBS, secondary antibodies diluted in blocking solution without Triton X-100 were applied to the neurons for 1 hr at room temperature. After removal of unbound antibodies, coverslips were mounted on slides with Mowiol (Calbiochem), including 1,4-diazobicyclo-[2.2.2]-octane (Merck, Darmstadt, Germany) to reduce fading. For some experiments, cells were stimulated with KCl or glutamate solution as described above before fixation. The culture medium was substituted against physiological buffer, and cells were stimulated for 3 min and subsequently fixed with paraformaldehyde. Fluorescence was visualized using a Leica DMR fluorescence microscope as described above or a TCS NT laser confocal microscope with a TCS software package (Leica). Images were recorded digitally and processed using Adobe Photoshop 5.5 (Adobe Systems, San Jose, CA) and NIH Image 1.62 software (National Institutes of Health, Bethesda, MD) (available at http://rsb.info.nih.gov/nih-image/).
Western blotting and immunodetection. NG108–15 cells or hippocampal neurons transfected with GFP,myrVILIP-1–GFP, andG2AVILIP-1–GFP were lysed with 1.5% SDS in Tris-buffered saline (TBS) (50 mm Tris, pH 7.6, and 150 mm NaCl). Lysates of two coverslips (NG108–15 cells, 2 d after transfection) and four coverslips (hippocampal neurons, 3 d after transfection) were pooled and separated on a 5–20% gradient SDS-polyacrylamide gel using the Laemmli buffer system (Laemmli, 1970) and blotted onto nitrocellulose. After blocking of unspecific binding sites for 2 hr with blocking buffer (5% low-fat milk powder and 0.1% Tween 20 in TBS), the nitrocellulose membranes were incubated overnight at 4°C with polyclonal anti-VILIP-1 antibody or monoclonal anti-GFP antibody (Clontech). The immunoreactivity was visualized using HRP-coupled goat anti-rabbit or goat anti-mouse secondary antibodies (Dianova) and the ECL detection system (Amersham Biosciences, Freiburg, Germany).
RESULTS
Expression of myristoylated and non-myristoylated VILIP-1–GFP in transiently transfected cell lines and primary hippocampal neurons
VILIP-1–GFP-fusion constructs were generated by cloning wild-type VILIP-1 cDNA (myrVILIP-1–GFP) and a myristoylation mutant cDNA with the N-terminal glycine replaced by an alanine (G2AVILIP-1–GFP) into the pEGFP-N1 vector. We first tested whether the fusion constructs are expressed as full-length proteins in transfected cell lines and primary neurons. Therefore, we analyzed extracts of transiently transfected NG108–15 cells and cultured hippocampal neurons on Western blots using a monoclonal GFP antibody and a polyclonal VILIP-1 antibody. As control, the pEGFP-N1 plasmid without VILIP-1 insert was transfected in parallel. As shown in Figure 1, NG108–15 cells and hippocampal neurons express a fusion protein with the correct size of 49 kDa consisting of myrVILIP-1 orG2AVILIP-1 (22 kDa) fused to GFP (27 kDa), as detected with both antibodies. Additionally, in extracts of cells expressing only GFP, the antibody against GFP detects a band with a molecular weight of 27 kDa. Note that no proteolytic cleavage can be observed in the extracts.
Fig. 1.

Transiently transfected NG108–15 cells and hippocampal neurons express GFP or VILIP-1–GFP fusion proteins. Extracts of NG108–15 cells transfected with GFP (lane 1), myrVILIP-1–GFP (lane 2), andG2AVILIP–1-GFP (lane 3) and extracts of hippocampal neurons transfected with GFP (lane 4) and myrVILIP-1–GFP (lane 5) were subjected to SDS-PAGE and Western blot analysis. Blots were incubated with either a monoclonal anti-GFP antibody or a polyclonal anti-VILIP-1 antibody. Both antibodies detect bands of 49 kDa in size, which represent the VILIP-1–GFP fusion proteins (lanes 2, 3, and 5, 22 kDa VILIP-1 plus 27 kDa GFP). The anti-GFP antibody detects a 27 kDa band (lanes 1,4) in cells transfected with GFP alone (lanes 1, 4). Molecular weights are indicated at the left in kilodaltons.
Real-time translocation of myristoylated VILIP-1–GFP to the plasma membrane and to intracellular membranes in living cells
To investigate the ability of VILIP-1 to perform a calcium–myristoyl switch and, therefore, to associate with cell membranes in living cells, we used transiently transfected COS-7 and NG108–15 cells for time-lapse microscopy. Real-time translocation experiments were performed 2 d after transfection, and the localization of the VILIP-1–GFP fusion proteins before and after raising the intracellular calcium concentration in cells cultured on glass coverslips was recorded with a digital camera taking images every 30 sec.
In unstimulated COS-7 cells, both proteins,myrVILIP-1–GFP andG2AVILIP-1–GFP, were diffusely distributed throughout the cell (Fig.2C,E) and were indistinguishable from GFP expressed without fusion partner (Fig.2A). To increase the intracellular calcium level in transfected cells, the calcium ionophore ionomycin was applied in the incubation buffer. In COS-7 cells transfected with the pEGFP plasmid without insert, no differences between untreated cells and cells incubated with ionomycin for 7 min could be observed (Fig.2A,B). In cells expressingmyrVILIP-1–GFP, a rapid redistribution of the protein to distinct sites of the plasma membrane (arrows), to the nuclear membrane, and to intracellular structures, presumably membranes of the Golgi apparatus (arrowheads), occurred within 4 min (Fig. 2D). Compared with untreated cells (Fig. 2C), there was a decrease of fluorescence intensity in cytosolic compartments and an increase at membranous structures (Fig.2D). In cells transfected with myristoylation-deficient G2AVILIP-1–GFP, no translocation could be observed (Fig.2E,F), indicating that myristoylation is necessary for a calcium-induced redistribution of VILIP-1 from cytosolic to membranous compartments.
Fig. 2.

Myristoylated VILIP-1–GFP but not myristoylation-deficient G2AVILIP-1–GFP or GFP alone shows a translocation in transfected COS-7 cells during addition of ionomycin. COS-7 cells were transiently transfected with GFP (A, B), myrVILIP-1–GFP (C, D), or G2AVILIP-1–GFP (E, F). Two days after transfection, cells were used for time-lapse microscopy. Living COS-7 cells expressing GFP before stimulation (A) and 7 min after raising the intracellular calcium concentration by addition of 2 μm ionomycin (B). COS-7 cells expressingmyrVILIP-1–GFP before (C) and 4 min after (D) addition of 2 μmionomycin. Note the decrease of fluorescence in the cytosol compared with untreated cells and the increase of fluorescence at the plasma membrane (arrows) and at intracellular structures (arrowheads). COS-7 cells expressing non-myristoylatedG2AVILIP-1–GFP before (E) and after (F) addition of 2 μm ionomycin. No redistribution of GFP fluorescence can be observed. Scale bars, 20 μm.
We next addressed the question whether the calcium-induced translocation of myrVILIP-1–GFP in COS-7 cells is reversible. Therefore, we lowered the calcium concentration in the incubation buffer by addition of EGTA immediately after a translocation was observed. Within 4 min after application of EGTA, 8 min after ionomycin application, the enrichment of fluorescence at the plasma membrane and intracellular sites disappeared (Fig.3, compare A, B), and a diffuse distribution of fluorescence comparable with untreated cells (Fig. 2C) reappeared (supplementary material, video 1) [for supplementary material, see the Journal of Neuroscience website (www.jneurosci.org)]. Higher magnification of a COS-7 cell clearly revealed the ionomycin-induced cell surface membrane association of myrVILIP-1–GFP (Fig.3C, arrows), which was completely reversible by EGTA treatment (Fig. 3D).
Fig. 3.

Ionomycin-induced translocation ofmyrVILIP-1–GFP can be completely reversed after depletion of calcium. COS-7 and NG108–15 cells were transiently transfected withmyrVILIP-1–GFP, and fluorescence was monitored as described in Materials and Methods. A, Living COS-7 cells expressing myrVILIP-1–GFP 4 min after addition of 2 μm ionomycin with increased fluorescence at the plasma membrane (arrows) and at intracellular structures (arrowheads). B, Reversal of the ionomycin-dependent localization of myrVILIP-1–GFP 8 min after addition of ionomycin and 4 min after addition of 5 mm EGTA to lower the calcium concentration in the incubation buffer (supplementary material, video 1). Magnification of a COS-7 cell expressing myrVILIP-1–GFP 4 min after addition of 2 μm ionomycin with increased fluorescence at the plasma membrane (arrows) and at intracellular structures (arrowheads) (C) and 8 min after addition of ionomycin and 4 min after addition of 5 mm EGTA showing reversal of myrVILIP-1–GFP localization (D). E, Living NG108–15 cells 4 min after addition of 2 μm ionomycin to the stimulation buffer. Note the strong fluorescence at the plasma membrane (arrows), especially at cell–cell contact sites.F, NG108–15 cells 8 min after addition of ionomycin and 4 min after addition of 5 mm EGTA. The plasma membrane localization of myrVILIP-1–GFP has completely disappeared. Scale bars: B, F, 25 μm;D, 12.5 μm.
Similarly, in the transiently transfected neuroblastoma × glioma hybrid cell line NG108–15, treatment with ionomycin led to a prominent plasma membrane localization of myrVILIP-1–GFP, especially at cell–cell contact sites (Fig. 3E). Membrane localization of VILIP-1 completely disappeared after addition of EGTA to the incubation medium comparable with the observation in COS-7 cells (Fig. 3D). These results indicate thatmyrVILIP-1–GFP can calcium-dependently localize to different subcellular compartments, including the cell surface membrane of COS-7 and neuroblastoma cells. Thus, the calcium–myristoyl switch of myrVILIP-1–GFP occurs in living cells and can function in both directions.
Real-time translocation of myristoylated VILIP-1–GFP in transfected hippocampal neurons after physiological stimulation
We next sought to examine whether a calcium-dependent translocation of VILIP-1 can be observed in cells that endogenously express the protein to create a more physiological basis for translocation experiments. It was shown recently by in situhybridization that VILIP-1 is highly expressed in all regions of the rat hippocampus (Paterlini et al., 2000). Primary hippocampal neurons in culture represent a simple and useful model system to study the localization and trafficking of heterologously expressed proteins. Therefore, we used this cell system as follows: (1) for translocation experiments after transfection of VILIP-1–GFP constructs, and (2) to study the subcellular localization of endogenous VILIP-1.
To test whether a translocation ofmyrVILIP-1–GFP can be observed after a physiological stimulus, we transiently transfected hippocampal neurons cultured for 6 d using the calcium phosphate precipitation method. A low expression of GFP–VILIP-1 fusion protein was observed as soon as after 4–6 hr (data not shown), and strong expression was seen 3 d after transfection (Fig. 1). Thus, for translocation experiments, neurons were used 3–6 d after transfection (9–12 d in culture). Hippocampal neurons, which at that time have reached stage 5 of differentiation according to Goslin and Banker (1998), were transferred into appropriate culture dishes for time-lapse image analysis as described for the cell lines.
The subcellular localization of myrVILIP-1–GFP and G2AVILIP-1–GFP in unstimulated cells did not differ from GFP alone, showing a diffuse distribution in all cellular compartments, including the soma, nucleus, axons, and dendrites (Fig.4A, formyrVILIP-1–GFP) (data not shown forG2AVILIP-1–GFP and GFP). To raise the intracellular calcium concentration in a more physiological manner compared with treatment with a calcium ionophore, 50 mm KCl or 100 μmglutamate/10 μm glycine (data not shown) was applied to the cells, and the fluorescence of the fusion proteins was monitored by taking images every 10 sec. In neurons transfected withmyrVILIP-1–GFP, depolarization with KCl induced an enrichment of fluorescence in intracellular structures, at the plasma membrane, and along dendritic processes. The translocation occurred rapidly, and clear changes in localization were seen within 20 sec. VILIP-1 reached a maximum redistribution within 2 min after depolarization (Fig. 4B).
Fig. 4.
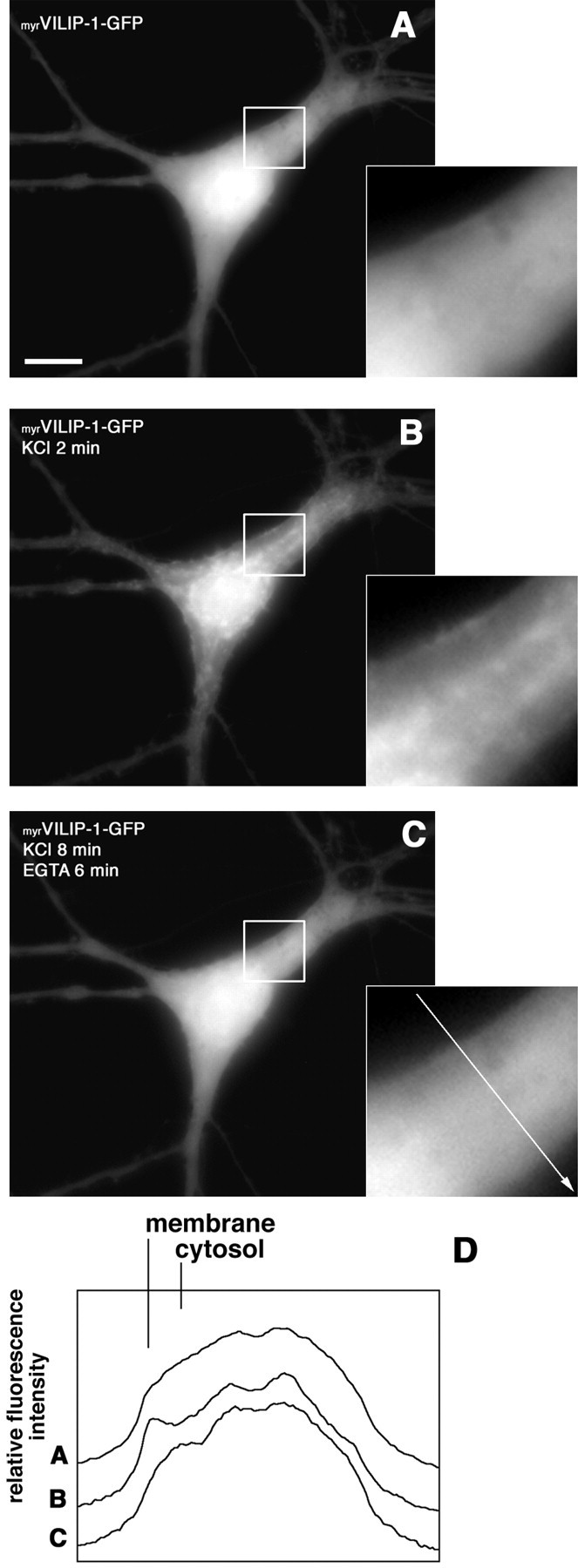
myrVILIP-1–GFP reversibly translocates to the plasma membrane and subcellular compartments in hippocampal neurons after a depolarizing stimulus. Hippocampal neurons transiently transfected with the calcium phosphate method were used for translocation experiments 6 d after transfection. A representative living neuron with diffusely distributed myrVILIP-1–GFP fluorescence before stimulation (A) and the same neuron 2 min after addition of 50 mm KCl to the stimulation buffer (B). The green fluorescence concentrates at specific sites of the plasma membrane and intracellular membranes. C, Reversibility ofmyrVILIP-1–GFP translocation after depletion of calcium from the stimulation buffer by addition of 5 mm EGTA (supplementary material, video 2). D, Comparison of the distribution for the fluorescence intensity ofmyrVILIP-1–GFP measured as pixel values (relative fluorescence intensity) from a cross section of the cell inA–C as indicated by the white arrow in the magnification in C. Scale bar, 10 μm.
Similar to the results obtained in transfected cell lines, the localization of myrVILIP–1-GFP could be reversed by chelating extracellular calcium with EGTA or by removal of KCl through an exchange of the incubation buffer. Figure4A shows a neuron before addition of KCl, Figure4B shows the same neuron after saturation of translocation 2 min after addition of 50 mm KCl, and, in Figure 4C, the same neuron is shown 6 min after removal of extracellular calcium when translocation was reversed again (supplementary material, video 2). In contrast, in cells expressingG2AVILIP-1–GFP or GFP, no translocation was observed (data not shown). Figure 4D compares the distribution of the fluorescence intensity before and after depolarization and EGTA treatment from a cross section of the cell, as indicated by the white arrow in the inserted magnification in Figure 4C. Note the peak of staining intensity at the plasma membrane and the reduced intensity in the cytosol of the stimulated cell (Fig. 4D, line B) compared with the unstimulated cell (Fig. 4D, line A).
These results indicate that an increase in the intracellular calcium concentration after neuronal activity can induce a rapid translocation of myrVILIP-1–GFP to membranes in hippocampal neurons, and this calcium-induced localization is reversible. Therefore, the calcium–myristoyl switch mechanism enables VILIP-1 to dynamically shuttle between different cellular compartments and probably to reversibly interact with putative interaction partners, such as signal effector proteins, in distinct membrane compartments.
Translocation of endogenously expressed VILIP-1 to Golgi membranes in cultured hippocampal neurons after treatment with glutamate as shown by colocalization with the Golgi-marker syntaxin-6
We examined the localization of endogenous VILIP-1 to different membrane compartments in cultured hippocampal neurons in greater detail. We addressed the question whether, in addition to the plasma membrane, endogenous VILIP-1 is able to translocate also to other compartments, such as Golgi membranes, as implicated by the observations in cell lines in response to a calcium stimulus. In COS-7 cells transiently transfected withmyrVILIP-1–GFP, we noticed a translocation of fluorescence to the plasma membrane and to intracellular sites such as the nuclear membrane and presumably the Golgi apparatus (Fig. 3). To detect endogenous VILIP-1, we used indirect immunofluorescence labeling with polyclonal VILIP-1 antibodies. The VILIP-1 antibody was shown recently to be highly specific for its antigen with a slight cross-reactivity to the 89% homologous protein VILIP-2 (Braunewell et al., 2001a). The specificity of the antibody was tested using GST-fusion proteins of four different NCS proteins, VILIP-1, VILIP-2, VILIP-3, and hippocalcin, expressed in E. coli and subjected to SDS-PAGE (Fig. 5A) and Western blot analysis (Fig. 5B,C). To prevent a cross-reaction with VILIP-2, which is also highly expressed in the hippocampus, we preabsorbed the VILIP-1 antibody with GST–VILIP-2 fusion proteins before incubating hippocampal neurons with the primary antibody. The anti-VILIP-1 antibody preabsorbed with GST–VILIP-2 was able to specifically recognize its antigen with barely detectable cross-reactivity to VILIP-2 (Fig. 5C), whereas preincubation with GST–VILIP-1 fusion protein as a control blocked the VILIP-1 immunoreactivity (Fig. 5B).
Fig. 5.

Preabsorbed VILIP-1 antibody is highly specific for its antigen. Equal amounts of GST fusion protein of four different NCS proteins (lane 1, VILIP-1; lane 2, VILIP-2; lane 3, VILIP-3; and lane 4, hippocalcin) were loaded on SDS gels and stained with Coomassie brilliant blue (A) or blotted onto nitrocellulose (B, C) and incubated with VILIP-1 antibodies. In B, VILIP-1 antibody was preabsorbed with a 100-fold excess of GST–VILIP-1. In C, the antibody was incubated with GST–VILIP-2. Molecular weights of marker proteins are indicated at the left in kilodaltons.
To investigate the activity-dependent subcellular localization of VILIP-1, hippocampal neurons were stimulated with 100 μmglutamate/10 μm glycine for 3 min before fixation. The cells were labeled with the preabsorbed polyclonal VILIP-1 antibody and a monoclonal antibody against syntaxin-6, which is a marker for thetrans-Golgi network (Bock et al., 1997). Endogenous VILIP-1 is highly expressed in hippocampal neurons and is distributed throughout the cell in the soma, as well as in neuritic processes (Fig.6A), comparable with the distribution of the GFP fusion protein (Fig. 4). Confocal images of nonstimulated (Fig. 6A–C, ctr) and stimulated hippocampal neurons (Fig. 6D–F, +glu) revealed a glutamate-induced colocalization (Fig. 6, compare C, F) of VILIP-1 (red fluorescence) with syntaxin-6 (green fluorescence), indicating that a calcium stimulus is able to localize endogenous VILIP-1 to an intracellular membrane compartment, the trans-Golgi network.
Fig. 6.

Endogenous VILIP-1 associates with the plasma membrane and with membranes of the Golgi apparatus after glutamate receptor activation. Hippocampal neurons cultured for 4 weeks were treated without (A–C, ctr) or with (D–F, +glu) 100 μmglutamate–10 μm glycine for 3 min before fixation. Neurons were incubated with polyclonal anti-VILIP-1 antibodies and secondary Cy3 anti-rabbit antibodies (A,D) and monoclonal anti-syntaxin-6 antibodies and secondary Alexa Fluor 488 anti-mouse antibodies (B,E), and fluorescence was monitored with confocal microscopy. C and F are merged images ofred (A, D; Cy3) andgreen (B, E; Alexa Fluor 488) fluorescence. Scale bar, 20 μm.
Localization of endogenously expressed VILIP-1 to the plasma membrane in cultured hippocampal neurons and translocation after physiological stimulation
In the next set of experiments, we investigated the cell surface membrane localization of endogenous VILIP-1 in more detail. Hippocampal neurons after 3 weeks in culture were fixed and stained for VILIP-1 (Fig. 7A, red fluorescence) and the dendritic marker protein MAP-2 (Fig.7B, green fluorescence). VILIP-1 was distributed in the soma, as well as in MAP-2-positive dendrites and MAP-2-negative axons (Fig. 7C, arrows mark an MAP-2-negative axon). A higher magnification (Fig. 7C, box) reveals the localization of VILIP-1 in dendrites with a typical punctate staining pattern in membranous structures of unstimulated neurons (Fig. 7G). This observation corresponds with results from cerebellar granule cell cultures and cerebellar slices in which VILIP-1 was found to be associated with the cell membrane at resting conditions (Spilker et al., 2000; Braunewell et al., 2001a).
Fig. 7.

Endogenous VILIP-1 is highly expressed in primary neurons and translocates to the plasma membrane after a physiological stimulus. Rat hippocampal neurons grown on glass coverslips were fixed after 4 weeks in culture and labeled with polyclonal anti-VILIP-1 antibodies and secondary Cy3 anti-rabbit antibodies (A) and monoclonal anti-MAP-2 antibodies and secondary Alexa Fluor 488 anti-mouse antibodies (B). C, Merged images ofred (A; Cy3) and green(B; Alexa Fluor 488) fluorescence (arrowsindicate MAP-2-negative axon). D, Double-stained primary neuron incubated with 50 mm KCl for 3 min before fixation shows strong labeling of the plasma membrane (arrows) around the soma with red VILIP-1 fluorescence (green MAP-2 fluorescence). E, Primary neuron incubated with 50 mm KCl for 3 min before fixation shows labeling of the soma plasma membrane and dendritic membranes with red VILIP-1 fluorescence.F, Enlarged section of the white box inE showing distinct punctate and additional patchy plasma membrane staining in a dendritic process after depolarization with KCl.G, Enlarged section of the white box inC showing punctate staining in dendritic processes without depolarization. Primary neurons without (H,I) and with (J,K) incubation with 50 mm KCl for 3 min before fixation were stained with polyclonal anti-VILIP-1 antibodies and secondary Alexa Fluor 488 anti-rabbit antibodies (H–K; pseudocolor red) and phalloidin-TRITC (H, J; pseudocolorgreen). After depolarization, a colocalization (J) of cortical actin (green) and VILIP-1 (red) at membranes of the soma and proximal dendrites can be found. Note that, in H–K, pseudocolors were used to match the red color for VILIP in the other figures. Scale bars, 10 μm.
Depolarization of neurons before fixation led to a prominent enrichment of VILIP-1 immunoreactivity at the plasma membrane around the soma (Fig. 7D, arrows, E), similar to the translocation observed for GFP–VILIP-1 (Fig. 4). Note that, after stimulation with KCl or glutamate, no clear localization with the Golgi can be observed because, in contrast to Figure 6, no confocal image analysis was applied. However, after depolarization VILIP-1 antibodies showed a clear enrichment of the protein in dendritic membranes (Fig.7E). At higher magnification (Fig. 7E,box), the localization of VILIP-1 in dendrites shows a still patchy but clear surface membrane staining pattern (Fig. 7F,arrows), most likely resembling dendritic membrane specializations. Interestingly, after glutamate stimulation, VILIP-1 colocalizes at the membrane around the soma and in proximal dendrites with the cortical actin cytoskeleton (Fig.7J,K) but shows no colocalization without stimulation (Fig.7H,I). These results indicate that an activity-dependent and calcium-mediated translocation to the cell surface membrane of hippocampal neurons around the soma and to distinct sites of dendritic membranes can be observed for endogenously expressed VILIP-1.
DISCUSSION
We used GFP as fluorescence tag for real-time imaging in living cell lines and primary hippocampal neurons to study the calcium–myristoyl switch properties and the activity-dependent subcellular localization of VILIP-1, a member of the NCS family of calcium binding proteins. Because several NCS proteins play roles in signal transduction pathways, we were interested whether the calcium-dependent membrane association of NCS proteins, the calcium–myristoyl switch, may play an active role as part of signal transduction processes in neurons. N-terminal myristoylation and calcium-dependent membrane association has been shown biochemically for a variety of NCS proteins (for review, see Braunewell and Gundelfinger, 1999). The three-dimensional structure of the photoreceptor protein recoverin has been analyzed in detail previously (Ames et al., 1995,1997), proving the concept of the calcium–myristoyl switch (Zozulya and Stryer, 1992). Binding of calcium triggers a conformational change within the protein, resulting in a rotation of the N-terminal part and exposure of the myristoyl moiety and hydrophobic residues, enabling the protein to associate with membranes and probably to activate their specific target molecules. Based on these data, it was hypothesized that calcium binding leads to a reversible translocation of NCS proteins to the plasma membrane.
However, this model may not be applicable for all NCS proteins. NMR and crystal structures of unmyristoylated guanylate cyclase-activating protein-2, neurocalcin δ, and frequenin (Ames et al., 1999;Vijay-Kumar and Kumar, 1999; Bourne et al., 2001) revealed a similar three-dimensional structure to that of recoverin, suggesting that the described calcium–myristoyl switch may occur in all NCS proteins. In contrast, biochemical data on myristoylated frequenin/NCS-1 show a constitutive membrane association independent of calcium binding (McFerran et al., 1999), which was explained by NMR studies on frequenin/NCS-1 (Freq1) from Saccharomyces cerevisiae, showing that the myristoyl group is exposed in both the calcium-free and the calcium-bound conformation (Hendricks et al., 1999; Ames et al., 2000b). In line with this notion, it has been reported that, in COS-7 cells that have been transfected with GFP–NCS-1, no effect on localization has been observed after ionomycin application (Zhao et al., 2001).
Based on the known structural and biochemical data, Meyer and York (1999) have proposed two different mechanism by which the cellular targets of calcium–myristoyl switch proteins are activated. In the translocation model, the myristoyl switch protein is located in the cytosol in the calcium-free state, and calcium binding leads to translocation to the membrane compartment and subsequent activation of the target molecule. This model applies to all NCS proteins showing a calcium–myristoyl switch. In the activation model, the myristoylated protein is constitutively membrane bound, and calcium binding triggers activation of the membrane-localized target enzyme. In this model, the myristoyl group may mediate the interaction between sensor and target protein. The second model applies for frequenin/NCS-1, which was shown to be constitutively localized at membranes (McFerran et al., 1999), but myristoylation was still essential for activity (Zhao et al., 2001).
These data implicate that, despite strong sequence and structure similarities among NCS proteins, the occurrence and exact mechanisms of the calcium–myristoyl switch still have to be elucidated for each protein. It may serve different functions, i.e., it may function in subcellular compartmentalization of a calcium signal and target activation or may rather be involved in direct calcium-dependent target activation. These different forms of a calcium–myristoyl switch most likely correlate to different types of signaling processes.
For VILIP-1, we could show that an increase in intracellular calcium concentration, by different means, such as depolarization or glutamate receptor activation, induces a translocation of myristoylated but not myristoylation-deficient VILIP-1 to specialized sites of the plasma membrane and to intracellular compartments, such as the Golgi complex, thus corresponding to the first calcium–myristoyl switch model. Using time-lapse microscopy, it was shown that the calcium–myristoyl switch is reversible and can occur within 10–20 sec after stimulation of a neuron. Thus, the switch fulfills several criteria of a fast signal transduction mechanism. It may reversibly shuttle calcium signals from or to cellular membranes, e.g., to sites close to the cortical actin cytoskeleton (this study; Lenz et al., 1996). However, VILIP-1 is already strongly associated with the plasma membrane at resting calcium levels in cerebellar granule cells (Spilker et al., 2000) and at least partly in hippocampal neurons (this study). It cannot be excluded that, in these neurons, calcium binding of VILIP-1 additionally triggers direct activation of membrane-localized target enzymes according to the second calcium–myristoyl switch model. Thus, different calcium–myristoyl switch mechanisms may be applicable for VILIP-1 depending on the signaling requirements of a cell type.
At resting calcium levels, we could observe small differences in localization of endogenous VILIP-1, which is already associated with the cell membrane, and myrVILIP-1–GFP, which shows a diffuse distribution. AlthoughmyrVILIP-1–GFP is still able to perform a calcium–myristoyl switch, as shown by biochemical experiments in transfected NG108–15 cells (data not shown), we cannot rule out that the GFP tag, which is larger than VILIP-1 itself, influences the calcium binding properties of the NCS protein by sterical interference or influencing proper folding during the calcium-induced conformational change. However myrVILIP-1–GFP is clearly able to localize to its intracellular targets similar to the endogenous protein, although it might be possible that higher calcium levels are necessary. We also saw an association ofmyrVILIP-1–GFP with nuclear membranes, which may be an artifact attributable to overexpression ofmyrVILIP-1–GFP. Nuclear association has not been observed for endogenous VILIP-1 in hippocampal cultures. However, under conditions of disturbed calcium homeostasis, e.g., in Alzheimer's disease, VILIP-1 shows a reduced expression level, an association with pathologic hallmarks, and an abnormal juxtanuclear localization (Braunewell et al., 2001b).
To evaluate the functional implications of the occurrence of the calcium–myristoyl switch in living cells, especially in hippocampal neurons, we investigated the subcellular localization of calcium-activated VILIP-1 in greater detail. A clear association of GFP–VILIP-1 and endogenous VILIP-1 with intracellular membranes such as the Golgi network was observed besides the association with the cell surface membrane. Interestingly, another member of the NCS protein family, NCS-1, was also shown to be localized to Golgi membranes. Furthermore, NCS-1 interacts and colocalizes with phosphatidylinositol 4-kinase at the Golgi and can influence membrane transport phenomena (Weisz et al., 2000; Zhao et al., 2001). In the future, it will be interesting to investigate whether the localization of VILIP-1 to Golgi membranes may also affect membrane trafficking.
Noteworthy in this context, cell surface membrane specializations, such as raft membranes, but also the cortical actin cytoskeleton are functionally interconnected with trans-Golgi and endoplasmic reticulum (ER) membranes, respectively, to form a membrane system implicated in membrane trafficking (Herreros et al., 2001;Lockwich et al., 2001). Neurocalcin α, the bovine counterpart of VILIP-1, was found recently to be enriched in raft compartments of rat brain synaptic membranes (Orito et al., 2001). These compartments are considered to be important in calcium signaling and were shown to contain many components of signal transducing pathways from receptor tyrosine kinases, G-proteins, glycosylphosphatidylinositol (GPI)-anchored proteins, and Src/tyrosine kinases to nitric oxide synthase (Galbiati et al., 2001). Because we observed localization of VILIP-1 to distinct membrane domains in resting and stimulated hippocampal neurons, these sites might be membrane rafts. Using the GPI-anchored raft membrane marker Thy-1 (Aarts et al., 1999), no strong overlap with VILIP-1 localization was observed (data not shown), indicating that VILIP-1 may localize to a yet unidentified lipid membrane compartment in dendrites. Especially in view of the described activation of signaling cascades by VILIP-1, it is likely that the calcium sensor localizes to subcompartments of the plasma membrane in which signal transduction events occur and a clustering of signaling proteins results in a cross talk and enhanced efficiency of signaling cascades (Galbiati et al., 2001). Recently, it was shown that olfactory knobs contain raft-like membrane specializations that can be found together with the olfactory adenylyl cyclase III (Schreiber et al., 2000). VILIP-1 is expressed in olfactory knobs containing the signaling machinery of olfactory neurons and influences olfactory adenylyl cyclase III (Boekhoff et al., 1997). Similarly, adenylyl cyclase VI (Fagan et al., 2000) and particulate guanylyl cyclase B (Doyle et al., 1997) have been found in membrane rafts of cells, and we showed previously that VILIP-1 can activate adenylyl cyclase VI (Braunewell et al., 1997), as well as different types of guanylyl cyclases, in a calcium- and myristoyl-dependent manner (Braunewell et al., 2001a). Another possible functional activity arises from the observation of Mathisen et al. (1999), who showed calcium-dependent interaction of VILIP-1 with double-stranded mRNA, such as the trkB receptor mRNA. Thus, it is possible that the calcium–myristoyl switch of VILIP-1 is a mechanism to translocate trkB mRNA to dendrites of hippocampal neurons and to subcellular compartments, such as ER membranes or cortical actin filaments, known to be involved in mRNA localization and protein translation.
In this study, it was shown that the calcium–myristoyl switch exists in living hippocampal neurons and can be influenced by neuronal activity and receptor activation. After stimulation, the switch from the cytosol to membranes, such as dendritic membrane substructures or Golgi membranes, occurs within a time window of 10–20 sec and is reversible. Thus, the calcium–myristoyl switch of the NCS protein VILIP-1 may provide a fast signaling mechanism to shuttle cellular signals and signaling molecules in a calcium-dependent manner to cellular compartments, e.g., to raft membranes, and thereby influence raft membrane-associated signaling effectors, such as adenylyl and guanylyl cyclases and, for example, to the cortical actin cytoskeleton and dendritic membranes and thereby influence dendritic protein translocation and translation. In general, the calcium–myristoyl switch may enable VILIP-1 to change the effective signaling properties of a neuron in a calcium-dependent manner. As hypothesized previously for a model cell line (Braunewell and Gundelfinger, 1997), a physiological role of VILIP-1 may be to influence the signaling and, thus, differentiation status of a neuron depending on the degree of VILIP-1 expression. Future studies with transgenic animals may shed light on the precise physiological role of VILIP-1 in the brain. It will be interesting to determine how the basal signaling mechanisms of VILIP-1 and its activity-dependent subcellular localization correlate with the known physiological functions of other NCS proteins (Gomez et al., 2001), e.g., as a molecular calcium switch for synaptic plasticity in the hippocampus.
Footnotes
This work was supported by Deutsche Forschungsgemeinschaft Grants Br-1579/2–1 and Br-1579/3–1 and Kultusministerium des Landes Sachsen-Anhalt Grant 2781A/0087G. We thank Dr. U. Kuchinke for advice with confocal microscopy.
Correspondence should be addressed to Karl-Heinz Braunewell, Humboldt-University Berlin, Neuroscience Research Center of the Charite, Signal Transduction Research Group, Tucholskystrasse 2, 10117 Berlin, Germany. E-mail: karl-heinz.braunewell@charite.de.
REFERENCES
- 1.Aarts LH, Verkade P, van Dalen JJ, van Rozen AJ, Gispen WH, Schrama LH, Schotman P. B-50/GAP-43 potentiates cytoskeletal reorganization in raft domains. Mol Cell Neurosci. 1999;14:85–97. doi: 10.1006/mcne.1999.0775. [DOI] [PubMed] [Google Scholar]
- 2.Ames JB, Tanaka T, Ikura M, Stryer L. Nuclear magnetic resonance evidence for Ca2+-induced extrusion of the myristoyl group of recoverin. J Biol Chem. 1995;270:30909–30913. doi: 10.1074/jbc.270.52.30909. [DOI] [PubMed] [Google Scholar]
- 3.Ames JB, Ishima R, Tanaka T, Gordon JI, Stryer L, Ikura M. Molecular mechanics of calcium-myristoyl switches. Nature. 1997;389:198–202. doi: 10.1038/38310. [DOI] [PubMed] [Google Scholar]
- 4.Ames JB, Dizhoor AM, Ikura M, Palczewski K, Stryer L. Three-dimensional structure of guanylyl cyclase activating protein-2, a calcium-sensitive modulator of photoreceptor guanylyl cyclases. J Biol Chem. 1999;274:19329–19337. doi: 10.1074/jbc.274.27.19329. [DOI] [PubMed] [Google Scholar]
- 5.Ames JB, Ikura M, Stryer L. Molecular structure of membrane-targeting calcium sensors in vision: recoverin and guanylate cyclase-activating protein 2. Methods Enzymol. 2000a;316:121–132. doi: 10.1016/s0076-6879(00)16720-5. [DOI] [PubMed] [Google Scholar]
- 6.Ames JB, Hendricks KB, Strahl T, Huttner IG, Hamasaki N, Thorner J. Structure and calcium-binding properties of Frq1, a novel calcium sensor in the yeast Saccharomyces cerevisiae. Biochemistry. 2000b;39:12149–12161. doi: 10.1021/bi0012890. [DOI] [PubMed] [Google Scholar]
- 7.Bock JB, Klumperman J, Davanger S, Scheller RH. Syntaxin 6 functions in trans-Golgi network vesicle trafficking. Mol Biol Cell. 1997;8:1261–1271. doi: 10.1091/mbc.8.7.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boekhoff I, Braunewell K-H, Andreini I, Breer H, Gundelfinger ED. The calcium-binding protein VILIP in olfactory neurons: regulation of second messenger signaling. Eur J Cell Biol. 1997;72:151–158. [PubMed] [Google Scholar]
- 9.Bourne Y, Dannenberg J, Pollmann V, Marchot P, Pongs O. Immunocytochemical localization and crystal structure of human frequenin (neuronal calcium sensor 1). J Biol Chem. 2001;276:11949–11955. doi: 10.1074/jbc.M009373200. [DOI] [PubMed] [Google Scholar]
- 10.Braunewell KH, Gundelfinger ED. Low level expression of calcium-sensor protein VILIP induces cAMP-dependent differentiation in rat C6 glioma cells. Neurosci Lett. 1997;234:139–142. doi: 10.1016/s0304-3940(97)00696-4. [DOI] [PubMed] [Google Scholar]
- 11.Braunewell KH, Gundelfinger ED. Intracellular neuronal calcium sensor proteins: a family of EF-hand calcium-binding proteins in search of a function. Cell Tissue Res. 1999;295:1–12. doi: 10.1007/s004410051207. [DOI] [PubMed] [Google Scholar]
- 12.Braunewell KH, Spilker C, Behnisch T, Gundelfinger ED. The neuronal calcium-sensor protein VILIP modulates cyclic AMP accumulation in stably transfected C6 glioma cells: amino-terminal myristoylation determines functional activity. J Neurochem. 1997;68:2129–2139. doi: 10.1046/j.1471-4159.1997.68052129.x. [DOI] [PubMed] [Google Scholar]
- 13.Braunewell KH, Brackmann M, Schaupp M, Spilker C, Anand R, Gundelfinger ED. Intracellular neuronal calcium sensor (NCS) protein VILIP-1 modulates cGMP signalling pathways in transfected neural cells and cerebellar granule neurones. J Neurochem. 2001a;78:1277–1286. doi: 10.1046/j.1471-4159.2001.00506.x. [DOI] [PubMed] [Google Scholar]
- 14.Braunewell KH, Riederer P, Spilker C, Gundelfinger ED, Bogerts B, Bernstein HG. Abnormal localization of two neuronal calcium sensor proteins, visinin-like proteins (vilips)-1 and -3, in neocortical brain areas of Alzheimer disease patients. Dement Geriatr Cogn Disord. 2001b;12:110–116. doi: 10.1159/000051244. [DOI] [PubMed] [Google Scholar]
- 15.Burgoyne RD, Weiss JL. The neuronal calcium sensor family of Ca2+-binding proteins. Biochem J. 2001;353:1–12. [PMC free article] [PubMed] [Google Scholar]
- 16.Casey PJ. Protein lipidation in cell signaling. Science. 1995;268:221–225. doi: 10.1126/science.7716512. [DOI] [PubMed] [Google Scholar]
- 17.Doyle DD, Ambler SK, Upshaw-Earley J, Bastawrous A, Goings GE, Page E. Type B atrial natriuretic peptide receptor in cardiac myocyte caveolae. Circ Res. 1997;81:86–91. doi: 10.1161/01.res.81.1.86. [DOI] [PubMed] [Google Scholar]
- 18.Fagan KA, Smith KE, Cooper DM. Regulation of the Ca2+-inhibitable adenylyl cyclase type VI by capacitative Ca2+ entry requires localization in cholesterol-rich domains. J Biol Chem. 2000;275:26530–26537. doi: 10.1074/jbc.M001369200. [DOI] [PubMed] [Google Scholar]
- 19.Galbiati F, Razani B, Lisanti MP. Emerging themes in lipid rafts and caveolae. Cell. 2001;106:403–411. doi: 10.1016/s0092-8674(01)00472-x. [DOI] [PubMed] [Google Scholar]
- 20.Gomez M, De Castro E, Guarin E, Sasakura H, Kuhara A, Mori I, Bartfai T, Bargmann CI, Nef P. Ca2+ signaling via the neuronal calcium sensor-1 regulates associative learning and memory in C. elegans. Neuron. 2001;30:241–248. doi: 10.1016/s0896-6273(01)00276-8. [DOI] [PubMed] [Google Scholar]
- 21.Goslin K, Banker G. Rat hippocampal neurons in low-density culture. In: Banker G, Goslin K, editors. Culturing nerve cells, Ed 2. MIT; Cambridge, MA: 1998. pp. 339–370. [Google Scholar]
- 22.Hendricks KB, Wang BQ, Schnieders EA, Thorner J. Yeast homologue of neuronal frequenin is a regulator of phosphatidylinositol-4-OH kinase. Nat Cell Biol. 1999;1:234–241. doi: 10.1038/12058. [DOI] [PubMed] [Google Scholar]
- 23.Herreros J, Ng T, Schiavo G. Lipid rafts act as specialized domains for tetanus toxin binding and internalization into neurons. Mol Biol Cell. 2001;12:2947–2960. doi: 10.1091/mbc.12.10.2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kennedy MT, Brockman H, Rusnak F. Contributions of myristoylation to calcineurin structure/function. J Biol Chem. 1996;271:26517–26521. doi: 10.1074/jbc.271.43.26517. [DOI] [PubMed] [Google Scholar]
- 25.Köhrmann M, Haubensak W, Hemraj I, Kaether C, Lessmann VJ, Kiebler MA. Fast, convenient, and effective method to transiently transfect primary hippocampal neurons. J Neurosci Res. 1999;58:831–835. [PubMed] [Google Scholar]
- 26.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 27.Lenz SE, Braunewell KH, Weise C, Nedlina-Chittka A, Gundelfinger ED. The neuronal EF-hand Ca2+-binding protein VILIP: interaction with cell membrane and actin-based cytoskeleton. Biochem Biophys Res Commun. 1996;225:1078–1083. doi: 10.1006/bbrc.1996.1298. [DOI] [PubMed] [Google Scholar]
- 28.Lockwich T, Singh BB, Liu X, Ambudkar IS. Stabilization of cortical actin induces internalization of transient receptor potential 3 (Trp3)-associated caveolar Ca2+ signaling complex and loss of Ca2+ influx without disruption of Trp3-inositol trisphosphate receptor association. J Biol Chem. 2001;276:42401–42408. doi: 10.1074/jbc.M106956200. [DOI] [PubMed] [Google Scholar]
- 29.Mathisen PM, Johnson JM, Kawczak JA, Tuohy VK. Visinin-like protein (VILIP) is a neuron-specific calcium-dependent double-stranded RNA-binding protein. J Biol Chem. 1999;274:31571–31576. doi: 10.1074/jbc.274.44.31571. [DOI] [PubMed] [Google Scholar]
- 30.McFerran BW, Weiss JL, Burgoyne RD. Neuronal Ca2+ sensor 1. Characterization of the myristoylated protein, its cellular effects in permeabilized adrenal chromaffin cells, Ca2+-independent membrane association, and interaction with binding proteins, suggesting a role in rapid Ca2+ signal transduction. J Biol Chem. 1999;274:30258–30265. doi: 10.1074/jbc.274.42.30258. [DOI] [PubMed] [Google Scholar]
- 31.McLaughlin S, Aderem A. The myristoyl-electrostatic switch: a modulator of reversible protein-membrane interactions. Trends Biochem Sci. 1995;20:272–276. doi: 10.1016/s0968-0004(00)89042-8. [DOI] [PubMed] [Google Scholar]
- 32.Meyer T, York JD. Calcium-myristoyl switches turn on new lights. Nat Cell Biol. 1999;1:E93–E95. doi: 10.1038/12090. [DOI] [PubMed] [Google Scholar]
- 33.Orito A, Kumanogoh H, Yasaka K, Sokawa J, Hidaka H, Sokawa Y, Maekawa S. Calcium-dependent association of annexin VI, protein kinase C alpha, and neurocalcin alpha on the raft fraction derived from the synaptic plasma membrane of rat brain. J Neurosci Res. 2001;64:235–241. doi: 10.1002/jnr.1071. [DOI] [PubMed] [Google Scholar]
- 34.Paterlini M, Revilla V, Grant AL, Wisden W. Expression of the neuronal calcium sensor protein family in the rat brain. Neuroscience. 2000;99:205–216. doi: 10.1016/s0306-4522(00)00201-3. [DOI] [PubMed] [Google Scholar]
- 35.Randazzo PA, Terui T, Sturch S, Fales HM, Ferrige AG, Kahn RA. The myristoylated amino terminus of ADP-ribosylation factor 1 is a phospholipid- and GTP-sensitive switch. J Biol Chem. 1995;270:14809–14815. doi: 10.1074/jbc.270.24.14809. [DOI] [PubMed] [Google Scholar]
- 36.Resh MD. Fatty acylation of proteins: new insights into membrane targeting of myristoylated and palmitoylated proteins. Biochim Biophys Acta. 1999;1451:1–16. doi: 10.1016/s0167-4889(99)00075-0. [DOI] [PubMed] [Google Scholar]
- 37.Schreiber S, Fleischer J, Breer H, Boekhoff I. A possible role for caveolin as a signaling organizer in olfactory sensory membranes. J Biol Chem. 2000;275:24115–24123. doi: 10.1074/jbc.M001876200. [DOI] [PubMed] [Google Scholar]
- 38.Spilker C, Richter K, Smalla KH, Manahan-Vaughan D, Gundelfinger ED, Braunewell KH. The neuronal EF-hand calcium-binding protein visinin-like protein-3 is expressed in cerebellar Purkinje cells and shows a calcium-dependent membrane association. Neuroscience. 2000;96:121–129. doi: 10.1016/s0306-4522(99)00536-9. [DOI] [PubMed] [Google Scholar]
- 39.Takasaki A, Hayashi N, Matsubara M, Yamauchi E, Taniguchi H. Identification of the calmodulin-binding domain of neuron-specific protein kinase C substrate protein CAP-22/NAP-22. Direct involvement of protein myristoylation in calmodulin-target protein interaction. J Biol Chem. 1999;274:11848–11853. doi: 10.1074/jbc.274.17.11848. [DOI] [PubMed] [Google Scholar]
- 40.Teruel MN, Meyer T. Translocation and reversible localization of signaling proteins: a dynamic future for signal transduction. Cell. 2000;103:181–184. doi: 10.1016/s0092-8674(00)00109-4. [DOI] [PubMed] [Google Scholar]
- 41.Towler DA, Gordon JI, Adams SP, Glaser L. The biology and enzymology of eukaryotic protein acylation. Annu Rev Biochem. 1988;57:69–99. doi: 10.1146/annurev.bi.57.070188.000441. [DOI] [PubMed] [Google Scholar]
- 42.Vijay-Kumar S, Kumar VD. Crystal structure of recombinant bovine neurocalcin. Nat Struct Biol. 1999;6:80–88. doi: 10.1038/4956. [DOI] [PubMed] [Google Scholar]
- 43.Weisz OA, Gibson GA, Leung SM, Roder J, Jeromin A. Overexpression of frequenin, a modulator of phosphatidylinositol 4-kinase, inhibits biosynthetic delivery of an apical protein in polarized madin-darby canine kidney cells. J Biol Chem. 2000;275:24341–24347. doi: 10.1074/jbc.M000671200. [DOI] [PubMed] [Google Scholar]
- 44.Zhao X, Varnai P, Tuymetova G, Balla A, Toth ZE, Oker-Blom C, Roder J, Jeromin A, Balla T. Interaction of neuronal calcium sensor-1 (NCS-1) with phosphatidylinositol 4-kinase beta stimulates lipid kinase activity and affects membrane trafficking in COS-7 cells. J Biol Chem. 2001;276:40183–40189. doi: 10.1074/jbc.M104048200. [DOI] [PubMed] [Google Scholar]
- 45.Zozulya S, Stryer L. Calcium-myristoyl protein switch. Proc Natl Acad Sci USA. 1992;89:11569–11573. doi: 10.1073/pnas.89.23.11569. [DOI] [PMC free article] [PubMed] [Google Scholar]