

Abstract
Neurodegenerative disorders in humans may be triggered or exacerbated by exposure to occupational or environmental agents. Here, we show that a brief exposure to methylisothiazolinone, a widely used industrial and household biocide, is highly toxic to cultured neurons but not to glia. We also show that the toxic actions of this biocide are zinc dependent and require the activation of p44/42 extracellular signal-regulated kinase (ERK) via a 12-lipoxygenase-mediated pathway. The cell death process also involves activation of NADPH oxidase, generation of reactive oxygen species, DNA damage, and overactivation of poly(ADP-ribose) polymerase, all occurring downstream from ERK phosphorylation. The toxic effects of methylisothiazolinone and related biocides on neurons have not been reported previously. Because of their widespread use, the neurotoxic consequences of both acute and chronic human exposure to these toxins need to be evaluated.
Keywords: neurotoxicity, isothiazolone, biocide, oxidation, necrosis, zinc, glutathione, ERK, lipoxygenase, NADPH oxidase, PARP
There is mounting evidence that a number of neurodegenerative disorders may be triggered or exacerbated by exposure to occupational or environmental agents (Lohmann et al., 1996; Gorell et al., 1999; Andersen et al., 2000; Betarbet et al., 2000; Olden and Guthrie, 2001). Unfortunately, we have little or no information about the potential negative impact on the brain for many commonly used substances. In this study, we have focused on 2-methyl-4-isothiazolin-3-one (MIT), a cyclic, sulfur-containing biocide for which there are no published data available regarding its neurotoxic properties, although we have found it to be highly toxic to neurons in vitro.
Isothiazolinone (or isothiazolone)-derived biocides, such as Kathon CG [a 3:1 mixture of 5-chloro-2-methyl-4-isothiazolin-3-one (CMIT) and MIT], are widely used for controlling microbial growth in water-containing solutions (Collier et al., 1990). The biocidal applications of these agents range from industrial water storage tanks to cooling units, in processes as varied as mining to energy production. Their widespread use has resulted in a large number of reported cases of human occupational exposure. This occurs primarily, but not exclusively, when workers come into contact with stock solutions (containing 15 gm/l or 0.1 m of the active ingredients) during the dilution process, usually resulting in caustic burns, contact dermatitis, and allergic sensitization (Ng and Tay, 1996; Primka and Taylor, 1997). Nonoccupational exposure to isothiazolinones by the general population also occurs, albeit at much lower concentrations. Because of their use in dehumidifiers, these compounds can be detected in air-conditioned indoor air (Nagorka et al., 1990) and are also present in a very large number of commonly used cosmetics (Rastogi, 1990). The long-term consequences of low-level chronic exposure to isothiazolinones on the CNS have not been investigated.
Isothiazolinones kill microorganisms by interacting and oxidizing accessible cellular thiols (Collier et al., 1990). We have shown previously that another thiol oxidant, 2,2′-dithiodipyridine (DTDP), induces neuronal cell death by liberating zinc from intracellular, thiol-containing stores, such as metal-binding proteins (Aizenman et al., 2000). The intracellular release of Zn2+ can lead to activation of p38 mitogen-activated protein (MAP) kinase (MAPK), subsequent enhancement of voltage-gated potassium currents, and caspase-dependent cell death (McLaughlin et al., 2001). A similar zinc-p38-K channel pathway can be activated by nitric oxide-derived species (Pal et al., 2001). We hypothesized that isothiazolinone biocides would be neurotoxic by activating similar signaling molecules. In this study, we describe the neurotoxic cascade induced by MIT, which, contrary to our expectations, proceeds via the activation of p44/42 extracellular signal-regulated kinase (ERK) MAPK, rather than p38, and culminates in the demise of neurons via a caspase-independent pathway.
MATERIALS AND METHODS
Materials.Bis[amino([1-aminophenyl]thio)methylene]butanedinitrile (U0126), 2′-amino-3′-methoxyflavone (PD98059), 8-hydroxy-2-methylquinazoline-4-one (NU1025), andN-(5-(3-dimethylaminobenzamido)-2-methylphenyl)-4-hydroxybenzamide (ZM336372) were purchased from Calbiochem (La Jolla, CA).trans-1-(4-hydroxycyclohexyl)-4-(fluorophenyl)-5-(2-methoxy-pyrimidin-4-yl)imidazole (SB239063) was a gift from GlaxoSmithKline Pharmaceuticals (King of Prussia, PA). ERK and p38 antibodies were from Cell Signaling Technology (Beverly, MA); 12-lipoxygenase (12-LOX) antibody was from Cayman Chemical Co. (Ann Arbor, MI). Unless specified, all other chemicals were purchased from Sigma (St. Louis, MO).
Cell culture and toxicity assay. Mixed cultures of rat cortical neurons and glia were prepared as described previously (Hartnett et al., 1997). In brief, cortices from embryonic day 16 Sprague Dawley rat fetuses were isolated and incubated in trypsin for 2 hr at 37°C. Cortices were dissociated in 10 ml of plating medium containing DMEM (Invitrogen, Gaithersburg, MD), 10% F12 nutrients, and 10% bovine calf serum (Hyclone, Logan, UT). Astrocyte cultures were prepared from postnatal day 5 Swiss-Webster mice as described by Noble and Mayer-Proschel (1998) and were a kind gift from C. Lagenaur (University of Pittsburgh). A few toxicity experiments were performed on neuron-enriched cultures (Aizenman et al., 2000; McLaughlin et al., 2001), but we found that in this system, MIT toxicity had a moderate excitotoxic component, likely because of the inability of these cultures to effectively take up glutamate released by injured or dying cells (Rosenberg and Aizenman, 1989). As such, proteins for immunoblots (see below) were generally harvested from MIT-treated, neuron-enriched cultures that had also been exposed to the NMDA receptor antagonist (+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate (MK-801; 10 μm). We found, however, that the expression profile of proteins of interest was unaffected by the presence of MK-801, suggesting that the excitotoxic component was a late event in cell death and did not impact on the upstream signaling pathways reported here. All toxicity experiments shown were performed on mixed 4-week-old cultures in which the excitotoxic component was absent and would not confound the interpretation of the results. Cells were exposed to MIT for 10 min in MEM (plus 0.01% BSA and 25 mm HEPES). Unless otherwise noted, cells were normally exposed to neuroprotective compounds 10 min before, during, and in the 18–20 hr after MIT exposure. In addition to the pre-exposure and coexposure period,N,N,N′,N′-tetrakis (2-pyridylmethyl) ethylenediamine (TPEN) was included for 10 min in the postexposure period. Cell viability was determined 18–20 hr after MIT exposure using a lactate dehydrogenase (LDH)-based in vitro toxicity assay kit (Sigma). Toxicity data were either represented as averaged raw LDH values or normalized to the toxicity induced by 100 μm MIT alone (100% toxicity) (Fig. 1). In the former case, an ANOVA followed by post hoc Bonferroni tests were used to assess a significant deviation from control, whereas in the latter case, one-sample tests (vs 100%) were performed.
Fig. 1.
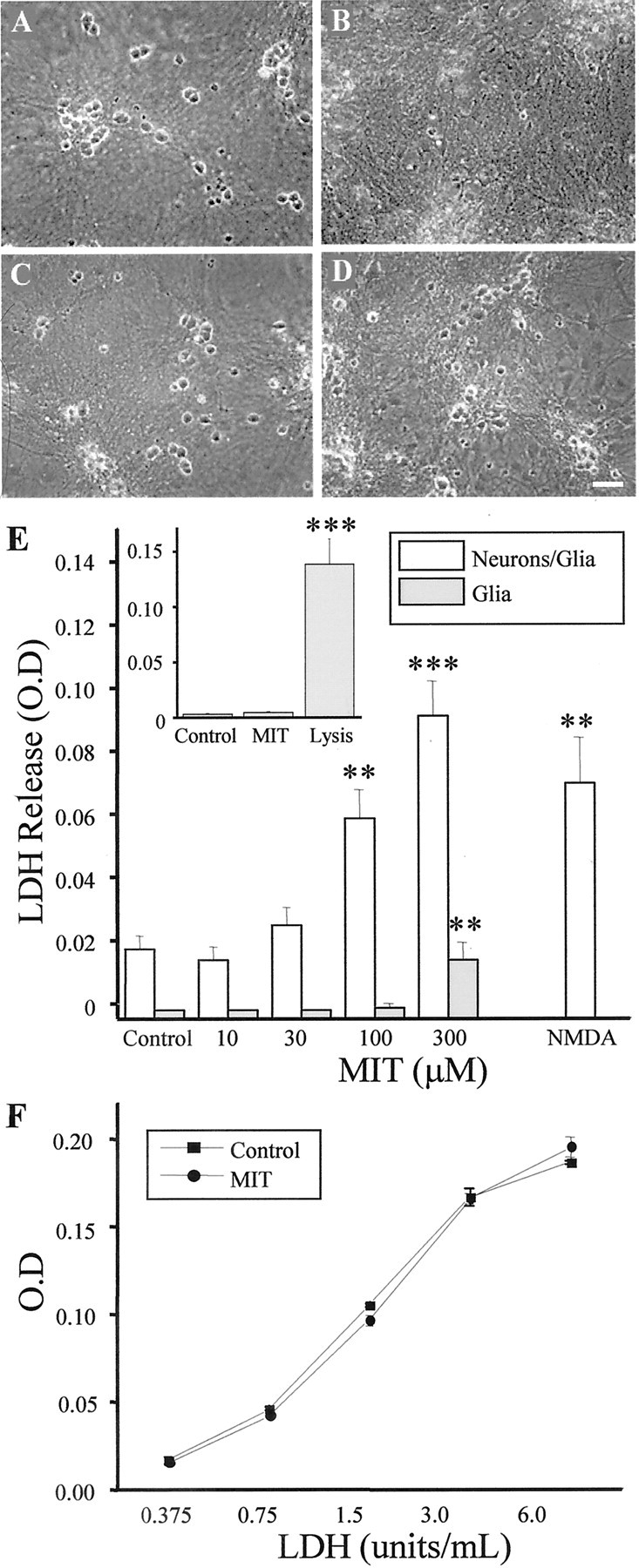
MIT is neurotoxic in vitro.A–D, Phase–contrast micrographs of rat cortical cultures 24 hr after being treated for 10 min with either vehicle (A), 100 μm MIT (B), 10 μm TPEN (C), or 100 μm MIT plus 10 μm TPEN (D). Note the relative absence of phase–bright (live) neurons in B and the neuroprotective actions of TPEN in D. E, Concentration–toxicity relationship for MIT in control mixed cultures (Neurons/Glia) and in sister cultures that had been treated 72 hr earlier with kainic acid (1 mm, 24 hr) to remove the neuronal component (Glia). MIT results in a large increase in LDH release (an index of cell death) in the mixed but not in the glial cultures. LDH release induced by a 1 hr exposure to 200 μm NMDA (a selective neuronal toxin) is included for comparison. Values represent the mean ± SD for a total of six experiments in the mixed cultures and three experiments in the kainate-treated cells; ∗∗p < 0.01; ∗∗∗p < 0.001. The inset shows a representative experiment, in triplicate (mean ± SD), performed on primary mouse astrocytes. A 10 min exposure to 100 μmMIT was not toxic to the cells. Total LDH in the culture was measured after cell lysis; ∗∗∗p < 0.001.F, LDH activity measured with known concentrations of the enzyme alone or in the continuous presence of 100 μmMIT. No differences were observed between the two standard curves. Values represent the mean ± SD of three independent measurements.
Western blot assessment of MAPK activation. Proteins were harvested from neuron-enriched cultures to prevent a potential glial contamination of the signal. All results were confirmed with immunostaining and with immunoblots using proteins harvested from mixed cultures. Total protein extract was prepared as described previously (McLaughlin et al., 2001). Equal protein concentrations were separated using 7.5% Criterion gels (Bio-Rad, Hercules, CA). Proteins were then transferred to polyvinylidene difluoride membranes (Amersham Biosciences, Piscataway, NJ) and blocked for 1 hr at room temperature with 5% milk in PBS/0.1% Tween 20. Membranes were then incubated overnight in primary antibodies against ERK and p38 MAPK diluted 1:1000 in blocking solution. Membranes were then treated with an HRP-conjugated anti-rabbit secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA) for 1 hr at room temperature, placed in 2 ml of ECL substrate (Amersham Biosciences) for 1 min at room temperature, and exposed to X-OMAT film (VWR Scientific, West Chester, PA).
Immunostaining. Cultures were fixed in 10% formaldehyde and then permeabilized with 0.3% Triton X-100. Cells were blocked with 1% BSA diluted in PBS and incubated in 12-LOX primary antibody (1:100) overnight. Cultures were then washed in PBS for a total of 20 min and incubated in cy-2 anti-rabbit secondary antibody for 60 min. After additional washes, coverslips were mounted, and fluorescence was visualized with an Olympus AX70 confocal microscope system. OlympusFluoview software was used to scan and view the resulting images.
Glutathione assay. Cultures were exposed to 100 μm MIT for 10 min and rinsed with MEM. Five minutes later, cells were rinsed, scraped off the dish, and resuspended in a lysis buffer on ice. The supernatant was collected and incubated with a solution containing monochlorobimane (MCB), a dye that has a high affinity for glutathione (GSH) (Biovision, Mountain View, CA). Free MCB is nonfluorescent, whereas the GSH-MCB adduct emits at 461 nm after excitation at 380 nm. Fluorescence measurements were performed in a fluorimetric plate reader and normalized to protein content (BCA; Pierce, Rockford, IL).
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling staining. Cells were fixed in 4% paraformaldehyde for terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) staining as per the manufacturer's specifications (Promega, Madison, WI). Briefly, cells were washed twice in PBS and permeabilized in 0.1% sodium citrate with 0.1% Triton X-100. Biotinylated nucleotide was incorporated at the 3′-OH DNA. Positive control slides were generated by incubating cells with DNase I, whereas adding labeling mix without terminal transferase to DNase-treated cells generated negative control slides. Horseradish peroxidase-labeled streptavidin was then bound to biotinylated nucleotides and reacted with diaminobenzidine. Cells were counterstained with 0.25% thionin.
Electrophysiological measurements of K+currents. Electrophysiological recordings were performed using the whole-cell configuration of the patch-clamp technique as described previously (McLaughlin et al., 2001). The extracellular solution contained (in mm): 115 NaCl, 2.5 KCl, 2 MgCl2, 10 HEPES, 0.1 1,2-bis(2-aminophenoxy)ethane-N,N,N,N-tetraacetic acid, and 10 d-glucose, pH 7.2; 0.1 μm tetrodotoxin was added to inhibit voltage-gated sodium channels. The intracellular (electrode) solution contained (in mm): 120 KCl, 1.5 MgCl2, 1 CaCl2, 2 Na2ATP, 1 BAPTA, and 10 HEPES, pH 7.2. Measurements were obtained under voltage clamp with an Axopatch 200 amplifier (Axon Instruments, Foster City, CA) and pClamp software (Axon Instruments) using 2 MΩ electrodes. Partial compensation (≥80%) for series resistance was performed in all cases. Currents were filtered at 2 kHz and digitized at 10 kHz (Digidata; Axon Instruments). Potassium currents were evoked with a series of incremental 80 msec voltage steps to 35 mV from a holding potential of −70 mV. Steady-state current amplitudes were measured relative to baseline 70 msec after the initiation of each voltage step and normalized to cell capacitance.
RESULTS
MIT is neurotoxic to neurons in culture
Rat cortical cultures exposed for 10 min to 100 μmMIT underwent widespread neuronal cell death within 24 hr. MIT toxicity spared the underlying glial cell layer (Fig. 1A,B). Exposure to increasing concentrations of MIT augmented the number of injured neurons, as measured by the release of LDH from the cultures (Fig. 1E). Sister cultures that had been exposed previously to 1 mm kainate overnight to remove the neuronal component of the cultures (Aizenman et al., 2000) showed a much less pronounced increase in LDH release on MIT exposure (Fig.1E), suggesting that gliotoxicity was relatively low even at high concentrations of this agent. This observation was confirmed in primary astrocyte cultures (Fig. 1E,inset). This argues that neurons are appreciably more sensitive than glial cells to the toxic actions of MIT. Although cells are exposed to MIT for only 10 min, and the LDH is assayed 24 hr after the MIT exposure, we confirmed that MIT (100 μm) does not interfere with the cell death assay itself by using known concentrations of the enzyme (Fig.1F). We also evaluated whether NMDA receptor-mediated excitotoxicity was indirectly responsible for the effects of MIT. Inclusion of 10 μm MK-801 during and after MIT exposure did not afford any neuroprotection to the cultures (data not shown). NMDA receptors are solely responsible for the induction of glutamate excitotoxicity in our system (Aizenman and Hartnett, 1992;Sinor et al., 2000).
A role for Zn2+ in MIT toxicity
Previous studies in our laboratory reported that thiol-oxidizing agents, such as DTDP, triggered neuronal cell death by inducing intracellular zinc release (Aizenman et al., 2000; McLaughlin et al., 2001). Thus, we sought to investigate whether MIT toxicity might also involve zinc. We observed that TPEN, a cell-permeable, metal-chelating agent with very high affinity for zinc and iron and only moderate affinity for calcium (Arslan et al., 1985), rescued MIT-induced neuronal death in a concentration-dependent manner (Figs.1C,D, 2A). The protective effects of 10 μm TPEN were confirmed with cell counts (Fig. 2A,inset). Pretreatment of 1 μm TPEN with equal molar zinc chloride abolished its neuroprotective properties (Fig. 2B). In contrast, TPEN pretreated with equal molar iron was still able to protect neurons from MIT toxicity (Fig.2B). A similar paradigm was used against DTDP toxicity where an identical outcome was observed (Fig. 2C). These results strongly suggest that zinc contributes to MIT neurotoxicity.
Fig. 2.

Role of Zn2+ in MIT toxicity.A, Cortical cultures were exposed to 100 μm MIT (10 min) in the absence or presence of various concentrations of TPEN. LDH release for each experimental group was normalized to that generated by MIT alone (100% relative toxicity) in this and subsequent figures. Note that the toxicity after MIT exposure decreases as a function of TPEN concentration. Values represent the mean ± SEM (n = 4); ∗∗p < 0.01; ∗∗∗p < 0.001; significantly different from MIT alone. The insetconfirms the neuroprotective actions of 1 μm TPEN, as determined by cell counting; ∗∗∗p < 0.001.B, The neuroprotective action of 1 μm TPEN was eliminated by preincubating the chelating agent with equimolar zinc (ZnCl2) but not iron (FeSO4); ∗p < 0.05; ∗∗p < 0.01 (n = 4). C, Similar toB above, except that neurons were killed by 100 μm DTDP instead of MIT (Aizenman et al., 2000; McLaughlin et al., 2001) (n = 3).
MIT-induced cell death is triggered by ERK activation
Because zinc appears to play a critical role in both DTDP and MIT toxicity, we anticipated that downstream-signaling pathways leading to cell death would be similar. In our previous study with DTDP, we observed that intracellularly liberated zinc activated p38 MAPK within 30 min, which in turn induced an enhancement of voltage-dependent K+currents and caspase cleavage, measured 3–6 hr later (McLaughlin et al., 2001). Therefore, we investigated whether p38 MAPK activation and K+ channel enhancement would follow MIT exposure. In contrast to our results with DTDP, phosphorylation of p38 was completely absent in cells treated with MIT (Fig.3A, top panels). In addition, SB239063, a specific p38 MAPK inhibitor that is effective in protecting neurons against DTDP toxicity (McLaughlin et al., 2001), did not afford any neuroprotection against MIT (Fig. 3B). Moreover, whole-cell patch-clamp recordings did not reveal any delayed enhancement of voltage-gated K+ currents after MIT treatment (Fig. 3C). Consistent with this finding, neither tetraethylammonium (TEA) nor high extracellular K+ concentrations (25 mm) had any significant effects on MIT-induced cell death (Fig. 3D), suggesting that K+ efflux (Yu et al., 1997) was not involved in the MIT-induced death pathway.
Fig. 3.
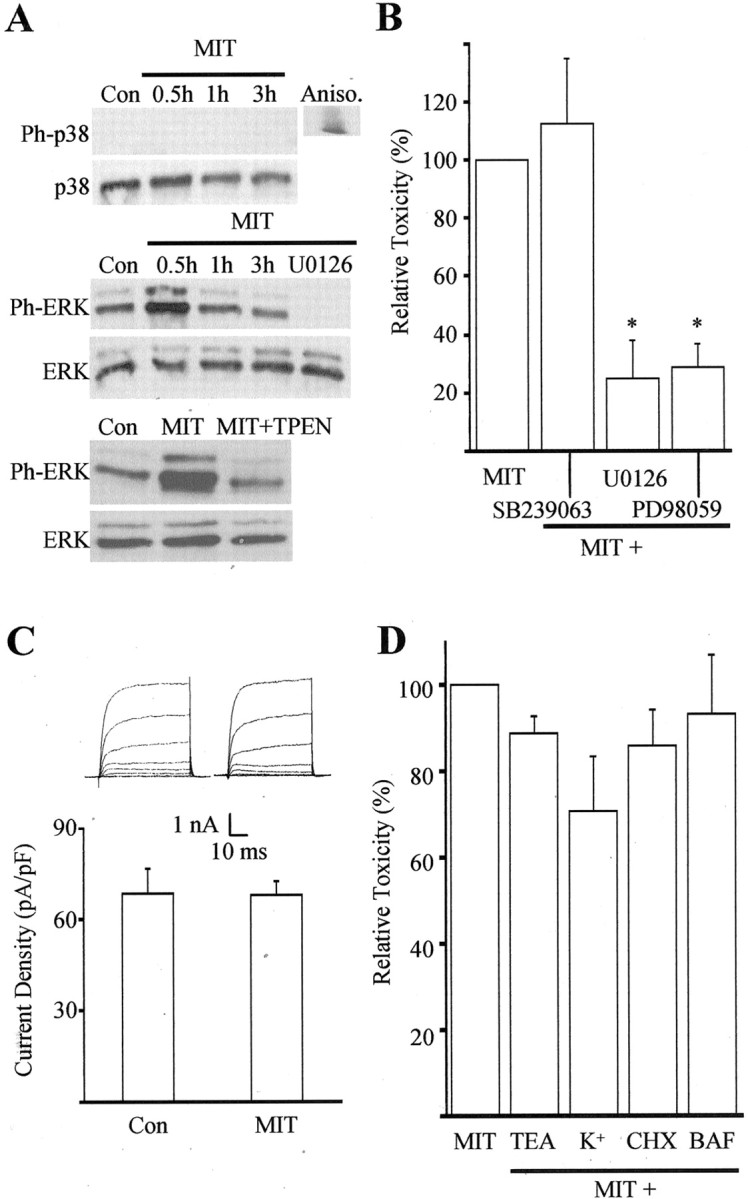
MIT toxicity involves ERK activation.A, Immunoblots of cell extracts from cortical cultures harvested at various time points after a 10 min exposure to 100 μm MIT. Proteins were separated by SDS-PAGE and probed with antibodies specific to the phosphorylated and nonphosphorylated forms of both p38 and p44/42 ERK. Note the absence of phosphorylated p38 (Ph-p38) at all time points and the early increase in phosphorylated ERK (Ph-ERK) after MIT exposure. The MEK inhibitor U0126 (10 μm) and the zinc chelator TPEN (1 μm) completely blocked ERK phosphorylation (30 min). Similar results were obtained in three additional experiments. Aniso, Anisomycin;Con, control. B, MIT toxicity was not blocked by the p38 inhibitor SB239063 (20 μm) but was significantly inhibited by the MEK inhibitors U0126 (10 μm) and PD98059 (40 μm). Results represent the mean ± SEM of three to four independent experiments; ∗p < 0.05. C, A lack of p38 involvement in MIT toxicity was confirmed by a lack of enhancement in potassium channel currents 3–4 hr after a 10 min MIT (100 μm) exposure (compare with McLaughlin et al., 2001). Results represent the mean ± SD current density (n = 6, 11) for potassium currents evoked in voltage-clamped cortical neurons by stepping the voltage to −10 mV from a holding voltage of −70 mV. Con, Control.Insets, Examples of whole-cell potassium currents obtained in two separate cortical neurons ∼3 hr after a 10 min exposure to vehicle or MIT. Currents were evoked by a series of steps to 35 mV from a holding voltage of −70 mV. D, Lack of neuroprotection against MIT by the following agents: TEA (10 mm), high extracellular potassium (25 mm), cyclohexamide (CHX, 3.5 μm), and BAF (20 μm); data represent the mean ± SEM (n = 4–9).
Although generally associated with cell survival and proliferation pathways (Xia et al., 1995; Derkinderen et al., 1999), activation of ERK, another member of the MAPK family, has been reported to be responsible for neuronal cell death after oxidative stimuli (Stanciu et al., 2000). ERK activation has also been implicated in neuronal cell death after focal cerebral ischemia (Alessandrini et al., 1999), seizures (Murray et al., 1998), and zinc exposure (Seo et al., 2001). Therefore, we examined whether ERK activation was present after MIT treatment and tested whether such activation was responsible for the ensuing neurotoxicity. A time course study of ERK phosphorylation revealed a transient activation of p44/42 ERK within 30 min of MIT exposure (Fig. 3A, middle panels). The levels of phosphorylated p44/42 ERK quickly returned to baseline 1 hr after treatment. U0126, a specific inhibitor of the ERK kinases MAP kinase kinase (MEK) 1/2, blocked MIT-induced ERK phosphorylation (Fig.3A) as well as MIT toxicity (Fig. 3B). PD98059, a structurally unrelated MEK 1/2 inhibitor, exhibited similar levels of protection against MIT toxicity (Fig. 3B). ERK activation was also prevented by TPEN (Fig. 3A, bottom panels), suggesting that the neuroprotective actions of the metal chelator occurred upstream from the phosphorylation of the MAPK. ERK-mediated cell death after MIT exposure was not affected by the protein synthesis inhibitor cycloheximide or by butoxy-carbonyl-aspartate-fluoromethyl ketone (BAF), a broad-spectrum cysteine protease inhibitor (Fig. 3D). This indicates that MIT neurotoxicity proceeds without the synthesis of new proteins and is independent of caspase activation.
MIT-induced ERK phosphorylation is mediated via activation of 12-lipoxygenase
It has been suggested that lipoxygenase metabolites of arachidonic acid (AA) can activate the ERK cascade (Rao et al., 1994; Chakraborti and Chakraborti, 1998; Alexander et al., 2001; Chang and Wang, 2001). Indeed, zinc can bind to and stimulate phospholipase A2 (PLA2) (Lindahl and Tagesson, 1996), an enzyme that releases arachidonic acid from lipids, and lipoxygenases have been widely associated with activation of cell death pathways (Maccarrone et al., 2001). Furthermore, 12-LOX, the predominant LOX present in the brain (Bendani et al., 1995), has been implicated in neuronal oxidative injury (Li et al., 1997; Stanciu et al., 2000). Based on this information, we tested whether a 12-LOX inhibitor could prevent MIT toxicity and block MIT-induced ERK activation. We used 5,6,7-trihydroxyflavone (baicalein) (Cho et al., 1991), a 12-LOX inhibitor that inhibits oxidative stress-induced ERK activation (Stanciu et al., 2000) and has been shown to be neuroprotective (Li et al., 1997). This drug effectively abrogated both MIT neurotoxicity (Fig. 4A) and MIT-initiated ERK activation (Fig. 4B). Similar protective effects were obtained with the broad-spectrum LOX inhibitor 2,3,5-trimethyl-6-(12-hydroxy-5-10-dodecadiynyl)-1, 4-benzoquinone (AA861) (Li et al., 1997) (Fig. 4A). The neuroprotective actions of two PLA2 inhibitors, bromoenol lactone (20 μm) and quinacrine (20 μm), could not be properly evaluated, because these substances were toxic to neurons on their own.
Fig. 4.

MIT toxicity and MIT-induced ERK activation requires 12-LOX activity. A, MIT (100 μm) toxicity was significantly inhibited by the 12-LOX inhibitor baicalein (20 μm) and by the less-specific LOX inhibitor AA861 (1 μm). Results represent the mean ± SEM (n = 3); ∗∗∗p < 0.001.B, Immunoblots demonstrate that MIT-induced ERK activation is blocked by baicalein. Baicalein had no effect on ERK activation when added alone (data not shown). Similar results were observed in a total of three independent experiments.Phospho-ERK, Phosphorylated ERK.C–F, 12-LOX immunostaining in control cultures (C) and 5 min after a 10 min exposure to 100 μm MIT alone (D) or in the presence of either 20 μm baicalein (E) or 1 μm TPEN (F). LOX activation is usually accompanied by its translocation to the cell membrane.
LOX activation has been associated with its translocation to the cell membrane (Hagmann et al., 1993). As such, activation of 12-LOX was confirmed by detecting said translocation MIT exposure (Hagmann et al., 1993; Li et al., 1997). We observed a dramatic redistribution of 12-LOX antibody staining to the cell membrane within 5 min after a 10 min exposure to MIT (Fig. 4A,B). Coexposure of the cells to MIT with either TPEN or baicalein completely prevented the translocation of 12-LOX to the cell membrane (Fig.4C,D).
MIT-induced decrease in GSH levels
Decreases in intracellular GSH can be directly correlated with increased 12-LOX activity, possibly as a consequence of eliminating the tonic inhibition of the enzyme by GSH itself (Hagmann et al., 1993;Shornick and Holtzman, 1993; Li et al., 1997). We speculated that MIT induction of 12-LOX activity could be partly attributable to the oxidation of GSH to oxidized glutathione (GSSG). Indeed, Fuller et al. (1985) reported that the interaction of benzothiazolone (BIT, a structural analog of MIT) with GSH resulted in the formation of GSSG and thiol dimers of the ring-opened form of BIT (mercaptobenzamide). Thus, we tested whether MIT exposure would result in a decrease in GSH levels in our cultures. Cells were exposed for 10 min to MIT, and cell lysates were collected 5 min later and assayed for GSH activity. MIT was able to rapidly decrease cellular GSH levels by ∼40% (Fig.5). This suggests that MIT-induced activation of 12-LOX activity might be mediated in part by a direct oxidation of GSH. However, TPEN did not block the MIT-induced effects on GSH levels, suggesting that the Zn2+-mediated component of 12-LOX activation occurs independently of this process. This result also suggests that GSH depletion is necessary but not sufficient for the MIT toxicity pathway to be activated.
Fig. 5.

MIT induces a decrease in GSH activity. Cultures were exposed to 100 μm MIT for 10 min and assayed for cellular GSH activity 5 min later. MIT induces a decrease in GSH levels, which can be prevented by coexposure to the cysteine reagent NAC (1 mm) but not by the antioxidant trolox (100 μm) or by TPEN (1 μm) (mean ± SEM;n = 3–4); ∗∗p < 0.01.
MIT-induced ERK activation and reactive oxygen species
Li et al. (1997) suggested that neuronal oxidative injury requires a 12-LOX-dependent production of reactive oxygen species (ROS). Because several reports have indicated that ROS are involved in ERK activation, including those that are generated after Zn2+ exposure (Seo et al., 2001), we tested whether ROS production downstream from 12-LOX was responsible for ERK activation and contributed to the MIT toxicity. We observed that two antioxidants, N-acetylcysteine (NAC) and trolox, almost completely abrogated MIT-induced cell death in our cultures (Fig. 6A). In initial experiments, the antioxidants were applied before, during, and after MIT exposure to ensure full neuroprotection. Surprisingly, NAC but not trolox prevented ERK activation (Fig. 6B). The simplest explanation to account for this observation is that ROS production occurs downstream from ERK phosphorylation. NAC, being a thiol-containing agent like GSH, can directly interact with MIT, whereas trolox, being a true radical scavenger, must be working at a point subsequent to GSH depletion. Indeed, exogenous GSH mimicked the effects of NAC in blocking MIT toxicity (Fig. 6A). To further test this hypothesis, we applied NAC and trolox during the post-MIT exposure period only. Under these circumstances, NAC was no longer neuroprotective (Fig. 6A), nor did it prevent MIT-induced ERK activation (Fig. 6B). Trolox, however, continued to effectively protect neurons when applied only after the MIT treatment (Fig. 6A). Finally, we observed that NAC, but not trolox, could block the effects of MIT on GSH depletion (Fig. 5). A glutathione peroxidase analog, ebselen (10 μm) (Mueller at al., 1984; Wendel et al., 1984), mimicked the actions of trolox. Together, these results strongly suggest that the injurious production of ROS occurs downstream from ERK activation.
Fig. 6.

Effect of antioxidants and PARP inhibitors on MIT-induced toxicity. A, MIT toxicity was abrogated by including NAC (1 mm), trolox (100 μm), and GSH (1 mm) before, in conjunction with, and after a 10 min MIT (100 μm) treatment. NAC, but not trolox, lost its neuroprotective properties when included in the postexposure period only (post). The PARP inhibitors NU1025 (5 μm) and DPQ (1 μm) were also protective. Results represent the mean ± SEM for three to four experiments; ∗∗p < 0.01; ∗∗∗p < 0.001. B, Immunoblots demonstrating that ERK phosphorylation after MIT exposure could be abolished by the NAC treatment but not by trolox (before exposure, during exposure, and after exposure). The effect of NAC was absent when the antioxidant was included in the postexposure period only. Trolox, NAC, and NU1025 had no effect on ERK activation when added alone (data not shown). Similar results were obtained in a total of three independent experiments. These results suggest that ROS production and PARP activation occur downstream from ERK phosphorylation. Phospho-ERK, Phosphorylated ERK.
Poly(ADP-ribose) polymerase activation is required for MIT toxicity
DNA is vulnerable to ROS attack during oxidative stress. Single-stranded DNA breaks activate the repair enzyme poly(ADP-ribose) polymerase (PARP) (de Murcia and Menissier de Murcia, 1994). Excessive DNA damage and PARP activation can deplete nicotine adenine dinucleotide (NAD+) and impair ATP production (Schraufstatter et al., 1986), a process that can lead to neuronal cell death (Szabo and Dawson, 1998; Pieper et al., 1999). Indeed, pharmacological and genetic disruption of PARP function has been shown to be neuroprotective in models of cerebral ischemia (Eliasson et al., 1997; Endres et al., 1997; Lo et al., 1998; Takahashi et al., 1999; Moroni et al., 2001). Thus, we sought to determine whether MIT-induced toxicity is accompanied by DNA damage and whether overactivation of PARP contributes to the cell death process. The appearance of DNA strand breaks was apparent in our cultures 4 hr after MIT exposure (Fig. 7). Virtually no TUNEL-positive cells were present when cells were exposed to MIT in conjunction with the MEK inhibitor U0126 or with trolox (Fig. 7). The importance of PARP overactivation in MIT toxicity was evident, because two inhibitors on this enzyme, NU1025 and 3,4-dihydro-5-[4-(1-piperidinyl)butoxy]-1(2H)-isoquinolinone (DPQ), substantially abrogated cell death (Fig. 6A). Moreover, ROS production, DNA damage, and PARP overactivation occur downstream from ERK activation, because NU1025 did not prevent MIT-induced ERK phosphorylation (Fig. 6B).
Fig. 7.

NADPH oxidase inhibitors are protective against MIT toxicity. A, MIT (100 μm) toxicity was significantly inhibited by the NADPH inhibitors AEBSF (100 μm) and DPI (100 nm). Results represent the mean ± SEM (n = 3); ∗p< 0.05. B, Immunoblots demonstrating that MIT-induced ERK activation was not blocked by AEBSF or DPI. Neither of these compounds had an effect on ERK phosphorylation when added alone (data not shown). Similar results were observed in a total of three independent experiments. Phospho-ERK, Phosphorylated ERK.
NADPH oxidase inhibitors prevent MIT toxicity by acting downstream from ERK activation
NADPH oxidase is a superoxide-generating enzyme system that is responsible for generating the respiratory burst in phagocytic cells (Babior, 1999) and has been shown recently to be present in neurons (Tammariello et al., 2000). NADPH oxidase has also been implicated in Zn2+ neuronal toxicity (Noh and Koh, 2000) and linked to ERK activity in several systems (Lu et al., 1993; Cui et al., 2000; Dewas et al., 2000; Karlsson et al., 2000). We hypothesized that MIT-induced production of ROS occurred downstream from ERK via activation of this NADPH oxidase. Therefore, we investigated whether NADPH oxidase inhibitors could block MIT toxicity without preventing MIT-induced ERK activation. Cultures were exposed to MIT in the presence and absence of the NADPH oxidase inhibitors 4-(2-aminoethyl)-benzenesulfonyl fluoride (AEBSF) and diphenyleneiodonium (DPI) (Diatchuk et al., 1997; Li and Trush, 1998; Holland et al., 2000). Both of these substances proved to be effective neuroprotective agents against MIT toxicity (Fig.8A). Moreover, neither compound blocked ERK activation (Fig. 8B), suggesting that NADPH oxidase is a probable source of ROS production in the MIT toxicity pathway and that its activation occurs downstream from ERK phosphorylation.
Fig. 8.

MIT induces ERK-dependent DNA damage. Four hours after a 10 min exposure to 100 μm MIT, cultures were stained for TUNEL and counterstained with thionin. Using this procedure, nuclei with damaged DNA are stained darkbrown (arrowheads in B), and cells are stained purple. A, Control.B, MIT-treated cultures. C, MIT-treated cultures in the presence of the MEK inhibitor U0126 (10 μm). D, MIT-treated cultures in the presence of the antioxidant trolox (100 μm). Note the absence of TUNEL staining in A, C, andD.
DISCUSSION
Molecular mechanism of MIT neurotoxicity
Neurons acutely exposed in vitro to MIT undergo caspase-independent cell death. We hypothesize that a molecular cascade that involves GSH depletion, zinc-dependent 12-LOX enzymatic activity, ERK phosphorylation, NADPH oxidase-dependent ROS production, DNA damage, and PARP overactivation mediates this process:
| Equation 1 |
The temporal ordering and interdependence of these events are strongly suggested by the fact that: (1) MIT induces a decrease in GSH levels, (2) TPEN can block translocation of 12-LOX to the membrane, (3) TPEN, thiol-containing agents, and a 12-LOX inhibitor can block ERK activation and cell death, (4) inhibition of ERK activation is neuroprotective and prevents DNA damage, and (5) inhibitors of NADPH oxidase and PARP, as well as a nonthiol-containing antioxidant, can block cell death without preventing ERK activation. We do not yet know, however, how these various components are linked or whether other cellular events are involved in this process.
The neurotoxic cascade of MIT is reminiscent, at least in part, of the proposed pathway mediating oxidative glutamate toxicity, a process whereby high concentrations of extracellular glutamate lead to a slow and relatively large depletion of glutathione (GSH) via inhibition of the cysteine transporter (Murphy et al., 1989). Based on results reported by Li et al. (1997) and Stanciu et al. (2000), the putative mechanism mediating oxidative glutamate toxicity can be summarized as follows:
| Equation 2 |
The term ([Ca2+]i/ERK/ROS) is listed in parentheses because the temporal ordering of these three events has not been established. Glutamate-induced decreases in intracellular GSH have been directly correlated with increased 12-LOX activity, perhaps as a consequence of eliminating the tonic inhibition of the enzyme by GSH itself (Hagmann et al., 1993; Shornick and Holtzman, 1993; Li et al., 1997). An examination of equations 1 and 2 reveals that several key components are shared by both cell death pathways, namely GSH depletion, 12-LOX activation, phosphorylation of ERK, and the production of ROS. There are, however, some important differences. In particular, MIT toxicity proceeds in a protein synthesis and caspase-independent manner, whereas oxidative glutamate toxicity depends on protein synthesis (Ratan et al., 1994a,b) and has several hallmark features of apoptosis (Ratan et al., 1994b). One possible reason for this difference may be that MIT and oxidative glutamate toxicity actually represent two ends of a necrotic–apoptotic spectrum that has an initial common cellular trigger (e.g., 12-LOX activity) but lead to activation of diverse downstream death effectors because of differences in the severity of the overall insult. Because MIT can theoretically oxidize GSH directly, one would expect the neurotoxic cascade be triggered very rapidly, and indeed, we do see GSH depletion and ERK activation occurring rather suddenly after MIT exposure. Oxidative glutamate toxicity requires prolonged applications of the amino acid, because GSH depletion occurs as a result of the inhibition of its synthesis (Murphy et al., 1989; Li et al., 1997).
Our neuroprotection, immunoblots, and immunostaining experiments with TPEN suggest that zinc is involved upstream from 12-LOX in the neurotoxic cascade of MIT. We do not believe that TPEN is protective by binding cellular iron and inhibiting Fenton generation of hydroxyl radicals, because the pretreatment of the chelating agent with this metal did not abolish MIT toxicity. Interestingly, Zn2+ can bind to and stimulate PLA2 and enzymes that lead to AA release from the membrane (Lindahl and Tagesson, 1996). Therefore, it is possible that an additional pathway may be activated by MIT that is concurrent with GSH depletion and leads to increased levels of AA. An increase in 12-LOX activity coupled with an increase in the availability of substrate for 12-LOX (i.e., AA) may also help explain the increased severity of the MIT insult when compared with oxidative glutamate toxicity. It is noteworthy, however, that very low concentrations of MIT have been shown to trigger apoptosis in HL60 cells after prolonged exposures (Anselmi et al., 2002); thus, we may indeed find a similar outcome in future chronic toxicity studies in neurons.
Our previous studies on the mechanism of DTDP toxicity, a disulfide-containing oxidizing agent, clearly demonstrate that a rise in intracellular Zn2+ was an early and critical signal for induction of neuronal apoptosis (Aizenman et al., 2000; McLaughlin et al., 2001). Activation of p38 rapidly followed Zn2+ liberation in this system. Nitric oxide-derived species can also effectively activate this pathway (Pal et al., 2001). In those studies, however, we observed that although ERK was phosphorylated, p38 inhibition but not MEK inhibition more effectively blocked cell death. In addition, we noted that caspase inhibitors were neuroprotective against DTDP toxicity, which is not the case for MIT. We have yet to fully characterize the point of divergence for the toxic actions of DTDP and MIT, but it is likely to be intimately dependent on the activation of p38, which in the case of DTDP appears to be also entirely Zn2+dependent (McLaughlin et al., 2001).
Environmental and occupational exposure concerns regarding MIT exposure
MIT and related biocides currently are being used in a large number of industrial settings (Collier et al., 1990). In addition, many cosmetics contain these compounds. The concentration of Kathon CG in these household products ranges from 15 to 30 ppm, which corresponds to ∼100–200 μm (for the combined CMIT and MIT) (Rastogi, 1990). There is no question that in addition to the many known cases of occupational exposure to these compounds (Ng and Tay, 1996; Primka and Taylor, 1997), a significant portion of the general population is being constantly exposed to low levels of these compounds, which are potent neurotoxins. Use of these substances has only escalated relatively recently. Therefore, it may be some time before potential adverse neurological consequences may surface in humans as a result of occupational or environmental exposure to these biocides. Our results suggest that the neurotoxic consequences of both acute and chronic exposure to MIT and related biocides in humans need to be investigated.
Footnotes
This work was supported in part by National Institutes of Health Grant NS29365 and by an American Heart Association grant-in-aid. We thank Drs. Paul Rosenberg, Ian Reynolds, and Don DeFranco and Daniel Leszkiewicz and Kirk Dineley for helpful comments and suggestions, Dr. Frank Barone for SB239063, Dr. Carl Lagenaur for the astrocyte cultures, and Karen Hartnett for assistance with some of the experiments.
Correspondence should be addressed to Dr. Elias Aizenman, Department of Neurobiology, University of Pittsburgh School of Medicine, E1456 Biomedical Science Tower, Pittsburgh, PA 15261. E-mail:redox@pitt.edu.
REFERENCES
- 1.Aizenman E, Hartnett KA. The action of CGS-19755 on the redox enhancement of NMDA toxicity in rat cortical neurons in vitro. Brain Res. 1992;585:28–34. doi: 10.1016/0006-8993(92)91187-j. [DOI] [PubMed] [Google Scholar]
- 2.Aizenman E, Stout AK, Hartnett KA, Dineley KE, McLaughlin B, Reynolds IJ. Induction of neuronal apoptosis by thiol oxidation: putative role of intracellular zinc release. J Neurochem. 2000;75:1878–1888. doi: 10.1046/j.1471-4159.2000.0751878.x. [DOI] [PubMed] [Google Scholar]
- 3.Alessandrini A, Namura S, Moskowitz MA, Bonventre JV. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc Natl Acad Sci USA. 1999;96:12866–12869. doi: 10.1073/pnas.96.22.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alexander LD, Cui XL, Falck JR, Douglas JG. Arachidonic acid directly activates members of the mitogen-activated protein kinase superfamily in rabbit proximal tubule cells. Kidney Int. 2001;59:2039–2053. doi: 10.1046/j.1523-1755.2001.00718.x. [DOI] [PubMed] [Google Scholar]
- 5.Andersen HR, Nielsen JB, Grandjean P. Toxicologic evidence of developmental neurotoxicity of environmental chemicals. Toxicology. 2000;144:121–127. doi: 10.1016/s0300-483x(99)00198-5. [DOI] [PubMed] [Google Scholar]
- 6.Anselmi C, Ettorre A, Andreassi M, Centini M, Neri P, Di Stefano A. In vitro induction of apoptosis vs. necrosis by widely used preservatives: 2-phenoxyethanol, a mixture of isothiazolinones, imidazolidinyl urea and 1,2-pentanediol. Biochem Pharmacol. 2002;63:437–453. doi: 10.1016/s0006-2952(01)00910-8. [DOI] [PubMed] [Google Scholar]
- 7.Arslan P, Di Virgilio F, Beltrame M, Tsien RY, Pozzan T. Cytosolic Ca2+ homeostasis in Ehrlich and Yoshida carcinomas. A new, membrane-permeant chelator of heavy metals reveals that these ascites tumor cell lines have normal cytosolic free Ca2+. J Biol Chem. 1985;260:2719–2727. [PubMed] [Google Scholar]
- 8.Babior BM. NADPH oxidase: an update. Blood. 1999;93:1464–1476. [PubMed] [Google Scholar]
- 9.Bendani MK, Palluy O, Cook-Moreau J, Beneytout JL, Rigaud M, Vallat JM. Localization of 12-lipoxygenase mRNA in cultured oligodendrocytes and astrocytes by in situ reverse transcriptase and polymerase chain reaction. Neurosci Lett. 1995;189:159–162. doi: 10.1016/0304-3940(95)11482-c. [DOI] [PubMed] [Google Scholar]
- 10.Betarbet R, Sherer TB, MacKenzie G, Garcia-Osuna M, Panov AV, Greenamyre JT. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat Neurosci. 2000;3:1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 11.Chakraborti S, Chakraborti T. Oxidant-mediated activation of mitogen-activated protein kinases and nuclear transcription factors in the cardiovascular system: a brief overview. Cell Signal. 1998;10:675–683. doi: 10.1016/s0898-6568(98)00014-x. [DOI] [PubMed] [Google Scholar]
- 12.Chang LC, Wang JP. Signal transduction pathways for activation of extracellular signal-regulated kinase by arachidonic acid in rat neutrophils. J Leukoc Biol. 2001;69:659–665. [PubMed] [Google Scholar]
- 13.Cho H, Ueda M, Tamaoka M, Hamaguchi M, Aisaka K, Kiso Y, Inoue T, Ogino R, Tatsuoka T, Ishihara T. Novel caffeic acid derivatives: extremely potent inhibitors of 12-lipoxygenase. J Med Chem. 1991;34:1503–1505. doi: 10.1021/jm00108a039. [DOI] [PubMed] [Google Scholar]
- 14.Collier PJ, Ramsey A, Waigh RD, Douglas KT, Austin P, Gilbert P. Chemical reactivity of some isothiazolone biocides. J Appl Bacteriol. 1990;69:578–584. doi: 10.1111/j.1365-2672.1990.tb01551.x. [DOI] [PubMed] [Google Scholar]
- 15.Cui YD, Inanami O, Yamamori T, Niwa K, Nagahata H, Kuwabara M. FMLP-induced formation of F-actin in HL60 cells is dependent on PI3-K but not on intracellular Ca2+, PKC, ERK or p38 MAPK. Inflamm Res. 2000;49:684–691. doi: 10.1007/s000110050647. [DOI] [PubMed] [Google Scholar]
- 16.de Murcia G, Menissier de Murcia J. Poly(ADP-ribose) polymerase: a molecular nick-sensor. Trends Biochem Sci. 1994;19:172–176. doi: 10.1016/0968-0004(94)90280-1. [DOI] [PubMed] [Google Scholar]
- 17.Derkinderen P, Enslen H, Girault JA. The ERK/MAP-kinases cascade in the nervous system. NeuroReport. 1999;10:R24–R34. [PubMed] [Google Scholar]
- 18.Dewas C, Fay M, Gougerot-Pocidalo MA, El-Benna J. The mitogen-activated protein kinase extracellular signal-regulated kinase 1/2 pathway is involved in formyl-methionyl-leucyl-phenylalanine-induced p47phox phosphorylation in human neutrophils. J Immunol. 2000;165:5238–5244. doi: 10.4049/jimmunol.165.9.5238. [DOI] [PubMed] [Google Scholar]
- 19.Diatchuk V, Lotan O, Koshkin V, Wikstroem P, Pick E. Inhibition of NADPH oxidase activation by 4-(2-aminoethyl)-benzenesulfonyl fluoride and related compounds. J Biol Chem. 1997;272:13292–13301. doi: 10.1074/jbc.272.20.13292. [DOI] [PubMed] [Google Scholar]
- 20.Eliasson MJ, Sampei K, Mandir AS, Hurn PD, Traystman RJ, Bao J, Pieper A, Wang ZQ, Dawson TM, Snyder SH, Dawson VL. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089–1095. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- 21.Endres M, Wang ZQ, Namura S, Waeber C, Moskowitz MA. Ischemic brain injury is mediated by the activation of poly(ADP-ribose)polymerase. J Cereb Blood Flow Metab. 1997;17:1143–1151. doi: 10.1097/00004647-199711000-00002. [DOI] [PubMed] [Google Scholar]
- 22.Fuller SJ, Denyer SP, Hugo WB, Pemberton C, Woodcock PM, Buckley AJ. The mode of action of 1,2-benzisothiazolin-3-one on Staphylococcus aureus. Lett Appl Microbiol. 1985;1:13–15. [Google Scholar]
- 23.Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Kortsha GX, Brown GG, Richardson RJ. Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of Parkinson's disease. Neurotoxicology. 1999;20:239–247. [PubMed] [Google Scholar]
- 24.Hagmann W, Kagawa D, Renaud C, Honn KV. Activity and protein distribution of 12-lipoxygenase in HEL cells: induction of membrane-association by phorbol ester TPA, modulation of activity by glutathione and 13-HPODE, and Ca(2+)-dependent translocation to membranes. Prostaglandins. 1993;46:471–477. doi: 10.1016/0090-6980(93)90066-g. [DOI] [PubMed] [Google Scholar]
- 25.Hartnett KA, Stout AK, Rajdev S, Rosenberg PA, Reynolds IJ, Aizenman E. NMDA receptor-mediated neurotoxicity: a paradoxical requirement for extracellular Mg2+ in Na+/Ca2+-free solutions in rat cortical neurons in vitro. J Neurochem. 1997;68:1836–1845. doi: 10.1046/j.1471-4159.1997.68051836.x. [DOI] [PubMed] [Google Scholar]
- 26.Holland JA, O'Donnell RW, Chang MM, Johnson DK, Ziegler LM. Endothelial cell oxidant production: effect of NADPH oxidase inhibitors. Endothelium. 2000;7:109–119. doi: 10.3109/10623320009072206. [DOI] [PubMed] [Google Scholar]
- 27.Karlsson A, Nixon JB, McPhail LC. Phorbol myristate acetate induces neutrophil NADPH-oxidase activity by two separate signal transduction pathways: dependent or independent of phosphatidylinositol 3-kinase. J Leukoc Biol. 2000;67:396–404. doi: 10.1002/jlb.67.3.396. [DOI] [PubMed] [Google Scholar]
- 28.Li Y, Trush MA. Diphenyleneiodonium, an NAD(P)H oxidase inhibitor, also potently inhibits mitochondrial reactive oxygen species production. Biochem Biophys Res Commun. 1998;253:295–299. doi: 10.1006/bbrc.1998.9729. [DOI] [PubMed] [Google Scholar]
- 29.Li Y, Maher P, Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997;19:453–463. doi: 10.1016/s0896-6273(00)80953-8. [DOI] [PubMed] [Google Scholar]
- 30.Lindahl M, Tagesson C. Zinc (Zn2+) binds to and stimulates the activity of group I but not group II phospholipase A2. Inflammation. 1996;20:599–611. doi: 10.1007/BF01488798. [DOI] [PubMed] [Google Scholar]
- 31.Lo EH, Bosque-Hamilton P, Meng W. Inhibition of poly(ADP-ribose) polymerase: reduction of ischemic injury and attenuation of N-methyl-d-aspartate-induced neurotransmitter dysregulation. Stroke. 1998;29:830–836. doi: 10.1161/01.str.29.4.830. [DOI] [PubMed] [Google Scholar]
- 32.Lohmann K, Prohl A, Schwarz E. Multiple chemical sensitivity disorder in patients with neurotoxic illnesses. Gesundheitswesen. 1996;58:322–331. [PubMed] [Google Scholar]
- 33.Lu DJ, Furuya W, Grinstein S. Involvement of multiple kinases in neutrophil activation. Blood Cells. 1993;19:343–349. [PubMed] [Google Scholar]
- 34.Maccarrone M, Melino G, Finazzi-Agro A. Lipoxygenases and their involvement in programmed cell death. Cell Death Differ. 2001;8:776–784. doi: 10.1038/sj.cdd.4400908. [DOI] [PubMed] [Google Scholar]
- 35.McLaughlin B, Pal S, Tran MP, Parsons AA, Barone FC, Erhardt JA, Aizenman E. p38 activation is required upstream of potassium current enhancement and caspase cleavage in thiol oxidant-induced neuronal apoptosis. J Neurosci. 2001;21:3303–3311. doi: 10.1523/JNEUROSCI.21-10-03303.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Moroni F, Meli E, Peruginelli F, Chiarugi A, Cozzi A, Picca R, Romagnoli P, Pellicciari R, Pellegrini-Giampietro DE. Poly(ADP-ribose) polymerase inhibitors attenuate necrotic but not apoptotic neuronal death in experimental models of cerebral ischemia. Cell Death Differ. 2001;8:921–932. doi: 10.1038/sj.cdd.4400884. [DOI] [PubMed] [Google Scholar]
- 37.Mueller A, Cadenas E, Graf, Sies H. A novel biologically active seleno-organic compound-1. Biochem Pharmacol. 1984;33:3235–3239. doi: 10.1016/0006-2952(84)90083-2. [DOI] [PubMed] [Google Scholar]
- 38.Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2:1547–1558. doi: 10.1016/0896-6273(89)90043-3. [DOI] [PubMed] [Google Scholar]
- 39.Murray B, Alessandrini A, Cole AJ, Yee AG, Furshpan EJ. Inhibition of the p44/42 MAP kinase pathway protects hippocampal neurons in a cell-culture model of seizure activity. Proc Natl Acad Sci USA. 1998;95:11975–11980. doi: 10.1073/pnas.95.20.11975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nagorka R, Rosskamp E, Seidel K. Evaluation of humidifier units within the scope of room climate modification. Offentliche Gesundheitswesen. 1990;52:168–173. [PubMed] [Google Scholar]
- 41.Ng CK, Tay P. Two case reports of delayed skin burns from methylisothiazolines used in water treatment. Singapore Med J. 1996;37:577–578. [PubMed] [Google Scholar]
- 42.Noble M, Mayer-Proschel M. Culture of astrocytes, oligodendrocytes, and A-2 progenitor cells. In: Banker G, Goslin K, editors. Culturing nerve cells. MIT; Cambridge: 1998. pp. 499–543. [Google Scholar]
- 43.Noh KM, Koh JY. Induction and activation by zinc of NADPH oxidase in cultured cortical neurons and astrocytes. J Neurosci. 2000;20:RC111:1–5. doi: 10.1523/JNEUROSCI.20-23-j0001.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olden K, Guthrie J. Genomics: implications for toxicology. Mutat Res. 2001;473:3–10. doi: 10.1016/s0027-5107(00)00161-5. [DOI] [PubMed] [Google Scholar]
- 45.Pal S, McLaughlin BA, Aizenman E. Peroxynitritate induces a zinc and p38 dependent enhancement of potassium currents. Soc Neurosci Abstr. 2001;27:1741. [Google Scholar]
- 46.Pieper AA, Verma A, Zhang J, Snyder SH. Poly (ADP-ribose) polymerase, nitric oxide and cell death. Trends Pharmacol Sci. 1999;20:171–181. doi: 10.1016/s0165-6147(99)01292-4. [DOI] [PubMed] [Google Scholar]
- 47.Primka EJ, III, Taylor JS. Three cases of contact allergy after chemical burns from methylchloroisothiazolinone/methylisothiazolinone: one with concomitant allergy to methyldibromoglutaronitrile/phenoxyethanol. Am J Contact Dermat. 1997;8:43–46. [PubMed] [Google Scholar]
- 48.Rao GN, Baas AS, Glasgow WC, Eling TE, Runge MS, Alexander RW. Activation of mitogen-activated protein kinases by arachidonic acid and its metabolites in vascular smooth muscle cells. J Biol Chem. 1994;269:32586–32591. [PubMed] [Google Scholar]
- 49.Rastogi SC. Kathon CG and cosmetic products. Contact Dermatitis. 1990;22:155–160. doi: 10.1111/j.1600-0536.1990.tb01551.x. [DOI] [PubMed] [Google Scholar]
- 50.Ratan RR, Murphy TH, Baraban JM. Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J Neurosci. 1994a;14:4385–4392. doi: 10.1523/JNEUROSCI.14-07-04385.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ratan RR, Murphy TH, Baraban JM. Oxidative stress induces apoptosis in embryonic cortical neurons. J Neurochem. 1994b;62:376–379. doi: 10.1046/j.1471-4159.1994.62010376.x. [DOI] [PubMed] [Google Scholar]
- 52.Rosenberg PA, Aizenman E. Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex. Neurosci Lett. 1989;103:162–168. doi: 10.1016/0304-3940(89)90569-7. [DOI] [PubMed] [Google Scholar]
- 53.Schraufstatter IU, Hinshaw DB, Hyslop PA, Spragg RG, Cochrane CG. Oxidant injury of cells. DNA strand-breaks activate polyadenosine diphosphate-ribose polymerase and lead to depletion of nicotinamide adenine dinucleotide. J Clin Invest. 1986;77:1312–1320. doi: 10.1172/JCI112436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seo SR, Chong SA, Lee SI, Sung JY, Ahn YS, Chung KC, Seo JT. Zn2+-induced ERK activation mediated by reactive oxygen species causes cell death in differentiated PC12 cells. J Neurochem. 2001;78:600–610. doi: 10.1046/j.1471-4159.2001.00438.x. [DOI] [PubMed] [Google Scholar]
- 55.Shornick LP, Holtzman MJ. A cryptic, microsomal-type arachidonate 12-lipoxygenase is tonically inactivated by oxidation-reduction conditions in cultured epithelial cells. J Biol Chem. 1993;268:371–376. [PubMed] [Google Scholar]
- 56.Sinor JD, Du S, Venneti S, Blitzblau RC, Rosenberg PA, Leszkiewicz DL, Aizenman E. NMDA and glutamate evoke excitotoxicity at distinct cellular locations in rat cortical neurons in vitro. J Neurosci. 2000;20:8831–8837. doi: 10.1523/JNEUROSCI.20-23-08831.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stanciu M, Wang Y, Kentor R, Burke N, Watkins S, Kress G, Reynolds I, Klann E, Angiolieri MR, Johnson JW, DeFranco DB. Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J Biol Chem. 2000;275:12200–12206. doi: 10.1074/jbc.275.16.12200. [DOI] [PubMed] [Google Scholar]
- 58.Szabo C, Dawson VL. Role of poly(ADP-ribose) synthetase in inflammation and ischaemia-reperfusion. Trends Pharmacol Sci. 1998;19:287–298. doi: 10.1016/s0165-6147(98)01193-6. [DOI] [PubMed] [Google Scholar]
- 59.Takahashi K, Pieper AA, Croul SE, Zhang J, Snyder SH, Greenberg JH. Post-treatment with an inhibitor of poly(ADP-ribose) polymerase attenuates cerebral damage in focal ischemia. Brain Res. 1999;829:46–54. doi: 10.1016/s0006-8993(99)01335-9. [DOI] [PubMed] [Google Scholar]
- 60.Tammariello SP, Quinn MT, Estus S. NADPH oxidase contributes directly to oxidative stress and apoptosis in nerve growth factor-deprived sympathetic neurons. J Neurosci. 2000;20:RC53:1–5. doi: 10.1523/JNEUROSCI.20-01-j0006.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wendel A, Fausel M, Safayhi H, Tiegs G, Otter R. A novel biologically active seleno-organic compound-2. Biochem Pharmacol. 1984;33:3241–3245. doi: 10.1016/0006-2952(84)90084-4. [DOI] [PubMed] [Google Scholar]
- 62.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 63.Yu SP, Yeh CH, Sensi SL, Gwag BJ, Canzoniero LM, Farhangrazi ZS, Ying HS, Tian M, Dugan LL, Choi DW. Mediation of neuronal apoptosis by enhancement of outward potassium current. Science. 1997;278:114–117. doi: 10.1126/science.278.5335.114. [DOI] [PubMed] [Google Scholar]