

Abstract
Transmissible spongiform encephalopathies often are caused by peripheral uptake of infectious prions, and the peripheral nervous system is involved in prion spread to the brain. Although the cellular prion protein is subjected to fast axonal transport, the mechanism of intranerval transport of infectious prions is unclear. Here we administered prions intranervally to transgenic mice overexpressing the four-repeat human tau protein, which exhibit defective fast axonal transport. These mice showed unaltered neuroinvasion, suggesting that transport mechanisms distinct from fast axonal transport effect prion neuroinvasion along peripheral nerves. Surprisingly, scrapie-sick tau transgenic mice accumulated intraneuronal deposits of hyperphosphorylated tau protein. The coincidence of tau and prion pathology resembled Gerstmann–Sträussler–Scheinker syndrome. These findings identify tau pathology as a possible end stretch of prion-induced neurodegeneration.
Keywords: transmissible spongiform encephalopathy, prion neuroinvasion, peripheral nervous system, impaired fast axonal transport, tau overexpression, tau hyperphosphorylation
Prion diseases are transmissible fatal neurodegenerative diseases occurring in genetic, infectious, and sporadic forms (Prusiner, 1991; Aguzzi et al., 2001). Although the infectious agent is propagated most efficiently via intracerebral inoculation, peripheral infection is the natural route of transmission in many prion diseases. A large body of evidence suggests that the lymphoreticular system (LRS) as well as the peripheral nervous system (PNS) are important for neuroinvasion (Lasmezas et al., 1996; Klein et al., 1997). Intraperitoneal inoculation of the infectious agent initially results in lymphatic prion accumulation and replication (Eklund et al., 1967; Kimberlin and Walker, 1986). Follicular dendritic cells (FDC), which express cellular prion protein (PrPC) and for which the maturation depends on lymphotoxin β-mediated signals, may be necessary for prion replication and may be important for neuroinvasion (Fraser et al., 1996; Brown et al., 1999; Montrasio et al., 2000; Klein et al., 2001). On the other hand, several studies have shown that neuroinvasion can be achieved in mice for which the LRS does not express PrPC (Race et al., 2000) and, in certain instances, even in mice that do not contain mature FDCs (Prinz et al., 2002). Finally, adoptive bone marrow transfer of PrPC-expressing cells into PrP knock-out mice results in the accumulation of prions in lymphoid organs, but not in the transport of prions to the brain, indicating that a nonhematopoetic PrPC-expressing tissue compartment is required for efficient neuroinvasion (Blättler et al., 1997; Kaeser et al., 2001).
Transport along peripheral nerves to the CNS was suggested by the finding that intranerval (i.n.) injection of prions bypasses the need for extraneural replication of the infectious agent (Kimberlin et al., 1983). Both the parasympathetic (Beekes et al., 1998) and the sympathetic nervous systems (Glatzel et al., 2001) appear to be involved in neuroinvasion. Whether axonal or nonaxonal mechanisms of transport underlie the progression of the infectious agent within the nervous system remains to be elucidated. Whereas PrPC was shown to be subjected to fast axonal transport (Borchelt et al., 1994), the transport of PrPSc has been investigated so far only indirectly, by comparing the incubation times of mice inoculated intraneurally, extraneurally, and intracerebrally (Kimberlin et al., 1983; Glatzel and Aguzzi, 2000).
Fast axonal transport is a microtubule-dependent mechanism in which tau, a member of microtubule-associated proteins known to stabilize microtubules, plays an important role (Mandelkow and Mandelkow, 1995). Here we show that transgenic mice with a defect in fast axonal transport attributable to the overexpression of the four-repeat tau protein isoform under a neuron-specific promoter (thy-1.1-tau) (Spittaels et al., 1999) do not exhibit prolonged incubation times after intranerval inoculation with prions when compared with control mice, suggesting that mechanisms distinct from fast axonal transport effect prion neuroinvasion along peripheral nerves. Surprisingly, thy-1.1-tau mice showed abundant deposits of hyperphosphorylated tau protein at advanced stages of prion disease, reminiscent of the neuropathological lesions observed in Gerstmann–Sträussler–Scheinker disease (GSS).
MATERIALS AND METHODS
Measurement of axonal transport in vivo. Retrograde fast axonal transport in thy-1.1-tau transgenic mice was studied by injecting 0.5 μl of Texas Red-labeled dextran (stock concentration 50 mg/ml, molecular weight 70,000, lysine-fixable, diluted 1:1 in PBS; Molecular Probes, Leiden, The Netherlands) in the sciatic notch of transgenic and control animals. For intranerval injections the mice were anesthetized with xylazine/ketamine, and the left sciatic nerve was exposed surgically by dislodging the gluteus superficialis muscle and the biceps femoris muscle. The nerve was placed onto a metal plate (20 × 5 × 0.5 mm), and 0.5 μl of Texas Red-labeled dextran was injected with a 34-gauge Hamilton syringe over a period of 2 min. To increase the efficiency of uptake, we gently squeezed the injected nerve with a microsurgical forceps (Vercelli et al., 2000). The nerve was repositioned anatomically, and the skin was closed with USP 4.0 nylon sutures. Six control mice and six tau transgenic mice were anesthetized with xylazine/ketamine and perfused transcardially with 2% paraformaldehyde/PBS [1 hr (n= 8) or 3 hr (n = 2) after injection]. Injected (ipsilateral) and noninjected (contralateral) sciatic nerves were isolated and postfixed for 10 sec in 2% paraformaldehyde/PBS; a 1 mm segment, located 10 mm proximally to the injection site, was fiber teased, embedded in fluorescent mounting medium (Dako, Glostrup, Denmark), and stored protected from light. Fluorescent vesicles were quantified via a fluorescence microscope (excitation filter, 530–585 nm) linked to a digital camera (Zeiss, AxioCam 072, Axiovision Software). Axons containing fluorescent vesicles were counted as positive; axons from the noninjected side served as negative controls. Additional negative controls included one mouse injected with dextran intranervally after transection of the injected nerve proximally to the injection site and mice killed immediately before (n = 1) or after (n = 1) dextran injection. We transformed the data logarithmically and analyzed them in a one-tailed Student's t test (p = 0.01).
Measurement of axonal transport in vitro. We performed time-lapse recordings of fluorescently labeled vesicles in axons from cultured dorsal root ganglia (DRGs) isolated from thy-1.1-tau transgenic mice and from control mice. For this, DRGs were isolated from postnatal day 0 (P0) to P3 mice and kept in culture for 5–11 d in DMEM/F-12 medium (Invitrogen, San Diego, CA) supplemented with l-glutamine (Invitrogen), 1% penicillin/streptomycin (Invitrogen), 10% fetal calf serum (Fluka, Buchs, Switzerland), sodium pyruvate (cfin = 1 mm; Invitrogen), and NGF (cfin = 10 ng/ml; Sigma, Deisenhofen, Germany). Endocytotic uptake of fluorescent dextran was achieved by incubating DRGs for 30, 45, 50, or 60 min with medium containing fluorescent dextran (cfin = 0.1 mg/ml, 70,000 MW, lysine-fixable; Molecular Probes). Cells were washed twice and transferred to a microscope chamber with phenol red-free NGF growth medium supplemented with horseradish peroxidase (HRP; cfin = 200 μg/ml), glucose oxidase (cfin = 200 μg/ml), glucose (cfin = 25 mm), and hemoglobin (cfin = 1–4 mg/ml). Analysis of fluorescent vesicles was performed according to established protocols (Nakano and Greber, 2000). The velocities of anterogradely and retrogradely transported vesicles were calculated if they were identifiable for at least 20 frames in a series and represented the mean of all average velocities of vesicles transported toward the nucleus or the cell periphery. Fluorescent vesicles with velocities <0.1 μm/sec were disregarded to exclude movements caused by Brownian motion, which produces trajectories with a variety of random patterns. The numbers of individual vesicle steps were obtained by tracing individual vesicles moving faster than 0.1 μm/min (n = 70 for controls;n = 48 for thy-1.1-tau). The calculated data of transport velocities were analyzed with a one-tailed Student'st test (p = 0.01).
Thin section electron microscopy. DRGs isolated from control mice (15 d in cell culture) were incubated with HRP (175 U/mg, 10 mg/ml in NGF growth medium; Sigma) at 37°C for 15 min, washed, and fixed in 2% glutaraldehyde/PBS for 20 min at room temperature. HRP activity was visualized by incubating DRGs with 0.5 mg/ml diaminobenzidine (DAB; Sigma) in 0.05 m Tris-HCl, pH 7.4 (Sigma), containing 0.01% hydrogen peroxide (Fluka) for 10 min; embedding and thin section electron microscopy (TEM) analysis of cells were performed according to established protocols (Suomalainen et al., 1999).
Scrapie inoculation. After anesthesia with xylazine/ketamine, the control and thy-1.1-tau mice were inoculated with a 1% (1 μl, i.n.) or 0.001% (30 μl, i.c.) brain homogenate prepared as described previously (Büeler et al., 1993) from mice infected with the Rocky Mountain Laboratory scrapie prions (RML 4.1) or with similarly prepared brain homogenate from uninfected mice (mock). Intranerval injections were performed as described for dextran injections. Inoculated mice were checked every third day. Scrapie was diagnosed according to standard clinical signs.
Infectivity bioassays. Homogenates of DRGs (2.5% in 0.32m sucrose) and sciatic nerves (10% in 0.32 msucrose) derived from terminally scrapie-sick control and tau transgenic mice were prepared by homogenizing the tissues with a pellet mixer (TreffLab), followed by sonication (Branson 450 for 5 min; constant output power, 400 W). Then 30 μl (diluted 1:10 in PBS and 1% BSA) of homogenates was inoculated intracerebrally in fourtga20 mice (Fischer et al., 1996) by using a 300 μl syringe (Becton Dickinson, Heidelberg, Germany). Incubation times until the development of terminal scrapie disease were determined, and infectivity titers were calculated (Prusiner et al., 1982) by using the relationship: y = 11.45 − 0.088x, where y is LD50 and x is the incubation time (days) to terminal disease (Brandner et al., 1996b).
Western blot analysis. Homogenates (10%, w/v) of brain and spleen were prepared as described previously (Büeler et al., 1993) and, where indicated, digested with 20 μg/ml proteinase K for 30 min at 37°C. Then 50 μg (brain) or 100 μg (spleen) of total protein was electrophoresed through a 12%-SDS polyacrylamide gel, transferred to a nitrocellulose membrane (BA 85, 0.45 μm pore size; Schleicher & Schuell, Dassel, Germany), probed with antibody ICSM18 (1:20,000), and developed by enhanced chemiluminescence (Amersham Biosciences, Braunschweig, Germany). Quantification of un-, mono-, and diglycosylated PrPSc was accomplished by scanning the membranes with a Kodak image station 440.
Histological studies. Brains from selected mice were fixed with 4% buffered formalin, inactivated for 1 hr with 98% formic acid, and embedded in paraffin. Sections of 3 μm thickness were subjected to conventional staining and to immunostaining to glial fibrillary acidic protein (GFAP; Dako) and hyperphosphorylated tau (AT8 and AT270, Innogenetics, Gent, Belgium) according to standard protocols. Selected sections were silver impregnated according to the Bielschowsky and Gallyas protocol.
For quantification of hyperphosphorylated tau deposits, AT270-positive axonal spheroids were counted in consecutive step sections of the brainstem (n = 10; distance, 25 μm) in sex and age-matched mock-inoculated tau transgenic (n = 4), terminally scrapie-sick tau transgenic (n = 4), and wild-type (n = 2 for mock; n = 2 for terminally scrapie-sick) mice. Counting was performed independently by two blinded researchers. The number of tau spheroid-containing neurons, averaged for each mouse, was analyzed statistically by the one-tailed Student's t test (p = 0.05).
RESULTS
Impaired axonal transport in tau transgenic mice
Previous morphological studies of thy-1.1-tau mice suggested a defect in fast axonal transport (Spittaels et al., 1999). This was studied in more detail, visualizing fast axonal transport of fluorescent dextran in vivo and in vitro. Quantification in a nerve segment located 10 mm proximally to the intranerval injection site demonstrated a significant decrease of fluorescent vesicles in tau transgenic mice [number of vesicles per unit area (n/a) = 0.90] when compared with controls (n/a = 1.58;p < 0.01) at 1 hr after injection. However, the quantification of fluorescent vesicles at 2 hr (n/a = 1.16 for tau; n/a = 1.68 for controls) or 3 hr (n/a = 1.69 for tau; n/a = 1.57 for controls) after dextran injection did not show any significant differences between transgenic and control nerves.
In an attempt to substantiate the in vivo data, we undertookin vitro experiments by taking advantage of cultured transgenic and wild-type DRGs. Incubation with fluorescent dextran resulted in strictly axonal transport of fluorescent vesicles, as evidenced by TEM of HRP-treated DRGs (Fig.1A).
Fig. 1.

Visualization of fast axonal transport.A–F, Recordings of fluorescent vesicles in thy-1.1-tau DRGs 15 min after dextran incubation. A single fluorescent vesicle (arrow) was traced in 153 subsequent frames. Shown are frames 7, 17, 103, 123, and 153. Scale bar, 10 μm. A more intuitive representation is available for download as a complete time-lapse video sequence at www.jneurosci.org. G, Transmission electron microscopic picture showing intra-axonal localization of HRP (arrow; original magnification, 30,000×).H, I, Density blots of traced vesicles in transgenic (H) and wild-type (I) DRGs display an asymmetrical shape and show only a slight vertical extension, indicating microtubule-dependent transport rather than random movement.
Time-lapse fluorescent microscopy enabled us to study both retrograde and anterograde vesicle transport in real time (Fig.1A–F, time-lapse video sequence, available at awww.jneurosci.org). For control DRGs 5255 individual vesicle steps (n = 1199 retrograde; n = 1032 anterograde) and for tau-DRGs 5292 vesicle steps (n = 753 retrograde; n = 610 anterograde) were traced. Statistical analysis revealed a significant reduction in the relative velocities of vesicles (i.e., the proportion of vesicles with any velocity >0.1 μm/min) in both retrograde and anterograde vesicle transport of DRGs that expressed transgenic tau. In contrast, the calculated mean of absolute velocities of fluorescent vesicles moving faster than 0.1 μm/min revealed that both retrograde and anterograde vesicle transport did not differ significantly between transgenic (48 traced vesicles resulting in 0.2 μm/sec for retrograde and 0.184 μm/sec for anterograde transport) and control mice (70 traced vesicles resulting in 0.21 μm/sec for anterograde and 0.196 μm/sec for retrograde transport; p > 0.05; one-tailed Student's t test). These results are consistent with the finding that the motility of vesicles in transgenic DRGs (25.93%) is reduced compared with control DRGs (42.16%).
Unhampered prion transport in peripheral nerves of tau transgenic mice
To determine whether the observed axonal transport defect of tau transgenic mice might decrease the efficiency of prion neuroinvasion along peripheral nerves, we inoculated tau transgenic (n = 8) and control mice (n = 8) intranervally with prions (4 logLD50). All animals developed scrapie at similar time points; therefore, expression of the transgene did not elicit any significant delay until the onset of terminal scrapie disease. The mean incubation time for tau transgenic mice was 222 d ± 37.89 and 201.6 d ± 30.41 for wild-type controls (Fig. 2). None of the mock-inoculated tau transgenic (n = 4) and control mice (n = 4) developed scrapie, and all were killed after 451 d. The severity of pathological lesions (spongiosis and gliosis) in tau and control mice was similar (Fig.3).
Fig. 2.

Survival plots of prion-inoculated mice. Incubation time (days) until terminal scrapie after administration of 4 logLD50 scrapie prions intranervally (A) and 3.5 logLD50 intracerebrally (B) to thy-1.1-tau transgenic mice and control mice. Incubation times were similar in the two groups.C, Infectivity levels in ipsilateral sciatic nerve (scn ip) of thy-1.1-tau transgenic mice (dark diamonds), controls (lighter diamonds), ipsilateral dorsal root ganglia (drg ip) of thy-1.1-tau transgenic (dark square), and control mice (lighter squares). Each symbol represents one individual assayed mouse; error bars reflect the variability of the infectivity bioassays. Prion titers are indicated in logLD50 units per milliliter.
Fig. 3.
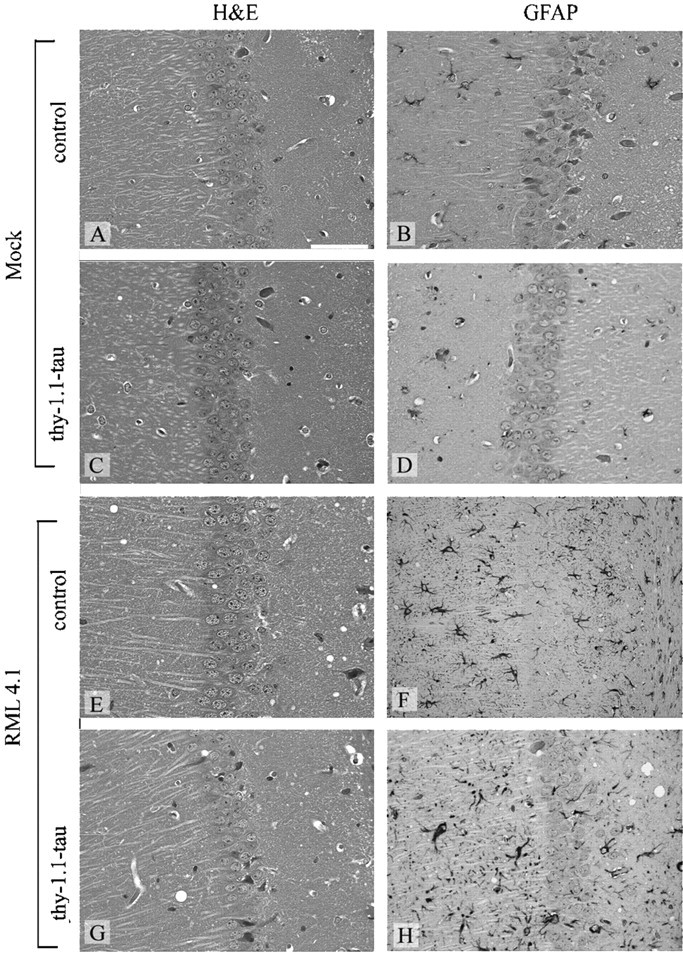
Histopathological findings in hippocampi of mice. Hematoxylin and eosin-stained (A, C, E, G) and GFAP-stained (B, D, F, H) brain sections of thy-1.1 tau (C, D, G, H) and control mice (A, B, E, F) after intranerval inoculation of scrapie prions (E–H) and mock inoculum (A–D). Spongiform changes and intense, diffuse gliosis were visible in brains of infected thy-1.1-tau and control mice. Scale bar, 50 μm.
Biochemical analysis of proteinase K-resistant PrPSc in brains and spleens by Western blotting demonstrated similar levels of PrPSc in transgenic and control mice (Fig.4). Analysis of the glycotype patterns (Collinge et al., 1996) of tau and control mice did not reveal significant differences (data not shown). Mock-inoculated mice contained native PrPC in brain and spleen, but no proteinase K-resistant PrPSc (Fig.4).
Fig. 4.

Western blot detection of PrPCand PrPSc. Top blot, Extracts of brain; bottom blot, extracts of spleen. Genotypes of mice, route of inoculation, and proteinase K digest are indicated above each lane. For control, we analyzed terminally sick, intracerebrally inoculated CD1 mice (control), and mock-injected wild-type mice (mock). The amounts of total protein loaded in each lane were 50 μg for brain and 100 μg for spleen. PrPSc, defined by the appearance of three partially protease-resistant immunoreactive bands (corresponding to unglycosylated, mono-, and diglycosylated forms of the prion protein), was readily detectable in brains and spleens of thy-1.1-tau and control mice.
No differences in scrapie incubation time of intracerebrally injected mice
Several studies have suggested a possible interaction of hyperphosphorylated tau protein with PrPCor PrPSc resulting in variations in disease progression or clinical presentation (Ghetti et al., 1996;Tranchant et al., 1997; Brown, 2000). To investigate this in more detail and to exclude any transport-unrelated effects on the incubation time of scrapie in tau transgenic mice, we inoculated both groups of mice intracerebrally with prions (3.5 logLD50). We were not able to detect differences until the onset of terminal scrapie in mice inoculated with prions intracerebrally. Incubation times were 148.5 d ± 1.73 for tau transgenic (n = 4) and 150.33 d ± 2.89 for control mice (n = 4) (Fig. 3). Histological analysis did not reveal differences in the degree of gliosis and spongiosis between transgenic and control mice.
Unaltered prion titers in sciatic nerves and DRGs of tau transgenic and control mice
Infectivity titers of specific portions of the PNS were examined by bioassay. Calculated prion titers in sciatic nerves and DRGs of thy-1.1-tau and control mice were similar (3.2 and 3.1 logLD50 × gm−1 for control sciatic nerves; 4.2 and 3.8 logLD50 × gm−1 for thy-1.1-tau sciatic nerves; 4.0 and 3.2 logLD50 × gm−1 for control DRGs; and 4.2 logLD50 × gm−1 for thy-1.1-tau DRGs) (Fig.2C).
PrPSc induces accumulation of hyperphosphorylated tau protein
In certain human prion diseases the accumulation of PrPSc is accompanied by the accumulation of hyperphosphorylated tau protein (Ghetti et al., 1996; Brown, 2000). A recent publication highlighted the possibility that protein–protein interactions might be involved in abnormal tau protein accumulation (Götz et al., 2001). We therefore studied whether the accumulation of PrPSc observed in terminally scrapie-sick mice might induce a similar phenomenon in tau transgenic mice. Quantitative analysis revealed a significantly (t test; p = 0.05) larger number of hyperphosphorylated tau-containing axonal spheroids per brainstem section (n = 10) in terminally sick thy-1.1-tau transgenic mice (n = 4) compared with age-matched noninfected thy-1.1-tau transgenic mice (n = 4) (Fig.5). The presence of hyperphosphorylated tau in thy-1.1-tau transgenic mice was confirmed by using the phosphorylation-dependent antibody AT8. No hyperphosphorylated tau containing axonal spheroids was observed in mock-inoculated and terminally scrapie-sick wild-type mice.
Fig. 5.

Histological analysis demonstrating deposition of abnormally phosphorylated tau protein. Shown are brainstem (A, C) and nucleus olivaris accessorius dorsalis (B, D) of terminally scrapie-sick thy-1.1-tau (A, B) and age-matched noninfected thy-1.1-tau mice (C, D). Sections are stained with the phosphorylation-dependent anti-tau antibody AT270, showing significantly increased accumulation of pathological tau in scrapie-sick thy-1.1-tau mice (B) when compared with controls (D). E, AT270-positive tau deposits in the brainstem of scrapie-sick (prion-inoculated,left four lanes) and age-matched control mice (mock-inoculated, right four lanes) were counted in 10 standardized brainstem sections. Each column represents one individual mouse. A statistically significant increase of abnormal tau deposits was observed in scrapie-sick thy-1.1-tau mice (Student'st test; p = 0.05).
DISCUSSION
Numerous publications have demonstrated the importance of the PNS for prion neuroinvasion (Kimberlin et al., 1983; Beekes et al., 1998;Race et al., 2000). These studies have contributed significantly to our current understanding of prion neuroinvasion along peripheral nerves: the favored model proposes that PrPCexpression on the PNS is essential for efficient neuroinvasion (Blättler et al., 1997; Glatzel and Aguzzi, 2000; Race et al., 2000) and that prion neuroinvasion occurs mainly along nerve fibers belonging to the sympathetic and the parasympathetic nervous systems (Beekes et al., 1998; Glatzel et al., 2001).
In contrast, relatively little is known about the molecular details of PrPSc transport within or along peripheral nerves. Axonal transport mechanisms are conceivable, including passive slow axonal transport and ATP-driven fast axonal transport, and even nonaxonal modes of transport. Determination of the velocity of PrPSc movement within peripheral nerves by comparing incubation times of mice inoculated in distal or proximal parts of the PNS suggested slow axonal transport for PrPSc (Kimberlin et al., 1983). Conversely, in vitro studies showing fast axonal transport for PrPC (Borchelt et al., 1994) led to the hypothesis that PrPSc might be transported in a similar manner. Alternatively, nonaxonal modes of transport for PrPSc have been put forth by a number of studies. The proposed mode of transport encompasses the membrane-bound progression of PrPSc along axons and Schwann cells in a domino manner (Brandner et al., 1996a;Glatzel and Aguzzi, 2000).
We decided to investigate peripheral prion transport in transgenic mice with defective axonal transport. The exact nature of this transport defect, which is caused by overexpression of the four-repeat human tau protein, has been described only indirectly (Spittaels et al., 1999). The results of in vivo and in vitro experiments substantiate previous morphological findings and confirm that overexpression of this human tau isoform results in impaired axonal transport. Furthermore, we were able to specify the exact nature of this defect as impairment of anterograde and retrograde fast axonal transport. The sequential events underlying the cause of this defect are unknown; an excess of tau protein may (1) prevent the initial attachment of proteins by competing for binding sites on the microtubules, (2) reduce the transport capacity of motor proteins by blocking the microtubule network, or (3) reduce the binding efficiency of vesicles to motor proteins.
Transgenic and appropriate control mice were then inoculated intranervally or intracerebrally with prions. We were not able to detect a significant difference in the incubation time until the onset of terminal scrapie disease, and the titers of infectious prions were similar in sciatic nerves and DRGs of transgenic and control mice.
Two conclusions can be drawn from the analysis of these data. First, transport of infectious prions exploits, in contrast to PrPC (Borchelt et al., 1994), mechanisms distinct from fast axonal transport. This is in accordance with previous studies (Brandner et al., 1996a; Glatzel and Aguzzi, 2000) and raises the possibility that infectious prions might be transported via noncanonical mechanisms, e.g., in a domino-like manner along PrPC-expressing nerve membranes. Sciatic nerves and DRGs of tau transgenic and control mice contained substantial amounts of infectious prions when assayed at the terminal stage of scrapie, indicating that affected nerves may function not only as conduits but also as reservoirs of the agent. If, conversely, prions were transported strictly in vesicles moving retrogradely, one would expect only the transient presence of infectious prions in affected nerves.
On the other hand, the moderate impairment of fast axonal transport observed in tau transgenic mice may not be sufficient to evoke drastic changes in scrapie incubation times. We feel that this is unlikely, because the incubation time after challenging with a defined inoculum is extremely sensitive to perturbations of pathogenesis.
Transgenes often display significant variegation of expression (Robertson et al., 1995); if fast axonal transport were blocked only in a small fraction of the relevant neurons, the above conclusions would be less strong. This was studied in detail earlier, and no evidence for strong variegation of expression of the tau transgene was found (Spittaels et al., 1999, 2000).
The entirely unexpected finding that the accumulation of prions leads to enhanced production of hyperphosphorylated tau protein in tau transgenic mice, but not in wild-type mice, has several important implications.
First, it may result from yet unidentified protein–protein interactions between PrPSc and tau. Such interactions may be direct or may be part of an “amyloid cascade,” analogous to the finding that neurofibrillary tangle formation is augmented by intracerebral administration of recombinant β-amyloid in tau transgenic mice (Götz et al., 2001). This could explain why neurodegenerative conditions like Alzheimer's or Creutzfeldt–Jakob disease are associated with a pathological accumulation of various proteins, including tau and Aβ, a finding that was interpreted as epiphenomenological in some studies (Hainfellner et al., 1998).
Second, the observed neuropathological features are highly reminiscent of GSS disease, indicating that prion-inoculated thy-1.1-tau mice are the first viable mouse model for this entity. The generation of mouse models for GSS was attempted both by transgenesis and by injecting synthetic PrP, yet the essential features of this disease, like the deposition of abnormal tau protein, could not be achieved (Manson et al., 1999; Kaneko et al., 2000). Prion-injected thy-1.1-tau mice will be useful for the study of temporal and spatial aggregation of PrPSc and hyperphosphorylated tau protein and might help to understand the role of tau aggregation in GSS disease (Ghetti et al., 1994).
In conclusion, this study provides insights in the transport of PrPSc along peripheral nerves and strengthens the notion that prions are not transported by any of the conventional axonal cargo systems. Additionally, scrapie-sick tau transgenic mice show pathological and biochemical features reminiscent of GSS disease, implying that these mice represent a useful model of this entity.
V.K and M.G. contributed equally to this work.
This work was supported by the National Center of Competence in Research for Neural Plasticity and Repair of the Swiss National Science Foundation, the Swiss National Science Foundation, and the Bundesamt für Bildung und Wissenschaft. We thank M. Peltola, K. Boucke, and M. König for technical help and Dr. J. Collinge for the gift of antibody ICSM18.
Correspondence should be addressed to Adriano Aguzzi, Institute of Neuropathology, University Hospital Zürich, Schmelzbergstrasse 12, CH-8091 Zürich, Switzerland. E-mail:adriano@pathol.unizh.ch.
REFERENCES
- 1.Aguzzi A, Montrasio F, Kaeser PS. Prions: health scare and biological challenge. Nat Rev Mol Cell Biol. 2001;2:118–126. doi: 10.1038/35052063. [DOI] [PubMed] [Google Scholar]
- 2.Beekes M, McBride PA, Baldauf E (1998) Cerebral targeting indicates vagal spread of infection in hamsters fed with scrapie. J Gen Virol 79[Pt 3]:601–607. [DOI] [PubMed]
- 3.Blättler T, Brandner S, Raeber AJ, Klein MA, Voigtländer T, Weissmann C, Aguzzi A. PrP-expressing tissue required for transfer of scrapie infectivity from spleen to brain. Nature. 1997;389:69–73. doi: 10.1038/37981. [DOI] [PubMed] [Google Scholar]
- 4.Borchelt DR, Koliatsos VE, Guarnieri M, Pardo CA, Sisodia SS, Price DL. Rapid anterograde axonal transport of the cellular prion glycoprotein in the peripheral and central nervous systems. J Biol Chem. 1994;269:14711–14714. [PubMed] [Google Scholar]
- 5.Brandner S, Raeber A, Sailer A, Blattler T, Fischer M, Weissmann C, Aguzzi A. Normal host prion protein (PrPC) is required for scrapie spread within the central nervous system. Proc Natl Acad Sci USA. 1996a;93:13148–13151. doi: 10.1073/pnas.93.23.13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brandner S, Isenmann S, Raeber A, Fischer M, Sailer A, Kobayashi Y, Marino S, Weissmann C, Aguzzi A. Normal host prion protein necessary for scrapie-induced neurotoxicity. Nature. 1996b;379:339–343. doi: 10.1038/379339a0. [DOI] [PubMed] [Google Scholar]
- 7.Brown DR. Altered toxicity of the prion protein peptide PrP106–126 carrying the Ala117→Val mutation. Biochem J. 2000;346:785–791. [PMC free article] [PubMed] [Google Scholar]
- 8.Brown KL, Stewart K, Ritchie DL, Mabbott NA, Williams A, Fraser H, Morrison WI, Bruce ME. Scrapie replication in lymphoid tissues depends on prion protein-expressing follicular dendritic cells. Nat Med. 1999;5:1308–1312. doi: 10.1038/15264. [DOI] [PubMed] [Google Scholar]
- 9.Büeler HR, Aguzzi A, Sailer A, Greiner RA, Autenried P, Aguet M, Weissmann C. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73:1339–1347. doi: 10.1016/0092-8674(93)90360-3. [DOI] [PubMed] [Google Scholar]
- 10.Collinge J, Sidle KC, Meads J, Ironside J, Hill AF. Molecular analysis of prion strain variation and the etiology of “new variant” CJD. Nature. 1996;383:685–690. doi: 10.1038/383685a0. [DOI] [PubMed] [Google Scholar]
- 11.Eklund CM, Kennedy RC, Hadlow WJ. Pathogenesis of scrapie virus infection in the mouse. J Infect Dis. 1967;117:15–22. doi: 10.1093/infdis/117.1.15. [DOI] [PubMed] [Google Scholar]
- 12.Fischer M, Rülicke T, Raeber A, Sailer A, Moser M, Oesch B, Brandner S, Aguzzi A, Weissmann C. Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knock-out mice to scrapie. EMBO J. 1996;15:1255–1264. [PMC free article] [PubMed] [Google Scholar]
- 13.Fraser H, Brown KL, Stewart K, McConnell I, McBride P, Williams A. Replication of scrapie in spleens of SCID mice follows reconstitution with wild-type mouse bone marrow. J Gen Virol. 1996;77:1935–1940. doi: 10.1099/0022-1317-77-8-1935. [DOI] [PubMed] [Google Scholar]
- 14.Ghetti B, Tagliavini F, Giaccone G, Bugiani O, Frangione B, Farlow MR, Dlouhy SR. Familial Gerstmann–Sträussler–Scheinker disease with neurofibrillary tangles. Mol Neurobiol. 1994;8:41–48. doi: 10.1007/BF02778006. [DOI] [PubMed] [Google Scholar]
- 15.Ghetti B, Piccardo P, Spillantini MG, Ichimiya Y, Porro M, Perini F, Kitamoto T, Tateishi J, Seiler C, Frangione B, Bugiani O, Giaccone G, Prelli F, Goedert M, Dlouhy SR, Tagliavini F. Vascular variant of prion protein cerebral amyloidosis with tau-positive neurofibrillary tangles: the phenotype of the stop codon 145 mutation in PRNP. Proc Natl Acad Sci USA. 1996;93:744–748. doi: 10.1073/pnas.93.2.744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glatzel M, Aguzzi A. PrPC expression in the peripheral nervous system is a determinant of prion neuroinvasion. J Gen Virol. 2000;81:2813–2821. doi: 10.1099/0022-1317-81-11-2813. [DOI] [PubMed] [Google Scholar]
- 17.Glatzel M, Heppner FL, Albers KM, Aguzzi A. Sympathetic innervation of lymphoreticular organs is rate limiting for prion neuroinvasion. Neuron. 2001;31:25–34. doi: 10.1016/s0896-6273(01)00331-2. [DOI] [PubMed] [Google Scholar]
- 18.Götz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301L tau transgenic mice induced by Aβ42 fibrils. Science. 2001;293:1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 19.Hainfellner JA, Wanschitz J, Jellinger K, Liberski PP, Gullotta F, Budka H. Coexistence of Alzheimer-type neuropathology in Creutzfeldt–Jakob disease. Acta Neuropathol (Berl) 1998;96:116–122. doi: 10.1007/s004010050870. [DOI] [PubMed] [Google Scholar]
- 20.Kaeser PS, Klein MA, Schwarz P, Aguzzi A. Efficient lymphoreticular prion propagation requires PrPC in stromal and hematopoietic cells. J Virol. 2001;75:7097–7106. doi: 10.1128/JVI.75.15.7097-7106.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaneko K, Ball HL, Wille H, Zhang H, Groth D, Torchia M, Tremblay P, Safar J, Prusiner SB, DeArmond SJ, Baldwin MA, Cohen FE. A synthetic peptide initiates Gerstmann–Sträussler–Scheinker (GSS) disease in transgenic mice. J Mol Biol. 2000;295:997–1007. doi: 10.1006/jmbi.1999.3386. [DOI] [PubMed] [Google Scholar]
- 22.Kimberlin RH, Walker CA. Pathogenesis of scrapie (strain 263K) in hamsters infected intracerebrally, intraperitoneally, or intraocularly. J Gen Virol. 1986;67:255–263. doi: 10.1099/0022-1317-67-2-255. [DOI] [PubMed] [Google Scholar]
- 23.Kimberlin RH, Hall SM, Walker CA. Pathogenesis of mouse scrapie. Evidence for direct neural spread of infection to the CNS after injection of sciatic nerve. J Neurol Sci. 1983;61:315–325. doi: 10.1016/0022-510x(83)90165-x. [DOI] [PubMed] [Google Scholar]
- 24.Klein MA, Frigg R, Flechsig E, Raeber AJ, Kalinke U, Bluethmann H, Bootz F, Suter M, Zinkernagel RM, Aguzzi A. A crucial role for B cells in neuroinvasive scrapie. Nature. 1997;390:687–690. doi: 10.1038/37789. [DOI] [PubMed] [Google Scholar]
- 25.Klein MA, Kaeser PS, Schwarz P, Weyd H, Xenarios I, Zinkernagel RM, Carroll MC, Verbeek JS, Botto M, Walport MJ, Molina H, Kalinke U, Acha-Orbea H, Aguzzi A. Complement facilitates early prion pathogenesis. Nat Med. 2001;7:488–492. doi: 10.1038/86567. [DOI] [PubMed] [Google Scholar]
- 26.Lasmezas CI, Cesbron JY, Deslys JP, Demaimay R, Adjou KT, Rioux R, Lemaire C, Locht C, Dormont D. Immune system-dependent and -independent replication of the scrapie agent. J Virol. 1996;70:1292–1295. doi: 10.1128/jvi.70.2.1292-1295.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mandelkow E, Mandelkow EM. Microtubules and microtubule-associated proteins. Curr Opin Cell Biol. 1995;7:72–81. doi: 10.1016/0955-0674(95)80047-6. [DOI] [PubMed] [Google Scholar]
- 28.Manson JC, Jamieson E, Baybutt H, Tuzi NL, Barron R, McConnell I, Somerville R, Ironside J, Will R, Sy MS, Melton DW, Hope J, Bostock C. A single amino acid alteration (101L) introduced into murine PrP dramatically alters incubation time of transmissible spongiform encephalopathy. EMBO J. 1999;18:6855–6864. doi: 10.1093/emboj/18.23.6855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Montrasio F, Frigg R, Glatzel M, Klein MA, Mackay F, Aguzzi A, Weissmann C. Impaired prion replication in spleens of mice lacking functional follicular dendritic cells. Science. 2000;288:1257–1259. doi: 10.1126/science.288.5469.1257. [DOI] [PubMed] [Google Scholar]
- 30.Nakano MY, Greber UF. Quantitative microscopy of fluorescent adenovirus entry. J Struct Biol. 2000;129:57–68. doi: 10.1006/jsbi.1999.4201. [DOI] [PubMed] [Google Scholar]
- 31.Prinz M, Montrasio F, Klein MA, Schwarz P, Priller J, Odermatt B, Pfeffer K, Aguzzi A. Lymph nodal prion replication and neuroinvasion in mice devoid of follicular dendritic cells. Proc Natl Acad Sci USA. 2002;99:919–924. doi: 10.1073/pnas.022626399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Prusiner SB. Molecular biology of prion diseases. Science. 1991;252:1515–1522. doi: 10.1126/science.1675487. [DOI] [PubMed] [Google Scholar]
- 33.Prusiner SB, Cochran SP, Groth DF, Downey DE, Bowman KA, Martinez HM. Measurement of the scrapie agent using an incubation time interval assay. Ann Neurol. 1982;11:353–358. doi: 10.1002/ana.410110406. [DOI] [PubMed] [Google Scholar]
- 34.Race R, Oldstone M, Chesebro B. Entry versus blockade of brain infection following oral or intraperitoneal scrapie administration: role of prion protein expression in peripheral nerves and spleen. J Virol. 2000;74:828–833. doi: 10.1128/jvi.74.2.828-833.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Robertson G, Garrick D, Wu W, Kearns M, Martin D, Whitelaw E. Position-dependent variegation of globin transgene expression in mice. Proc Natl Acad Sci USA. 1995;92:5371–5375. doi: 10.1073/pnas.92.12.5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spittaels K, Van den Haute C, Van Dorpe J, Bruynseels K, Vandezande K, Laenen I, Geerts H, Mercken M, Sciot R, Van Lommel A, Loos R, Van Leuven F. Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. Am J Pathol. 1999;155:2153–2165. doi: 10.1016/S0002-9440(10)65533-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Spittaels K, Van den Haute C, Van Dorpe J, Geerts H, Mercken M, Bruynseels K, Lasrado R, Vandezande K, Laenen I, Boon T, Van Lint J, Vandenheede J, Moechars D, Loos R, Van Leuven F. Glycogen synthase kinase-3β phosphorylates protein tau and rescues the axonopathy in the central nervous system of human four-repeat tau transgenic mice. J Biol Chem. 2000;275:41340–41349. doi: 10.1074/jbc.M006219200. [DOI] [PubMed] [Google Scholar]
- 38.Suomalainen M, Nakano MY, Keller S, Bouke K, Stidwill RP, Greber UF. Microtubule-dependent plus- and minus end-directed motilities are competing processes for nuclear targeting of adenovirus. J Cell Biol. 1999;144:657–672. doi: 10.1083/jcb.144.4.657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tranchant C, Sergeant N, Wattez A, Mohr M, Warter JM, Delacourte A. Neurofibrillary tangles in Gerstmann–Sträussler–Scheinker syndrome with the A117V prion gene mutation. J Neurol Neurosurg Psychiatry. 1997;63:240–246. doi: 10.1136/jnnp.63.2.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vercelli A, Repici M, Garbossa D, Grimaldi A. Recent techniques for tracing pathways in the central nervous system of developing and adult mammals. Brain Res Bull. 2000;51:11–28. doi: 10.1016/s0361-9230(99)00229-4. [DOI] [PubMed] [Google Scholar]