

Abstract
Alteration in the intracellular signal transduction pathway in primary afferent neurons may contribute to pain hypersensitivity. We demonstrated that very rapid phosphorylation of extracellular signal-regulated protein kinases (pERK) occurred in DRG neurons that were taking part in the transmission of various noxious signals. The electrical stimulation of Aδ fibers induced pERK primarily in neurons with myelinated fibers. c-Fiber activation by capsaicin injection induced pERK in small neurons with unmyelinated fibers containing vanilloid receptor-1 (VR-1), suggesting that pERK labeling in DRG neurons is modality specific. Electrical stimulation at the c-fiber level with different intensities and frequencies revealed that phosphorylation of ERK is dependent on the frequency. We examined the pERK in the DRG after application of natural noxious stimuli and found a stimulus intensity-dependent increase in labeled cell size and in the number of activated neurons in the c- and Aδ-fiber population. Immunohistochemical double labeling with phosphorylated ERK/VR-1 and pharmacological study demonstrated that noxious heat stimulation induced pERK in primary afferents in a VR-1-dependent manner. Capsaicin injection into the skin also increased pERK labeling significantly in peripheral fibers and terminals in the skin, which was prevented by a mitogen-activated protein kinase/ERK kinase inhibitor, 1,4-diamino-2,3-dicyano-1,4-bis(2-aminopheylthio)butadiene (U0126). Behavioral experiments showed that U0126 dose-dependently attenuated thermal hyperalgesia after capsaicin injection and suggested that the activation of ERK pathways in primary afferent neurons is involved in the sensitization of primary afferent neurons. Thus, pERK in primary afferents by noxious stimulationin vivo showed distinct characteristics of expression and may be correlated with the functional activity of primary afferent neurons.
Keywords: extracellular signal-regulated kinase, phosphorylation, dorsal root ganglion, pain stimuli, MAP kinase, peripheral sensitization
Primary sensory neurons are highly specialized to transduce and transmit sensory information from the periphery to the CNS and are selectively equipped to detect different kinds of stimuli (Snider and McMahon, 1998). The initial step in pain perception is that noxious thermal, mechanical, or chemical stimuli excite specialized nociceptive transducer receptor/ion channel complexes in peripheral terminals of nociceptors. Vanilloid receptor-1 (VR-1), one of the transducer proteins, can generate depolarizing currents in response to noxious thermal stimuli (Caterina et al., 1997;Tominaga et al., 1998). VR-1 is a nociceptor-specific cation channel that is the molecular target of capsaicin and is essential for selective modalities of pain sensation and for tissue injury-induced thermal hyperalgesia. Action potentials that are transmitted from the periphery may activate the intracellular signaling pathway and regulate gene expression in dorsal root ganglion (DRG) neurons (Fields et al., 1997; Fields, 1998). The alteration in gene expression and the resultant changes in the excitability of DRG neurons may be involved in peripheral and central sensitization in acute and chronic pain conditions (Dubner and Ruda, 1992; Woolf and Salter, 2000).
Much attention has focused on the signal transduction mechanisms of primary afferent neurons responsible for the modulation of pain transmission. Recent articles reported that inflammatory mediators, such as prostaglandin E2, serotonin, epinephrine, and nerve growth factor, produce hyperalgesia through activation of protein kinase A (PKA), protein kinase C (PKC), or extracellular signal-regulated kinases (ERKs) in the primary afferent neurons (Gold et al., 1998; Khasar et al., 1999; Aley et al., 2001). ERKs are mitogen-activated protein kinases (MAPKs) that are activated by membrane depolarization and calcium influx (Rosen et al., 1994), activated by an upstream kinase, MAPK/ERK kinase (MEK) (Chang and Karin, 2001), and known to be one of the intracellular signaling pathways involved in neuronal plasticity (Fields et al., 1997; Martin et al., 1997; Fields, 1998; Impey et al., 1999). Physiological and pathological activity-dependent activation of ERK occurs in the CNS (Baraban et al., 1993; English and Sweatt, 1996; Atkins et al., 1998;Obrietan et al., 1998). However, there have been few studies of signal transduction involved in the activity-dependent plasticity of primary afferent neurons (Fields et al., 1997; Fitzgerald, 2000).
Here, we report modality-specific and intensity-dependent ERK phosphorylation in primary afferent neurons by pain stimuli, an event that may be involved in peripheral sensitization.
MATERIALS AND METHODS
Stimulation. Adult male Sprague Dawley rats (220–250 gm) were used. All procedures were performed under sodium pentobarbital anesthesia (50 mg/kg, i.p.). Capsaicin (8-methyl-N-vanillyl-6-noneamide; Sigma, St. Louis, MO) (10 mm) was dissolved in 10% Tween 80 and injected into the plantar surface of the left hindpaw (5–200 μl). A brief force (∼47.0N/cm2 for the low intensity of pinch, 94.1N/cm2 for the high intensity of pinch) was applied (six times in 10 sec; interval of 10 sec; total of 2 min) to the plantar surface of the middle of the left hindpaw with a surgical bulldog clamp. Thermal (42–60°C) stimuli were produced by immersion of the hindpaw into a water bath (six times in 10 sec; interval of 10 sec; total of 2 min).
Electrophysiological recordings and stimulations.Electrophysiological recordings were applied to distinguish the kinds of nerve fiber that were stimulated. Procedures were performed initially under anesthesia with pentobarbital sodium (50 mg/kg, i.p.), followed by maintenance with halothane (2–3%) and air. Rats were immobilized with pancuronium bromide (1 mg · kg−1 · hr−1, i.v.) for compound action potential recording. The left sciatic nerve was exposed and completely isolated from the surrounding connective tissue. Bipolar platinum wire electrodes were placed under the isolated sciatic nerve just distal to the level of the joint of the three terminal branches (the sural, common peroneal, and tibial nerves). Paraffin oil was pooled, and recording electrodes were placed 25 mm proximal to the stimulation electrodes. To see c-fiber responses, the recordings of 100 trials were averaged. On the basis of the data from these recordings, electrical stimulation was used for activation of each type of nerve fiber. A train of 60 pulses of 0.1 mA, 0.2 msec, and 100 Hz for Aβ fibers; 0.3 mA, 0.2 msec, and 100 Hz for A fibers; and 1–5 mA, 2 msec, and 0.5–100 Hz for c-fiber stimulation was delivered. Sham operations without electrical stimulation were also performed. The electrical stimulations were done 20 min after the incision of skin in the lower leg.
Immunohistochemistry. After appropriate survival times (the survival time after stimulation in all experiments was 2 min, except in the time course study), rats were perfused transcardially with 1% paraformaldehyde in 0.1 m phosphate buffer followed by 4% paraformaldehyde in 0.1 mphosphate buffer, pH 7.4. The lumbar (L) 4/5 DRGs or the skin ∼0.6 × 0.6 cm2 around the injection point were removed and processed for phosphorylated ERK (pERK) immunohistochemistry according to our previous studies (Noguchi et al., 1995; Fukuoka et al., 2001). The polyclonal primary antibody for pERK (New England BioLabs, Beverly, MA) at 1:1000 was used for DAB staining. For double immunofluorescent staining, the tyramide signal amplification (TSA; NEN, Boston, MA) fluorescence procedures (Michael et al., 1997) were used for pERK (1:10,000) staining. Subsequently, the pERK antibody was combined with one of the following antibodies: monoclonal anti-neurofilament 200 (NF200, 1:400; Sigma), rabbit polyclonal protein gene product 9.5 (PGP 9.5) antibody (1:1000; Ultra Clone Ltd., Isle of Wight, UK), or rabbit polyclonal VR-1 antibody (1:1000, gift from Dr. M. Tominaga, Mie University, Mie, Japan). The characteristics and staining specificity of these antibodies have been reported previously (Trojanowski et al., 1986; Day and Thompson, 1987; Tominaga et al., 1998). When two primary antisera raised in rabbits were combined, nonspecific double labeling was not observed. A similar protocol has been used by many other workers (Hunyady et al., 1996; Shindler and Roth, 1996; Michael et al., 1997;Bennett et al., 1998; Amaya et al., 2000), and the lack of cross-reactivity is thought to be attributable to the fact that the TSA procedure allows the first series primary antibody to be used at a dilution that is too high to be detected by the second reagent set (Michael et al., 1997). Our data support this explanation. In control single labeling using indirect labeled immunofluorescence, we were unable to visualize the pERK antiserum at the dilutions used for the TSA procedure.
Quantitative and statistical analysis for immunohistochemistry. For DRG neuron quantification, 8–12 sections of the L4/5 DRG were selected randomly from each rat. The total number and the number of labeled neurons per section were counted to determine changes in pERK expression in the L4/5 DRG after various stimuli. The proportion of pERK-expressing DRG neurons was determined by counting the neuronal profiles that showed distinctive pERK labeling compared with background labeling in DRG sections. The total number of DRG neurons was obtained by the background staining of neurons and the Nomarski differential interference contrast image. An average number of labeled neurons per total neurons was obtained for each animal across the different tissue sections; then the mean ± SEM across animals was determined. The cross-sectional area of pERK-labeled neuron profiles was quantified using a computerized image analysis system (NIH Image, version 1.62; W. Rasband, National Institutes of Health, Bethesda, MD). Because a stereological approach was not used in this study, quantification of the data may represent a biased estimate of the actual number of neurons. For the quantification in the skin, 8–12 sections of skin were selected randomly in each rat, and the relative immunostained area of labeled fiber profiles in a outlined profile (50.25 × 428.81 μm2) crossing the epidermis and dermis per section was quantified using the computerized image analysis system (NIH Image, version 1.62). An assistant who was unaware of the treatment group of the tissue sections performed all counting. A significance of difference was analyzed using at test or ANOVA followed by Fisher's PLSD test.
Western blot analysis. To examine the specificity of the pERK antibody, Western blot analysis for rat DRG neurons was performed. Animals were deeply anesthetized with diethyl ether and killed by decapitation. The L4/5 DRG was rapidly removed and lysed with 20 mm Tris-HCl buffer, pH 8.0, containing 1% NP-40, 150 mm NaCl, 1 mm EDTA, 10% glycerol, 0.1% β-mercaptoethanol, 0.5 mmdithiothreitol, and a mixture of proteinase inhibitors. Briefly, the samples were homogenized in 0.2 ml of lysis buffer reagent and centrifuged at 4°C. The supernatant, containing 15 μg of protein, was electrophoresed in a 10–20 SDS-polyacrylamide gel (Bio-Rad, Hercules, CA) and blotted onto Hybond P membrane (Amersham Biosciences, Arlington Heights, IL) using Multiphore II (Amersham Biosciences, Uppsala, Sweden) for 30 min. The blotted membrane was incubated first with the antibody rabbit anti-phospho-ERK (1:1000; New England BioLabs) overnight at 4°C. The membrane was then incubated with the alkaline phosphatase-conjugated second antibody (goat anti-rabbit IgG; Jackson ImmunoResearch, West Grove, PA) for 2 hr at room temperature. The membrane was rinsed and treated with nitroblue tetrazolium (Sigma) and 5-bromo-4-chloro-3-indolyl phosphate (Sigma) to visualize protein bands.
Behavioral studies. All tests were performed on male rats weighing 220–250 gm. The MEK inhibitor 1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio)butadiene (U0126; Calbiochem, Darmstadt, Germany) (diluted in 2.5% DMSO; dose 7.5, 0.75, and 0.075 μg in 10 μl per paw) was first injected intradermally into the middle of the hindpaw using a 10 μl microsyringe (Hamilton, Reno, NV); 2 min later, capsaicin (15 μg in 5 μl per paw) was injected by the same procedure. Primary thermal hyperalgesia to the capsaicin injection site was measured using the Hargreaves method (Hargreaves et al., 1988) before and 10, 30, 60, and 120 min after injection. The heat stimulus was terminated with a withdrawal response or at 20 sec to avoid skin damage. Two latencies were recorded and averaged for the ipsilateral hindpaw in each test session. Secondary mechanical allodynia was assessed with calibrated von Frey filaments using an up–down paradigm (Chaplan et al., 1994). Briefly, animals were placed in plastic cages with a wire mesh floor and allowed to acclimate for 15 min before each test session. To determine the 50% response threshold, the von Frey filaments were applied to the uninjured area surrounding the capsaicin injection site for 8 sec or until a withdrawal response occurred. Testing was initiated with the 4.0 gm intensity of filament, and the cutoffs of 0.6 gm and 26 gm intensities of filament were selected as the lower and upper limits for testing, respectively. When a positive response was noted, we tested a weaker stimulus. If there was no response to a stimulus, then a stronger stimulus was presented. After the initial threshold crossing, this procedure was repeated for five stimulus presentations per animal per test session. An assistant who was unaware of the treatment group performed all of the behavioral experiments.
RESULTS
To clarify the in vivo activation of ERK in DRG neurons, we stimulated the receptive fields in a variety of ways and used an antibody that recognizes pERK. We found very few neurons labeled for pERK in the naive DRG (Fig.1A). However we found that selective stimulation of c fibers by capsaicin injection into the plantar surface of the hindpaw induced a number of small DRG neurons labeled for pERK (Fig. 1B). To confirm the specificity of the antibody, we used Western blotting with the pERK antibody (Fig. 1B, inset) and found that this antibody could recognize two bands (p42/44 ERK) (n= 3), which is consistent with previous reports (Fields et al., 1997;Impey et al., 1998). We examined the time course of phosphorylation of ERK in small DRG neurons after capsaicin injection and found that this response was very quick: the peak was 2 min after stimulation and the number of labeled cells rapidly declined for 10 min (Fig.1C). The pERK-labeled small neurons were not labeled for NF200 (Trojanowski et al., 1986), a marker of neurons with myelinated fibers, indicating that these neurons had unmyelinated fibers (Fig.1D). The double-labeling experiment with pERK and VR-1, which is a capsaicin receptor, showed that 91.7 ± 2.1% (mean ± SEM; n = 4) of pERK-labeled cells were also labeled for VR-1 after activation of c fibers by capsaicin injection into the plantar surface of the hindpaw (Fig.1E). The topical application of lidocaine (1%) around the sciatic nerve significantly prevented pERK labeling in small DRG neurons 2 min after capsaicin injection (n = 4) (Fig. 1F,G).
Fig. 1.

Stimulus-evoked ERK phosphorylation in rat DRG neurons. A, L4 DRG section (30 μm) immunostained for pERK in the naive rat. B, pERK labeling in many small L4 DRG neurons 2 min after intraplantar injection of capsaicin (10 mm, 200 μl). The inset shows a Western blot of capsaicin-stimulated DRG tissue with pERK antibody.C, Time course of capsaicin-evoked pERK expression in L4/5 DRG neurons (n = 5 at each time point; *p < 0.01; **p < 0.001 compared with naive). D, L4 DRG section double-immunostained for NF200 (red) and pERK (green) after intraplantar capsaicin injection.E, Colocalization of pERK (green) and VR-1 (red) in L4 DRG neurons after capsaicin injection. Double staining appears yellow. F, G, Capsaicin-induced pERK was inhibited by the lidocaine block to the sciatic nerve. Cap, Capsaicin group;Lid, capsaicin plus lidocaine group (**p < 0.001 compared with capsaicin group).H, Example of 0.1 mA (a) and 0.3 mA (b, c) evoked action potentials at the sciatic nerve.a, Aβ response; b, c, Aβ and Aδ responses with no c response (c; 100 sweeps were averaged). I, L4 DRG section immunostained for pERK 2 min after 0.3 mA of stimulation to the sciatic nerve. J, L4 DRG section immunostained for NF200 (a marker of myelinated neurons,red) and pERK (green) after a 0.3 mA electrical stimulation. Double staining appearsyellow. Scale bars, 100 μm.
Subsequently, we examined the pERK labeling in DRG neurons after A-fiber stimulation. We used 0.1 mA of electrical stimulation to the sciatic nerve and found that this Aβ stimulation (no Aδ response) (Fig. 1H,a) induced very few neurons labeled for pERK in the DRG (data not shown). The sham operation induced no pERK labeling in the DRG. We then used 0.3 mA of stimulation and found that this stimulation produced Aδ-fiber activation and no c-fiber activation (Fig. 1H,b,c). The electrical stimulation at the Aδ-fiber level induced labeling for pERK in some medium-to-large DRG neurons (Fig. 1I). Double labeling with pERK and NF200 showed that 90.9 ± 2.9% (mean ± SEM; n = 4) of pERK-labeled neurons contained NF200 (Fig. 1J), which was in clear contrast to the effects of c-fiber stimulation (Fig. 1B,D). These data indicated that the electrical stimulation of Aδ fibers induced pERK, primarily in neurons with myelinated fibers. Thus, these data clearly indicate that the action potentials in c and Aδ fibers of primary afferents could rapidly induce an activation of the MAPK cascade in individual neurons. These findings demonstrate that the modality of stimulation was highly correlated with the expression pattern of pERK in DRG neurons.
We examined the changes in pERK labeling after different frequencies and intensities of c-fiber electrical stimulation. First, we found that c-fiber stimulation at 0.5 Hz, 3 mA, and 2 msec to the sciatic nerve induced pERK labeling in small DRG neurons (Fig.2A). When the frequency of electrical stimulation increased (10 Hz, 3 mA, and 2 msec), the number of pERK-labeled DRG neurons was clearly increased (Fig.2B). The total number of pERK-labeled neurons after c-fiber stimulation at 10 Hz was significantly increased compared with the 0.5 Hz stimulation (Fig. 2E). However, a greater frequency of stimulation (100 Hz) induced less pERK labeling (Fig.2C,E). We measured the cross-sectional areas of neurons labeled for pERK after electrical stimulation at different frequencies (Fig. 2D). Each size-distribution curve after different frequencies had a peak of ∼500 μm2, and the pERK labeling after higher frequencies (50 and 100 Hz) increased the number of medium- to large-sized neurons (>1000 μm2). We also examined the different intensities of c-fiber electrical stimulation. Three intensities (1, 3, and 5 mA) to the sciatic nerve showed no significant difference in the size distribution and the total number of pERK-labeled neurons (Fig. 2F,G). The 1, 3, and 5 mA electrical stimulations to the sciatic nerve activate both A and C fibers (Fig. 2H). These data suggested that the pERK labeling after electrical stimulation to the sciatic nerve is partly dependent on the frequency of the stimulation.
Fig. 2.

Electrical stimulation-induced pERK expression.A–C, Photomicrographs of pERK-labeled neurons in L4 DRG 2 min after 60 pulses of 3 mA/0.5 Hz (A), 3 mA/10 Hz (B), and 3 mA/100 Hz (C) electrical stimulation to the sciatic nerve. D, Size distribution of pERK-labeled neurons in the L4/5 DRGs in the rats that received 60 pulses of 3 mA/0.5 Hz, 3 mA/10 Hz, 3 mA/50 Hz, and 3 mA/100 Hz (n = 3 for each frequency) electrical stimulation to the sciatic nerve. E, The percentage of pERK-labeled neurons in L4/5 DRG neurons (n = 3 for each frequency; ANOVA test shows ap < 0.05 significant change among the groups; *p < 0.01 compared with the 0.5 Hz group by Fisher's PLSD test). F, Size distribution of pERK-labeled neurons in the L4/5 DRGs in the rats that received 60 pulses of 1 mA/0.5 Hz, 3 mA/0.5 Hz, and 5 mA/0.5 Hz (n = 3 for each intensity) electrical stimulation to the sciatic nerve. G, The percentage of pERK-labeled neurons in L4/5 DRG neurons (n = 3 for each intensity; no significant change by ANOVA). H, Electrical stimulation (3 mA) to the sciatic nerve showed both A- and C-fiber responses (100 sweeps were averaged). Scale bars, 100 μm.
Subsequently, we examined the pERK labeling after natural stimulation. We first examined the relationship between thermal stimuli at different temperatures and the induction of pERK in DRG neurons. We applied thermal stimuli by immersion of the hindpaw into warm to hot water (42–60°C). The thermal stimulus of 42°C induced pERK only in very small cells (Fig. 3A). To determine whether the very small cells labeled for pERK at 42°C were neurons, double labeling was performed with pERK and PGP 9.5 (Day and Thompson, 1987), which is a neuronal marker. All pERK-labeled cells were also labeled for PGP 9.5 (Fig. 3B), indicating that the innocuous warm stimulus of 42°C activates a subpopulation of DRG neurons with very small cell bodies. In contrast, noxious heat stimulation at higher temperatures was found to induce pERK in more and larger neurons (Fig. 3C). The double-labeling experiment revealed that some labeled neurons at 60°C showed colocalization with NF200 (Fig. 3D), indicating the activation of neurons with A fibers at higher temperatures. The size distribution of pERK-labeled neurons at graded stimulus temperatures shows a clear rightward shift of the distribution curve (Fig. 3E) and an increased number of labeled neurons (Fig. 3F). The mean areas of pERK-labeled neurons at 50, 54, and 60°C were significantly increased compared with that at 42°C (p < 0.05; one-way ANOVA).
Fig. 3.

Thermal stimulus intensity-dependent pERK expression. A, pERK labeling in L4 DRG neurons 2 min after thermal stimulation at 42°C. B, Colocalization of pERK (green) and PGP 9.5 (red) in L4 DRG neurons after the 42°C stimulus. Double staining appearsyellow. C, pERK labeling in L4 DRG neurons 2 min after thermal stimulation at 60°C. D, Colocalization of pERK (green) and NF200 (red) in L4 DRG neurons after the 60°C stimulus. Double staining appears yellow. E, Size distribution of pERK-labeled neurons in the L4/5 DRGs 2 min after thermal stimulation at 42, 46, 50, 54, and 60°C. F, The percentage of pERK-labeled neurons in L4/5 DRG neurons (n = 5 for each temperature; *p< 0.01; **p < 0.001 compared with 38°C). Scale bars, 100 μm.
We also examined pERK labeling after mechanical stimulation of the peripheral tissue. We applied high and low intensities of pinch stimulation to the plantar surface of the hindpaw (Fig.4A,B). The high-intensity pinch, in contrast to the low-intensity pinch, produced a greater number of DRG neurons that were labeled for pERK, and these neurons tended to be larger. These tendencies were clearly apparent in the size distribution of pERK neurons (Fig. 4C): low-intensity pinch induced pERK almost exclusively in very small neurons (mean, 392.9 ± 37.7 μm2;n = 5), whereas high-intensity stimulation induced pERK in larger-sized neurons, remarkably in 400–700 μm2 neurons (mean, 495.1 ± 74.2 μm2; n = 5). The change in mean size of pERK-labeled neurons was statistically significant (p < 0.05). The total number of pERK-labeled neurons after high-intensity stimulation was significantly increased compared with that after low-intensity stimulation (Fig.4D). The innocuous tactile stimulation of brushing the rat hindpaw did not induce pERK in DRG neurons (data not shown). These data from Figures 3 and 4 suggest that the increase in thermal and mechanical stimulus intensity was highly correlated with the number and size of DRG neurons in which the ERK cascade was activated.
Fig. 4.

Mechanical stimulus intensity-dependent pERK expression. A, B, Photomicrographs of pERK-labeled neurons in L4 DRG 2 min after low-intensity (A) and high-intensity (B) mechanical stimulation of the plantar surface of the hindpaw. Scale bars, 100 μm. C, Size-frequency histogram illustrating the distribution of the profiles 2 min after low-intensity (white) and high-intensity (black) mechanical stimulation. D, The percentage of labeled neurons in L4/5 DRG neurons (n = 5 for each intensity; **p< 0.001).
These characteristics of pERK labeling in primary afferent neurons suggest that phosphorylation of ERK may reflect the activation of primary afferent neurons. Therefore, we subsequently examined the relationship between the VR-1 and thermal–mechanical stimulation using pERK labeling. We found that many neurons were double labeled for VR-1 and pERK after noxious heat stimulation (Fig.5A), and that the percentage of VR-1-labeled neurons with pERK labeling increased with stimulus temperature (Fig. 5B). In contrast, 75–80% of pERK-labeled neurons were also labeled for VR-1 (Fig. 5C) at all stimulus temperatures (42–60°C). To ascertain whether ERK activation in neurons by different stimuli is specifically mediated through this VR-1, we used the competitive VR-1 antagonist capsazepine (Bevan et al., 1992) to block VR-1 activation in primary afferent fibers after natural stimulation, such as heat or mechanical stimulation. The pERK labeling in DRG neurons after capsaicin injection into the hindpaw was dose-dependently inhibited by topical injection of capsazepine (0.1–10 mm, 100 μl) given 2 min before capsaicin (10 mm, 50 μl) injection (Fig. 5D). The increase in pERK-labeled neurons produced by 54°C stimulation of the hindpaw was also dose-dependently suppressed by the capsazepine pretreatment (Fig. 5E). However, the mechanical stimulation-induced pERK in DRG neurons was not affected by this treatment (Fig. 5F). These data clearly demonstrate that noxious heat stimulation but not mechanical stimulation induced ERK phosphorylation of DRG neurons through VR-1 and are consistent with the behavioral data from VR-1 knock-out mice (Caterina et al., 2000;Davis et al., 2000).
Fig. 5.
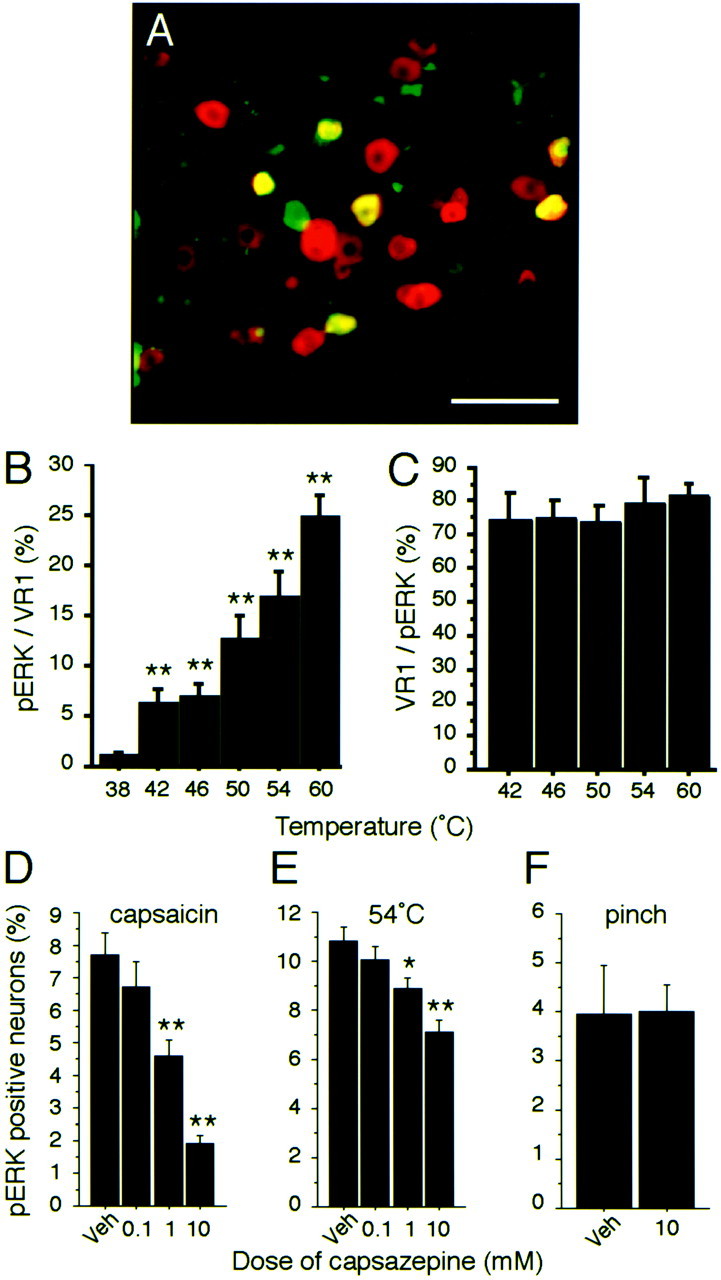
Thermal stimulus-specific phosphorylation of ERK in DRG neurons with VR-1. A, Colocalization of pERK (green) and VR-1 (red) in L4 DRG neurons after thermal stimulation (54°C) of the plantar surface of the hindpaw. Double staining appears yellow.B, Percentages of VR-1-labeled neurons also labeled for pERK after different thermal stimuli (**p < 0.001 compared with 38°C). C, Percentage of pERK-labeled neurons also labeled for VR-1 after different thermal stimuli (n = 4 at each temperature). D–F, Percentage of VR-1-labeled neurons in L4/5 DRG neurons after capsazepine treatment combined with capsaicin injection (D), thermal stimulation (54°C) (E), or mechanical stimulation (F) of the plantar surface of the hindpaw (n = 5 for each group; *p < 0.05; **p < 0.001 compared with vehicle). All data show pERK expression 2 min after stimulation. Veh, Vehicle. Scale bar, 100 μm.
To determine the functional roles of phosphorylation of ERK in primary afferents in peripheral tissues, we examined the changes in pERK in skin and pain behaviors after capsaicin injection into the plantar surface of the hindpaw. First, we examined pERK immunoreactivity in primary afferent terminals in the skin of the plantar surface of the hindpaw. In the skin of control rats, the pERK-labeled fibers and terminals were scattered in the dermis and epidermis (Fig.6A). At 2 min after capsaicin injection (5 μl, 10 mm), the pERK-labeled nerve bundles in the dermis and terminals penetrating into the epidermis were clearly increased compared with controls (Fig.6B). To ascertain whether the pERK labeling was on the nerve terminals, we used double labeling with PGP 9.5 and found that most pERK labeling in the dermis and epidermis was also labeled for PGP 9.5 (Fig. 6C). This dose of capsaicin induced pERK in very few DRG cell bodies. Pretreatment with a selective MEK inhibitor, U0126, 2 min before capsaicin injection clearly prevented the increase in the pERK immunoreactivity in the skin (Fig.6D). Quantification of pERK-labeled fibers and terminals in the dermis and epidermis revealed that pretreatment with the MEK inhibitor U0126 before capsaicin injection significantly suppressed the increase in pERK by capsaicin injection (Fig.6E). These data indicate that activation of the ERK signal transduction pathway occurred in the nerve terminals and fibers in the skin, as well as cell bodies in the DRG, in response to stimulation of the receptor in the periphery.
Fig. 6.

ERK phosphorylation in peripheral nerve terminals and fibers and behavioral effects after capsaicin injection.A–D, Immunostaining of pERK labeling of the plantar surface of the hindpaw. A, Control rats showed the pERK-labeled nerve bundle in the dermis and weak labeling in nerve terminals that penetrate into the epidermis. B, Increased labeling of pERK in nerve terminals and fibers was observed in rats 2 min after capsaicin injection (5 μl, 10 mm) into the plantar surface of the hindpaw. C, Colocalization of pERK (green) and PGP 9.5 (red) in the plantar surface of the rat's hindpaw 2 min after capsaicin injection. Double staining appearsyellow. D, U0126 injection 2 min before capsaicin injection prevented the increase in pERK labeling in peripheral nerve terminals and fibers. E, Relative immunostaining analysis of pERK in the nerve of plantar skin tissue. Each bar represents the relative pERK immunostaining area in the skin tissue. veh, Vehicle;cap, capsaicin. n = 5 for each group (*p < 0.05 compared with veh+cap).F, G, Behavioral changes after the injection of capsaicin and the MEK inhibitor U0126 into the plantar surface of the hindpaw. F, Thermal hyperalgesia was examined using the plantar test. U0126 injections (7.5 and 0.75 μg) significantly changed the time course of withdrawal latency to noxious heat after capsaicin injection into the plantar surface of the hindpaw (n = 6 for each group; *p < 0.05; **p < 0.001). G, Mechanical allodynia was examined using von Frey filaments. U0126 did not show any effect on the 50% threshold of response to mechanical stimuli into the plantar surface of the hindpaw (n = 6 each group). Scale bars, 100 μm.
We subsequently examined whether the phosphorylation of ERK contributes to capsaicin-induced thermal hyperalgesia and secondary mechanical allodynia. The capsaicin injection induced a rapid thermal hyperalgesia at the site of injection that recovered almost completely by 2 hr after injection. The MEK inhibitor U0126 dose-dependently inhibited thermal hyperalgesia (Fig. 6F). The time courses of withdrawal latency with 7.5 and 0.75 μg of U0126-treated rats were significantly different from those of vehicle-treated rats (p < 0.001; two-way repeated ANOVA). U0126 did not affect the withdrawal latencies in naive rats that received no capsaicin treatment (data not shown). In contrast to the effect of U0126 against the thermal hyperalgesia, there was no effect of U0126 on secondary mechanical allodynia (Fig. 6G). These behavioral data indicate that the phosphorylation of ERK through MEK activity in primary afferent neurons is involved in the formation of thermal hyperalgesia (i.e., sensitization of primary afferent neurons).
DISCUSSION
In this article, we show the activity-dependent phosphorylation of ERK in primary afferent neurons. Activity-dependent activation of ERK has been reported in the CNS, especially in the hippocampus (Baraban et al., 1993; English and Sweatt, 1996; Atkins et al., 1998; Durek and Fields, 2001). Recently, several studies have reported ERK phosphorylation in the nociceptive pathway; for example, nociceptive stimuli induce ERK phosphorylation in the spinal dorsal horn (Ji et al., 1999, 2002; Karim et al., 2001), and this event may be related to the hypersensitivity of spinal neurons in inflammatory pain. The activation of metabotropic glutamate receptors in dorsal horn neurons by peripheral inflammation is shown to activate a downstream ERK pathway and result in enhanced pain sensitivity. With respect to the primary afferent neurons, it has been shown that the ERK cascade acts in epinephrine-induced hyperalgesia: the Ras-MEK-ERK1/2 pathway is activated independently of PKA or PKCε (Aley et al., 2001). Nerve growth factor injected into the peripheral tissue increased pERK labeling in tyrosine kinase A-containing DRG neurons (Averill et al., 2001). However, there has been no study examining the ERK activation after application of noxious stimuli to normal tissue.
Action potentials that are transmitted from the periphery and associated Ca2+ transients could activate the specific intracellular signaling pathway and regulate gene expression in DRG neurons (Fields et al., 1997; Fields, 1998). The inhibition of capsaicin-induced pERK labeling by lidocaine block in the present study demonstrates a role of action potentials in the pERK activation. An in vitro study of mouse DRG neurons showed that the phosphorylation of cAMP-responsive element binding protein and ERK/MAP kinase was increased by action potentials with specific temporal patterns (Fields et al., 1997). Other studies suggest that activation of the MAPK (ERK) pathway modulates the voltage-dependent calcium channel (Fitzgerald and Dolphin, 1997; Fitzgerald, 2000), which is in turn involved in the activity of sensory neurons.
Aδ-level electrical stimulation induced pERK in large neurons with NF200. In contrast, c-fiber stimulation by capsaicin injection induced pERK in small neurons containing VR-1 (Fig.1B,I). These data indicated that action potentials transmitted through the primary afferent neurons by stimuli to the periphery induced pERK in DRG cell bodies in a modality-specific way. We found that the electrical stimulation-induced phosphorylation of ERK was dependent on the frequency of stimulation. The frequency may be more effective for inducing pERK in DRG neurons compared with the intensity of the electrical stimulation at the c-fiber level. Actually, as natural stimuli, the intensity of the stimulation of the receptive field was transmitted to the CNS as a function of the frequency of the action potentials evoked by the stimuli (Willis and Coggeshall, 1991). More intense natural stimuli induced more frequent impulses transmitted by the primary afferents (Raja et al., 1999). These characteristics of the primary afferents might be relevant to the data in the present study, in which the 10 Hz electrical stimulation was the most effective in the induction of pERK in DRG neurons. Higher-frequency stimuli (50 or 100 Hz) showed decreased pERK labeling, because these high frequencies may result in the failure to induce full generation of action potentials in the DRG (Djouhri et al., 2001).
An interesting finding in the present study is that the natural stimulation of the receptive field with different intensities resulted in changes in a subpopulation of pERK-labeled neurons. The increase in stimulus intensity resulted in a rightward shift of the size-distribution curve of pERK-labeled neurons (Figs. 3, 4). This tendency was observed in experiments using both mechanical and thermal stimuli, and the number of pERK-labeled neurons also increased as stimulus intensity increased. The percentage of pERK-labeled neurons after natural noxious stimuli to the receptive field was smaller than after electrical stimulation at the c-fiber level. The reason for this discrepancy is that natural stimulation of the receptive field (mechanical or thermal) may activate a part of the L4/5 DRG neurons innervating the hindpaw. Most pERK-labeled neurons are presumably C-polymodal nociceptors. Our data suggest that smaller neurons have lower thresholds in terms of pERK activation in C-polymodal receptors. The intense stimuli induced pERK in NF200-containing neurons, probably in Aδ mechanothermal nociceptors (Fig. 3D). The threshold of the Aδ mechanothermal nociceptor is higher than that of C-polymodal nociceptors (LaMotte et al., 1983), which is consistent with our data that higher temperatures induce double labeling with pERK and NF200.
The characteristics of pERK labeling in DRG neurons after noxious stimuli clearly indicated that the pERK labeling is correlated with the activation state of primary afferent neurons. Therefore, we believe that examination of pERK is very useful as an indicator of the activated DRG neurons after noxious stimuli in vivo. Different noxious stimuli, such as capsaicin injection, mechanical stimuli, and thermal stimuli, induced a variable number of DRG neurons labeled for pERK, suggesting that each noxious stimulus may have a different threshold for activation of the ERK pathway. In addition, non-noxious stimuli, such as the 42°C stimuli, induced pERK labeling in a small number of small neurons. These data suggest that not only noxious stimuli but also innocuous stimuli may induce pERK labeling in DRG neurons. The direct relationship between phosphorylation of ERK and electrophysiological activity in each neuron was not demonstrated here, because the histochemical analysis of the DRG tissue used here did not give us the relationship with electrophysiological activity in individual neurons. Additional study using single-cell recording and analysis of phosphorylation of proteins in the DRG neurons may be required.
As a useful application of pERK labeling by natural stimuli, we examined the relationship between natural stimuli and the function of VR-1. The double-labeling experiments with pERK and VR-1 after natural stimuli showed a high double-labeling ratio after thermal stimuli. The VR-1 antagonist injected into the peripheral tissue just before natural stimulation inhibited the pERK activation in primary afferent neurons evoked by thermal stimuli but not mechanical stimuli. These findings were very consistent with the previous electrophysiological studies and gene knock-out studies of VR-1 function (Caterina et al., 1997, 2000;Tominaga et al., 1998; Davis et al., 2000). The primary afferent activation through VR-1 by capsaicin injection or thermal stimuli may produce action potentials, which in turn result in the phosphorylation of ERK in DRG neurons. The inhibition of heat stimulation-induced pERK labeling by capsazepine was less effective than that of capsaicin-induced pERK labeling (Fig. 5D,E). One possible reason for this may be a methodological problem. The receptive fields that received heat stimuli were larger than the area injected with capsazepine in this experiment. Another possible explanation is that unidentified heat receptors, other than the VR-1, may be working to induce pERK labeling in DRG neurons.
We also found pERK activation in peripheral nerve terminals and fibers as well as DRG cell bodies (Fig. 6). The inhibition of the MEK inhibitor of pERK labeling in peripheral terminals revealed that the signal transduction pathway through MEK and ERK is working in peripheral nerve terminals after activation of VR-1 by capsaicin injection with the same time course as DRG cell bodies. Because the application of the MEK inhibitor prevented thermal hyperalgesia after capsaicin injection, the dense labeling in the epidermis preparation of these fibers and the nerve bundle in the dermis suggested nociceptor activation (i.e., peripheral sensitization). The changes in ERK phosphorylation in peripheral terminals might affect the sensitivity of the primary afferent itself and consequently affect the pain behavior. In a human study, after the injection of capsaicin into the skin, primary hyperalgesia and secondary hyperalgesia appeared around the injection site (Torebjork et al., 1992). In the present study, the secondary mechanical allodynia was not changed by application of MEK inhibitor to the peripheral tissue. Because the secondary mechanical allodynia is a result of the central sensitization after capsaicin injection (Raja et al., 1999), we believe that the phosphorylation of ERK in peripheral nerve is not involved in the central sensitization. A number of recent studies using isolated DRG neurons in culture have shed light on the cellular and molecular mechanisms of nociception and sensitization (for review, see Julius and Basbaum, 2001). The signaling pathway mediating hyperalgesia produced by inflammatory agents has been examined extensively (Gold et al., 1998; Khasar et al., 1999; Aley et al., 2001; Ji et al., 2002). The molecular mechanisms by which some chemicals, such as bradykinin, modulate VR-1 activity have been reported to show the involvement of PKC and phospholipase C signaling pathways (Premkumar and Ahern, 2000; Chuang et al., 2001). Our data indicating that the ERK pathway was activated shortly after capsaicin injection and was involved in the thermal hyperalgesia might account for the important signaling mechanism of peripheral sensitization.
We demonstrated in vivo that very rapid phosphorylation of ERK occurred in DRG neurons that were taking part in the transmission of various noxious signals. Moreover, intensity-dependent changes in subpopulations showing pERK labeling are novel findings that would have been difficult to obtain using electrophysiological techniques. The phosphorylation of ERK in DRG neurons after noxious stimulation might be useful for examining the activation state of each neuron that contains various pain-related molecules. Moreover, using this pERK activation, we could confirm the physiological role of the VR-1 in nociception and found that the ERK signaling pathway plays an important role in peripheral sensitization after noxious stimulation to the peripheral tissues.
Footnotes
This study was supported in part by grants-in-aid for Scientific Research and a grant for the Open Research Center of the Hyogo College of Medicine from the Japanese Ministry of Education, Science, and Culture. We thank Dr. Tominaga for providing the VR-1 antibody. We gratefully acknowledge technical assistance from Kimiko Kobayashi and Nobumasa Ushio. We thank Dr. D. A. Thomas for correcting the English usage in this manuscript.
Correspondence should be addressed to Koichi Noguchi, Department of Anatomy and Neuroscience, Hyogo College of Medicine, 1-1 Mukogawa-cho, Nishinomiya, Hyogo 663-8501, Japan. E-mail:noguchi@hyo-med.ac.jp.
REFERENCES
- 1.Aley KO, Martin A, McMahon T, Mok J, Levine JD, Messing RO. Nociceptor sensitization by extracellular signal-regulated kinases. J Neurosci. 2001;21:6933–6939. doi: 10.1523/JNEUROSCI.21-17-06933.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amaya F, Decosterd I, Samad TA, Plumpton C, Tate S, Mannion RJ, Costigan M, Woolf CJ. Diversity of expression of the sensory neuron-specific TTX-resistant voltage-gated sodium ion channels SNS and SNS2. Mol Cell Neurosci. 2000;15:331–342. doi: 10.1006/mcne.1999.0828. [DOI] [PubMed] [Google Scholar]
- 3.Atkins CM, Selcher JC, Petraitis JJ, Trzaskos JM, Sweatt JD. The MAPK cascade is required for mammalian associative learning. Nat Neurosci. 1998;1:602–609. doi: 10.1038/2836. [DOI] [PubMed] [Google Scholar]
- 4.Averill S, Delcroix JD, Michael GJ, Tomlinson DR, Fernyhough P, Priestley JV. Nerve growth factor modulates the activation status and fast axonal transport of ERK 1/2 in adult nociceptive neurons. Mol Cell Neurosci. 2001;18:183–196. doi: 10.1006/mcne.2001.1015. [DOI] [PubMed] [Google Scholar]
- 5.Baraban J, Fiore RS, Sanghera JS, Paddon HB, Pelech SL. Identification of p42 mitogen-activated protein kinase as a tyrosine kinase substrate activated by maximal electroconvulsive shock in hippocampus. J Neurochem. 1993;60:330–336. doi: 10.1111/j.1471-4159.1993.tb05855.x. [DOI] [PubMed] [Google Scholar]
- 6.Bennett DL, Michael GJ, Ramachandran N, Munson JB, Averill S, Yan Q, McMahon SB, Priestley JV. A distinct subgroup of small DRG cells express GDNF receptor components and GDNF is protective for these neurons after nerve injury. J Neurosci. 1998;18:3059–3072. doi: 10.1523/JNEUROSCI.18-08-03059.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bevan S, Hothi S, Hughes G, James IF, Rang HP, Shah K, Walpole CS, Yeats JC. Capsazepine: a competitive antagonist of the sensory neurone excitant capsaicin. Br J Pharmacol. 1992;107:544–552. doi: 10.1111/j.1476-5381.1992.tb12781.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- 9.Caterina MJ, Leffler A, Malmberg AB, Martin WJ, Trafton J, Petersen-Zeitz KR, Koltzenburg M, Basbaum AI, Julius D. Impaired nociception and pain sensation in mice lacking the capsaicin receptor. Science. 2000;288:306–313. doi: 10.1126/science.288.5464.306. [DOI] [PubMed] [Google Scholar]
- 10.Chang L, Karin K. Mammalian MAP kinase signaling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 11.Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- 12.Chuang HH, Prescott ED, Kong H, Shields S, Jordt SE, Basbaum AI, Chao MV, Julius D. Bradykinin and nerve growth factor release the capsaicin receptor from Ptdlns(4,5)P2-mediated inhibition. Nature. 2001;411:957–962. doi: 10.1038/35082088. [DOI] [PubMed] [Google Scholar]
- 13.Davis JB, Gray J, Gunthorpe MJ, Hatcher JP, Davey PT, Overend P, Harries MH, Latcham J, Clapham C, Atkinson K, Hughes SA, Rance K, Grau E, Harper AJ, Pugh PL, Rogers DC, Bingham S, Randall A, Sheardown SA. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature. 2000;405:183–187. doi: 10.1038/35012076. [DOI] [PubMed] [Google Scholar]
- 14.Day IN, Thompson RJ. Molecular cloning of cDNA coding for human PGP 9.5 protein: a novel cytoplasmic marker for neurones and neuroendocrine cells. FEBS Lett. 1987;210:157–160. doi: 10.1016/0014-5793(87)81327-3. [DOI] [PubMed] [Google Scholar]
- 15.Djouhri L, Dawbarn D, Robertson A, Newton R. Time course and nerve growth factor dependence of inflammation-induced alterations in electrophysiological membrane properties in nociceptive primary afferent neurons. J Neurosci. 2001;15:8722–8733. doi: 10.1523/JNEUROSCI.21-22-08722.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dubner R, Ruda MA. Activity-dependent neuronal plasticity following tissue injury and inflammation. Trends Neurosci. 1992;15:96–103. doi: 10.1016/0166-2236(92)90019-5. [DOI] [PubMed] [Google Scholar]
- 17.Durek SM, Fields RD. Mitogen-activated protein kinase/extracellular signal-regulated kinase activation in somatodendritic compartments: roles of action potentials, frequency, and model of calcium entry. J Neurosci. 2001;21:RC122:1–5. doi: 10.1523/JNEUROSCI.21-02-j0002.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.English JD, Sweatt JD. Activation of p42 mitogen-activated protein kinase in hippocampal long-term potentiation. J Biol Chem. 1996;271:24329–24332. doi: 10.1074/jbc.271.40.24329. [DOI] [PubMed] [Google Scholar]
- 19.Fields RD. Effects of ion channel activity on development of dorsal root ganglion neurons. J Neurobiol. 1998;37:158–170. doi: 10.1002/(sici)1097-4695(199810)37:1<158::aid-neu12>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- 20.Fields RD, Eshete F, Stevens B, Itoh K. Action potential-dependent regulation of gene expression: temporal specificity in Ca2+, cAMP-responsive element binding proteins, and mitogen-activated protein kinase signaling. J Neurosci. 1997;17:7252–7266. doi: 10.1523/JNEUROSCI.17-19-07252.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fitzgerald EM. Regulation of voltage-dependent calcium channels in rat sensory neurones involves a Ras-mitogen-activated protein kinase pathway. J Physiol (Lond) 2000;527:433–444. doi: 10.1111/j.1469-7793.2000.00433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fitzgerald EM, Dolphin AC. Regulation of rat neuronal voltage-dependent calcium channels by endogenous p21-ras. Eur J Neurosci. 1997;9:1252–1261. doi: 10.1111/j.1460-9568.1997.tb01480.x. [DOI] [PubMed] [Google Scholar]
- 23.Fukuoka T, Kondo E, Dai Y, Hashimoto N, Noguchi K. Brain-derived neurotrophic factor increases in the uninjured dorsal root ganglion neurons in selective spinal nerve ligation model. J Neurosci. 2001;21:4891–4900. doi: 10.1523/JNEUROSCI.21-13-04891.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gold MS, Levine J, Correa AM. Modulation of TTX-R Ina by PKC and PKA and their role in PGE2-induced sensitization of rat sensory neurons in vitro. J Neurosci. 1998;18:10345–10355. doi: 10.1523/JNEUROSCI.18-24-10345.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- 26.Hunyady B, Krempels K, Harta G, Mezey E. Immunohistochemical signal amplification by catalyzed reporter deposition and its application in double immunostaining. J Histochem Cytochem. 1996;44:1353–1362. doi: 10.1177/44.12.8985127. [DOI] [PubMed] [Google Scholar]
- 27.Impey S, Obrietan K, Wong ST, Poser S, Yano S, Wayman G, Deloulme JC, Chan G, Storm DR. Cross talk between ERK and PKA is required for Ca2+ stimulation of CREB-dependent transcription and ERK nuclear translocation. Neuron. 1998;21:869–883. doi: 10.1016/s0896-6273(00)80602-9. [DOI] [PubMed] [Google Scholar]
- 28.Impey S, Obrietan K, Storm DR. Making new connections: role of ERK/MAP kinase signaling in neuronal plasticity. Neuron. 1999;23:11–14. doi: 10.1016/s0896-6273(00)80747-3. [DOI] [PubMed] [Google Scholar]
- 29.Ji RR, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci. 1999;2:1114–1119. doi: 10.1038/16040. [DOI] [PubMed] [Google Scholar]
- 30.Ji RR, Befort K, Brenner GJ, Woolf CJ. ERK MAP kinase activation in superficial spinal cord neurons induces prodynorphin and NK-1 upregulation and contributes to persistent inflammatory pain hypersensitivity. J Neurosci. 2002;22:478–485. doi: 10.1523/JNEUROSCI.22-02-00478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001;413:203–210. doi: 10.1038/35093019. [DOI] [PubMed] [Google Scholar]
- 32.Karim F, Wang CC, Gereau RW., IV Metabotropic glutamate receptor subtypes 1 and 5 are activators of extracellular signal-regulated kinase signaling required for inflammatory pain in mice. J Neurosci. 2001;21:3771–3779. doi: 10.1523/JNEUROSCI.21-11-03771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khasar SG, Lin YH, Martin A, Dadgar J, McMahon T, Wang D, Hundle B, Aley KO, Isenberg W, McCarter G, Green PG, Hodge CW, Levine JD, Messing RO. A novel nociceptor signaling pathway revealed in protein kinase C ε mutant mice. Neuron. 1999;24:253–260. doi: 10.1016/s0896-6273(00)80837-5. [DOI] [PubMed] [Google Scholar]
- 34.LaMotte RM, Thalhamme JG, Robinson CJ. Peripheral neural correlates of magnitude of cutaneous pain and hyperalgesia: a comparison of neural events in monkey with sensory judgments in human. J Neurophysiol. 1983;50:1–26. doi: 10.1152/jn.1983.50.1.1. [DOI] [PubMed] [Google Scholar]
- 35.Martin KC, Michael D, Rose JC, Barad M, Casadio A, Zhu H, Kandel ER. MAP kinase translocates into the nucleus of the presynaptic cell and is required for long-term facilitation in Aplysia. Neuron. 1997;18:899–912. doi: 10.1016/s0896-6273(00)80330-x. [DOI] [PubMed] [Google Scholar]
- 36.Michael GJ, Averill S, Nitkunan A, Rattray M, Bennett DL, Yan Q, Priestley JV. Nerve growth factor treatment increases brain-derived neurotrophic factor selectively in TrkA-expressing dorsal root ganglion cells and in their central terminations within the spinal cord. J Neurosci. 1997;17:8476–8490. doi: 10.1523/JNEUROSCI.17-21-08476.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Noguchi K, Kawai Y, Fukuoka T, Senba E, Miki K. Substance P induced by peripheral nerve injury in primary afferent sensory neurons and its effect on dorsal column nucleus neurons. J Neurosci. 1995;15:7633–7643. doi: 10.1523/JNEUROSCI.15-11-07633.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Obrietan K, Impey S, Storm DR. Light and circadian rhythmicity regulate MAP kinase activation in the suprachiasmatic nuclei. Nat Neurosci. 1998;1:693–700. doi: 10.1038/3695. [DOI] [PubMed] [Google Scholar]
- 39.Premkumar LS, Ahern GP. Induction of vanilloid receptor channel activity by protein kinase C. Nature. 2000;408:985–990. doi: 10.1038/35050121. [DOI] [PubMed] [Google Scholar]
- 40.Raja SN, Meyer RA, Ringkamp M, Campbell JN. Peripheral neural mechanisms of nociception. In: Wall PD, Melzack R, editors. Textbook of pain. Churchill Livingstone; New York: 1999. pp. 11–57. [Google Scholar]
- 41.Rosen LB, Ginty DD, Weber MJ, Greenberg ME. Membrane depolarization and calcium influx stimulate MEK and MAP kinase via activation of Ras. Neuron. 1994;12:1207–1221. doi: 10.1016/0896-6273(94)90438-3. [DOI] [PubMed] [Google Scholar]
- 42.Shindler KS, Roth KA. Double immunofluorescent staining using two unconjugated primary antisera raised in the same species. J Histochem Cytochem. 1996;44:1331–1335. doi: 10.1177/44.11.8918908. [DOI] [PubMed] [Google Scholar]
- 43.Snider WD, McMahon SB. Tackling pain at the source: new ideas about nociceptors. Neuron. 1998;20:629–632. doi: 10.1016/s0896-6273(00)81003-x. [DOI] [PubMed] [Google Scholar]
- 44.Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum AI, Julius D. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 1998;21:531–543. doi: 10.1016/s0896-6273(00)80564-4. [DOI] [PubMed] [Google Scholar]
- 45.Torebjork HE, Lundberg LER, LaMotte RH. Central changes in processing of mechanoreceptive input in capsaicin-induced secondary hyperalgesia in humans. J Neurophysiol. 1992;448:765–780. doi: 10.1113/jphysiol.1992.sp019069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trojanowski JQ, Walkenstein N, Lee VM. Expression of neurofilament subunits in neurons of the central and peripheral nervous system: an immunohistochemical study with monoclonal antibodies. J Neurosci. 1986;6:650–660. doi: 10.1523/JNEUROSCI.06-03-00650.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Willis WD, Coggeshall RE. Sensory mechanisms of the spinal cord, Ed 2. Plenum, New York; 1991. Peripheral nerves and sensory receptors. pp. 13–45. [Google Scholar]
- 48.Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]