

Abstract
Genetic and pharmacological perturbations suggest that tyrosine receptor kinase B (trkB) receptor activation promotes limbic epileptogenesis, but whether or where trkB activation occurs during epileptogenesis is uncertain. Because activation of trk receptors involves phosphorylation of specific tyrosine residues (Segal et al., 1996), the availability of antibodies that selectively recognize the phosphorylated form of trk receptors at the Shc site permits an immunohistochemical assessment of trk receptor activation. We reported previously increased phospho-specific trk (p-trk) immunoreactivity in the mossy fiber pathway of the hippocampus during epileptogenesis in rats (Binder et al., 1999b). Because the p-trk antibody does not distinguish among trkA, trkB, and trkC, the identity of the neurotrophin receptor(s) undergoing phosphorylation was uncertain. The development of mice carrying a point mutation of the Shc binding site (Y515F) in the trkB gene (trkBshc) provided an opportunity to test the hypothesis that trkB is the neurotrophin receptor undergoing phosphorylation. Epileptogenesis in wild-type (WT) mice was associated with increased p-trk immunoreactivity in both the mossy fiber pathway and CA3 stratum oriens of hippocampus. In contrast, the epileptogenesis-associated increase of p-trk immunoreactivity was reduced in trkBshc mutant mice. The development of epileptogenesis as measured by electrophysiological and behavioral indices did not differ betweentrkBshc mutant and WT mice. These data demonstrate that the neurotrophin receptor trkB undergoes phosphorylation in the mossy fiber pathway and CA3 stratum oriens of the hippocampus during limbic epileptogenesis. In addition, the signaling pathways activated by the Shc site of trkB exert no detectable regulatory effects on limbic epileptogenesis.
Keywords: epileptogenesis, kindling, seizures, neurotrophin, trkB receptor, Shc, hippocampus, phosphorylation
Elucidating the mechanisms of limbic epileptogenesis in cellular and molecular terms may provide novel therapeutic approaches to prevent the disease. In the kindling model, repeated induction of brief, focal seizures by application of initially subconvulsive electrical stimuli eventually results in intense focal and tonic–clonic seizures (Goddard et al., 1969). Once established, the enhanced sensitivity to electrical stimulation is lifelong. The cellular and molecular events by which seizures beget more intense seizures are incompletely understood. The idea emerged that extracellular signaling molecules might be pivotal in the transduction process. Two criteria for candidate signals emerged: that seizure activity regulates the production and release of the molecules and that the molecules effect structural and functional changes that could result in hyperexcitability. One class of molecules that met these criteria was the neurotrophins, in particular brain-derived neurotrophic factor (BDNF) and its cognate tyrosine kinase receptor trkB. Limbic seizures increase the mRNA content of BDNF in multiple neuronal populations of forebrain (Isackson et al., 1991). The development of kindling is inhibited in mice heterozygous for BDNF (Kokaia et al., 1995). Intraventricular infusion of trk receptor bodies sequester and limit the action of distinct neurotrophins; infusion of trkB-Fc inhibited epileptogenesis, whereas infusion of trkA-Fc or trkC-Fc did not (Binder et al., 1999a). The occurrence of epileptiform spiking on electroencephalogram (EEG) long after kainate-induced status epilepticus is impaired in transgenic mice overexpressing truncated trkB, presumably by limiting activation of trkB (Lähteinen et al., 2002). Finally, although conflicting results emerged from intracerebral infusions of BDNF (Larmet et al., 1995; Scharfman et al., 2002), transgenic mice overexpressing BDNF exhibit an enhanced response to epileptogenic stimuli (Croll et al., 1999).
Together with the seizure-induced expression of BDNF, these findings led to the hypothesis that enhanced activation of trkB occurs in the hippocampus during limbic epileptogenesis. Because activation of trk receptors involves phosphorylation of specific tyrosine residues (Schlessinger and Ullrich, 1992; Segal and Greenberg, 1996), the availability of antibodies that selectively recognize the phosphorylated form of trk receptors (Segal et al., 1996) permits immunohistochemical assessment of trk receptor activation. We reported increased phospho-specific trk immunoreactivity (p-trk IR) in the hippocampal mossy fiber pathway during epileptogenesis in rats (Binder et al., 1999b). Because the p-trk antibody does not distinguish among trkA, trkB, and trkC, the precise neurotrophin receptor(s) undergoing phosphorylation was uncertain. The temporal and spatial concordance of seizure-induced increases of p-trk immunoreactivity and BDNF protein suggested that increased phosphorylation of trkB was responsible for the increased p-trk immunoreactivity. The development of mice carrying a point mutation of the Shc binding site in the trkB gene (trkBshc mutant mice) permitted testing this hypothesis. That is, substitution of phenylalanine for tyrosine at residue 515 of trkB (Y515F) eliminates the binding site for the phospho-trk antibody in trkB but not trkA or trkC. This mutation of trkB disrupts the binding of Shc adaptor protein to trkB and abolishes Shc site-mediated downstream signaling events (Minichiello et al., 1998). If our hypothesis is correct, induction of epileptogenesis should produce increased phospho-trk immunoreactivity in the mossy fiber pathway of wild-type (WT) but nottrkBshc mutant mice.
MATERIALS AND METHODS
Mice. trkBshc mutant mice were generated as described previously (Minichiello et al., 1998). In brief, PCR-aided mutagenesis was used to introduce a single point mutation (A to T, position 2055) in trkB receptor that resulted in a change of tyrosine 515 into phenylalanine (Y515F). Nonphosphorylatable F515 disrupted the binding of adaptor proteins to the Shc site in trkB and abolished Shc site-mediated down-stream signaling events. The amount of autophosphorylated trkB was reduced intrkBshc/shc neurons after BDNF or neurotrophin-4 (NT-4) stimulation. In the present study, the genotype of each animal was assessed twice using PCR of genomic DNA isolated from tail (before experiment) and liver (after being killed). Nine wild-type (+/+), 14 heterozygous (+/−), and 13 homozygous (−/−) trkBshc mice (20–35 gm) were used in this experiment.
Antibody. A peptide affinity-purified polyclonal trk antibody (pY490) (New England Biolabs, Beverly, MA) directed against a synthetic phospho-tyr 490 peptide corresponding to residues 485–493 (IENPQY*FSD) of human trkA was used for immunohistochemistry as we described previously (Binder et al., 1999b). The sequence of this peptide is highly conserved among trkA, trkB, and trkC receptors and also among human, rat, and mouse. Antibody pY490 is phospho-specific, recognizing the phosphorylated but not the nonphosphorylated form of peptide 490. Additionally, pY490 is phosphorylation site specific, recognizing phosphorylated 490 but not a trk peptide phosphorylated at residues 674/675 (Binder et al., 1999b).
Surgery and kindling. Under pentobarbital anesthesia (60 mg/kg, i.p.), a stimulation–recording bipolar electrode was stereotactically implanted in the right amygdala using the following coordinates, with bregma as the reference: 1.0 mm posterior, 2.9 mm lateral, and 4.6 mm below dura. A wire secured to the skull overlying the left frontal cortex was used as the ground electrode. After a postoperative recovery period of at least 1 week, the electrographic seizure threshold (EST) was determined by application of 1 sec train of 1 msec biphasic rectangular pulses at 60 Hz beginning at 60 μA; additional stimulations increasing by 20 μA were administered at 1 min intervals until an electrographic seizure lasting at least 5 sec was detected on the EEG recorded from the amygdala. Subsequently, experimental animals were stimulated twice each day at a stimulus intensity 100 μA above the EST, with an interstimulus interval of at least 4 hr until three consecutive seizures of class 4 or greater with limb clonus and/or tonus lasting at least 12 sec were evoked. The behavioral manifestations of seizures were classified according to a modification of Racine's classification (Racine, 1972): 1, facial clonus; 2, head nodding; 3, unilateral forelimb clonus; 4, rearing with bilateral forelimb clonus; 5, rearing and falling (loss of postural control); 6, running or bouncing seizures; and 7, tonic hindlimb extension. The surgery and kindling procedures were performed by an individual blinded to genotype of the animals. Control animals included unimplanted as well as implanted but unstimulated mice.
Perfusion and histology. After completion of kindling and a subsequent stimulation-free period of 2 weeks, the persistence of the hyperexcitability was determined by administration of an additional stimulation of the same intensity used in kindling procedure. Twenty-four hours after stimulation, the experimental and control animals were perfused transcardially under pentobarbital anesthesia with ice-cold 4% paraformaldehyde in 1× PBS containing 1 mm sodium orthovanadate (PBSV) for 5 min at a flow rate 7.5 ml/min. The brains were removed, postfixed in the same solution overnight at 4°C, and cryoprotected in 20% sucrose in 1× PBSV until they sank. Coronal sections of 40 μm were cut in a cryostat at −20°C, and two sections per slide were mounted in PBSV on slides, air dried, frozen, and stored at −70°C until use. The sections were stained with methyl green pyronine-Y for determination of electrode placement. Only animals with correct electrode placement in the amygdaloid complex were included in the statistical analysis.
Phospho-trk immunohistochemistry. To facilitate quantitative comparisons, slide-mounted sections isolated from stimulated and unstimulated animals of all three genotypes (WT, heterozygotes, and homozygotes) were processed in batches using the identical solutions and durations of incubations, washes, etc. Antibody pY490 was used to detect the phosphorylated form of trk receptor using the protocol described in detail previously (Binder et al., 1999b). Briefly, after quenching of endogenous peroxidase activity with 0.3% H2O2/MeOH for 30 min, sections were blocked and permeabilized in 5% normal goat serum (NGS) and 0.5% NP-40 in 1× PBSV for 1 hr. The primary antibody, pY490 (1:10 in 1× PBSV and 5% NGS), was incubated with sections and covered by coverslips in a humidified chamber at room temperature for 1 hr. The sections were subsequently incubated in biotin-conjugated goat anti-rabbit IgG (1:100 in PBSV and 5% NGS; Jackson ImmunoResearch, West Grove, PA) for 1 hr. The ABC method (1:100 in PBSV with 5% NGS, applied twice; Vectastain Elite; Vector Laboratories, Burlingame, CA) was used for detection of immunoreactivity. Biotinyl tyramide (1:100 in PBSV with 5% NGS, applied between two ABC incubations; Bio-Rad, Richmond, CA) was used to enhance detection of the immunoreactivity. The sections were preincubated with DAB solution (Sigma Fast; Sigma, St. Louis, MO) containing 0.04% nickel ammonium sulfate for 15 min and then developed for 10–30 min in the same DAB solution but also containing 0.03% H2O2. The slides were rinsed in PBS, dehydrated in ethanol, cleared in xylene, and coverslipped. The slides were subsequently coded and analyzed blinded.
Peptide competition. The immunogen of antibody pY490 (phosphopeptide 490) and the unphosphorylated form of the same peptide were used for peptide competition. For other controls, four additional peptides of trkA, the phospho- and unphospho-peptides of sites 674/675 and 785, respectively (New England Biolabs), were included in the experiments. Each peptide used at 300 nm was incubated at room temperature with the primary antibody solution (1:10 in PBSV with 5% NGS) for 30 min to 1 hr. After slides were quenched, blocked, and permeabilized, the primary antibody solution coincubated with the relevant peptide was applied to the sections. The remaining steps followed the standard protocol of phospho-trk immunohistochemistry.
Densitometry quantification. Images of the immunoreactivity in the CA3 and dentate gyrus (DG) of hippocampus were captured using a high-resolution CCD camera interfaced with a light microscope (Axiovert 135; Zeiss, Oberkochen, Germany) and measured using a computer-assisted image analyzer (MetaMorph 3D; Universal Imaging, West Chester, PA). For densitometric analysis, a square box of fixed size was placed in distinct regions within CA3 and dentate gyrus to measure the average gray value for each location. For CA3, the gray value was measured in three locations: strata radiatum, lucidum, and oriens. For dentate gyrus, gray values were measured in five locations: outer molecular layer (OML), middle molecular layer (MML), inner molecular layer (IML), hilar border with granule cell layer (hilus-GCL border), and deep hilus at the midpoint of granule cell layer (see Fig. 3). Because the corpus callosum had less variation and a higher gray value reflecting less immunoreactivity, it was chosen as the reference. The results from measured locations are presented as the percentage of reduction in gray value compared with corpus callosum, a greater reduction in gray value reflecting more phospho-trk immunoreactivity. The data for each animal are expressed as the average from four hippocampi (one slide per animal containing two sections, each with two hippocampi). All results were analyzed by one-way ANOVA.
Fig. 3.

Locations at which densitometric measures of p-trk immunoreactivity were performed: 1, strata radiatum;2, lucidum; 3, oriens; 4, deep hilus; 5, hilus-GCL border; 6, IML;7, MML; 8, OML.
RESULTS
Increased p-trk IR in WT mice after kindling
Our recent studies provided immunohistochemical evidence that trk receptors undergo phosphorylation in a spatially specific pattern in the hippocampus during the development of kindling of rats (Binder et al., 1999b). To determine whether trk receptor phosphorylation also occurs in mice, p-trk immunohistochemistry was performed in wild-type mice killed 24 hr after the last seizure evoked by stimulation of the right amygdala. A spatially selective increase of p-trk IR in the hippocampal formation was detected in each of the four stimulated WT mice compared with unstimulated WT mice (Fig.1, compare A, B,arrows). The increased immunoreactivity was evident in the hippocampal formation bilaterally, mainly in the mossy fiber pathway. In addition, a moderately high level but more diffuse pattern of p-trk IR was apparent throughout stratum oriens of CA3. In contrast, no overt changes of p-trk IR were evident in CA1, in stratum radiatum of CA3, or in the molecular layer of the dentate gyrus. Quantitative analyses of these data are presented below. Importantly, omission of the primary antibody eliminated immunoreactivity in these WT animals (data not shown).
Fig. 1.
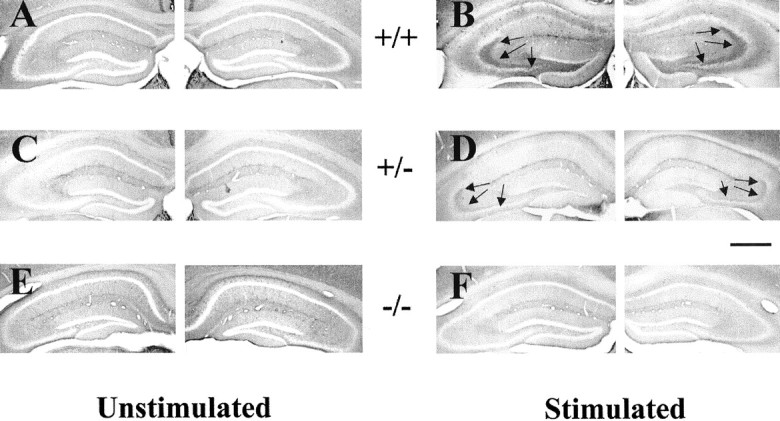
Reduction of seizure-induced increase of p-trk immunoreactivity in trkBshcmutant mice.A, C, E, p-trk immunoreactivity in unstimulated +/+, +/−, and −/− mice. Note the absence of detectable immunoreactivity in dentate hilus and CA3 stratum lucidum of hippocampus. B, D,F, p-trk immunoreactivity in +/+, +/−, and −/− mice 24 hr after a seizure evoked by stimulation of right amygdala.Arrows denote immunoreactivity in dentate hilus and stratum lucidum in stimulated +/+ and +/− mice. Despite some differences in background among individual animals, no systematic variation in background attributable to genotype or stimulation status was present (data not shown). Note that quantitation was controlled for individual differences in background by use of a control region (corpus callosum) intrinsic to each section. Scale bar, 650 μm.
To verify the specificity of p-trk IR in kindled mice, peptide competition experiments were performed. Preincubation of pY490 antibody with its phosphopeptide immunogen (300 nm) eliminated p-trk IR in sections from kindled mice (Fig. 2, compare A, C) and unstimulated controls (data not shown). Importantly, preincubation with the unphosphorylated form of peptide 490 had no effect on the immunoreactivity (Fig. 2, compareA, B). Moreover, preincubation of pY490 antibody with either phospho- or unphospho-peptides from other sites of the trk receptor, 674/675 and 785, did not reduce p-trk IR (data not shown). Together, the results from these competition experiments support the assertion that the p-trk IR detected by antibody pY490 in wild-type mice reflects the phosphorylated form of a trk receptor at a tyrosine residue within a sequence similar to that of pY490 of human trkA.
Fig. 2.
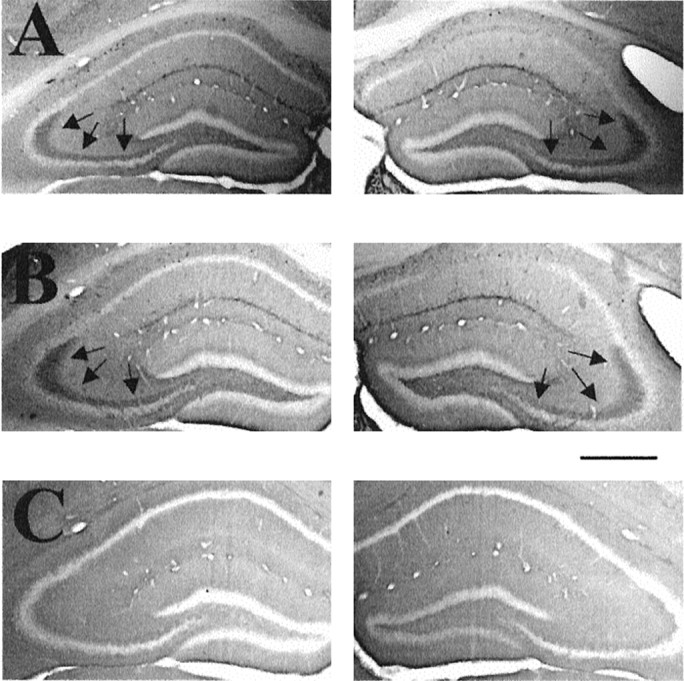
Reduction of p-trk immunoreactivity by phosphopeptide but not by nonphosphopeptide. Immunoreactivity to p-trk is shown in representative coronal sections of hippocampus from a WT mouse killed 24 hr after a kindled seizure in which pY490 antibody is coincubated with the following: A, no peptide;B, 300 nm nonphosphorylated form of peptide 490; C, 300 nm phosphorylated form of peptide 490. Scale bar, 650 μm.
Analyses of brain regions outside the hippocampal formation in unstimulated controls disclosed p-trk immunoreactivity in multiple areas of forebrain, including neocortex, lateral habenula, and some hypothalamic nuclei (data not shown). In contrast to the hippocampal formation, no marked increases were noticed in any of these regions after seizures evoked in WT mice. Thus, subsequent analyses were confined to the hippocampal formation in these studies.
Reduction of seizure-induced increased phospho-trk immunoreactivity in trkBshc mutant mice
Because the pY490 antibody can detect the phosphorylated tyrosine residue in trkA, trkB, or trkC (Segal et al., 1996), the precise trk receptor undergoing phosphorylation in the hippocampus during epileptogenesis is uncertain. To determine whether phosphorylation of trkB in particular accounts for the increased immunoreactivity in the hippocampus during epileptogenesis, p-trk immunohistochemistry was performed in WT and trkBshc mice, which were either not stimulated or were killed 1 d after a stimulation-evoked seizure. As described in the preceding section, a marked increase of p-trk IR was evident in stratum lucidum and dentate hilus with a moderate increase in stratum oriens of CA3 after a seizure evoked in WT mice (Fig. 1, compare A, B). ThetrkBshc mutation reduced the seizure-evoked increase of immunoreactivity. That is, seizures evoked in trkBshc null mice triggered no significant increase of immunoreactivity in stratum lucidum, dentate hilus, or stratum oriens of CA3 (Fig. 1, compare B,F). Results with heterozygoustrkBshc were intermediate between the WT and trkBshc nulls (Fig. 1, compareB, D, F, in lucidum, dentate hilus, and stratum oriens of CA3).
The immunoreactivity within hippocampus was quantified in distinct subregions (Fig. 3) and confirmed the findings evident by visual inspection. First, analyses of WT mice confirmed the spatially selective, twofold to threefold increase of phospho-trk immunoreactivity in stratum lucidum, the dentate hilus, and hilus border after a seizure compared with unstimulated controls (Fig.4A,B). Significant (p < 0.05; ANOVA) increases of a smaller magnitude were also evident in stratum oriens of CA3 (Fig.4A). In contrast, comparison of stimulated with unstimulated trkBshc null mutants disclosed no significant seizure-induced increases of immunoreactivity in stratum lucidum, the dentate hilus or hilus border, or CA3 stratum oriens (Fig. 4A,B). In addition to the above comparisons of unstimulated versus stimulated mice of a given genotype, comparisons of seizure-treated mice of distinct genotypes revealed significant increases of immunoreactivity in the mossy fiber pathway and stratum oriens of CA3 in WT compared withtrkBshc null mutants (Fig.4A,B). Similar analyses oftrkBshc heterozygotes disclosed results intermediate between wild-type and null mutants, but no significant increases of immunoreactivity were detected in the mossy fiber pathway and stratum oriens of CA3 after a kindled seizure compared with unstimulated controls of the same genotype (Fig.4A,B). Interestingly, the increases of p-trk IR after seizures exhibited striking heterogeneity among thetrkBshc heterozygotes (data described but not shown). Robust staining was seen in three of nine heterozygous mice after a seizure with intensity and distribution similar to WT mice after a seizure. However, no detectable increase was evident in four heterozygous mice and a slight increase in the remaining two mice. Together, these quantitative analyses demonstrate that substitution of phenylalanine for tyrosine at residue 515 of trkB reduces the increased p-trk IR after limbic seizures.
Fig. 4.

Quantitative analyses of p-trk immunoreactivity in hippocampal subregions of stimulated and unstimulated WT, heterozygous, and null (trkBshc) mutant mice. The data are the mean ± SEM of the percentage reduction in gray value in the given subregions compared with corpus callosum (see Materials and Methods); high values reflect more intense immunoreactivity. *p < 0.05 compared with unstimulated WT mice; φp < 0.05 compared with kindled and stimulated WT mice (one-way ANOVA with post hoc Bonferroni's test).
Development and persistence of kindling intrkBshc mutant mice
No significant differences in the development of kindling were detected among WT (+/+), heterozygous, and nulltrkBshc mutants, as evident in the following observations. First, the current required to evoke an afterdischarge (i.e., EST) was similar in the three groups (180.0 ± 40.1 μA for +/+, n = 4; 151.1 ± 22.6 μA for +/−, n = 9; 180 ± 24.8 μA for −/−,n = 8; mean ± SEM; p > 0.05; one-way ANOVA). Second, no significant differences were detected in the numbers of stimulations required to evoke the initial clonic motor seizures (class 4 or greater) in WT (5.5 ± 0.9 stimulations) compared with heterozygous (6.4 ± 0.5) or null (6.8 ± 0.7) mutants. Third, no significant differences were found in the numbers of stimulations required to evoke three consecutive class 4 (or greater) seizures, the numbers of stimulations being 9.8 ± 1.5, 10.3 ± 0.5, and 10.8 ± 0.9 in WT, heterozygous, and null mutants, respectively (Fig. 5A). Finally, no significant differences were noted in the duration of electrographic seizures during kindling development among the three experimental groups (Fig. 5B). In summary, kindling development appears to proceed normally in trkBshc mutants.
Fig. 5.
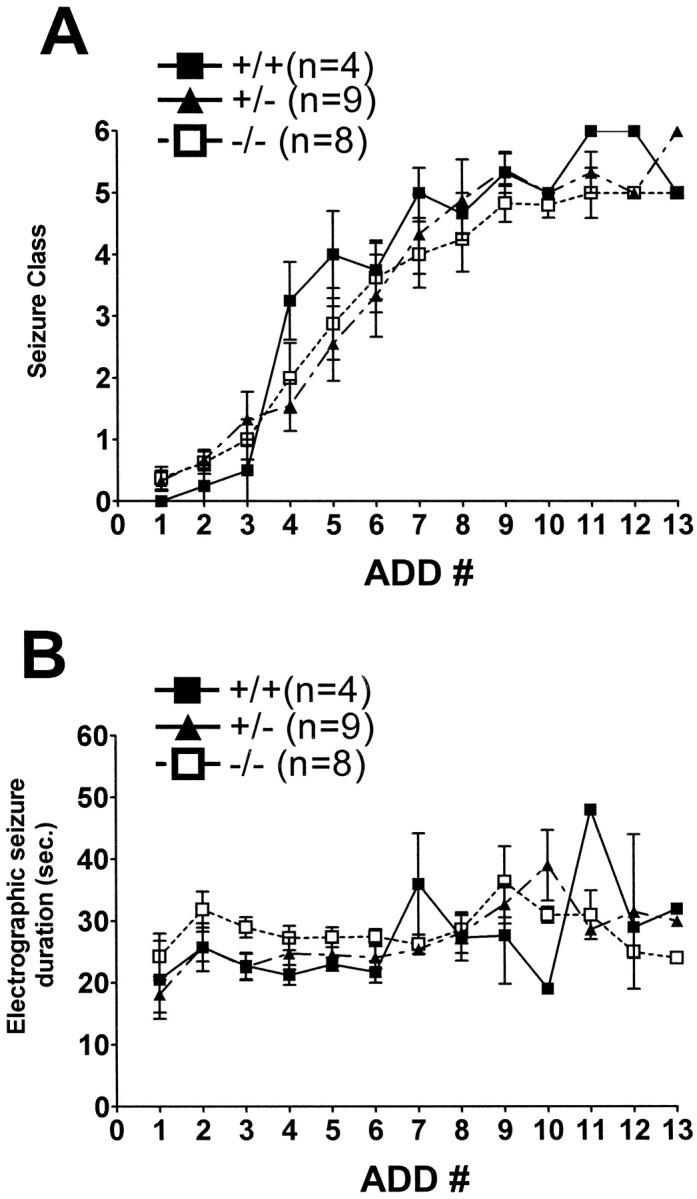
Behavioral (A) and electrographic seizure (B) measures of kindling development in WT and trkBshcmutant mice. Kindling development is presented as behavioral seizure class (A) and electrographic seizure duration as a function of the number of stimulations that evoked electrographic seizures (y-axis). Wild type (+/+;n = 4), heterozygous (+/−; n = 9), and homozygous (−/−; n = 8) mutant mice.ADD# (x-axis) refers to the number of stimulations that evoked an afterdischarge with duration of at least 5 sec. Data are mean ± SEM. Note that neither seizure class nor electrographic seizure duration was significantly different among the three groups (one-way ANOVA).
To determine whether the Shc binding site of the trkB receptor influenced the persistence of the hyperexcitability after completion of kindling, WT and trkBshc mutants were maintained for a stimulation-free period of 2 weeks after kindling and then subjected to a single stimulation. The stimulation evoked a generalized seizure in all animals from each of the three groups (+/+, +/−, and −/−). There was no significant difference among the groups in duration of electrographic seizure (41.5 ± 8.1, 41.4 ± 4.0, and 35.4 ± 2.1 sec, respectively) or seizure class (5.5 ± 0.5, 5.8 ± 0.2, and 5.3 ± 0.2, respectively;p > 0.05; one-way ANOVA). Thus, the persistence of kindling was unaffected in the trkBshcmutant mice.
DISCUSSION
Our previous ex vivo analyses of limbic epileptogenesis in rats demonstrated increased pY490 immunoreactivity in the hippocampal mossy fiber pathway, providing immunohistochemical evidence for activation of trk receptor tyrosine kinase. Which trk receptor(s) in particular accounted for the immunoreactivity was unclear. To test the hypothesis that phosphorylation of trkB contributed to the increased immunoreactivity, we studied mutant mice (trkBshc) in which phenylalanine was substituted for tyrosine at residue 515 of trkB, a site homologous to residue 490 of human trkA. Three principal findings emerge. First, the previous results in rats were confirmed and extended by the studies of mice. A single, brief seizure induced by stimulation of the right amygdala in a kindled mouse in a WT background induced increased pY490 immunoreactivity bilaterally in the hippocampal mossy fiber pathway. In addition, increased pY490 immunoreactivity was evident in stratum oriens of CA3 bilaterally. Second, in contrast to WT mice, seizures evoked in null mutant (trkBshc) mice induced no significant increase of pY490 immunoreactivity in either the mossy fiber pathway or CA3 stratum oriens. Third, despite the reduction of the seizure-induced increase of pY490 immunoreactivity, the development and persistence of kindling as measured by electrophysiological and behavioral indices did not differ betweentrkBshc mutant and WT mice. These data demonstrate that the neurotrophin receptor trkB undergoes phosphorylation in the mossy fiber pathway and CA3 stratum oriens of the hippocampus during limbic epileptogenesis. In addition, the signaling pathways activated by the Shc site of trkB exert no detectable regulatory effects on limbic epileptogenesis.
The present study revealed that the increased p-trk immunoreactivity evident in the hippocampus during limbic epileptogenesis was reduced intrkBshc mutant mice, implying that phosphorylation of trkB underlies the increased p-trk immunoreactivity. Whether the increased p-trkB immunoreactivity is attributable to a post-translational modification of a fixed number of constitutively expressed trkB molecules, to post-translational modification of increased numbers of trkB molecules, or some combination of the two is uncertain. The presence of the increased p-trk immunoreactivity in WT mice adds to previous findings in rats and demonstrates that trkB activation occurs during limbic epileptogenesis in multiple species. Moreover, the induction of the immunoreactivity by a single, brief seizure evoked in a kindled mouse extends the previous studies using chemoconvulsant-induced status epilepticus or massed electrical stimulations and demonstrates that activation of trkB is common to limbic epileptogenesis induced by diverse methods. In light of evidence implicating a causal role for trkB activation in limbic epileptogenesis (Kokaia et al., 1995; Binder et al., 1999a; Croll et al., 1999;Lähteinen et al., 2002), knowing where and when trkB activation occurs will facilitate elucidating its structural and functional consequences and, in turn, provide insight into mechanisms of limbic epileptogenesis.
The light microscopic distribution provides a clue to the cellular localization of the increased p-trkB immunoreactivity. Its presence in the dentate hilus and stratum lucidum of CA3 corresponds to the mossy fiber axons of the dentate granule cells, providing the most parsimonious explanation for its cellular localization. Whereas our studies of rats disclosed increased p-trk immunoreactivity confined to the mossy fiber pathway (Binder et al., 1999b), the present study revealed increased p-trkB immunoreactivity not only in the mossy fiber pathway but also throughout stratum oriens of CA3 extending to the junction with CA2 (Fig. 1B). Whether this p-trkB immunoreactivity is localized to an infrapyramidal projection of mossy fibers, axons of CA3 pyramidal cells, and/or postsynaptic targets such as interneurons or basilar dendrites of CA3 pyramidal cells is uncertain. Ultrastructural studies will be required to establish the cellular locale of the p-trkB immunoreactivity in both the mossy fiber pathway and stratum oriens of CA3.
Circumstantial evidence implicates increased release of BDNF as the proximate cause for the increased p-trkB immunoreactivity during limbic epileptogenesis. The binding of neurotrophin to the trk receptor induces dimerization and trans-autophosphorylation of a subset of tyrosine residues (Schlessinger and Ullrich, 1992; Guiton et al., 1994), thereby suggesting that the binding of a neurotrophin to trkB underlies its increased phosphorylation. The occurrence of the increased p-trkB immunoreactivity after seizures suggests the possibility that seizures induced increased expression of a neurotrophin that is translated, transported, released, and thereby activates trkB. Analysis of the temporal and spatial patterns of seizure-mediated regulation of neurotrophins supports the candidacy of BDNF. The seizure-induced increase of BDNF protein peaks at ∼24 hr, the time point corresponding to increased p-trkB immunoreactivity (Nawa et al., 1995; Elmér et al., 1998; Rudge et al., 1998; Binder et al., 1999b). Concordance of the anatomic distribution of seizure-induced increases of BDNF and the p-trkB immunoreactivity further implicate BDNF; that is, immunohistochemical studies disclosed increased BDNF in the granule cell soma within 2 hr after seizures and in the mossy fiber pathway 1 to 2 d thereafter (Yan et al., 1997; Vezzani et al., 1999). We favor the idea that BDNF is anterogradely transported to the mossy fiber axons (Conner et al., 1997; Smith et al., 1997), from which it is released and activates trkB on the mossy fibers by a paracrine or autocrine mechanism. Concordance of the anatomic distribution of seizure-induced BDNF (Yan et al., 1997, their Fig. 7D) and p-trkB immunoreactivity in stratum oriens of CA3 implicates BDNF in this site as well. Together with persistence of seizure-induced p-trk immunoreactivity in NT-4 null mutants (He et al., 1999), this circumstantial evidence favors BDNF as the neurotrophin activating p-trkB in hippocampus.
What consequence(s) of trkB activation, presumably by BDNF, might contribute to the increased excitability evident as limbic epileptogenesis? The fact that at least part of the increased p-trkB immunoreactivity may reside in dentate granule cells refines the question: what functional and structural consequences of enhanced activation of trkB in granule cells by BDNF might promote increased hippocampal excitability? The fact that activation of trkB by BDNF can enhance excitatory synaptic transmission (Lohof et al., 1993; Kang and Schuman, 1995; Levine et al., 1995, Stoop and Poo, 1996) and reduce inhibitory synaptic transmission (Tanaka et al., 1997) in hippocampus raises the interesting possibility that enhanced synaptic activation of CA3 pyramidal cells by mossy fibers, either directly or indirectly by disinhibition of the mossy fiber–interneuron–CA3 pyramidal cell circuit is one consequence of trkB activation. The increased excitability of CA3 pyramidal cells in kindled animals as detected by increased epileptiform bursting induced by elevated K+ or lowered Mg2+ in isolated hippocampal slices (King et al., 1985; Behr et al., 1998) is consistent with this idea. Another possibility relates to the increased recurrent excitatory synapses that have been identified among dentate granule cells in the epileptic brain, synapses that would be expected to promote repetitive firing of the granule cells in response to an isolated excitatory synaptic input (Wuarin and Dudek, 2001). Two distinct morphological plasticities of the granule cells have been identified in the epileptic brain, either of which could underlie increased recurrent excitatory synapses: sprouting of the mossy fiber axons and formation of basal dendrites (Nadler et al., 1980; Spigelman et al., 1998). We found that particle-mediated gene transfer of BDNF, but not NGF, into the dentate granule cells in hippocampal explant cultures is sufficient to induce increases in both axonal branching and basilar dendrite number (Danzer et al., 2001). Whether BDNF-mediated activation of trkB during limbic epileptogenesis in vivo mediates these functional and/or structural effects remains to be elucidated.
Despite the evidence that trkB activation promotes limbic epileptogenesis, the signaling pathways mediating the epileptogenic consequences of trkB activation are obscure. After binding a ligand, trk receptors autophosphorylate on cytoplasmic tyrosine residues, which themselves become docking sites for intracellular signaling proteins. Shc adaptor proteins associate directly with a specific site in trk receptors and activate a signaling pathway involving Ras, Raf, MAPK1 [for mitogen-activated protein (MAP) kinase 1], MAPK2, Mek 1 (for MAP kinase kinase 1), and Mek 2, one consequence of which is activation of transcription factors and regulation of gene expression. The present studies reveal that introduction of the Y515F mutation into the germline of the trkBshc/shc mice did not modify either electrophysiological or behavioral indices of epileptogenesis, despite previous evidence that BDNF- and NT-4-mediated activation of ERKs (for extracellular signal-regulated kinase) were attenuated in neurons isolated from Shc mutant mice (Minichiello et al., 1998). These results imply that trkB-mediated activation of the Ras/ERK pathway does not mediate epileptogenesis or that this pathway is redundant with other pathways, such as phospholipase Cγ pathway. The absence of detectable differences in epileptogenesis between WT and trkBshc/shc is reminiscent of the absence of detectable differences in long-term potentiation between WT and trkBshc/shcmice (Korte et al., 2000), despite defects in long-term potentiation in BDNF and trkB null mutant mice (Korte et al., 1995; Minichiello et al., 1999; Xu et al., 2000). The trkB signaling pathways mediating epileptogenesis remain elusive, and solving this question will require the generation of additional signaling alleles of the trkBgene.
Footnotes
This work was supported by National Institutes of Health Grant NS17771 (J.O.M.).
Correspondence should be addressed to Dr. James O. McNamara, 401 Bryan Research Building, Durham, NC 27705. E-mail: jmc@neuro.duke.edu.
REFERENCES
- 1.Behr J, Lyson KJ, Mody I. Enhanced propagation of epileptiform activity through the kindled dentate gyrus. J Neurophysiol. 1998;79:1726–1732. doi: 10.1152/jn.1998.79.4.1726. [DOI] [PubMed] [Google Scholar]
- 2.Binder DK, Routbort MJ, Ryan TE, Yancopoulos GD, McNamara JO. Selective inhibition of kindling development by intraventricular administration of trkB receptor body. J Neurosci. 1999a;19:1424–1436. doi: 10.1523/JNEUROSCI.19-04-01424.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Binder DK, Routbort MJ, McNamara JO. Immunohistochemical evidence of seizure-induced activation of trk receptors in the mossy fiber pathway of adult rat hippocampus. J Neurosci. 1999b;19:4616–4626. doi: 10.1523/JNEUROSCI.19-11-04616.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J Neurosci. 1997;17:2295–2313. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Croll SD, Surl C, Compton DL, Simmons MV, Yancopoulos GD, Lindsay RM, Wiegand SJ, Rudge JS, Scharfman HE. Brain-derived neurotrophic factor transgenic mice exhibit passive avoidance deficits, increased seizure severity and in vitro hyperexcitability in the hippocampus and entorhinal cortex. Neuroscience. 1999;93:1491–1506. doi: 10.1016/s0306-4522(99)00296-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Danzer SC, Crooks KRC, Lo DC, McNamara JO. Increased basal dendrites and axonal branching of BDNF-transfected dentate granule cells in hippocampal explant cultures. Soc Neurosci Abstr. 2001;31:137.19. doi: 10.1523/JNEUROSCI.22-22-09754.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elmér E, Kokaia Z, Kokaia M, Carnahan J, Nawa H, Lindvall O. Dynamic changes of brain-derived neurotrophic factor protein levels in the rat forebrain after single and recurring kindling-induced seizures. Neuroscience. 1998;83:351–362. doi: 10.1016/s0306-4522(97)00387-4. [DOI] [PubMed] [Google Scholar]
- 8.Goddard GV, McIntyre DC, Leech CK. A permanent change in brain function resulting from daily electrical stimulation. Exp Neurol. 1969;25:295–330. doi: 10.1016/0014-4886(69)90128-9. [DOI] [PubMed] [Google Scholar]
- 9.Guiton M, Gunn-Moore FJ, Stitt TN, Yancopoulos GD, Tavare JM. Identification of in vivo brain-derived neurotrophic factor-stimulated autophosphorylation sites on the trkB receptor tyrosine kinase by site-directed mutagenesis. J Biol Chem. 1994;269:30370–30377. [PubMed] [Google Scholar]
- 10.He XP, Butler L, Liu X, McNamara JO. Activation of TRK receptor in stratum lucidum of hippocampus during epileptogenesis does not require NT-4. Soc Neurosci Abstr. 1999;25:2024. [Google Scholar]
- 11.Isackson PJ, Huntsman MM, Murray KD, Gall CM. BDNF mRNA expression is increased in adult rat forebrain after limbic seizures: temporal patterns of induction distinct from NGF. Neuron. 1991;6:937–948. doi: 10.1016/0896-6273(91)90234-q. [DOI] [PubMed] [Google Scholar]
- 12.Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- 13.King GL, Dingledine R, Giacchino JL, McNamara JO. Abnormal neuronal excitability in hippocampal slices from kindled rats. J Neurophysiol. 1985;54:1295–1304. doi: 10.1152/jn.1985.54.5.1295. [DOI] [PubMed] [Google Scholar]
- 14.Kokaia M, Ernfors P, Kokaia Z, Elmér E, Jaenisch R, Lindvall O. Suppressed epileptogenesis in BDNF mutant mice. Exp Neurol. 1995;133:215–224. doi: 10.1006/exnr.1995.1024. [DOI] [PubMed] [Google Scholar]
- 15.Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proc Natl Acad Sci USA. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Korte M, Minichiello L, Klein R, Bonhoeffer T. Shc-binding site in the trkB receptor is not required for hippocampal long-term potentiation. Neuropharmacology. 2000;39:717–724. doi: 10.1016/s0028-3908(99)00273-7. [DOI] [PubMed] [Google Scholar]
- 17.Lähteinen S, Pitkänen A, Saarelainen T, Nissinen J, Koponen E, Castrén E. Decreased BDNF signaling in transgenic mice reduces epileptogenesis. Eur J Neurosci. 2002;15:721–734. doi: 10.1046/j.1460-9568.2002.01897.x. [DOI] [PubMed] [Google Scholar]
- 18.Larmet Y, Reibel S, Carnahan J, Nawa H, Marescaux C, Depaulis A. Protective effects of brain-derived neurotrophic factor on the development of hippocampal kindling in the rat. NeuroReport. 1995;6:1937–1941. doi: 10.1097/00001756-199510020-00027. [DOI] [PubMed] [Google Scholar]
- 19.Levine ES, Dreyfus CF, Black IB, Plummer MR. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptors. Proc Natl Acad Sci USA. 1995;92:8074–8077. doi: 10.1073/pnas.92.17.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lohof AM, Ip NY, Poo MM. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353. doi: 10.1038/363350a0. [DOI] [PubMed] [Google Scholar]
- 21.Minichiello L, Casagranda F, Tatche RS, Stucky CL, Postigo A, Lewin GR, Davies AM, Klein R. Point mutation in trkB causes loss of NT4-dependent neurons without major effects on diverse BDNF responses. Neuron. 1998;21:335–345. doi: 10.1016/s0896-6273(00)80543-7. [DOI] [PubMed] [Google Scholar]
- 22.Minichiello L, Korte M, Wolfer D, Kühn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, Klein R. Essential role for TrkB receptors in hippocampus-mediated learning. Neuron. 1999;24:401–414. doi: 10.1016/s0896-6273(00)80853-3. [DOI] [PubMed] [Google Scholar]
- 23.Nadler JV, Perry BW, Cotman CW. Selective reinnervation of hippocampal area CA1 and the fascia dentate after destruction of CA3-CA4 afferents with kainic acid. Brain Res. 1980;182:1–9. doi: 10.1016/0006-8993(80)90825-2. [DOI] [PubMed] [Google Scholar]
- 24.Nawa H, Carnahan J, Gall C. BDNF protein measured by a novel enzyme immunoassay in normal brain and after seizure: partial disagreement with mRNA levels. Eur J Neurosci. 1995;7:1527–1535. doi: 10.1111/j.1460-9568.1995.tb01148.x. [DOI] [PubMed] [Google Scholar]
- 25.Racine RJ. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol. 1972;32:281–294. doi: 10.1016/0013-4694(72)90177-0. [DOI] [PubMed] [Google Scholar]
- 26.Rudge JS, Mather PE, Pasnikowski EM, Cai N, Corcoran T, Acheson A, Anderson K, Lindsay RM, Wiegand SJ. Endogenous BDNF protein is increased in adult rat hippocampus after a kainic acid induced excitotoxic insult but exogenous BDNF is not neuroprotective. Exp Neurol. 1998;149:398–410. doi: 10.1006/exnr.1997.6737. [DOI] [PubMed] [Google Scholar]
- 27.Scharfman HE, Goodman JH, Sollas A, Croll SD. Spontaneous limbic seizures after intrahippocampal infusion of brain-derived neurotrophic factor. Exp Neurol. 2002;174:201–214. doi: 10.1006/exnr.2002.7869. [DOI] [PubMed] [Google Scholar]
- 28.Schlessinger J, Ullrich A. Growth factor signaling by receptor tyrosine kinases. Neuron. 1992;9:383–391. doi: 10.1016/0896-6273(92)90177-f. [DOI] [PubMed] [Google Scholar]
- 29.Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- 30.Segal RA, Bhattacharyya A, Rua LA, Alberta JA, Stephens RM, Kaplan DR, Stiles CD. Differential utilization of trk autophosphorylation sites. J Biol Chem. 1996;271:20175–20181. doi: 10.1074/jbc.271.33.20175. [DOI] [PubMed] [Google Scholar]
- 31.Smith MA, Zhang LX, Lyons WE, Mamounas LA. Anterograde transport of endogenous brain-derived neurotrophic factor in hippocampal mossy fibers. NeuroReport. 1997;8:1829–1834. doi: 10.1097/00001756-199705260-00008. [DOI] [PubMed] [Google Scholar]
- 32.Spigelman I, Yan XX, Obenaus A, Lee EY-S, Wasterlain CG, Ribak CE. Dentate granule cells form novel basal dendrites in a rat model of temporal lobe epilepsy. Neuroscience. 1998;86:109–120. doi: 10.1016/s0306-4522(98)00028-1. [DOI] [PubMed] [Google Scholar]
- 33.Stoop R, Poo MM. Synaptic modulation by neurotrophic factors: differential and synergistic effects of brain-derived neurotrophic factor and ciliary neurotrophic factor. J Neurosci. 1996;16:3256–3264. doi: 10.1523/JNEUROSCI.16-10-03256.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tanaka T, Saito H, Matsuki N. Inhibition of GABAA synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. J Neurosci. 1997;17:2959–2966. doi: 10.1523/JNEUROSCI.17-09-02959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vezzani A, Ravizza T, Moneta D, Conti M, Borroni A, Rizzi M, Samanin R, Maj R. Brain-derived neurotrophic factor immunoreactivity in the limbic system of rats after acute seizures and during spontaneous convulsions: temporal evolution of changes as compared to neuropeptide Y. Neurosci. 1999;90:1445–1461. doi: 10.1016/s0306-4522(98)00553-3. [DOI] [PubMed] [Google Scholar]
- 36.Wuarin JP, Dudek FE. Excitatory synaptic input to granule cells increases with time after kainate treatment. J Neurophysiol. 2001;85:1067–1077. doi: 10.1152/jn.2001.85.3.1067. [DOI] [PubMed] [Google Scholar]
- 37.Xu B, Gottschalk W, Chow A, Wilson RI, Schnell E, Zang K, Wang D, Nicoll RA, Lu B, Reichardt LF. The role of brain-derived neurotrophic factor receptors in the mature hippocampus: modulation of long-term potentiation through a presynaptic mechanism involving trkB. J Neurosci. 2000;20:6888–6897. doi: 10.1523/JNEUROSCI.20-18-06888.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yan Q, Rosenfeld RD, Matheson CR, Hawkins N, Lopez OT, Bennett L, Welcher AA. Expression of brain-derived neurotrophic factor protein in the adult rat central nervous system. Neuroscience. 1997;78:431–448. doi: 10.1016/s0306-4522(96)00613-6. [DOI] [PubMed] [Google Scholar]