

Abstract
Dystonia is a relatively common neurological syndrome characterized by twisting movements or sustained abnormal postures. Although the basal ganglia have been implicated in the expression of dystonia, recent evidence suggests that abnormal cerebellar function is also involved. In these studies, a novel mouse model was developed to study the role of the cerebellum in dystonia. Microinjection of low doses of kainic acid into the cerebellar vermis of mice elicited reliable and reproducible dystonic postures of the trunk and limbs. The severity of the dystonia increased linearly with kainate dose. Kainate-induced dystonia was blocked by the glutamatergic antagonist 1,2,3,4-tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide and reproduced by domoic acid microinjection, suggesting that the induction of dystonia is dependent on glutamatergic activation in this model. The abnormal movements were not associated with kainate-induced seizures, because EEG recordings showed no epileptiform activity during the dystonic events. Neuronal activation, as assessed by in situ hybridization for c-fos, revealed c-fos mRNA expression in the cerebellum, locus ceruleus, and red nucleus. In contrast, regions associated with epileptic seizures, such as the hippocampus, did not exhibit increased c-fos expression after cerebellar kainate injection. Furthermore, in transgenic mice lacking Purkinje cells, significantly less dystonia was induced after kainic acid injection, implicating Purkinje cells and the cerebellar cortex in this model of dystonia. Together, these data suggest that abnormal cerebellar signaling produces dystonia and that the cerebellum should be considered along with the basal ganglia in the pathophysiology of this movement disorder.
Keywords: dystonia, cerebellum, red nucleus, kainic acid, Purkinje cell, glutamate, movement disorder, transgenic, c-fos
Dystonia is a neurological disorder broadly characterized by simultaneous and sometimes sustained contractions of agonist and antagonist muscles. These co-contractions result in twisting movements and postures that have a wide range of speed, amplitude, and rhythmicity that varies among patients (Fahn and Marsden, 1994; Jankovic and Fahn, 1998). Although dystonia is a relatively common disorder, the underlying pathophysiology is poorly understood.
The various forms of dystonia reflect the heterogeneous biological basis of the disorder. Like epilepsy, dystonia may result from brain injury or insult (secondary or acquired) or occur as a sporadic or inherited disorder (primary or idiopathic); most cases of dystonia are idiopathic. In acquired dystonia, lesions are most often identified in the basal ganglia (Marsden and Quinn, 1990). By extension, it has been presumed that defects associated with primary dystonia also affect the basal ganglia. However, no consistent abnormalities of the basal ganglia have been identified in idiopathic dystonia; these inconsistencies are not surprising given the heterogeneous nature of the disorder (Jankovic and Fahn, 1998). Recently, a well circumscribed group of patients carrying the DYT1 gene defect, which is associated with early-onset idiopathic torsion dystonia, was analyzed using positron emission tomography (Eidelberg et al., 1995, 1998). As expected, an increase in metabolism was observed in the caudate, lentiform nuclei, and supplementary motor area. Surprisingly, hypermetabolism was also identified in the cerebellum. Because the cerebellum exhibited the highest relative increase in metabolism,Eidelberg et al. (1998) concluded that, in addition to the basal ganglia, cerebellar networks may play a role in DYT1 pathophysiology. Additional functional imaging studies performed in other patient populations similarly implicate the cerebellum in the pathophysiology of dystonia (Ceballos-Baumann and Brooks, 1998; Kluge et al., 1998;Mazziotta et al., 1998; Odergren et al., 1998; Playford et al., 1998;Hutchinson et al., 2000).
Animal models of dystonia are of considerable interest because they provide experimental paradigms for elucidating the mechanisms underlying this movement disorder. Dystonia is uncommon in rodents, with only a few well characterized inherited models. These include dystonic strains of mice (Campbell et al., 1999; Messer and Strominger, 1980; Sprunger et al., 1999), rats (Lorden et al., 1988), and hamsters (Richter and Loscher, 1998). These rodent models implicate the cerebellum and related regions, such as the red nucleus and thalamus, in the dystonic phenotype (Messer and Strominger, 1980; LeDoux et al., 1993; Gernert et al., 1997; Campbell and Hess, 1998; Richter and Loscher, 1998). Indeed, lesions of the cerebellum in both rat and mouse models abolish the dystonia (LeDoux et al., 1993; Campbell et al., 1999), suggesting that abnormal cerebellar signaling plays a role in the expression of dystonia. However, it can be argued that an abnormal developmental program and wiring anomalies specific to the species and gene mutation(s) cause the dystonia. To test the hypothesis that abnormal cerebellar signaling induces dystonia, low doses of the excitatory glutamate agonist kainate were microinjected into the cerebella of normal mice to disrupt signaling and produce dystonia in an otherwise intact cerebellum.
MATERIALS AND METHODS
Mice. Wild-type C57BL/6J mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and bred at the Pennsylvania State University College of Medicine. Both male and female mice (2–4 months of age) were used in the experiments. SV4 transgenic mice on an FVB background were originally obtained as a generous gift from Dr. R. Feddersen (University of Minnesota, St. Paul, MN). These mice carry a transgene that consists of the pcp-2 promoter coupled to the coding region of the SV40 T antigen. The SV40 T antigen is expressed exclusively in Purkinje cells resulting in virtually total Purkinje cell death after 5 months of age (Feddersen et al., 1992). SV4 mice were backcrossed onto a C57BL/6J background for five generations before use. Genotyping for the presence of the SV40 T antigen transgene was performed by PCR as described previously (Feddersen et al., 1992). Transgenic mice were at least 6 months of age before experimentation to ensure maximal Purkinje cell death. All mice were housed on a 12 hr light/dark cycle and had access to food and water ad libitum. Animal procedures were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Kainic acid microinjection. The goal of these experiments was to stimulate the cerebellum using the excitatory glutamate agonist kainate. The short-acting inhalant anesthetic methoxyflurane (Metofane; Mallinckrodt Veterinary, Mundelein, IL) or isoflurane (IsoSol; Vedco, St. Joseph, MO) was used because rapid recovery after surgery was required to observe the behavioral effects of the drug. Behavioral outcomes were compared with each anesthetic and found to be identical. Dose–response and transgenic experiments were performed with methoxyflurane; isoflurane was used in all other experiments. To limit the amount of time mice remained under and recovered from anesthesia, all injections were made freehand using a defined stylus for reproducible injections. Kainic acid was obtained from Tocris Cookson (Ellisville, MO) and dissolved in 0.9% saline. 1,2,3,4-Tetrahydro-6-nitro-2,3-dioxo-benzo[f]quinoxaline-7-sulfonamide (NBQX) (Sigma, St. Louis, MO) and domoic acid (Sigma) were also dissolved in 0.9% saline. A midline incision was made over the skull, and a small hole was drilled above the midline cerebellum. A Hamilton syringe with a needle cut to 2 mm was used to deliver 0.5 μl of kainic acid over 5 sec. The dose–response consisted of 0.5 μl injections of saline vehicle plus 10, 25, 50, and 100 μg/ml kainate, equivalent to 25, 60, 115, and 235 pmol. The wound was reapproximated and sealed with Nexaband S/C topical skin closure (Veterinary Products Laboratories, Phoenix, AZ), and mice generally recovered within 10 min of wound closure. Lateral cerebellar injections were made to the right or left of midline (anteroposterior, −6.5 mm bregma; lateral, 1–2 mm; vertical, −2 mm from skull). The lateral ventricle (anteroposterior, −0.5 mm bregma; lateral, 1.25 mm; vertical, −3 mm from skull) and striatum (anteroposterior, +1 mm bregma; lateral, 2 mm; vertical, −3 mm from skull) also received kainate injections in a dose–response paradigm similar to that perform for the midline cerebellar injections. To identify injection sites, 1:10 v/v of Trypan Blue (0.4%; Sigma) was added to the kainic acid solution. In situ hybridization for c-fos mRNA and standard hematoxylin and eosin-stained sections from all mice receiving regional kainic acid injections were also used to locate the injection site.
Behavioral observations. After wound closure, animals were immediately placed in an empty cage and observed for 60 sec every 10 min for 2 hr; during the observation period, mice received a score using a disability rating system (Table1) modified from Jinnah et al. (2000). It should be noted that a score of D1 represents behavioral slowing with no dystonia, whereas a score of D2–D5 denotes dystonia with increasing severity. If no spontaneous dystonia was seen, mice were disturbed by touch. If no escape or dystonia resulted, mice were lifted by the tail and placed down again to encourage movement, which most often preceded the dystonia. The observer scoring the mice for the kainate dose–response experiment was blinded to dose. Tonic-clonic seizures were rarely observed after kainate injection. This was invariably attributable to misplacement of the injection (too far anterior) or attributable to a high dose of kainic acid (>100 μg/ml). When tonic-clonic seizures were observed, these mice were excluded from the behavioral analysis. After the 2 hr observation period, mice were killed by carbon dioxide asphyxiation, and the brain was removed and frozen in isopentane (−40°C).
Table 1.
Disability scores
| Score | Behavioral phenotype |
|---|---|
| D0 | No motor abnormalities |
| D1 | Slightly slowed or abnormal motor behavior; no dystonia |
| D2 | Mild impairment; sometimes limited ambulation; dystonic postures when disturbed |
| D3 | Moderate impairment; frequent spontaneous dystonic postures |
| D4 | Severe impairment; sustained dystonic postures and limited ambulation |
| D5 | Prolonged immobility in dystonic postures |
In situ hybridization. Twenty micrometer coronal sections were cut using a cryostat and thaw mounted on Superfrost Plus glass slides (Fisher, Pittsburgh, PA). After drying, the slide-mounted sections were stored at −70°C. The template for c-fos was a full-length murine cDNA [2.2 kb; J. G. Belasco (Harvard Medical School, Boston, MA) and M. E. Greenberg (Harvard Medical School)] subcloned into pBluescript II SK+. Plasmid was linearized to generate either sense or antisense cRNA probes. In situ hybridization was performed as described previously (Campbell and Hess, 1999). Hybridized sections were exposed to x-ray film (DuPont Cronex; DuPont, Billerica, MA) and analyzed using the microcomputer imaging device (MCID) M5+ package (Imaging Research, St. Catherines, Ontario, Canada). After correction for film background, density of c-fos mRNA hybridization signal was quantified for several sections per animal.
EEG recordings. Under isofluorane anesthesia, two small machine screws ( inch diameter by inch length; Small Parts, Logansport, IN) were placed in the skull over the cortex ∼2 mm anterior to bregma with the recording electrode ∼2 mm lateral to the sagittal suture and the ground ∼1 mm lateral from midline. The screws were wrapped once with 30 gauge silver-plated copper wire attached to a plug or pin connector. Colloidal silver paint was applied over the screw and wire to ensure good electrical conductivity, and then the apparatus was cemented to the cleaned skull surface using Loctite glue and accelerator (Loctite, Rocky Hill, CT). After a minimum of 24 hr after surgery, baseline EEGs were recorded. The animal was then anesthetized, and 0.5 μl of kainic acid of 100 μg/ml kainic acid was injected into the midline anterior cerebellum at a depth of 2 mm. EEGs were collected for 10 sec at 5 min intervals for 90 min while behavioral data were also collected. In addition, EEGs were recorded when particularly gross dystonic postures were observed. After the 90 min recording period, mice were injected with 60 mg/kg pentylenetetrazol (intraperitoneally) to induce generalized seizure activity.
RESULTS
Induction of dystonia after cerebellar kainate injection
Normal mice reliably and reproducibly displayed a dystonic phenotype in response to kainic acid injection into the cerebellum. Mice typically displayed the first sign of dystonia 10–20 min after injection when they had recovered from the anesthetic. Most often a hindlimb was held up tonically against the trunk as the mouse was exploring. Within a few minutes, the entire trunk and all four limbs were involved, with the mouse flattened against the cage bottom with an arched back and the perineum pressed down. The hindlimbs were abducted at the hip and knee and held out above the base of the tail, often paddling in the air. The forelimbs were typically held tightly against the trunk or exhibited paddling. The neck often flexed or extended, and the ears were held back against the fur and the eyes were closed. This was the most common dystonic posture observed (Fig.1A), although other sustained abnormal positions were observed (Fig.1B–D). In general, dystonic postures occurred after being disturbed and attempting to escape or on volitional movement; dystonia was consistently preceded by a change in movement. A sudden noise that startled the mice would also frequently incite a dystonic attack. Mice immobilized in severe prolonged dystonic postures exhibited exacerbations defined by additional tensing of the muscles in the posture.
Fig. 1.

Dystonic postures after microinjection of 0.5 μl of 100 μg/ml kainic acid into the medial cerebellum of wild-type mice. A, The most common dystonic posture after kainate injection. B–D, Other dystonic postures after kainate injection.
Severity of dystonia is correlated with kainate dose
Saline injection did not produce motor abnormalities, indicating that the injury caused by the injection itself is not sufficient to produce dystonia. Low doses of kainic acid produced mild dystonia whereby mice walked or rested normally between dystonic attacks that lasted 2–15 sec. The most mildly affected animals displayed dystonic postures only after being disturbed by the experimenter. At higher doses, mice were immobilized by tensely held dystonic postures for 2–20 min. The severity of dystonia increased linearly with dose with a correlation coefficient of 0.97 (Fig.2A). The behavior returned to normal or near-normal in all dystonic animals within 2 hr of injection (Fig. 2B). In the vast majority of mice, doses up to 100 μg/ml in the cerebellum produced dystonia without seizures, but seizures were observed if the injection site was too anterior. Mice receiving 0.5 μl of 150 μg/ml kainic acid developed wild and erratic running, as well as tonic-clonic seizures; this behavior was not observed at lower doses.
Fig. 2.

Dose–response (A) and dose–recovery (B) curves after microinjection of kainic acid into the cerebellum of wild-type mice. The dose–response graph (A) represents the disability score 30 min after injection; these data are also presented in the dose–recovery graph (B) at the 30 min time point. Data represent means ± SEM (n = 5–6 per dose). The same mice presented in the dose–response graph (A) recovered ∼2 hr after injection of kainic acid (B). Data in B represent means. Error bars were omitted in the dose–recovery graph (B) for clarity of presentation, but SEMs were consistent with those presented in the dose–response graph (A).
Cerebellar injection sites
The location of the microinjection site for mice in the dose–response experiment was determined to verify the accuracy of the freehand injections. Although a stereotaxic apparatus was not used, microinjections for all mice tested in the kainic acid dose–response experiment were extremely consistent; all injection sites were located in the cerebellar vermis within 1 mm anteroposterior (Fig.3).
Fig. 3.
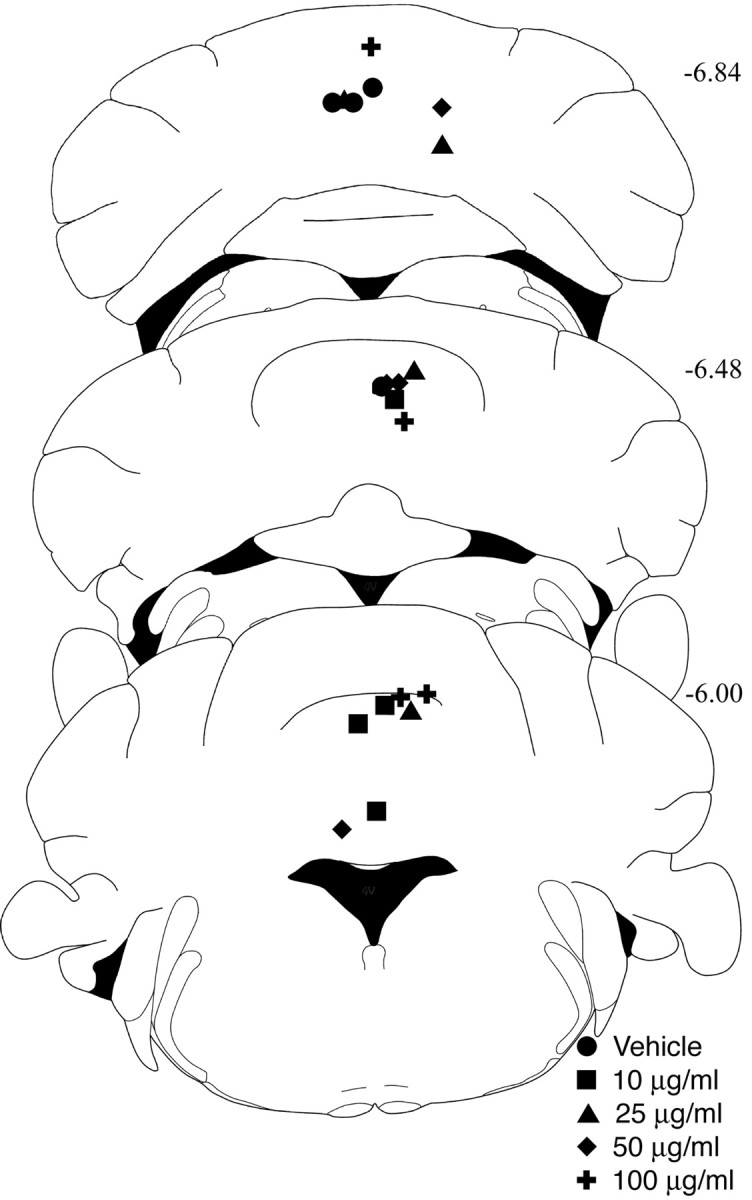
Injection site localization. The most ventral points of the needle tracks from kainate-injected mice in Figure 2 are depicted in schematics of the cerebellum modified fromFranklin and Paxinos (1997). Posterior position in millimeters from bregma appears at the right of each section.
EEGs in dystonic mice
Because kainic acid is frequently used to induce seizures in animals, EEG recordings were collected during kainate-induced dystonia (100 μg/ml) to determine whether the dystonia was associated with epileptiform EEGs (n = 5). Baseline EEGs were recorded before kainate injection. A total of 80 EEGs, which were 10 sec each, were recorded while mice were exhibiting dystonia. EEGs recorded during dystonic posturing were not associated with epileptiform activity. In fact, dystonic EEGs could not be distinguished from the baseline EEGs. In contrast, pentylenetetrazol administered 90 min after kainate injection induced spike and wave activity consistent with seizures. Representative EEGs of baseline, kainate-induced dystonia, and pentylenetetrazol-induced seizures are shown in Figure4.
Fig. 4.
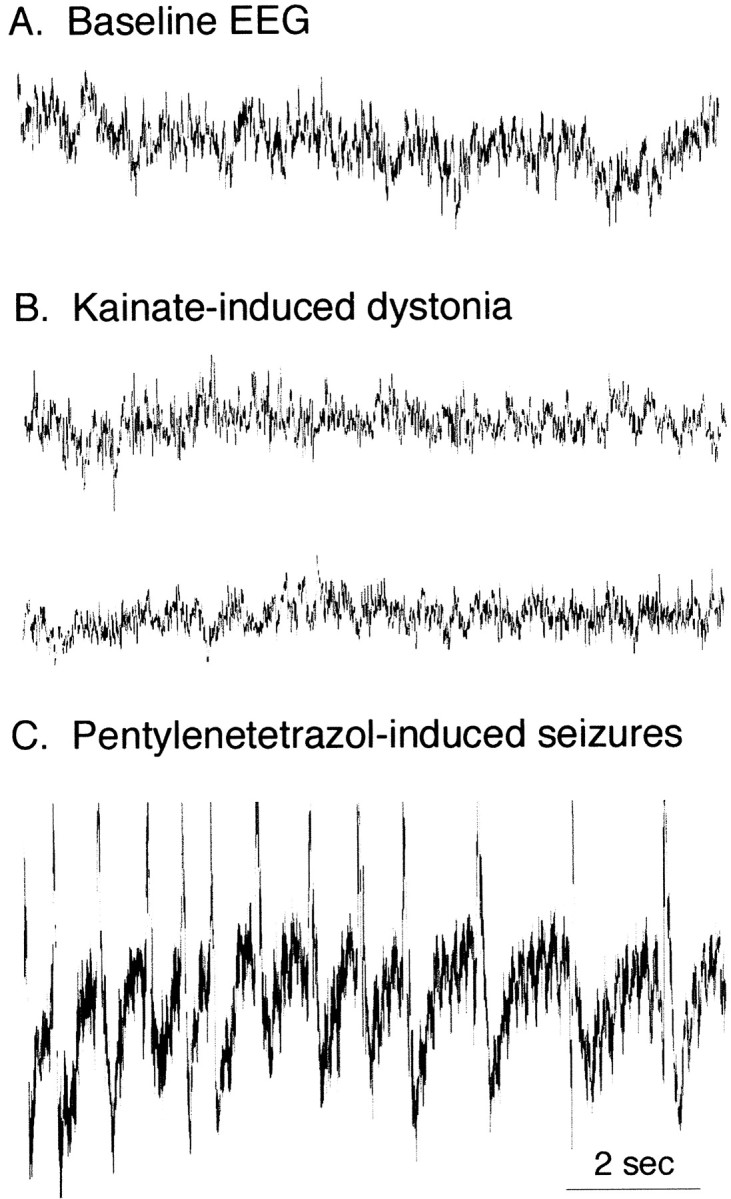
Representative EEG recordings. A, Baseline EEG recording from a wild-type C57BL/6J mouse.B, EEG after microinjection of 100 μg/ml kainic acid into the anterior cerebellar vermis of a wild-type mouse.C, EEG of a generalized seizure after pentylenetetrazol treatment (60 mg/kg, i.p.).
c-fos mRNA expression after cerebellar kainate injection
To identify the neuroanatomical substrate(s) of the kainate-induced dystonia, c-fos mRNA expression was assessed 2 hr after microinjection. c-fos expression maps functional polysynaptic pathways and appears to be an accurate marker of neuronal activation. As expected, kainate microinjection into the cerebellar vermis induced c-fos mRNA expression within the cerebellum (Fig. 5); this expression was primarily confined to the vermis, with little activation of the lateral cerebellum at 25 and 50 μg/ml, attesting to the limited dispersion of the kainate. Low doses of kainate produced modest c-fosexpression. At the highest dose, robust c-fos expression was observed throughout the cerebellum. In fact, c-fosexpression was dose dependent in the cerebellum (Fig.6). Significant increases in c-fos mRNA expression were also observed in the red nucleus and the locus ceruleus (Fig. 6). c-fos mRNA expression was not induced in the motor cortex, and there was a small but significant reduction in the striatum (Fig. 6). c-fos expression was also slightly reduced in the hippocampus, suggesting that the dystonia was not likely related to kainate-induced seizure. In contrast, intense c-fos expression was observed in the hippocampus and motor cortex after kainate-induced seizures (data not shown), as described previously (Morgan et al., 1987). The circumscribed pattern of c-fos induction without extraneous activation of brain regions near the cerebellum suggested that there was little diffusion of the kainic acid outside the cerebellum.
Fig. 5.

In situhybridization for c-fos mRNA expression 2 hr after microinjection of kainic acid into the cerebellum of wild-type mice. Representative sections from regions implicated in dystonia and from areas of c-fos induction are shown, including striatum and cortex (STR), hippocampus (HPC), red nucleus (RN), locus ceruleus (LC), and cerebellum (CB). Induction of c-fosmRNA expression was observed in the cerebellum, red nucleus, and locus ceruleus.
Fig. 6.

Quantification of c-fosmRNA induction after microinjection of kainic acid into the cerebellum of wild-type mice. In situ hybridization of c-fos mRNA expression 2 hr after injection was quantified using an MCID image analysis system. Data represent means ± SEM (n = 4 per dose). There was a significant dose-dependent increase in c-fos mRNA expression in the locus ceruleus (p < 0.01), red nucleus (p < 0.05), and cerebellum (p < 0.0001) but a significant decrease in the striatum (p < 0.05) and hippocampus (p < 0.05). There was no effect of dose in the cortex.
Region-specific effects
To verify that the cerebellum was responsible for the dystonic phenotype observed after kainate injection, several other sites were also injected. The cerebellum was injected 1–2 mm lateral from the midline (n = 5). At early time points, mild to moderate dystonia (D2–D3) was restricted to the ipsilateral side of the injection site. As the dystonic attacks progressed to more severe stages (D4–D5), the body became bilaterally affected, suggesting that some diffusion of the kainate may have occurred within the cerebellum. Alternatively, it is possible that the kainate injection produced a propagating signal, which ultimately produced bilateral activation.
If the kainic acid injected into the cerebellum acted by diffusing to other regions, injections of kainic acid into the lateral ventricle (n = 5–6 per dose) should also result in dystonia. No behavioral effect was observed after either vehicle or 25 μg/ml kainic acid was injected into the lateral ventricle. Mice receiving 50 or 100 μg/ml exhibited little movement, sitting still in the corner of the test cage with a few displaying epileptic seizures. At 100 μg/ml, three of five mice had numerous 5–15 sec seizures. Dystonic postures were never observed after ventricular injection of any dose of kainic acid.
Because the basal ganglia have been implicated in dystonia, kainic acid was also injected into the striatum of wild-type mice (n = 5–6/dose). Mice injected with vehicle typically explored for the first hour and then rested in the second hour. Striatal injections caused an increase in locomotor activity and grooming in a dose-dependent manner. At the highest dose of kainic acid, 100 μg/ml, two mice had brief seizures. Striatal injections never resulted in dystonic posturing at any dose.
Cerebellar kainate injections in mice lacking Purkinje cells
If kainic acid acts in the cerebellar cortex to produce dystonia, mice without Purkinje cells, the sole output of the cerebellar cortex, should not display a dystonic phenotype after kainate injection. Therefore, aged SV4 transgenic mice (n = 15), which have lost virtually all Purkinje cells, were injected with 100 μg/ml kainic acid into the midline cerebellum. Because the loss of Purkinje cells in SV4 mice results in a slight reduction in cerebellar size, injection sites were mapped and found to be consistent with wild-type mice. Vehicle-injected SV4 mice exhibited mild ataxia, accounting for the increase in disability scores over wild-type mice. Dystonia was significantly reduced (ANOVA with Scheffe's post hocanalysis; p < 0.0005) in kainate-injected SV4 mice compared with control mice receiving kainic acid (Fig.7). Furthermore, kainate-injected SV4 mice were not significantly different from saline-injected SV4 mice (ANOVA with Scheffe's post hoc analysis; p> 0.1).
Fig. 7.

Kainic acid cerebellar microinjection in SV4 transgenic mice lacking Purkinje cells. Transgenic mice lacking Purkinje cells (n = 15) and wild-type mice (n = 6) were injected with kainic acid into the midline cerebellum, and disability scores were recorded every 10 min for 2 hr; total disability score represents the sum of the scores for the entire 2 hr observation period. Data represent means ± SEM. Transgenic mice lacking Purkinje cells (n = 6) and wild-type mice (n = 5) were also injected with saline to demonstrate the basal level of motor performance in each genotype based on the disability scoring paradigm. ****p < 0.0001, indicates significantly reduced kainate-induced dystonia in SV4 transgenic mice compared with kainate-induced dystonia in wild-type mice (one-factor ANOVA; Scheffe's post hoc analysis). Kainate-injected SV4 transgenic mice were not significantly different from saline-injected SV4 transgenic mice.
Glutamatergic regulation of kainate-induced dystonia
Kainic acid activates both kainate and AMPA ionotropic glutamate receptors. To determine whether kainate is acting specifically at these sites to produce dystonia, NBQX, a glutamate antagonist at both kainate and AMPA receptors, was coadministered with kainic acid to block the induction of dystonia. The administration of NBQX alone was also performed to determine whether a derangement in glutamate signaling was sufficient to produce dystonia or whether the excitation produced by kainate was required for the induction of dystonia. Mice injected with 2.35 nmol of NBQX in the midline cerebellum displayed normal (two of five), hyperactive (two of five), or decreased (one of five) locomotor activity. Coinjection of 2.35 nmol of NBQX and 235 pmol of kainic acid (100 μg/ml) significantly reduced the occurrence of dystonia (p < 0.01) compared with kainic acid injection alone (Fig. 8). Furthermore, the response to coadministration of NBQX and kainic acid was comparable with NBQX alone.
Fig. 8.

Blockade of kainate-induced dystonia by coadministration of NBQX. Mice were microinjected with the glutamate receptor antagonist NBQX alone and with 100 μg/ml kainic acid. Disability scores were recorded every 10 min for 2 hr; total disability score represents the sum of the scores for the entire 2 hr observation period. Data represent means ± SEM (n = 5 per group). **p < 0.01, indicates a significant reduction in dystonia after NBQX coadministration with kainic acid compared with kainic acid alone (one-factor ANOVA; Scheffe'spost hoc analysis). NBQX plus kainic acid coadministration was not significantly different from administration of NBQX alone.
To determine whether the induction of dystonia was specific to kainate or is a general outcome of glutamate receptor activation, domoic acid, another agonist at AMPA and kainate receptors, was microinjected into the midline cerebellum of wild-type mice. Domoate-injected mice displayed reproducible generalized dystonia within 10–20 min after injection. Mice injected with 7.5 pmol (4.6 μg/ml) of domoate exhibited flattened trunks with the hindlimbs abducted and externally rotated. Forelimb paddling and facial movements were also consistently noted. These dystonic postures were comparable with those observed in mice after kainate injection into the cerebellum (Fig.9). Similarly, domoate-injected mice recovered to near-normal behavior within 2 hr after injection (data not shown).
Fig. 9.

Domoic acid microinjection into the cerebellum induced dystonia in normal mice. Domoate was injected into the medial cerebellum, and disability scores were recorded every 10 min for 2 hr; total disability score represents the sum of the scores for the entire 2 hr observation period. Data represent means ± SEM (n = 5 per group). ***p < 0.001, indicates a significant increase in dystonia over vehicle microinjection in domoate- and kainate-treated mice (one-factor ANOVA; Scheffe's post hoc analysis). Domoate-induced dystonia was not significantly different from kainate-induced dystonia.
DISCUSSION
Kainic acid microinjection into the cerebellum resulted in generalized dystonia that was associated with increasing kainic acid dose. Abnormalities of the cerebellum are generally associated with hypotonia, tremor, and ataxia but not with dystonia. The classical cerebellar signs are typically caused by stroke, injury, or heredodegenerative disorders, that is, loss of function. Dystonia is a movement disorder associated with an increase in and disordered timing of muscle contractions. This is in marked contrast to the behaviors associated with loss of cerebellar function, suggesting that the action of kainate in this model was likely cerebellar excitation resulting in abnormal signaling. Domoic acid injections into the cerebellum reproduced the effects of kainic acid, and the effects of kainic acid were blocked by NBQX; this strongly implicates an excess activation of cerebellar AMPA or kainate receptors in the induction and maintenance of the dystonic episodes. Indeed, blockade of glutamate receptors by NBQX did not induce dystonia, suggesting that an increase in glutamatergic tone, not simply the distortion or loss of glutamatergic signaling, is necessary to induce dystonia. Although dystonia is not conventionally associated with the cerebellum, cerebellar pathophysiology is routinely studied in terms of loss of function, whereas the behavioral outcome of gross alterations or increases in signaling is generally not considered. In this model, an abnormal increase in signaling superimposed on a normal cerebellum produced dystonia.
Although kainate is used extensively to induce seizures in rodents, the behavior described here is not attributable to epileptic seizures. First, the asynchronous, sustained twisting movements and postures are more typical of dystonia than seizures. Next, the absence of obvious abnormal activity on EEG argues against a seizure. Finally, the lack of c-fos induction in the hippocampus and cortex, regions that typically express c-fos with seizure, coupled with c-fos expression in the cerebellum, red nucleus, and locus ceruleus is atypical for seizures (Morgan et al., 1987). This behavior is best described as dystonia.
Several lines of evidence suggest that dystonia resulted from the local action of kainate in the cerebellum and not from kainate acting outside the cerebellum. Whereas midline microinjections into the cerebellum produced bilateral dystonia, lateral cerebellar injections resulted in unilateral dystonia ipsilateral to the site of injection. This ipsilateral response is typical of cerebellar connectivity but atypical for other central motor control systems, which influence contralateral movement. Injections of kainate into the striatum and the lateral ventricles never resulted in dystonia, refuting the notion that nonspecific global CNS activation by kainate diffusion could account for the dystonia observed after cerebellar microinjection. Finally, the marked reduction of dystonia in transgenic mice lacking cerebellar Purkinje cells provides the most compelling argument for the role of the cerebellum in kainate-induced dystonia. These results implicate the cerebellar cortex in the production of dystonia and suggest that kainate-induced dystonia occurs through activation of a circuit involving cerebellar Purkinje cells.
Cerebellar dysfunction also occurs in rodent models of dystonia. In both dystonic hamsters and rats, functional imaging studies using 2-deoxyglucose uptake demonstrate metabolic abnormalities in the cerebellum in response to dystonic attacks (Brown and Lorden, 1989;Richter et al., 1998). Similarly, the induction of c-fosexpression in the tottering mouse, a model of paroxysmal dystonia, is observed specifically in cerebellar circuits in response to a dystonic attack (Campbell and Hess, 1998), and abnormalities in Purkinje cell complex spiking attributable to defects in the sodium channel Scn8a are observed in dystonic mice (Raman et al., 1997; Sprunger et al., 1999). In support of the notion that the cerebellum is necessary for the expression of dystonia, cerebellectomy in the dystonic rat eliminates the dystonia (LeDoux et al., 1993). Consistent with the abolition of dystonia in mice lacking Purkinje cells in the kainate model, selective destruction of Purkinje cells in the tottering mutant mouse results in the loss of the dystonic phenotype (Campbell et al., 1999). It appears that cerebellar dysfunction is a consistent feature of animal models of dystonia.
This novel animal model is also consistent with findings in humans with idiopathic dystonia. Although recent research has emphasized the role of the basal ganglia, an association between cerebellar abnormalities and focal dystonia was noted over a decade ago (Fletcher et al., 1988). More recently, functional imaging studies demonstrated hypermetabolism of the cerebellum and premotor cortex in focal dystonias, such as writer's cramp (Ceballos-Baumann et al., 1997; Odergren et al., 1998;Preibisch et al., 2001) and cervical dystonia (Galardi et al., 1996). Blepharospasm, a focal dystonia of the upper face, is also associated with an increase in metabolic activity in the cerebellum (Hutchinson et al., 2000). In hemidystonia, which affects only one side of the body, increased activity is observed in the ipsilateral cerebellum, similar to unilateral cerebellar kainate injections (Ceballos-Baumann et al., 1995). Cerebellar overactivity also appears to be a common feature of generalized dystonia. In patients with exercise-induced paroxysmal dystonia, cerebellar perfusion is increased whereas cortical and basal ganglia perfusion is reduced during motor attacks (Kluge et al., 1998). Likewise, DYT1 dystonia (early-onset idiopathic torsion dystonia) is characterized by an increase in metabolic activity in the cerebellum, thalamus, and midbrain (Eidelberg et al., 1998). Overall, an increase in cerebellar activity is associated with dystonia in humans, similar to kainate-induced dystonia in mice.
The abnormal cerebellar signal must ultimately impinge on skeletal muscles to generate the twisting movements and posturing characteristic of dystonia. Because the cerebellum has few, if any, direct spinal projections, an intermediate nucleus is necessary to process and relay the signal. The red nucleus is a likely intermediate because the rubrospinal tract mediates cerebellar-induced motor activation (Perciavalle et al., 1978; Giuffrida et al., 1980; Levy et al., 1986;Keifer and Houk, 1994). The magnocellular red nucleus is a major efferent target of the deep cerebellar nuclei, receiving excitatory input from the contralateral interpositus and dentate nuclei, and is the only deep cerebellar nuclei target demonstrating c-fosinduction after cerebellar kainate injection. The magnocellular neurons of the red nucleus give rise to the rubrospinal tract that descends in the contralateral spinal cord and terminates predominantly on interneurons in the ventral gray column at all levels of the spinal cord. These interneurons, which also receive convergent input from corticospinal, tectospinal, and bulbospinal neurons (Illert et al., 1975a,b, 1978), then convey the processed information to motoneurons. At the same time, some rubrospinal axons, like corticospinal neurons, synapse directly on motoneurons (Holstege, 1987;Holstege et al., 1988; Ralston et al., 1988; Fujito et al., 1991). Activation of motoneurons directly via the rubrospinal tract or indirectly through propriospinal neurons may occur in discordance with corticospinal input, resulting in poor coordination of agonist and antagonist muscle contractions (dystonia) as a direct consequence of the abnormal cerebellar signal. Indeed, disruption of the red nucleus can result in dystonia in humans and rodents (Walker et al., 1988;Matsumoto et al., 1990; Vidailhet et al., 1999). Given the complexity of motor commands and the tremendous information processing that occurs with each movement, there are likely several mechanisms for distorting the timing between agonist and antagonist muscles.
Similar to the red nucleus, the locus ceruleus demonstrated marked c-fos expression after cerebellar kainate injection. The locus ceruleus response may represent an overall increase in arousal in response to the novel motor phenomenon. Alternatively, it has been suggested that the locus ceruleus may play a role in dystonia. Marked changes in norepinephrine brain concentrations have been noted in patients with dystonia musculorum deformans (Hornykiewicz et al., 1986). In an experimental model of dystonia, microinjection of adrenocorticotropin hormone into the locus ceruleus of rats caused ipsilateral leaning and postures reminiscent of human dystonia resulting from the direct action of norepinephrine at β-adrenergic receptors on cerebellar Purkinje cells (Jacquet and Abrams, 1982;Jacquet, 1988). Furthermore, the dystonic rat, dystonia musculorum mouse, tottering mouse, and dystonic hamster all exhibit abnormal norepinephrine concentrations (Levitt and Noebels, 1981; Riker et al., 1981; Richter and Loscher, 1998). It is unlikely that the locus ceruleus alone is responsible for the production of dystonia. However, locus ceruleus activity may modulate motor control functions as a primary component of the dystonia program or a secondary reaction to dystonic activity.
Dystonia is basically a disruption in the timing and coordination of agonist and antagonist muscle contractions. Although the focus of research has been on the basal ganglia, the massive convergence of information required to orchestrate the firing of motoneurons suggests that abnormalities in other motor systems, including the cerebellum, could result in such distorted signaling. In fact, both the basal ganglia and cerebellum subserve similar functions of planning, initiation, coordination, and termination of volitional movements. Thus, it is reasonable to suggest that both the basal ganglia and cerebellum play a role in the expression of dystonia. Each individual system may be implicated in specific patient populations, and there are examples in which both systems are deranged (Karbe et al., 1992;Galardi et al., 1996). The aggregate of the results presented here, results from other animal models, and recent imaging studies in patients suggests that the cerebellum should not be overlooked in the pathophysiology of dystonia.
Footnotes
This work was supported by United States Public Health Service Grants NS33592, ES05450, NS01985, and NS40470. We thank Drs. Steven Dear and Corey Hart for help with the EEG recordings and Dr. Rod Feddersen for the generous gift of the SV4 mice. We also thank Bryan Lee and Me Yeon Shin for technical assistance.
Correspondence should be addressed to Ellen J. Hess, Department of Neurology, Meyer, 6-181, Johns Hopkins University School of Medicine, 600 North Wolfe Street, Baltimore, MD 21287. E-mail: ehess@jhmi.edu.
REFERENCES
- 1.Brown LL, Lorden JF. Regional cerebral glucose utilization reveals widespread abnormalities in the motor system of the rat mutant dystonic. J Neurosci. 1989;9:4033–4041. doi: 10.1523/JNEUROSCI.09-11-04033.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campbell DB, Hess EJ. Cerebellar circuitry is activated during convulsive episodes in the tottering (tg/tg) mutant mouse. Neuroscience. 1998;85:773–783. doi: 10.1016/s0306-4522(97)00672-6. [DOI] [PubMed] [Google Scholar]
- 3.Campbell DB, Hess EJ. L-type calcium channels contribute to the tottering mouse dystonic episodes. Mol Pharmacol. 1999;55:23–31. doi: 10.1124/mol.55.1.23. [DOI] [PubMed] [Google Scholar]
- 4.Campbell DB, North JB, Hess EJ. Tottering mouse motor dysfunction is abolished on the Purkinje cell degeneration (pcd) mutant background. Exp Neurol. 1999;160:268–278. doi: 10.1006/exnr.1999.7171. [DOI] [PubMed] [Google Scholar]
- 5.Ceballos-Baumann AO, Brooks DJ. Activation positron emission tomography scanning in dystonia. Adv Neurol. 1998;78:135–152. [PubMed] [Google Scholar]
- 6.Ceballos-Baumann AO, Passingham RE, Marsden CD, Brooks DJ. Motor reorganization in acquired hemidystonia. Ann Neurol. 1995;37:746–757. doi: 10.1002/ana.410370608. [DOI] [PubMed] [Google Scholar]
- 7.Ceballos-Baumann AO, Sheean G, Passingham RE, Marsden CD, Brooks DJ. Botulinum toxin does not reverse the cortical dysfunction associated with writer's cramp. A PET study. Brain. 1997;120:571–582. doi: 10.1093/brain/120.4.571. [DOI] [PubMed] [Google Scholar]
- 8.Eidelberg D, Moeller JR, Ishikawa T, Dhawan V, Spetsieris P, Przedborski S, Fahn S. The metabolic topography of idiopathic torsion dystonia. Brain. 1995;118:1473–1484. doi: 10.1093/brain/118.6.1473. [DOI] [PubMed] [Google Scholar]
- 9.Eidelberg D, Moeller JR, Antonini A, Kazumata K, Nakamura T, Dhawan V, Spetsieris P, deLeon D, Bressman SB, Fahn S. Functional brain networks in DYT1 dystonia. Ann Neurol. 1998;44:303–312. doi: 10.1002/ana.410440304. [DOI] [PubMed] [Google Scholar]
- 10.Fahn S, Marsden CD. The paroxysmal dyskinesias. In: Marsden CD, Fahn S, editors. Movement disorders 3. Butterworth-Heinemann; Oxford: 1994. pp. 310–347. [Google Scholar]
- 11.Feddersen RM, Ehlenfeldt M, Yunis WS, Clark HB, Orr HT. Disrupted cerebellar cortical development and progressive degeneration of Purkinje cells in SV40 T antigen transgenic mice. Neuron. 1992;9:955–966. doi: 10.1016/0896-6273(92)90247-b. [DOI] [PubMed] [Google Scholar]
- 12.Fletcher NA, Stell R, Harding AE, Marsden CD. Degenerative cerebellar ataxia and focal dystonia. Mov Disord. 1988;3:336–342. doi: 10.1002/mds.870030410. [DOI] [PubMed] [Google Scholar]
- 13.Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Academic; New York: 1997. [Google Scholar]
- 14.Fujito Y, Imai T, Aoki M. Monosynaptic excitation of motoneurons innervating forelimb muscles following stimulation of the red nucleus in cats. Neurosci Lett. 1991;127:137–140. doi: 10.1016/0304-3940(91)90913-e. [DOI] [PubMed] [Google Scholar]
- 15.Galardi G, Perani D, Grassi F, Bressi S, Amadio S, Antoni M, Comi GC, Canal N, Fazio F. Basal ganglia and thalamo-cortical hypermetabolism in patients with spasmodic torticollis. Acta Neurol Scand. 1996;94:172–176. doi: 10.1111/j.1600-0404.1996.tb07049.x. [DOI] [PubMed] [Google Scholar]
- 16.Gernert M, Richter A, Loscher W. The electrical activity is impaired in the red nucleus of dt(sz) mutant hamsters with paroxysmal dystonia: an EEG power spectrum analysis of depth electrode recordings. Brain Res. 1997;760:102–108. doi: 10.1016/s0006-8993(97)00310-7. [DOI] [PubMed] [Google Scholar]
- 17.Giuffrida R, Li Volsi G, Panto MR, Perciavalle V, Sapienza S, Urbano A. Single muscle organization of interposito-rubral projections. Exp Brain Res. 1980;39:261–267. doi: 10.1007/BF00237115. [DOI] [PubMed] [Google Scholar]
- 18.Holstege G. Anatomical evidence for an ipsilateral rubrospinal pathway and for direct rubrospinal projections to motoneurons in the cat. Neurosci Lett. 1987;74:269–274. doi: 10.1016/0304-3940(87)90308-9. [DOI] [PubMed] [Google Scholar]
- 19.Holstege G, Blok BF, Ralston DD. Anatomical evidence for red nucleus projections to motoneuronal cell groups in the spinal cord of the monkey. Neurosci Lett. 1988;95:97–101. doi: 10.1016/0304-3940(88)90639-8. [DOI] [PubMed] [Google Scholar]
- 20.Hornykiewicz O, Kish SJ, Becker LE, Farley I, Shannak K. Brain neurotransmitters in dystonia musculorum deformans. N Engl J Med. 1986;315:347–353. doi: 10.1056/NEJM198608073150602. [DOI] [PubMed] [Google Scholar]
- 21.Hutchinson M, Nakamura T, Moeller JR, Antonini A, Belakhlef A, Dhawan V, Eidelberg D. The metabolic topography of essential blepharospasm: a focal dystonia with general implications. Neurology. 2000;55:673–677. doi: 10.1212/wnl.55.5.673. [DOI] [PubMed] [Google Scholar]
- 22.Illert M, Lundberg A, Tanaka R. Integration in a disynaptic cortico-motoneuronal pathway to the forelimb in the cat. Brain Res. 1975a;93:525–529. doi: 10.1016/0006-8993(75)90193-6. [DOI] [PubMed] [Google Scholar]
- 23.Illert M, Lundberg A, Padel Y, Tanaka R. Convergence on propriospinal neurones which may mediate disynaptic corticospinal excitation to forelimb motoneurones in the cat. Brain Res. 1975b;93:530–534. doi: 10.1016/0006-8993(75)90194-8. [DOI] [PubMed] [Google Scholar]
- 24.Illert M, Lundberg A, Padel Y, Tanaka R. Integration in descending motor pathways controlling the forelimb in the cat. V. Properties of and monosynaptic excitatory convergence on C3–C4 propriospinal neurones. Exp Brain Res. 1978;33:101–130. doi: 10.1007/BF00238798. [DOI] [PubMed] [Google Scholar]
- 25.Jacquet Y. A dystonia-like syndrome after neuropeptide (MSH/ACTH) stimulation of the rat locus ceruleus. Adv Neurol. 1988;50:299–311. [PubMed] [Google Scholar]
- 26.Jacquet YF, Abrams GM. Postural asymmetry and movement disorder after unilateral microinjection of adrenocorticotropin 1–24 in rat brainstem. Science. 1982;218:175–177. doi: 10.1126/science.6289433. [DOI] [PubMed] [Google Scholar]
- 27.Jankovic J, Fahn S. Dystonic disorders. In: Jankovic J, Tolosa E, editors. Parkinson's disease and movement disorders. Williams and Wilkins; Baltimore: 1998. pp. 513–551. [Google Scholar]
- 28.Jinnah HA, Sepkuty JP, Ho T, Yitta S, Drew T, Rothstein JD, Hess EJ. Calcium channel agonists and dystonia in the mouse. Mov Disord. 2000;15:542–551. doi: 10.1002/1531-8257(200005)15:3<542::AID-MDS1019>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 29.Karbe H, Holthoff VA, Rudolf J, Herholz K, Heiss WD. Positron emission tomography demonstrates frontal cortex and basal ganglia hypometabolism in dystonia. Neurology. 1992;42:1540–1544. doi: 10.1212/wnl.42.8.1540. [DOI] [PubMed] [Google Scholar]
- 30.Keifer J, Houk JC. Motor function of the cerebellorubrospinal system. Physiol Rev. 1994;74:509–542. doi: 10.1152/physrev.1994.74.3.509. [DOI] [PubMed] [Google Scholar]
- 31.Kluge A, Kettner B, Zschenderlein R, Sandrock D, Munz DL, Hesse S, Meierkord H. Changes in perfusion pattern using ECD-SPECT indicate frontal lobe and cerebellar involvement in exercise-induced paroxysmal dystonia. Mov Disord. 1998;13:125–134. doi: 10.1002/mds.870130124. [DOI] [PubMed] [Google Scholar]
- 32.LeDoux MS, Lorden JF, Ervin J. Cerebellectomy eliminates the motor syndrome of the genetically dystonic rat. Exp Neurol. 1993;120:302–310. doi: 10.1006/exnr.1993.1064. [DOI] [PubMed] [Google Scholar]
- 33.Levitt P, Noebels JL. Mutant mouse tottering: selective increase of locus ceruleus axons in a defined single-locus mutation. Proc Natl Acad Sci USA. 1981;78:4630–4634. doi: 10.1073/pnas.78.7.4630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Levy WJ, McCaffrey M, Goldman D, York DH. Nonpyramidal motor activation produced by stimulation of the cerebellum, direct or transcranial: a cerebellar evoked potential. Neurosurgery. 1986;19:163–176. doi: 10.1227/00006123-198608000-00001. [DOI] [PubMed] [Google Scholar]
- 35.Lorden JF, Oltmans GA, Stratton S, Mays LE. Neuropharmacological correlates of the motor syndrome of the genetically dystonic (dt) rat. In: Fahn S, editor. Dystonia 2. Raven; New York: 1988. pp. 277–297. [PubMed] [Google Scholar]
- 36.Marsden CD, Quinn NP. The dystonias. Br Med J. 1990;300:139–144. doi: 10.1136/bmj.300.6718.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsumoto RR, Hemstreet MK, Lai NL, Thurkauf A, De Costa BR, Rice KC, Hellewell SB, Bowen WD, Walker JM. Drug specificity of pharmacological dystonia. Pharmacol Biochem Behav. 1990;36:151–155. doi: 10.1016/0091-3057(90)90141-4. [DOI] [PubMed] [Google Scholar]
- 38.Mazziotta JC, Hutchinson M, Fife TD, Woods R. Advanced neuroimaging methods in the study of movement disorders: dystonia and blepharospasm. Adv Neurol. 1998;78:153–160. [PubMed] [Google Scholar]
- 39.Messer A, Strominger NL. An allele of the mouse mutant dystonia musculorum exhibits lesions in red nucleus and striatum. Neuroscience. 1980;5:543–549. doi: 10.1016/0306-4522(80)90051-2. [DOI] [PubMed] [Google Scholar]
- 40.Morgan JI, Cohen DR, Hempstead JL, Curran T. Mapping patterns of c-fos expression in the central nervous system after seizure. Science. 1987;237:192–197. doi: 10.1126/science.3037702. [DOI] [PubMed] [Google Scholar]
- 41.Odergren T, Stone-Elander S, Ingvar M. Cerebral and cerebellar activation in correlation to the action-induced dystonia in writer's cramp. Mov Disord. 1998;13:497–508. doi: 10.1002/mds.870130321. [DOI] [PubMed] [Google Scholar]
- 42.Perciavalle V, Santangelo F, Sapienza S, Serapide MF, Urbano A. Motor responses evoked by microstimulation of restiform body in the cat. Exp Brain Res. 1978;33:241–255. doi: 10.1007/BF00238063. [DOI] [PubMed] [Google Scholar]
- 43.Playford ED, Passingham RE, Marsden CD, Brooks DJ. Increased activation of frontal areas during arm movement in idiopathic torsion dystonia. Mov Disord. 1998;13:309–318. doi: 10.1002/mds.870130218. [DOI] [PubMed] [Google Scholar]
- 44.Preibisch C, Berg D, Hofmann E, Solymosi L, Naumann M. Cerebral activation patterns in patients with writer's cramp: a functional magnetic resonance imaging study. J Neurol. 2001;248:10–17. doi: 10.1007/s004150170263. [DOI] [PubMed] [Google Scholar]
- 45.Ralston DD, Milroy AM, Holstege G. Ultrastructural evidence for direct monosynaptic rubrospinal connections to motoneurons in Macaca mulatta. Neurosci Lett. 1988;95:102–106. doi: 10.1016/0304-3940(88)90640-4. [DOI] [PubMed] [Google Scholar]
- 46.Raman IM, Sprunger LK, Meisler MH, Bean BP. Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron. 1997;19:881–891. doi: 10.1016/s0896-6273(00)80969-1. [DOI] [PubMed] [Google Scholar]
- 47.Richter A, Loscher W. Pathology of idiopathic dystonia: findings from genetic animal models. Prog Neurobiol. 1998;54:633–677. doi: 10.1016/s0301-0082(97)00089-0. [DOI] [PubMed] [Google Scholar]
- 48.Richter A, Brotchie JM, Crossman AR, Loscher W. [3H]-2-deoxyglucose uptake study in mutant dystonic hamsters: abnormalities in discrete brain regions of the motor system. Mov Disord. 1998;13:718–725. doi: 10.1002/mds.870130419. [DOI] [PubMed] [Google Scholar]
- 49.Riker DK, Messer A, Roth RH. Increased noradrenergic metabolism in the cerebellum of the mouse mutant dystonia musculorum. J Neurochem. 1981;37:649–654. doi: 10.1111/j.1471-4159.1982.tb12536.x. [DOI] [PubMed] [Google Scholar]
- 50.Sprunger LK, Escayg A, Tallaksen-Greene S, Albin RL, Meisler MH. Dystonia associated with mutation of the neuronal sodium channel Scn8a and identification of the modifier locus Scnm1 on mouse chromosome 3. Hum Mol Genet. 1999;8:471–479. doi: 10.1093/hmg/8.3.471. [DOI] [PubMed] [Google Scholar]
- 51.Vidailhet M, Dupel C, Lehericy S, Remy P, Dormont D, Serdaru M, Jedynak P, Veber H, Samson Y, Marsault C, Agid Y. Dopaminergic dysfunction in midbrain dystonia: anatomoclinical study using 3-dimensional magnetic resonance imaging and fluorodopa F 18 positron emission tomography. Arch Neurol. 1999;56:982–989. doi: 10.1001/archneur.56.8.982. [DOI] [PubMed] [Google Scholar]
- 52.Walker JM, Matsumoto RR, Bowen WD, Gans DL, Jones KD, Walker FO. Evidence for a role of haloperidol-sensitive sigma-“opiate” receptors in the motor effects of antipsychotic drugs. Neurology. 1988;38:961–965. doi: 10.1212/wnl.38.6.961. [DOI] [PubMed] [Google Scholar]