

Abstract
Doublecortin (DCX) is a microtubule-associated protein that is required for normal neocortical and hippocampal development in humans. Mutations in the X-linked human DCX gene cause gross neocortical disorganization (lissencephaly or “smooth brain”) in hemizygous males, whereas heterozygous females show a mosaic phenotype with a normal cortex as well as a second band of misplaced (heterotopic) neurons beneath the cortex (“double cortex syndrome”). We created a mouse carrying a targeted mutation in theDcx gene. Hemizygous male Dcx mice show severe postnatal lethality; the few that survive to adulthood are variably fertile. Dcx mutant mice show neocortical lamination that is largely indistinguishable from wild type and show normal patterns of neocortical neurogenesis and neuronal migration. In contrast, the hippocampus of both heterozygous females and hemizygous males shows disrupted lamination that is most severe in the CA3 region. Behavioral tests show defects in context and cued conditioned fear tests, suggesting that deficits in hippocampal learning accompany the abnormal cytoarchitecture.
Keywords: doublecortin, knock-out, mouse, cerebral cortex, hippocampus, lissencephaly, neuronal migration
The mammalian cerebral cortex is a remarkably complex and elegantly organized structure that demonstrates a relatively stereotyped six-layered pattern of neuronal lamination. During development, the process of neurogenesis occurs at a distance from the definitive cerebral cortex in a region adjacent to the ventricular system known as the ventricular zone. Although recent studies have demonstrated that tangentially migrating neuroblasts derived from subcortical regions can give rise to many of the inhibitory interneurons of the neocortex (Marin and Rubenstein, 2001), the majority of cortical neurons reach the cortical plate via radial migration along glial fibers (Rakic, 1972). The cortex forms in an “inside-out” manner in which the neuroblasts that arrive earliest at the cortical plate populate the deeper layers of the definitive cortex and later-born neurons cross through these deeper layers to give rise to successively more superficial layers of cortex (Angevine and Sidman, 1961).
Lissencephaly is one of the most severe human cerebral cortical malformations (Dobyns and Truwit, 1995). It is thought to result from a failure of neuronal migration in which the majority of neurons stop short of their normal destination in the cortical plate. This migrational arrest results in an abnormally thickened and disorganized cerebral cortex, which shows poor formation of gyri and sulci. The resultant disorganization is thought to be the cause of the profound mental retardation and epilepsy that are associated with lissencephaly.
Mutations in two different genes, Lissencephaly-1(LIS1 or PAFA1B1) and doublecortin(DCX), are thought to cause the majority of cases of classical type I lissencephaly in humans (Pilz et al., 1998).Lis1 (Reiner et al., 1993) encodes a brain-specific, noncatalytic subunit of platelet-activating factor acetylhydrolase-1b, an enzyme that inactivates platelet-activating factor. In addition, theLis1 gene product has been shown to bind microtubules (Sapir et al., 1997); it is thought that this latter function may be central to the role of Lis1 in neuronal migration. A mouse knock-out ofLis1 has been engineered and shown to have abnormalities of neuronal migration (Hirotsune et al., 1998).
Doublecortin was first identified as the causative gene in X-linked lissencephaly/double cortex syndrome (des Portes et al., 1998;Gleeson et al., 1998). It encodes a microtubule-associated protein that is expressed in migrating neuroblasts (Francis et al., 1999; Gleeson et al., 1999a). Male patients with no functional doublecortin demonstrate a lissencephalic phenotype similar to that seen in patients with mutations in LIS1. In contrast, female patients carrying only a single mutant allele of DCX show a double cortex phenotype that consists of a heterotopic band of neurons in the white matter underlying the normal cortex. This heterotopia is thought to arise as a result of the early migrational arrest of the neuroblasts that experienced random X-inactivation of their only remaining normal allele of DCX. Because of the rarity of human cases of X-linked lissencephaly/double cortex syndrome and the impossibility of conducting a full developmental analysis of the phenotype in humans, we created a mouse model of this disease by engineering a mutation in the murine doublecortin gene.
MATERIALS AND METHODS
Gene targeting and generation of Dcxmutant mice
A mouse 129/SvJ λ phage library (Stratagene, La Jolla, CA) was screened with a probe derived from exon 2 of the humanDoublecortin (DCX) gene, and two partially overlapping clones were identified (Dcx λ clone 2.3 and Dcx λ clone 4.3) that covered exons 2 and 3 of mouse Dcx and the surrounding regions. The starting vector used, pSAGalpgkneolox2PGKDTA (gift from Sheila Thomas, Beth Israel Deaconess Medical Center, Boston, MA), contains a splice acceptor lacZ and a phosphoglycerate kinase (PGK)-neo cassette flanked by short polylinker sites. Downstream of the 3′ polylinker is a PGK-diphtheria toxin cassette. Upstream of the 5′ polylinker is a NotI site used for linearization of the final construct before transfection.
A 5 kb NheI fragment (containing the region immediately downstream of exon 3) was isolated from λ clone 4.3 and subcloned into the XbaI site of pBluescript SK−. From this fragment a 4.7 kb EcoRV–SmaI fragment was removed (the EcoRV site being 200 bp downstream of the end of exon 3) and was cloned into the blunted NheI site of the starting vector. Subsequently, a 3.6 kb NheI fragment from λ clone 2.3 (containing the sequence that begins 500 bp upstream from the beginning of exon 2) was subcloned into the XbaI site of pBluescript SK−. This 3.6 kb fragment was then removed as a NotI–SmaI fragment and cloned into the NotI–SmaI-digested starting vector plus downstream fragment to generate the final targeting vector.
This vector was linearized with NotI and transfected into TC1 embryonic stem (ES) cells as described previously (Deng et al., 1994, 1996). Correct targeting of the Dcx locus was identified in 1 (clone 28) of 115 clones by Southern blotting ofEcoRI-digested genomic DNA using a 380 bp probe (probe A) derived by PCR from a subcloned genomic fragment upstream of the 5′ arm of the targeting construct using a primer internal to the genomic fragment (5′-AAGAATAGGTATTTGGTTTGC) and the T3 primer (which binds to the T3 site in the subcloning vector pBluescript SK) (Fig.1B). Correct configuration of the targeted locus was further confirmed by Southern blotting of ApaI-digested genomic DNA from clone 28 and a control probed with a 460 bp XbaI–EcoRI fragment (probe B) derived from the region downstream of the 3′ arm of the targeting construct (data not shown).
Fig. 1.
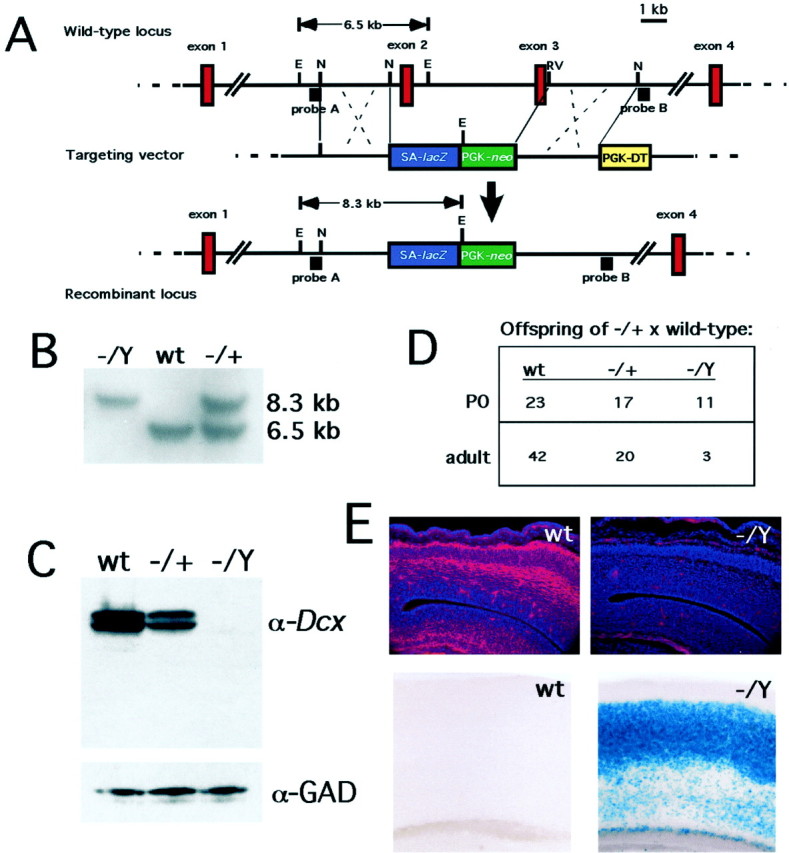
A targeted mutation in theDcx gene eliminates expression of the Dcx protein and results in increased postnatal lethality. A, Targeting strategy. SA-lacZ represents the lacZcoding sequence with an upstream splice acceptor. PGK-neo and PGK-DT are the neomycin and diphtheria toxin genes driven by a phosphoglycerokinase enhancer. Animals were typed by Southern blotting using a PCR product (probe A). The correct configuration of the targeted locus was also confirmed by Southern blotting using a second DNA fragment (probe B; data not shown). E, EcoRI;N, NheI; EV,EcoRV (only the relevant restriction endonuclease sites are indicated). B, Southern blot analysis of knock-out animals. Probe A was hybridized to genomic DNA from Dcxhemizygous mutant (−/Y), wild-type (wt), and heterozygous (−/+) animals (offspring of a heterozygous female and a wild-type male) that was digested with EcoRI. The 6.5 and 8.3 kb bands represent the wild-type and mutant alleles, respectively (see A).C, Western blot analysis of P0 mutant brains. Note the total absence of reactivity with the anti-Dcx antibody (11) in the hemizygous brain and approximately half normal quantity in the heterozygous brain. The blot was stripped and reprobed with an antibody against glutamate decarboxylase (α-GAD) as a loading control. D, Progeny of crosses between a Dcx −/+ female and a wild-type male. Note that the hemizygous males are present in an approximately Mendelian ratio (2:1:1) at P0 but that their relative numbers are reduced by adulthood. The P0 and adult figures represent two separate cohorts. E, Immunostaining of E15 brains with anti-Dcx antibody (top) and histochemical reaction of 5-week-old brains with X-gal (bottom). Note the total absence of Dcx reactivity in the mutant brain. The focal red signal in the mutant represents nonspecific background staining of blood vessels and leptomeninges. X-gal staining demonstrates perdurant β-galactosidase activity predominantly in layers 2–4 in the mutant brain but none in the wild type.
ES cell clone 28 was injected into C57BL/6J blastocysts, and germline transmission was obtained. We established the Dcx mutant allele in both a pure 129/SvJ and a mixed (129/SvJ × NIH Black Swiss) background. Routine genotyping was subsequently performed by PCR using pairs of primers specific for exon 2 ofDcx (Dcx-ex2-1, 5′-AAATATGAGAGGGTCACGGATG; Dcx-ex2-2, 5′-CTTCCAGTTCATCCATGCTTC) and lacZ (lacZ3, 5′-GAATCAGGCCACGGCGCTAATC; lacZ4, 5′-GCAAAGACCAGACCGTTCATACAG), which generate 313 and 440 bp fragments, respectively.
Animals were maintained in a virus-free mouse colony using standard procedures and treated according to protocols reviewed and approved by the institutional animal care and use committees of Harvard Medical School, Beth Israel Deaconess Medical Center, and the University of California, San Diego.
Histologic and immunohistochemical analysis
For routine histology, the offspring of female Dcx−/+ mice (in a mixed 129/SvJ × NIH Black Swiss background) mated to NIH Black Swiss males were raised to ∼6–8 weeks of age and deeply anesthetized with pentobarbital. The brains were then fixed via transcardiac perfusion with 4% paraformaldehyde, removed, processed through paraffin embedding, sectioned at 8 μm, and stained with either hematoxylin and eosin (H&E) or cresyl violet. The sections were then mounted on Superfrost/Plus glass slides (Fisher Scientific, Houston, TX) with Permount, coverslipped, and photographed.
Immunohistochemical staining with anti-Dcx (see Gleeson et al., 1999a, for details of antibody production) and anti-class III β-tubulin (TuJ1; Sigma, St. Louis, MO) antibodies was performed as follows. Embryonic day 15 (E15) litters derived from timed matings betweenDcx −/+ females and NIH Black Swiss males were drop-fixed in 4% paraformaldehyde overnight at 4°C, cryoprotected with 30% sucrose in 1× PBS, embedded in optimal cutting temperature compound, and sectioned in the coronal plane on a Leica (Nussloch, Germany) CM3000 cryostat. Sections were air-dried on Superfrost/Plus glass slides overnight at room temperature and then stored at −20°C.
For fluorescence immunohistochemistry, mounted sections were rinsed for 20 min in 1× PBS and 0.1% Triton X-100, blocked for 1 hr in blocking solution (1× PBS, 0.1% Triton X-100, and 5% heat-inactivated goat serum), incubated either for 2 hr at room temperature or overnight at 4°C with the primary antibody (anti-Dcx antibody, rabbit IgG, 1:300; TuJ1 antibody, rabbit IgG, 1:2000) diluted in blocking solution, rinsed three times for 5 min each with 1× PBS, incubated with the secondary antibody (Cy3-conjugated goat anti-rabbit; 1:300) diluted in blocking solution for 1 hr at room temperature, rinsed three times for 5 min each with 1× PBS, incubated 1–3 min with Hoechst 33342 (Molecular Probes, Eugene, OR), rinsed with 1× PBS, and coverslipped with Crystal/Mount (Biomeda, Foster City, CA). The sections were visualized on an Olympus AX70 fluorescence microscope (Olympus Optical, Tokyo, Japan) using a standard rhodamine filter set for Cy3 and a 4′,6′-diamidino-2-phenylindole (DAPI) filter set for the Hoechst stain. Images were captured in black and white using a SPOT camera, false-colored (rhodamine channel red and DAPI channel blue), and superimposed. Confocal images were captured using a Bio-Rad (Hercules, CA) MRC600 confocal microscope (60× oil immersion objective) and imported into Adobe Photoshop (Adobe Systems, San Jose, CA).
Histochemical staining for β-galactosidase activity was performed as follows. Fifty micrometer sections obtained by vibratome sectioning of brains from 6-week-old animals (for procedural details, see the section that discusses fluorescent layer-specific marker strain) were stained overnight at 37°C with 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) at a final concentration of 1 mg/ml in staining solution (0.1m phosphate buffer, pH 7.3, 2 mmMgCl2, 0.01% sodium deoxycholate, 0.02% NP-40, 5 mm potassium ferrocyanide, and 5 mm potassium ferricyanide). The stained sections were rinsed with 1× PBS, mounted with Crystal/Mount on Superfrost/Plus glass slides, and coverslipped. Images were captured with a SPOT camera.
Analysis of postnatal mortality
These data were derived from crosses between Dcx −/+ female mice (mixed 129/SvJ × NIH Black Swiss background) and NIH Black Swiss males. For the postnatal day 0 (P0; day of birth) cohort, all offspring of several litters were collected on the day of birth, tailed, and typed by PCR (see the section that discusses gene targeting and generation of Dcx mutant mice). The expected Mendelian ratio for wild type, Dcx −/+, and Dcx−/Y is 2:1:1 (note that wild type includes both +/+ females and +/Y males). For the adult cohort, all of the surviving offspring of several litters were tailed and typed by PCR. Again, the expected Mendelian ratio is 2:1:1.
Western blot analysis
Total brain protein from P0 littermates (the same animals from which liver genomic DNA was extracted for Fig. 1B) was boiled with SDS loading buffer, run on an 8% SDS-PAGE gel (50 mV for 2 hr), and transferred (150 mA overnight) to an Immobilon-P (Millipore, Bedford, MA) membrane. The blot was blocked in 5% milk/TBST (0.01 m Tris, pH 7.5, 0.15m NaCl, 0.1% Tween 20) for 1 hr, incubated with anti-Dcx primary antibody (rabbit IgG; 1:5000 dilution) in 5% milk/TBST at 4°C overnight, rinsed with TBST, incubated with a horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (1:3000 dilution; Bio-Rad), and developed with the ECL reaction (Kirkegaard & Perry, Gaithersburg, MD). The blot was stripped and reprobed with a rabbit anti-glutamate decarboxylase antibody (1:5000; Chemicon, Temecula, CA) to control for protein loading.
5-bromo-2′-deoxyuridine birth dating studies
Female Dcx −/+ mice pregnant from timed matings with NIH Black Swiss males were injected intraperitoneally at E12 and E14 with 300 μl of a 5 mg/ml solution of 5-bromo-2′-deoxyuridine (BrdU) in 0.9% NaCl. The brains of newborn (P0) littermates were removed, drop-fixed in 4% paraformaldehyde, embedded in paraffin, cut in 8 μm coronal sections. Further processing was performed as described previously (Gonzalez et al., 1997). The stained sections were mounted without counterstaining and photographed.
Fluorescent layer-specific marker strain
Female Dcx −/+ mice were mated to male mice heterozygous for a thy1–yellow fluorescent protein (YFP) transgene with expression predominantly in a subset of cerebral cortical layer 5 pyramidal cells as well as in a subset of hippocampal pyramidal cells and cells of the fascia dentata (YFP-G inFeng et al., 2000). In addition, we noted that the transgene labels a variable number of layer 3 pyramidal neurons in both wild-type and mutant backgrounds. These mice were obtained from The Jackson Laboratory (Bar Harbor, ME) [strain name: B6.Cg-TgN(Thy1–YFP-H)2Jrs]. Typing was performed by PCR as described previously (Feng et al., 2000).
Offspring of the above cross were raised to ∼6–8 weeks of age and then killed. The brains were fixed via transcardiac perfusion with 4% paraformaldehyde, removed, and embedded in 10% gelatin (225 bloom calf skin; Aldrich, Milwaukee, WI). Once the gelatin hardened (∼30 min at 4°C), the embedded brains were cut into appropriately sized blocks and fixed overnight in 4% paraformaldehyde at 4°C. The blocks were subsequently rinsed in 1× PBS, mounted with super glue on metal chucks, and cut with a vibratome into 50-μm-thick coronal sections. The sections were counterstained with the nuclear dye Hoechst 33342. These sections were then mounted with Crystal/Mount on Superfrost/Plus glass slides and visualized on an Olympus AX70 fluorescence microscope using an FITC filter set for YFP and a DAPI filter set for the Hoechst stain. Images were captured in black and white using a SPOT camera, false-colored (FITC channel green and DAPI channel blue), and superimposed.
Behavioral testing
The behavioral test battery was modified from that used byMcIlwain et al. (2001).
Gross physical assessment
Individual mice were placed in a clean shoebox cage. During a 2 min observation period, several behavioral (e.g., presence of wild running, excessive grooming, freezing, rearing, jumping, defecation, urination, cage exploration, hunched body posture) and physical (e.g., presence of dirty fur; ulcerated skin; bald spots; thinning fur; trimmed whiskers; labored breathing; piloerection; exophthalmos; palpebral closure; condition of teeth, nails, nose, genital/rectum area; fur color) features of the mice were noted. Body temperature and weight were also recorded.
Sensorimotor reflexes
With the use of a cotton-tipped applicator, each mouse was assessed for several sensorimotor reflexes (e.g., eye blink, ear twitch, whisker-orienting, sound orienting) and its response to an approaching object. The pupil constriction/dilation reflex was then assessed. Several postural reflexes were also assessed: (1) Latency to return to upright posture after turning the mouse onto its back, (2) ability to maintain upright balance in a rapidly moving cage (the normal reflex is the extension of all four legs and the ability to maintain balance), and (3) tail suspension response. (When lifting a mouse by its tail, the normal response is a raised head and outward extension of the forelimbs and hindlimbs. When lowered toward the edge of a tabletop, the forelimbs should reach for the ledge.). (4) Footprint analysis was also performed. To assess locomotor gait, each paw was coated with a different colored ink, and the mouse was allowed to walk through a 9 cm wide × 35 cm long × 6 cm high opaque tunnel placed on a sheet of paper.
Motor activity
Initiation of movement. The latency to move from the center to the edge of a 20-cm-diameter circular platform was recorded.
Open field. Exploratory locomotor activity in a 30 min test period was measured in an open field (45 × 45 cm) by a Digiscan apparatus (Accuscan Electronics, Columbus, OH). Horizontal activity (locomotor activity), vertical activity (rearing) total distance (in centimeters), and center distance were recorded. The center distance divided by the total distance is an indicator of anxiety-related behavior.
Rotarod. Locomotor coordination and balance were measured by placing mice on an accelerating, 3-cm-diameter, rotating drum (Ugo Basile, Comerio, Italy) for three trials with a minimum 15 min interval between trials. The rotarod started at 4 rpm and increased to 40 rpm over a 5 min period. The mean latency to fall over the three trials was the dependent measure.
Wire hang. Forelimb strength was assessed by suspending a mouse by the tail and gently lowering it until it grasped a 1-mm-diameter wire with its front paws. The mean latency to fall (maximum, 60 sec) over three trials is the dependent measure.
Grip strength. In a separate assessment of forelimb strength, a mouse was suspended by the tail and lowered until it grasped the loop of a mouse grip-strength meter (Ugo Basile). The mouse was then gently pulled away from the loop and the maximum grip force exerted by the mouse before losing its grip was recorded. Five trials were run, and the mean of the middle three scores was used in the analysis.
Cage-top hang test. This test allows the mouse to use both its forelimbs and its hindlimbs to maintain its grip. The mouse was placed on a modified cage lid (with duct tape placed around the edges), and the lid was inverted. The latency to fall is the dependent measure. The trial maximum is 60 sec.
Pole test. The mouse was placed at the end of a 3-cm-diameter pole lying in a horizontal position. The pole was gradually lifted to a vertical position, with the latency to fall off the pole being the dependent measure. These values were converted to a pole test score (fell before the pole reached a 45° or 90° angle = 0 or 1; fell in 0–10 sec = 2; fell in 11–20 sec = 3; fell in 21–30 sec = 4; fell in 31–40 sec = 5; fell in 41–50 sec = 6; fell in 51–60 sec = 7; stayed on 60 sec and climbed halfway down the pole = 8; climbed to the lower half of the pole = 9; climbed down and off the pole in 51–60 sec = 10; climbed down and off the pole in 41–50 sec = 11; climbed down and off the pole in 31–40 sec = 12; climbed down and off the pole in 21–30 sec = 13; climbed down and off the pole in 11–20 sec = 14; or climbed down and off the pole in 1–10 sec = 15).
Nociception
Hot plate. The mouse is placed on a hot plate analgesia meter (Columbus Instruments, Columbus, OH) set at 55°C. The latency for the mouse to either jump, lick the hindpaw, or shake the hindpaw is recorded and the mouse is removed from the apparatus. Trial maximum is 30 sec.
Tail flick. The intensity of a light beam (tail flick analgesia meter; Columbus Instruments) is adjusted to produce a tail flick of 4–6 sec. The mouse is wrapped in a soft towel with its tail extending over the light path. Three trials are run on different areas of the tail with at least a 5 min intertrial interval. Cutoff time is 10 sec.
Acoustic startle and prepulse inhibition of the acoustic startle
Acoustic startle and prepulse inhibition of the acoustic startle responses were measured using two SR-Lab Systems (San Diego Instruments, San Diego, CA). A test session was begun by placing a subject in the Plexiglas cylinder, where it was left undisturbed for 5 min. A test session consisted of seven trial types. One trial type was a 40 msec, 120 dB sound burst used as the startle stimulus. There were five different acoustic prepulse plus acoustic startle stimulus trials. The prepulse sound was presented 100 msec before the startle stimulus. The 20 msec prepulse sounds were 72, 74, 76, 78, or 80 dB. Finally, to measure baseline movement in the cylinders there were trials during which no stimulus was presented. Six blocks of the seven trial types were presented in pseudorandom order such that each trial type was presented once within a block of seven trials. The average intertrial interval was 15 sec (range, 10–20 sec). The startle response was recorded for 65 msec (measuring the response every 1 msec) starting with the onset of the startle stimulus. The background noise level in each chamber was 70 dB. The maximum startle amplitude recorded during the 65 msec sampling window was used as the dependent variable. The percentage prepulse inhibition of a startle response was calculated as 100 − [(startle response on acoustic prepulse and startle stimulus trials/startle response alone trials) × 100].
Learning and memory
Conditioned fear. A fear-conditioning shock chamber (26 × 22 × 18 cm high) made of clear Plexiglas and surrounded by photobeams was placed inside a sound attenuated chamber (internal dimensions: 56 × 38 × 36 cm; Med Associates Inc., East Fairfield, VT) and connected to the Freeze Monitor system (San Diego Instruments). Mice were observed through windows in the front of the sound-attenuated chamber. The conditioned stimulus (CS) was an 85 db, 2800 Hz, 20 sec tone; the unconditioned stimulus (US) was a scrambled foot shock at 0.75 mA presented during the last 3 sec of the CS. Mice were placed in the test chamber for 3 min before the CS, and freezing behavior was recorded. Freezing was defined as a lack of movement other than respiration. Three CS/US pairings were given with 1 min spacing; freezing during the CS was also recorded. After 24 hr, each mouse was placed back into the shock chamber and the freezing response was recorded for 3 min (context test). Two hours later the mouse was placed in a novel chamber and the freezing behavior was recorded for 3 min before and during three CS presentations (cued conditioning). The time spent freezing was converted to a percentage of freezing value.
Water maze
Pretraining. All mice were first tested for 2 d in a straight-swim pretraining protocol. Mice received 16 trials (8 trials on each of 2 consecutive days). A platform located 1 cm below the water was located opposite the start location. Latency to climb onto the platform was the dependent measure. Mice had to complete six of eight trials in under 10 sec on the second day to move on to water maze testing.
Hidden platform testing. Extra-maze visual cues were hung from a curtain located around a 1.26-m-diameter circular tank. The water was made opaque by the addition of nontoxic paint. A 10-cm-diameter escape platform was located 1 cm below the surface of the water, and a Polytrack (San Diego Instruments) video-tracking system was used to collect mouse movement (location, distance, and latency) data during training and probe trials. Each mouse was given eight trials per day in two blocks of four trials for 4 consecutive days. After trial 36, each animal was given a 60 sec probe trial. During the probe test, the platform was removed and quadrant search times and platform crossings were measured.
Visual cued testing. One day after the last hidden-platform training trial, mice were trained to locate a visible-cued platform. The visible cue was a gray plastic cube (9 cm) attached to a pole such that it was 10 cm above the platform. On each trial of the visible platform test, the platform was randomly located in one of the four quadrants. Mice were given eight trials in blocks of four trials, and the latency to find the platform was recorded for each trial.
All statistical analyses of behavioral data were performed using a Student's t test.
RESULTS
Targeted mutagenesis of the mouse Dcx locus
To create a mutation in the Dcx gene we targeted its second and third coding exons in ES cells. These exons encode the first and most of the second Dcx repeats, which are required for the microtubule binding and bundling properties of the protein (Taylor et al., 2000). The targeting construct creates a fusion between the first exon of Dcx and a lacZ transgene, so thatlacZ expression is driven in neurons that would ordinarily express the Dcx gene (Fig. 1A,E). Targeted ES cells were implanted into pseudopregnant females to produce highly agouti chimeric males that, when mated to wild-type females, gave rise to females heterozygous for the targeted allele. Mating of these females to wild-type males produced both hemizygous male and heterozygous female offspring (Fig. 1B), thus confirming the X-linked transmission of the mutation.
Hemizygous Dcx mutant males express no detectable Dcx protein when evaluated either by Western blotting or by immunohistochemistry. Although the antibody was raised against the C-terminal 16 aa of the Dcx protein, which are encoded by an exon not targeted by the induced mutation (Gleeson et al., 1999a), Western blot analysis of the brains of hemizygous Dcx mutant male mice showed no full-length or truncated Dcx (Fig. 1C). In addition, brains from Dcx heterozygous females showed approximately one-half the wild-type amount of Dcx protein (Fig.1C). The absence of Dcx protein from the brains of hemizgyous mutant males was confirmed by immunolabeling E15 mouse brain sections using the same Dcx antiserum. Despite the abundant expression of Dcx immunoreactivity in the intermediate zone and cortical plate of wild-type embryos (Fig. 1E, top left), we found no detectable Dcx immunoreactivity in the brain sections of hemizygous male embryos (Fig. 1E, top right). These data strongly suggest successful mutation of theDcx gene and an absence of residual Dcx protein.
Dcx hemizygous mice show decreased postnatal viability
Heterozygous Dcx mutant females were born in approximately Mendelian ratios (Fig. 1D) and survived and bred indistinguishably from control mice, whereas hemizygous mutant males showed severely decreased viability both in the neonatal period and beyond (Fig. 1D). The majority of the increased mortality in mutant males occurred in the first few days after birth. Although the cause of death in these animals is not clear, the fact that it tends to occur more frequently in larger litters and that some of the surviving mutant males are runted relative to their littermates suggests that they may not compete as well for milk. The small proportion of mutant males that did survive to adulthood were variably fertile (data not shown).
Dcx mutant mice demonstrate preserved neocortical architecture and development
Because humans with spontaneous mutations in the DCXgene show gross malformation of the cerebral cortex and profound neurological disability (Berg et al., 1998; des Portes et al., 1998;Gleeson et al., 1998), we were surprised that Dcxheterozygotes and hemizygotes showed remarkably normal overall brain morphology. The size of the forebrain, cerebellum, and other major gray matter structures was indistinguishable from normal (Fig.2), as were the size and gross organization of major white matter tracts, including the corpus callosum, anterior commissure, optic chiasm and tract, corticospinal and pyramidal tracts, cerebellar peduncles, and cranial nerves. Microscopic examination of the cerebellum (Fig. 2C) and retina (Fig. 2D) showed gray and white matter organization that was indistinguishable from normal mice.
Fig. 2.

Dcx mutant mice show normal brain morphology. A, The adult Dcx mutant brain is strikingly normal, with the exception of abnormal layering in the hippocampus. B, The Dcx mutant neocortex shows the normal six-layered pattern. Note the similarity in thickness of the laminas and the density of cell populations between mutant and wild type (wt). C, Dcxmutant cerebellum is indistinguishable from wild type. Note the normal overall morphology of the cerebellum (insets) as well as the normal layering pattern of the cerebellar cortex: molecular layer, Purkinje cell layer, and internal granular layer. D,Dcx mutant retina shows normal layering.
In contrast to the profound defects in cortical layering in humans withDCX mutations, heterozygous female and hemizygous maleDcx mutant mice showed the usual six layers of the cerebral cortex (Fig. 2B) (data for heterozygotes not shown). Although subtle layering abnormalities cannot be absolutely ruled out, overall cortical thickness was indistinguishable between mutant and wild type, as was the thickness of the individual layers. There was no evidence of periventricular or white matter neuronal heterotopia in the mutant mice by routine histology or immunostaining with the neuronal marker, neuronal-specific nuclear protein (NeuN) (data not shown).
Layer-specific markers also confirmed that cortical layering was preserved. Reelin immunoreactivity in layer 1 was indistinguishable from wild type, suggesting that layer 1 is normally formed in the mutant (data not shown). Lamination was further examined by using a transgenic mouse that expresses YFP under the control of athy1 enhancer, predominantly in a subset of layer 5 neurons (Feng et al., 2000). In male hemizgyous mutants, layer 5 neurons showed a sharp laminar organization that was indistinguishable from normal (Fig. 3A,B). YFP expression in the neuronal processes of hemizygous mutant mice revealed well developed apical and basal dendrites that were studded with well developed spines with a typical morphology (data not shown). These data suggest that the neocortex of Dcx mutant mice has a remarkably normal overall cytoarchitecture and dendritic structure.
Fig. 3.

Analysis of cortical layering in theDcx mutant mouse using the thy1–YFP marker. Top, The brains of adult wild-type (wt) and Dcx mutant (−/Y) mice into which has been crossed a thy1–YFP transgene (YFP-H in Feng et al., 2000) that is expressed predominantly in a subset of layer 5 neurons as well as in pyramidal cells of the hippocampus and cells of the fascia dentata. Close-up images of the mutant neocortex (bottom) show a distribution of YFP-expressing cells in layer 5 similar to that in the wild type. Note that scattered YFP-expressing cells are apparent in layer 3 (arrowhead) in the wild type. Such cells were identified with similar frequency in the mutant as well but are not present in the particular field pictured.
Patterns of neuronal migration in the neocortex were also indistinguishable between normal mice and mutant Dcx mice (Fig. 4). Routine histology and immunostaining with an antibody against TuJ1 showed normal neuronal morphology and organization at embryonic ages (Fig.4A). To assess neurogenesis and neuronal migration more carefully in vivo, we labeled cohorts of neurons with BrdU injections at E12 and E14 and examined the localization of labeled neurons on the day of birth (P0) (Fig. 4B) because of the postnatal lethality of Dcx mutant males. The distribution of BrdU-labeled neurons was indistinguishable in wild-type and Dcx mutant mice, indicating normal patterns of neurogenesis and neuronal migration in the mutant neocortex.
Fig. 4.
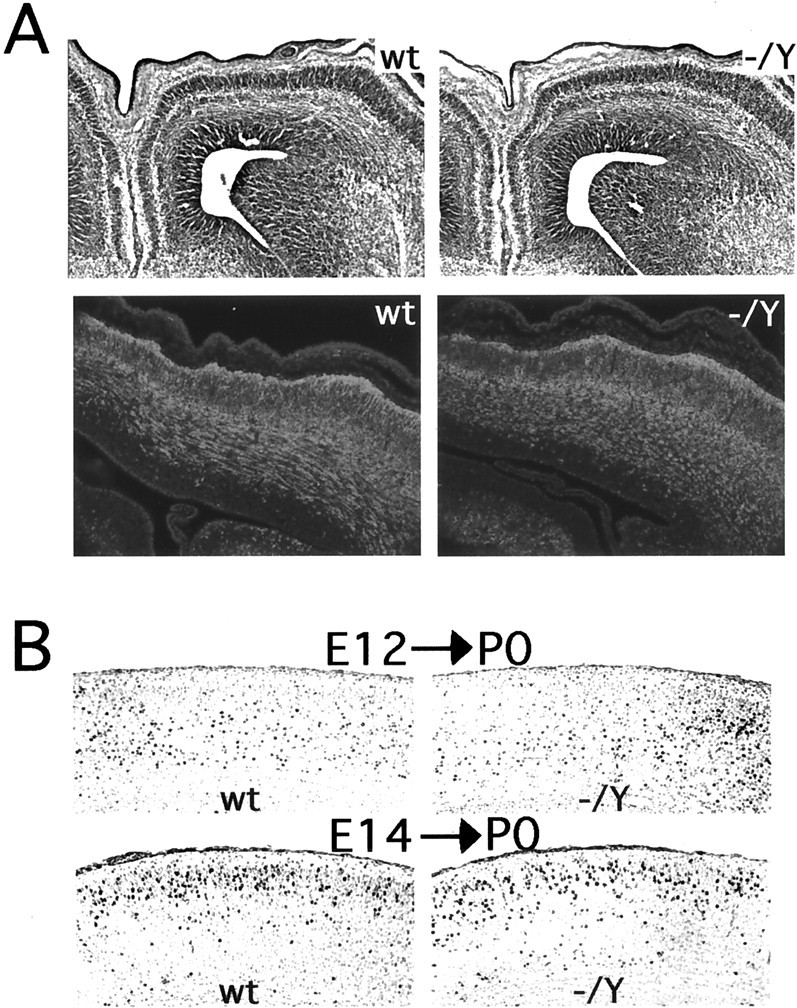
Embryonic development of the neocortex inDcx mutant mice. A, Cresyl violet-stained coronal sections of E15 brains (top). Note that theDcx mutant (−/Y) brain is indistinguishable from wild type (wt). Coronal sections of E15 brains immunostained with an antibody against TuJ1 are shown at the bottom. This antibody labels postmitotic migrating and differentiating neurons and is coexpressed with Dcx (Francis et al., 1999). Note the very similar pattern of staining between mutant and wild type. B, BrdU birth dating of cortical neurons in Dcx mutants. Brains were harvested at P0 from mice born to dams that had received intraperitoneal injections of BrdU either on E12 (E12→P0) or E14 (E14→P0) and were stained for BrdU. Note that in the brains from animals injected at E12 the labeled cells are more concentrated in the lower layers but are present throughout the cortex. In contrast, in brains from animals injected at E14 there is a tighter clustering of labeled cells in the upper layers of the cortex. The distribution of cells does not differ significantly between wild type and mutant.
Abnormal hippocampal development in Dcxmutant mice
In contrast to the relatively normal organization and development of the neocortex in Dcx mutant mice, the hippocampal formation was consistently malformed in both hemizygous and heterozygous mutant animals. The dentate gyrus showed indistinguishable organization in mutant and wild-type mice, but the various CA fields all showed a greater or lesser degree of disorganization in the mutant (Fig. 5). CA3 showed heterotopic neurons in the stratum radiatum as well as in the stratum oriens on either side of the pyramidal layer (Fig. 5A,B). In some regions the pyramidal layer of CA3 was partially split (Fig. 5B), whereas in other regions it was characterized by more loosely arrayed pyramidal neurons than usual and irregular inner and outer borders to the pyramidal cell layer (Fig. 5A,B). Disruptions of the architecture of the pyramidal layer continued in the CA2 and CA1 regions (Fig. 5A, middle) but were generally milder, without frank splitting of the pyramidal layer in either of these regions. In some cases a looser, more disordered packing of the neurons extended into the proximal portion of the subiculum (Fig.5A). Female mice heterozygous for the Dcxmutation showed similar disruptions of the hippocampus, particularly in CA3, with the overall extent of disorganization being somewhat milder than in hemizygous males (Fig. 5B, top). Evaluation of the disruption in CA3 with the thy1–YFP marker reveals an even greater degree of disorganization in the lamination of cell bodies of both heterozygotes and hemizygotes than can be appreciated with H&E alone (Fig. 5B,bottom). High-power confocal images of the CA3 neurons show a relative preservation of dendritic arbors in the mutant (Fig.5C,D), suggesting that the observed abnormalities in the hippocampus are primarily related to defective somal positioning.
Fig. 5.

Abnormal hippocampal lamination in theDcx mutant. A, Wild-type (wt) and Dcx mutant hippocampi stained with the neuronal marker NeuN. Note the greater degree of disorganization in the pyramidal layer of the Dcx mutant (−/Y) compared with the wild type. Although the disorder is greatest in area CA3 (bottom), a looser layering of cells is also readily appreciable in area CA1 (middle). B, Area CA3 in wild type,Dcx heterozygotes (−/+), andDcx hemizygotes (−/Y). Note the partial splitting of the pyramidal cell layer (top,black arrowhead) and disorganized lamination (white arrowhead) in the hemizygous brain. The heterozygote demonstrates milder but similar defects. Analysis of thethy1–YFP transgene in the heterozygote and homozygote shows a progressively greater disorganization of the YFP-expressing cells relative to wild type in area CA3 of the hippocampus (bottom). C, D, Confocal images of hippocampal area CA3. C, In the areas of CA3 in which lamination is relatively preserved in the mutant, dendritic processes appear to be indistinguishable from wild type. D, This is also true in areas that show a greater degree of disorganization in the mutant. The wider spacing between labeled neurons in theDcx mutant is secondary to greater scattering of the cell bodies.
Abnormalities of learning and memory in Dcxmutant mice
The anatomic abnormalities of the hippocampus identified in the mutant correlate with learning and memory deficits. Ten wild-type (i.e., mixed NIH Black Swiss and 129/SvJ background) female and 10 heterozygous Dcx female mice were assessed in a battery of behavioral tasks. No differences between experimental and control mice were observed with regard to gross physical appearance, sensorimotor reflexes, analgesia-related responses, sensorimotor gating (prepulse inhibition), startle response, and open-field activity (data not shown). Statistically significant deficits in heterozygous mice were observed in the cage-top hang test (t(18) = 2.75; p < 0.01) (Fig. 6A) and the cued conditioned fear test (t(16) = 4.94; p < 0.0001) (Fig. 6C). The observation that heterozygous mice had shorter fall latencies in the cage-top hang test suggests limb weakness. An impaired ability to associate the conditioned stimulus (tone) with the unconditioned stimulus (shock) in the fear test suggests impaired function of the amygdala (Phillips and LeDoux, 1992). Interestingly, despite this latter finding, no obvious morphologic abnormalities of the amygdala were identified by routine histologic evaluation (data not shown).
Fig. 6.

Dcx mutant mice show defects in strength and hippocampal-based learning.A, Cage-top hang test in a mixed (129/SvJ × NIH Black Swiss) background (t(18) = 2.75;p < 0.01). B, Wire hang test in a 129/SvJ background (t(10) = 9.7;p < 0.001). C, Cued conditioned fear test in a mixed background (t(16)= 4.94; p < 0.0001). D, Context conditioned fear test in a 129/SvJ background (t(10) = 3.5; p < 0.01).
Similar observations were made on female Dcx heterozygous mice in a pure 129/SvJ background. These mice demonstrated abnormalities in another test of limb strength, the wire hang test (t(10) = 9.7; p < 0.001) (Fig. 6B) and in both the cued conditioned fear test (t(10) = 3.85;p < 0.01; data not shown) and the context conditioned fear test (t(10) = 3.5;p < 0.01) (Fig. 6D). The animals in the 129/SvJ background demonstrated mild deficits in latency to find the hidden platform in the Morris water maze test as well (group effect: F(1,10) = 5.7;p < 0.03), with no significant impairment in the probe trials, however (data not shown). The overall pattern replicates the limb weakness and impaired amygdala function observed in the mixed background and also suggests that mice in the 129/SvJ background have hippocampal dysfunction. The fact that similar results were obtained in both mixed and inbred lines suggests that these findings are robust. Because of the postnatal lethality of hemizygous mutant Dcxmales, very few survived long enough to be tested behaviorally. However, a small group of hemizygous mutant males (four wild-type and four mutant) were also tested and showed comparable deficits (data not shown).
DISCUSSION
In this report, we find that an engineered mutation in the murineDcx gene causes disruption of hippocampal architecture and hippocampus-based learning. Surprisingly, despite the severe abnormalities in cortical architecture in humans with DCXmutations, the mouse neocortex develops remarkably normally in the absence of detectable Dcx protein. Although we cannot rule out subtle differences in lamination, BrdU studies and histological analysis during the major period of neuronal migration showed no obvious abnormalities of neocortical development. This finding suggests thatDcx is not essential in mice for the radial migration of projection neurons from the ventricular zone into the cortical plate.
In addition to its expression during neuronal migration, Dcx is also highly expressed in growing axons and dendrites (Francis et al., 1999;Gleeson et al., 1999a), suggesting that it could have essential roles beyond just directing neuronal migration. For example, the heterotopic neurons observed in humans with DCX mutations show very abnormal morphology as well as position (Berg et al., 1998). Although a potential role for Dcx in axonal and dendritic outgrowth represents a plausible explanation for the severe behavioral phenotype of these mice, which display increased early postnatal mortality despite the negligible effects on neocortical neuronal migration, the dendrites, dendritic spines, and axons visualized with thethy1–YFP transgenic construct in this study showed no obvious abnormalities. Still, the possibility of subtle derangements of process formation, particularly within subsets of neurons not visualized by this construct, cannot be excluded.
In contrast to the neocortex, the hippocampus of the Dcxmutant mice showed abnormalities suggestive of a role forDcx in neuronal migration. Neuronal lamination in the CA fields of the hippocampus was quite disrupted, suggesting abnormal neuronal migration during initial hippocampal development. Areas CA3 and CA1/subiculum showed the most significant degree of laminar disruption, with a lesser degree of disruption noted in areas CA1 and CA2. Perhaps the unique migratory path of hippocampal projection neurons (Stanfield and Cowan, 1979a,b) makes them more dependent onDcx function, or perhaps there are regional differences in the expression of other potentially compensatory genes. Despite the lamination defects within the hippocampus, the structure of the dendrites, dendritic spines, and axons of hippocampal neurons was relatively preserved. This finding suggests a preferential effect of the Dcx mutation on neuronal cell body positioning (i.e., migration) and raises the possibility that whatever subtle abnormalities of dendrite or axon orientation are present in the hippocampus may be secondary to the heterotopic location of the corresponding cell bodies.
Why is the Dcx mutant phenotype in mice so much milder than in humans? One possibility is that other microtubule regulatory proteins such as LIS1, tau, or doublecortin calcium/calmodulin-dependent protein kinase-like kinase 1 (DCAMKL1) might be upregulated in a compensatory manner in mice. However, Western blot analysis of hemizygous Dcx mutant mice showed no changes in the expression levels of any of these proteins (data not shown). A second possibility is that migrating neurons in the human brain have a quantitatively greater dependence on DCXfunction than in the mouse. Perhaps in mice, in which cortical neurons traverse a much shorter absolute distance than in humans during brain development, Dcx is simply not required for migration. Alternatively, the levels of proteins homologous to Dcx, such as DCAMKL1, may suffice to support relatively normal migration in the mouse Dcx mutant. DCAMKL1 (Burgess et al., 1999; Burgess and Reiner, 2000; Lin et al., 2000), which contains both a Dcxhomology domain and a calmodulin kinase-like domain, appears to be the evolutionarily more ancient of the two genes, because bothCaenorhabditis elegans and Drosophila have homologs of DCAMKL1 but not of Dcx (Gonczy et al., 2001;Reiter et al., 2001). Moreover, DCAMKL1 is coexpressed with Dcx in migrating cerebral cortical neurons and shows microtubule-stabilizing activities that are indistinguishable from Dcx (Burgess and Reiner, 2000; Lin et al., 2000). Therefore, it is possible that DCAMKL1 may be playing a partially redundant role in mice but is insufficient to support normal migration in humans who lack DCX function. The engineering of DCAMKL1 mutants and the creation of Dcx;DCAMKL1 double mutants will be necessary to test this possibility.
In addition to the obvious quantitative differences in the distance neuroblasts must migrate in humans compared with in mice, there may be subtle but significant differences in the molecular terrain these cells must traverse. For example, it is possible that there are important differences between rodents and primates in the structure or molecular constitution of the radial glia that support radial migration of cortical neurons (Schmechel and Rakic, 1979; Rakic, 1988).
Finally, given the fact that the mouse Dcx allele we have created appears to be protein-null, it is formally possible that human cases of X-linked lissencephaly/double cortex syndrome are the result of a dominant negative effect of aberrant, truncated forms of DCX protein, which might neutralize the functions of proteins such as DCAMKL1. Under such a hypothesis, one might expect that a truly protein-null human allele would be associated with a mild phenotype such as that of the mouse. However, the fact that numerous mutant human alleles of DCX have been sequenced, some of which represent severe truncations that are likely to be protein-null (Gleeson et al., 1999b), argues against such a possibility.
Whereas hemizygous DCX mutations and heterozygousLIS1 mutations produce profound lissencephaly and neocortical disorganization in humans (Pilz et al., 1998), analogous mutations in mice cause more subtle (in the case of Lis1) or negligible (in the case of Dcx) neocortical defects but remarkably similar hippocampal defects (Fleck et al., 2000). These strongly parallel mutant phenotypes in both mammalian species further support closely related biochemical roles for Dcx and Lis1. In addition, at least one report suggests a direct physical interaction between Dcx and Lis1 protein (Caspi et al., 2000). However, other genes that more severely affect hippocampal neuronal migration (e.g., Cdk5, Reelin, and mDab1) (Stanfield and Cowan, 1979a,b; Ohshima et al., 1996;Gonzalez et al., 1997) also profoundly affect neocortical organization in mice, suggesting that this latter set of genes may not share a simple biochemical pathway with Dcx and Lis1. The creation of doubly mutant mice will be necessary to test for genetic interactions between the various neuronal migration genes.
Footnotes
This work was supported by National Institute of Neurological Disorders and Stroke Grants PO1 NS40043 (C.A.W.) and PO1 NS39404 (C.A.W. and A.W.B.) and by National Institute of Environmental Health Sciences Superfund Center Grant P42 ES10337 (A.W.B.). We thank Sheila Thomas for the generous gift of the pSABGalpgkneolox2PGKDTA vector and Lourdes Madrigal for technical assistance.
Correspondence should be addressed to Dr. Christopher A. Walsh, Department of Neurology, Beth Israel Deaconess Medical Center, Room 816, Harvard Institutes of Medicine, 5 Blackfan Circle, Boston, MA 02115. E-mail: cwalsh@caregroup.harvard.edu.
REFERENCES
- 1.Angevine JB, Jr, Sidman RL. Autoradiographic study of the cell migration during histogenesis of cerebral cortex in the mouse. Nature. 1961;192:766–768. doi: 10.1038/192766b0. [DOI] [PubMed] [Google Scholar]
- 2.Berg MJ, Schifitto G, Powers JM, Martinez-Capolino C, Fong CT, Myers GJ, Epstein LG, Walsh CA. X-linked female band heterotopia-male lissencephaly syndrome. Neurology. 1998;50:1143–1146. doi: 10.1212/wnl.50.4.1143. [DOI] [PubMed] [Google Scholar]
- 3.Burgess A, Martinez S, Reiner O. KIAA0369, doublecortin-like kinase, is expressed during brain development. J Neurosci Res. 1999;58:567–575. [PubMed] [Google Scholar]
- 4.Burgess HA, Reiner O. Doublecortin-like kinase is associated with microtubules in neuronal growth cones. Mol Cell Neurosci. 2000;16:529–541. doi: 10.1006/mcne.2000.0891. [DOI] [PubMed] [Google Scholar]
- 5.Caspi M, Atlas R, Kantor A, Sapir T, Reiner O . Interactions between LIS1 and doublecortin, two lissencephaly gene products. Hum Mol Genet. 2000;9:2205–2213. doi: 10.1093/oxfordjournals.hmg.a018911. [DOI] [PubMed] [Google Scholar]
- 6.Deng CX, Wynshaw-Boris A, Shen MM, Daugherty C, Ornitz DM, Leder P. Murine FGFR-1 is required for early postimplantation growth and axial organization. Genes Dev. 1994;8:3045–3057. doi: 10.1101/gad.8.24.3045. [DOI] [PubMed] [Google Scholar]
- 7.Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84:911–921. doi: 10.1016/s0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- 8.des Portes V, Pinard JM, Billuart P, Vinet MC, Koulakoff A, Carrie A, Gelot A, Dupuis E, Motte J, Berwald-Netter Y, Catala M, Kahn A, Beldjord C, Chelly J. A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell. 1998;92:51–61. doi: 10.1016/s0092-8674(00)80898-3. [DOI] [PubMed] [Google Scholar]
- 9.Dobyns WB, Truwit CL. Lissencephaly and other malformations of cortical development: 1995 update. Neuropediatrics. 1995;26:132–147. doi: 10.1055/s-2007-979744. [DOI] [PubMed] [Google Scholar]
- 10.Feng G, Mellor RH, Bernstein M, Keller-Peck C, Nguyen QT, Wallace M, Nerbonne JM, Lichtman JW, Sanes JR. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000;28:41–51. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- 11.Fleck MW, Hirotsune S, Gambello MJ, Phillips-Tansey E, Suares G, Mervis RF, Wynshaw-Boris A, McBain CJ. Hippocampal abnormalities and enhanced excitability in a murine model of human lissencephaly. J Neurosci. 2000;20:2439–2450. doi: 10.1523/JNEUROSCI.20-07-02439.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Francis F, Koulakoff A, Boucher D, Chafey P, Schaar B, Vinet MC, Friocourt G, McDonnell N, Reiner O, Kahn A, McConnell SK, Berwald-Netter Y, Denoulet P, Chelly J. Doublecortin is a developmentally regulated microtubule-associated protein expressed in migrating and differentiating neurons. Neuron. 1999;23:247–256. doi: 10.1016/s0896-6273(00)80777-1. [DOI] [PubMed] [Google Scholar]
- 13.Gleeson JG, Allen KM, Fox JW, Lamperti ED, Berkovic S, Scheffer I, Cooper EC, Dobyns WB, Minnerath SR, Ross ME, Walsh CA. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling molecule. Cell. 1998;92:63–72. doi: 10.1016/s0092-8674(00)80899-5. [DOI] [PubMed] [Google Scholar]
- 14.Gleeson JG, Lin PT, Flanagan LA, Walsh CA. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 1999a;23:257–271. doi: 10.1016/s0896-6273(00)80778-3. [DOI] [PubMed] [Google Scholar]
- 15.Gleeson JG, Minnerath SR, Fox JW, Allen KM, Luo RF, Hong SE, Berg MJ, Kuzniecky R, Reitnauer PJ, Borgatti R, Mira AP, Guerrini R, Holmes GL, Rooney CM, Berkovic S, Scheffer I, Cooper EC, Ricci S, Cusmai R, Crawford TO. Characterization of mutations in the gene doublecortin in patients with double cortex syndrome. Ann Neurol. 1999b;45:146–153. doi: 10.1002/1531-8249(199902)45:2<146::aid-ana3>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- 16.Gonczy P, Bellanger JM, Kirkham M, Pozniakowski A, Baumer K, Phillips JB, Hyman AA. zyg-8, a gene required for spindle positioning in C. elegans encodes a doublecortin-related kinase that promotes microtubule assembly. Dev Cell. 2001;1:363–375. doi: 10.1016/s1534-5807(01)00046-6. [DOI] [PubMed] [Google Scholar]
- 17.Gonzalez JL, Russo CJ, Goldowitz D, Sweet HO, Davisson MT, Walsh CA. Birthdate and cell marker analysis of scrambler: a novel mutation affecting cortical development with a reeler-like phenotype. J Neurosci. 1997;17:9204–9211. doi: 10.1523/JNEUROSCI.17-23-09204.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hirotsune S, Fleck MW, Gambello MJ, Bix GJ, Chen A, Clark GD, Ledbetter DH, McBain CJ, Wynshaw-Boris A. Graded reduction of Pafah1b1 (Lis1) activity results in neuronal migration defects and early embryonic lethality. Nat Genet. 1998;19:333–339. doi: 10.1038/1221. [DOI] [PubMed] [Google Scholar]
- 19.Lin PT, Gleeson JG, Corbo JC, Flanagan L, Walsh CA. DCAMKL1 encodes a protein kinase with homology to doublecortin that regulates microtubule polymerization. J Neurosci. 2000;20:9152–9161. doi: 10.1523/JNEUROSCI.20-24-09152.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marin O, Rubenstein JL. A long, remarkable journey: tangential migration in the telencephalon. Nat Rev Neurosci. 2001;2:780–790. doi: 10.1038/35097509. [DOI] [PubMed] [Google Scholar]
- 21.McIlwain KL, Merriweather MY, Yuva-Paylor LA, Paylor R. The use of behavioral test batteries: effects of training history. Physiol Behav. 2001;73:705–717. doi: 10.1016/s0031-9384(01)00528-5. [DOI] [PubMed] [Google Scholar]
- 22.Ohshima T, Ward JM, Huh CG, Longenecker G, Veeranna, Pant HC, Brady RO, Martin LJ, Kulkarni AB. Targeted disruption of the cyclin-dependent kinase 5 gene results in abnormal corticogenesis, neuronal pathology, and perinatal death. Proc Natl Acad Sci USA. 1996;93:11173–11178. doi: 10.1073/pnas.93.20.11173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phillips RG, LeDoux JE. Differential contribution of amygdala and hippocampus to cued and contextual fear conditioning. Behav Neurosci. 1992;106:274–285. doi: 10.1037//0735-7044.106.2.274. [DOI] [PubMed] [Google Scholar]
- 24.Pilz DT, Matsumoto N, Minnerath S, Mills P, Gleeson JG, Allen KM, Walsh CA, Barkovich AJ, Dobyns WB, Ledbetter DH, Ross ME. LIS1 and XLIS (DCX) mutations cause most classical lissencephaly, but different patterns of malformation. Hum Mol Genet. 1998;7:2029–2037. doi: 10.1093/hmg/7.13.2029. [DOI] [PubMed] [Google Scholar]
- 25.Rakic P. Mode of cell migration to the superficial layers of the fetal monkey neocortex. J Comp Neurol. 1972;145:61–84. doi: 10.1002/cne.901450105. [DOI] [PubMed] [Google Scholar]
- 26.Rakic P. Specification of cerebral cortical areas. Science. 1988;241:170–176. doi: 10.1126/science.3291116. [DOI] [PubMed] [Google Scholar]
- 27.Reiner O, Carrozzo R, Shen Y, Wehnert M, Faustinella F, Dobyns WB, Caskey CT, Ledbetter DH. Isolation of a Miller-Dieker lissencephaly gene containing G protein beta-subunit-like repeats. Nature. 1993;364:717–721. doi: 10.1038/364717a0. [DOI] [PubMed] [Google Scholar]
- 28.Reiter LT, Potocki L, Chien S, Gribskov M, Bier E. A systematic analysis of human disease-associated gene sequences in Drosophila melanogaster. Genome Res. 2001;11:1114–1125. doi: 10.1101/gr.169101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sapir T, Elbaum M, Reiner O. Reduction of microtubule catastrophe events by LIS1, platelet-activating factor acetylhydrolase subunit. EMBO J. 1997;16:6977–6984. doi: 10.1093/emboj/16.23.6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmechel DE, Rakic P. Arrested proliferation of radial glial cells during midgestation in rhesus monkey. Nature. 1979;277:303–305. doi: 10.1038/277303a0. [DOI] [PubMed] [Google Scholar]
- 31.Stanfield BB, Cowan WM. The morphology of the hippocampus and dentate gyrus in normal and reeler mice. J Comp Neurol. 1979a;185:393–422. doi: 10.1002/cne.901850302. [DOI] [PubMed] [Google Scholar]
- 32.Stanfield BB, Cowan WM. The development of the hippocampus and dentate gyrus in normal and reeler mice. J Comp Neurol. 1979b;185:423–459. doi: 10.1002/cne.901850303. [DOI] [PubMed] [Google Scholar]
- 33.Taylor KR, Holzer AK, Bazan JF, Walsh CA, Gleeson JG. Patient mutations in doublecortin define a repeated tubulin-binding domain. J Biol Chem. 2000;275:34442–34450. doi: 10.1074/jbc.M007078200. [DOI] [PubMed] [Google Scholar]