

Abstract
Mints/X11s are neuron-specific (Mints 1 and 2) and ubiquitous (Mint 3) adaptor proteins composed of isoform-specific N-terminal sequences and common C-terminal phosphotyrosine-binding (PTB) and PDZ domains. We now show that all three Mints bind to the cytoplasmic tail of amyloid-β precursor protein (APP) and presenilins and strongly increase the levels of cellular APP in transfected cells. Immunocytochemistry revealed that in neurons, Mints 1 and 2 were colocalized with APP in the trans-Golgi network, with lower levels throughout the cell body and neurites. Using an APP-dependent transactivation assay that uses a fusion protein of APP coupled to the potent transcription factor Gal4/VP16, we examined the effects of Mints on the proteolytic processing and putative transcriptional function of APP. Although all Mints were biochemically similar, only Mints 1 and 2 but not Mint 3 strongly inhibited transactivation by APP–Gal4/VP16. Inhibition was enhanced by a mutation of the first PDZ domain and by deletion of the PDZ domains or the N-terminal sequences but abolished by inactivation of the PTB- and PDZ domains. Mint 1 also inhibited transactivation by the “precleaved” cytoplasmic tail of APP fused to Gal4/VP16, whereas Fe65 (which binds to APP as strongly as Mints) enhanced transactivation. Our data suggest that Mints 1 and 2 but not Mint 3 have a specific effect on APP function that cannot be explained simply by their interaction with presenilins and occurs at least partly after cleavage of APP. In view of their biochemical similarity, the functional differences among Mints are unexpected, suggesting that Mints 1 and 2 have a brain-specific function related to APP that is not executed by the ubiquitous Mint 3.
Keywords: APP, Alzheimer's disease, synapse, Mint, X11, Lin-10, Fe65, CASK, Munc18–1
Amyloid-β precursor protein (APP) of Alzheimer's disease is a ubiquitous type 1 membrane protein (Kang et al., 1987; Kitaguchi et al., 1988; Tanzi et al., 1988). APP is physiologically processed by proteolytic cleavage (for review, seeSelkoe, 1998; Bayer et al., 1999; Haass and De Strooper, 1999; Wolfe and Haass, 2001). First, cleavage by α- or β-secretases releases the large extracellular part of APP. Subsequently, the remaining sequences of APP composed of a small extracellular stub, the transmembrane region (TMR), and the cytoplasmic tail are digested by γ-secretase at multiple positions (Sastre et al., 2001; Yu et al., 2001). γ-Cleavage liberates an intracellular cytoplasmic fragment that may be translocated to the nucleus (Cupers et al., 2001; Kimberly et al., 2001) and function as a transcriptional activator (Cao and Südhof, 2001; Gao and Pimplikar, 2001), In addition, γ-cleavage generates small peptides derived from the TMR and adjacent extracellular sequences that include Aβ40 and Aβ42, which form the amyloid fibrils in Alzheimer's disease (Glenner and Wong, 1984;Masters et al., 1985) (for review, see Selkoe, 1998; Haass and De Strooper, 1999).
The short cytoplasmic tail of APP contains an NPTY sequence that binds to phosphotyrosine-binding (PTB) domains in multiple proteins, including Fe65 and Mints/X11s (Fiore et al., 1995; Borg et al., 1996; Guenette et al., 1996; McLoughlin and Miller, 1996; Zhang et al., 1997). Fe65 is an adaptor protein that forms a transcriptionally active complex with the released APP tail and a nuclear histone acetyltransferase, Tip60 (Cao and Südhof, 2001). The genes for Mints 1 and 2 were identified as candidates for Friedreich's ataxia and, on the basis of partial sequences, were thought to be orthologs (Duclos and Koenig, 1995). However, sequencing full-length cDNAs showed that these proteins were products of distinct genes (Okamoto et al., 1997). To prevent confusion among different types of X11s, we called these proteins Mints 1 and 2, and we named a third isoform Mint 3 (Okamoto et al., 1997; Okamoto and Südhof, 1998). Subsequent recloning of the same proteins led to further renaming, and they are now also variably referred to as X11α/β/γ, mLin-10s, X11a/b/c, or X11L1/L2.
Mints/X11s are composed of a long isoform-specific N-terminal sequence, a central PTB domain, and two C-terminal PDZ domains. Mints interact with several other proteins in addition to APP. Mint 1 (but not Mints 2 and 3) binds to CASK (Butz et al., 1998), another adaptor protein (Hata et al., 1996). In Caenorhabditis elegans, CASK and Mint 1 homologs are encoded by the Lin-2 and Lin-10 genes whose mutation causes similar vulvaless phenotypes, suggesting that the Mint 1/CASK complex is evolutionarily conserved (Borg et al., 1998b, 1999; Butz et al., 1998; Kaech et al., 1998;). Mints 1 and 2 also bind to Munc18–1, an essential fusion protein at the synapse (Okamoto et al., 1997; Biederer and Südhof, 2000;Verhage et al., 2000), and to presenilins, which are intrinsic components of the γ-secretase (Lau et al., 2000).
The functions of Mints remain obscure. In C. elegans, Lin-10 (Mint 1) mediates the correct targeting of EGF-like receptors to the basolateral membrane of vulval precursor cells (Whitfield et al., 1999) and is necessary for delivery of AMPA-like glutamate receptors to synapses (Rongo et al., 1998). These data suggest that Lin-10/Mint 1 functions in membrane traffic of proteins to specific plasma membrane domains. In vertebrates, however, various somewhat contradictory functions for Mints have been proposed. Transfection experiments revealed that Mints alter production of Aβ peptides, indicating a role in APP cleavage (Borg et al., 1998a; Sastre et al., 1998; Mueller et al., 2000). In contrast, an interaction of Mint 1 with KIF17in vitro led to the proposal that Mint 1 functions in trafficking neuronal NMDA- but not AMPA-type glutamate receptors in vertebrates (Setou et al., 2000). This study renamed Mint 1 “mLin-10” in analogy to the C. elegans gene but did not reference the previous finding that in nematodes Lin-10 affects only AMPA receptors and not NMDA receptors (Rongo et al., 1998).
In the present study, we compared all three Mint isoforms in the same experiments to test their functional relation to APP. APP was chosen because in vertebrates this appears to be the functionally best validated interaction (Borg et al., 1998a; Sastre et al., 1998), because the relation of APP to Alzheimer's disease makes understanding its biology imperative and because recent insights into a possible transcriptional function of APP have opened new avenues to address this question. Our data reveal that various Mints have similar biochemical properties but different functional effects and that these effects cannot be explained only by regulating APP cleavage.
MATERIALS AND METHODS
Plasmid construction
Gal4-containing transactivation plasmids. Most of the plasmids used for the transactivation experiments described here were reported previously (Cao and Südhof, 2001). All eukaryotic expression vectors containing Gal4 or Gal4/VP16 were based on pMst (Gal4) and pMst-GV (Gal4/VP16), which are derived from the SV40 promoter-based mammalian expression vector pM (Clontech) (Cao and Südhof, 2001). In addition to the previously described vectors, the following vectors were constructed: pMst-GV-APPICF(APPICF-Gal4/VP16), generated by cloning the intracellular fragment of human APP695(APPICF, residues 652–695) into theBamHI–SalI sites of pMst-GV; pMst-GV-APP (APP-Gal4/VP16), by cloning the extracellular and TMR fragments of human APP695 (APPe, residues 1–651) into theNheI site of pMst-GV-APPICF; pMst-GV-APPγ (APPγ-Gal4/VP16), by cloning residues 639–651 of human APP695 preceded by a methionine into theBglII–NheI sites of pMst-GV-APPICF. pMst-GV-APPICF* (APPICF*-Gal4/VP16), pMst-GV-APP* (APP*-Gal4/VP16), and pMst-GV-APPγ* (APPγ*-Gal4/VP16) were generated from their respective parent plasmid by site-specific mutagenesis replacing NPTY(684–687) with NATA(684–687) using the QuikChange kit (Stratagene, La Jolla, CA). pMst-GV-NRX (NRX-Gal4/VP16) was generated by cloning the intracellular fragment of rat Neurexin 1β (NRXICF, residues 414–468) into theBamHI–SalI sites of pMst-GV, followed by cloning the extracellular and TMR fragments (NRXe, residues 1–417) into theNheI site. pMst-GV-NA (NRXe-Gal4/VP16-APPICF) was generated by cloning NRXe into the NheI site of pMst-GV-APPICF, and pMst-GV-AN (APPe-Gal4/VP16-NRXICF) was generated by cloning the intracellular fragment of Neurexin 1β (NRXICF) into theBamHI–SalI sites of pMst-GV, followed by cloning of APPe into the NheI site.
Mint plasmids. The eukaryotic pCMV5 expression vectors for full-length rat Mints 1, 2, and 3 have been described previously (Okamoto and Südhof, 1997, 1998). Mutations were inserted in these parent vectors by site-specific mutagenesis as described above. pCMV-Mint 1-PDZ1* was constructed by mutating GV(670,671) to AA(670,671)in the carboxylate binding loop of the first PDZ domain, and pCMV-Mint 1-PDZ2* was created by mutating GF(762,763) to AA(762,763) in the carboxylate binding loop of the second PDZ domain. pCMV-Mint 1-ΔPDZ was generated by introducing a stop codon after residue 659 in pCMV-Mint 1. A hydrophobic pocket of the PTB domain of Mint 1 was altered at positions YQEF(613–616) to SQES(613–616) to generate the pCMV-Mint 1-PTB* construct. pCMVmyc-Mint 1 was generated by cloning the full-length Mint 1 coding sequence from pEGFP-Mint 1 (a gift from Dr. Anton Maximov, University of Texas Southwestern, Dallas, TX) into theEcoRI–KpnI sites of pCMVmyc. To construct pCMVmyc-Mint 1-ΔNterm, the C-terminal part of Mint 1 encoding residues 451–839 was cloned into the KpnI–XbaI sites of pCMVmyc. pCMV-Mint 2-ΔPDZ was generated by introducing a stop codon after residue 570 in pCMV-Mint 2, and pCMV-Mint 3-ΔPDZ was generated by introducing a stop codon after residue 391 in pCMV-Mint 3.
Other plasmids. The eukaryotic expression vectors for CASK (Hata et al., 1996) and Fe65 (Cao and Südhof, 2001) were described previously.
Antibodies
Most antibodies have been described previously (Biederer and Südhof, 2000, 2001; Cao and Südhof, 2001). The APP antibody used was a polyclonal rabbit serum (U955) raised against the cytosolic, extreme C-terminal 15 residues of APP coupled to keyhole-limpet hemocyanin. Monoclonal antibodies to Mints 1, 2, and 3, TGN 38, and early endosome antigen (EEA1) were from Transduction Laboratories, antibodies to calnexin were from Chemicon, and antibodies to Golgi 58K protein were from Sigma. Polyclonal Mint 1 antibodies have been described previously (P932) (Okamoto and Südhof, 1997), as were antibodies against Velis (T813) (Butz et al., 1998). Polyclonal anti-myc antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA), and polyclonal antibodies directed against Mint 3 were from Affinity Bioreagents (Denver, CO). For all Mint antibodies, specificity was confirmed using preparations from COS cells transfected with expression vectors for Mints 1–3 (see Fig. 1A and data not shown).
Fig. 1.

Similar tight binding of Mints 1, 2, and 3 to the cytoplasmic tail of APP. A, Immunoblot analysis of Mint binding to the immobilized cytoplasmic tail of APP. Lanes 1–4 were loaded with proteins extracted from COS cells transfected with a control vector (lane 1) or Mint 1–3 expression vectors (lanes 2–4) to demonstrate specificity of the antibodies used for the respective Mint isoforms. Proteins from rat forebrain homogenates (lane 5) were bound to an immobilized cytoplasmic peptide derived from APP in the absence of added soluble peptide (lane 6) or in the presence of 0.1 or 1.0 mg/ml of a soluble peptide containing the wild-type sequence of the APP cytoplasmic tail (lanes 7,8), or of this soluble APP tail peptide point-mutated in the NPTY binding sequence for Mints (lane 9). As a further control, binding of Mints to a control column with an immobilized peptide derived from gp41 was examined (lane 10). Binding was performed at 600 mm salt. The samples tested in lanes 6–10 are proteins bound to beads after the respective binding and peptide competition experiments. Immunoblotting was performed using monoclonal antibodies against Mints 1 and 2 and polyclonal antibodies against Mint 3. Fractions were also analyzed by immunoblotting for the negative control proteins Rab GDP-dissociation inhibitor protein (GDI), a soluble protein, and for synaptophysin 1 (Syp), a membrane protein of synaptic vesicles (bottom panels).B, Quantitation of the amount of Mints bound to the immobilized cytoplasmic tail of APP as percentage of Mints in the starting brain extract. Note that all Mints specifically bind only to the immobilized APP but not to the control column and that their binding can be inhibited only by high concentrations of wild-type APP peptide but not by mutant peptide.
Biochemical preparations
All steps were performed on ice or at 4°C. Rat forebrains (Pel Freez, Rogers, AR) were homogenized in a pestle tissue grinder using a slow-speed stirrer at a tissue to buffer ratio of 10% (w/v) in buffer RMP (20 mm HEPES-KOH, pH 7.4, 125 mmK-acetate, 5 mm MgCl2, 320 mm sucrose) adjusted to 1.0% Triton X-100 in the presence of protease inhibitors. For preparation of membrane proteins, rat forebrains were homogenized in buffer RMP, the samples were centrifuged in an Eppendorf microcentrifuge at 600 × g for 10 min to obtain the postnuclear supernatant, and membranes were pelleted in a Sorvall S80-AT3 rotor at 280,000 × g for 20 min. The membrane pellet was extracted in buffer RMP adjusted to 1.0% Triton X-100 using a pestle tissue grinder and centrifuged again at 280,000 × g for 20 min to yield the solubilized membrane proteins.
Peptide bead affinity chromatography
Peptides were synthesized on an ABI synthesizer with an added N-terminal cysteine for coupling to SulfoLink Beads (Pierce, Rockford, IL) according to the manufacturer's instructions at 1.0 mg peptide per 1 ml beads. For binding to the APP NPTY motif, a peptide corresponding to the APP-derived sequence CGYENPTYKFFEQMQN (human APP, residues 398–412) was immobilized on SulfoLink beads. For binding to presenilin C-terminal sequences, peptides corresponding to the extreme C termini of human Presenilin 1 (sequence CMDQLAFHQFYI), human Presenilin 2 (sequence CMDTLASHQLYI), or DrosophilaPresenilin (sequence CMEDLSAKQVFI) were immobilized. As negative controls, peptides corresponding to the extreme C terminus of human HPV2 (poliovirus receptor-related protein 2; sequence CGSLISRRAVYV) and a peptide derived from the gp41 glycoprotein (sequence CWFSITNWLWYI) were used. Extracts of transfected eukaryotic cells or proteins solubilized from rat brain were incubated with 20 μg peptide immobilized on SulfoLink beads for 12–16 hr at 4°C under mild agitation. Binding was performed in buffer RMP adjusted to 1.0% Triton X-100 and 600 mm potassium acetate. For APP competition experiments, the soluble peptide QNGYENPTYKFFEQ or QNGYENATAKFFEQ, corresponding to the native or mutated APP NPTY motif, was added during the binding incubation at the concentrations of 0.1 mg/ml and 1.0 mg/ml. Bound proteins were eluted with 2% SDS.
Immunoprecipitations
Human embryonic kidney (HEK) 293 cells were cotransfected for APP and the individual Mints 1, 2, or 3, respectively, and after 2 d they were collected in IP buffer [25 mm HEPES-KOH, pH 7.4, 125 mm K-acetate, 5 mmMgCl2, 1.0% IGEPAL CA-630 (Sigma), 10% glycerol] in the presence of protease inhibitors. After the cell suspension was passed through a 28 gauge syringe, the lysate was centrifuged in an Eppendorf microcentrifuge at 21,000 ×g for 20 min, and the detergent-extracted material was subjected for 2 hr to immunoprecipitation using antibodies directed against APP (U955) or the respective preimmune serum.
Miscellaneous biochemical procedures
SDS-PAGE and immunoblotting were performed as described (Laemmli, 1970; Towbin et al., 1979). For standard immunodetection on Western blots, enhanced chemiluminescence (ECL; Amersham) was applied. Quantitative immunoblotting was performed using radiolabeled 125I secondary antibodies (Amersham), and the signals were quantitated on a PhosphorImager (Molecular Dynamics) using ImageQuant software. To determine levels of expressed Mint 1 protein, signals of cell lysates were compared with those from known amounts of purified glutathioneS-transferase–Mint 1. Protein concentrations were determined using the BCA protein assay (Pierce).
Immunocytochemistry
Adult mice were perfusion-fixed in 4% paraformaldehyde in PBS, pH 7.4. Vibratome sections (35 μm) from brain were blocked for 1 hr in 10% normal goat serum containing 0.1% Triton X-100 and incubated with the various primary antibodies overnight at 4°C and with the biotinylated secondary antibody for 1 hr. Sections were processed using a VectaStain ABC Elite Kit (Vector, Burlingame, CA) according to the manufacturer's instruction. The final immunosignal was developed using 3′3-diaminobenzidine tetrahydrochloride. For immunofluorescence labeling, primary hippocampal cells on coverslips were fixed in situ for 10 min with absolute methanol at −20°C, permeabilized in 0.1% saponin/PBS, and blocked in 3% milk/PBS. The primary incubation was performed in blocking buffer for 1 hr at room temperature. After washing with PBS, the cells were incubated with goat anti-rabbit or goat anti-mouse secondary antibodies that were coupled with Alexa Fluor 488 and Alexa Fluor 546 (Molecular Probes). Labeled cells were viewed with a Leica TCS SP2 confocal microscope or a Zeissfluorescent microscope with a Hamamatsu ORCA-100 digital camera. Final images were processed by MetaMorph (Universal Imaging) and Adobe Photoshop.
Transactivation assays
PC12, COS, HeLa, and HEK293 cells were cotransfected with three or four plasmids: (1) pG5E1B-luc (HEK293 cells, HeLa cells, and COS cells = 0.2–0.5 μg DNA; PC12 cells = 1.0 μg); (2) pCMV-LacZ (HEK293 cells, HeLa cells, and COS cells = 0.05 μg DNA; PC12 cells = 0.5 μg DNA); (3) pMst (Gal4), pMst-GV-APP (APP-GV), pMst-GV (GV), pMst-GV-APPICF(APPICF-GV), pMst-APPICF(APPICF-Gal4), pMst-GV-APP* (APP*-GV), pMst-GV-APPICF* (APPICF*-GV), pMst-APPICF* (APPICF*-Gal4), pMst-GV-APPγ (APPγ-GV), pMst-GV-NRX (NRX-GV), pMst-GV-NA (NRXe-GV-APPc), pMst-GV-AN (APPe-GV-NRXc)(HEK293, HeLa, and COS cells = 0.1–0.3 μg DNA; PC12 cells = 1.0 μg DNA). Where indicated, a fourth plasmid was cotransfected: pcDNA3.1-PS2D366A (kind gift of Dr. C. Haass, Ludwig-Maximilians-Universität, Munich); pCMV-Mint1; pCMV-Mint 1-PDZ1*; pCMV-Mint 1-PDZ2*; pCMV-Mint 1-PTB*; pCMVmyc-Mint 1; pCMVmyc-Mint 1-ΔNterm; pCMV-Mint 1-ΔPDZ; pCMV-Mint 2; pCMV-Mint 2-ΔPDZ; pCMV-Mint 3; pCMV-Mint 3-ΔPDZ; pCMV5-Fe65; pCMV-CASK (HEK293, HeLa, and COS cells = 0.1–0.3 μg DNA or as described in the Figure Legends; PC12 cells = 0.5 μg DNA). For negative controls, the expression vector pCMV5 was used without insert. Cells were harvested 48 hr after transfection in 0.2 ml per well reporter lysis buffer (Promega), and their luciferase and β-galactosidase activities were determined with the Promegaluciferase assay kit using a chemiluminescence reader (Lucy2, Anthos Labtec, Salzburg, Austria) and the standardO-nitrophenyl-d-galacto-pyranoside (Sigma) method, respectively. The luciferase activity was standardized by the β-galactosidase activity to control for transfection efficiency and general effects on transcription and further normalized for the transactivation observed in cells expressing Gal4 alone where indicated. Values shown are averages of transactivation assays performed in duplicate and repeated at least three times for each cell type and constructs. All constructs were assayed in three or four cell lines, but usually only representative results for one cell line are shown.
Transfections were performed at 50–80% confluency in six-well plates using Fugene6 (Roche).
RESULTS
Comparison of the binding of Mints/X11 1, 2, and 3 to APP
Previous results have separately examined the ability of Mints to bind to APP or presenilins and to stabilize APP in cotransfected cultured cells (Borg et al., 1996, 1998; McLoughlin and Miller, 1996;Zhang et al., 1997; Sastre et al., 1998; Lau et al., 2000; Okamoto et al., 2001). These experiments established a potentially important connection between Mints, presenilins, and APP, but the relative activities of the three Mints and the generality of these putative targets were not analyzed. As an initial step toward understanding the common versus unique properties of Mint isoforms, we compared the binding of different Mints to APP and presenilin and their effect on APP in transfected cells. Because these and subsequent experiments critically depended on the specificity of the Mint antibodies used, we first validated the specificity of these antibodies using transfected COS cells that express individual Mints. Immunoblotting confirmed the monospecificity of the antibodies (Fig.1A, lanes 1–4), which could thus be applied to detect each Mint isoform separately in complex solutions containing multiple Mints, such as brain homogenates.
We then used affinity chromatography of rat brain proteins on the immobilized cytoplasmic tail of APP to examine the binding of endogenous Mints to APP. Proteins bound to a peptide corresponding to the cytoplasmic C-terminal tail of APP were analyzed by immunoblotting. Recovery of the three Mints from the brain homogenates was quantitated with 125I-labeled secondary antibodies (Fig. 1A, lanes 5–10). All three Mints tightly bound to APP (lane 6), and binding was inhibited by high concentrations of the corresponding wild-type but not mutant APP tail peptide (Fig. 1A, lanes 7–9). Quantitation revealed that Mints from the brain homogenate were recovered efficiently on the affinity column with yields of 25–90% (Fig. 1B); for example, the immobilized APP tail extracted almost all of the brain Mint 3, and even 1 mg/ml of competing peptide was unable to completely block it from binding to the column. In contrast, only 25% of brain Mint 1 was bound. No Mint binding to a control peptide was observed (Fig. 1A,lane 10).
To test whether Mints interact with APP in vivo, we measured the levels of transfected APP by quantitative immunoblotting using125I-labeled secondary antibodies (Fig.2A,B). As a control, we cotransfected Fe65, which also binds to the cytoplasmic tail of APP (Fiore et al., 1995). Cotransfection of all three Mints dramatically increased APP levels (up to threefold) (Fig.2, lanes 3–5), extending previous observations that Mint 1 stabilizes transfected APP and increases its steady-state levels (Borg et al., 1998a; Sastre et al., 1998). As a control, Fe65 (which also binds to APP) slightly decreased the steady-state levels of transfected APP, although this was not necessarily a specific effect as cotransfection of any protein usually decreases expression because it dilutes the transcription/translation machinery. The stabilization of APP in the transfected cells could be attributable to a direct or indirect interaction of Mints with APP. Although the affinity chromatography experiments already suggested a direct interaction (Fig. 1), we examined this question further by testing whether Mints were coimmunoprecipitated with APP from the transfected cells (Fig. 2C). Indeed, Mints 1, 2, and 3 were coimmunoprecipitated with antibodies to APP, but not with control antibodies (Fig. 2C), supporting the notion that Mints directly bind to APP.
Fig. 2.

Cotransfection of Mints 1, 2, or 3 increases the steady-state levels of APP. A, Immunoblot analysis of HEK293 cells cotransfected with expression plasmids encoding APP695, Mints 1–3, and Fe65 as indicated. Under control conditions (lane 1), little APP695 is expressed in the cells because the predominant splice variants in peripheral tissues include the Kunitz domain (Kitaguchi et al., 1988; Tanzi et al., 1988). The migration position of the endogenous APP containing the Kunitz domain and the transfected APP695 lacking this domain are indicated on theright. The quantities of loaded lysates were normalized to equal amounts of transfected cells based on the activity of cotransfected β-galactosidase. B, Quantitation of the levels of APP695 in transfected HEK293 cells shown in A. Note that Fe65 slightly decreases APP695 levels, whereas all Mints increase it several fold. The levels of endogenous APP do not change significantly because most of the cells are not transfected and thus are not exposed to the transfected Mints. C, Immunoprecipitation assay of Mint binding to APP. APP695 and Mints 1, 2, or 3 were coexpressed in transfected HEK293 cells (Start, lane 1). Cellular proteins were then immunoprecipitated with an antibody to the C terminus of APP (IP APP, lane 2) or a control antibody (IP Control, lane 3). Samples were analyzed by immunoblotting with monoclonal antibodies to the indicated proteins. The double band for Mint 2 is probably caused by a hypersensitive proteolytic site in the N terminus of Mint 2. For detection of APP, the quantity loaded in lanes 2 and3 was fivefold less than for detection of Mints.
Binding of Mints to presenilins
We next examined the potential interaction of Mints with presenilins using the same affinity chromatography approach used for APP binding. Immunoblotting showed that all three Mints bound to the cytoplasmic C-terminal sequence of presenilin 1 in agreement with previous observations (Fig.3A) (Lau et al., 2000). Binding was specific because Mints were not bound to control beads, and control proteins such as dissociation inhibitor protein (GDI) and synaptophysin were not retained on the presenilin 1 column. However, quantitations revealed that presenilin binding differed among Mints (Fig. 3B) (and data not shown). Of endogenous Mints 1 and 3 from brain, 14–16% were recovered on the presenilin column, but only 3% of Mint 2 was bound. Mints bind to the cytoplasmic tail of APP via an interaction of the Mint PTB domain with the NPTY sequence in APP (Borg et al., 1996; McLoughlin and Miller, 1996; Zhang et al., 1997). Presenilin 1 does not include an NPxY sequence but features a C-terminal sequence that resembles that of neurexins, which bind to the PDZ domains of Mints (Biederer and Südhof, 2000), suggesting that the binding of Mints to presenilin 1 may be mediated by one or both Mint PDZ domains. To test whether other PDZ domain proteins also bind to the C-terminal sequence of presenilin 1, we examined the presenilin binding of PSD-95, an abundant component of the postsynaptic density that contains three PDZ domains (Cho et al., 1992; Kistner et al., 1993), and of Velis, a family of three proteins that contain a single PDZ domain (Butz et al., 1998). Both PDZ domain proteins were bound; although binding of PSD-95 was weak, Velis were captured to the same extent as Mints 1 and 3 (Fig.3A,B).
Fig. 3.

Binding of both PDZ domains of Mints to the cytoplasmic C-terminal sequence of presenilins. A, Proteins solubilized from a rat forebrain membrane preparation (Start, lane 1) were bound to an affinity column containing a peptide corresponding to the C terminus of presenilin 1 (Pres1, lane 2) or a control peptide derived from gp41 (Control, lane 3). Bound proteins were analyzed by immunoblotting with antibodies to the indicated proteins, which included the negative control proteins GDI and synaptophysin (Syp). Note that the antibody to Velis that was used (Butz et al., 1998) recognizes all three Veli isoforms, which migrate as two distinct bands.B, Percentage of Mints solubilized from a rat forebrain membrane preparation that bound to an affinity column containing a peptide corresponding to the C terminus of presenilin 1 or a control peptide derived from gp41 (n = 3).C, Binding of Mint 1 solubilized from a rat forebrain preparation to the immobilized C-terminal sequences of human presenilins 1 and 2, Drosophila presenilin, and control peptides derived from HPV2 and gp41 (n = 3).D, Binding of various Mint 1 mutants prepared from transfected COS cells to the immobilized cytoplasmic C-terminal sequence of presenilin 1. Mint 1 mutants containing inactivating point mutations in either the first (Mint 1 PDZ1*), the second (Mint 1 PDZ2*), or both PDZ domains (Mint 1 PDZ1*/2*), or a truncation mutant of Mint 1 lacking the two PDZ domains (Mint 1ΔPDZ) were analyzed by affinity chromatography on immobilized presenilin 1 or gp41 control peptides.
The fact that multiple unrelated PDZ domain proteins bind to the C-terminal sequence of presenilin 1 but various Mints exhibit large differences in binding raises concerns about the specificity of the interaction of Mints with presenilins, prompting us to examine it further. The C-terminal presenilin 1 sequence is conserved in vertebrate presenilin 2 and in Drosophila presenilin. C-terminal peptides from all of these presenilins captured Mint 1, whereas control peptides did not, suggesting that all of these presenilins potentially interact with PDZ domain proteins (Fig.3C). To identify which of the two PDZ domains in Mints binds to presenilins, we mutated the PDZ1 and PDZ2 domains of Mint 1 separately or together, or deleted them both. Presenilin binding assays with wild-type and mutant Mint proteins produced in transfected COS cells revealed that mutations in each of the two Mint 1 PDZ domains resulted in an approximately twofold decrease in binding (Fig.3D). Mutations in both PDZ domains or deletion of both PDZ domains almost completely abolished binding. Together these data suggest that each of the two Mint PDZ domains individually binds to presenilins in vitro.
Localization of endogenous Mints in neurons
In vertebrates, Mints 1 and 2 are detectable only in brain, whereas Mint 3 is expressed ubiquitously (Okamoto et al., 1997; Okamoto and Südhof, 1998). To study the localization of Mints, we stained rat brain sections with antibodies to Mints 1 and 2. Mint 3 could not be investigated because the available antibodies were not suitable for immunocytochemistry (data not shown). Abundant labeling of the neuronal cell bodies, with less staining of the dendrites, was observed in the hippocampus (Fig.4a,b,d,e) [see also McLoughlin et al. (1999)]. Mint 1 was particularly strongly expressed in interneurons. APP exhibited a very similar distribution to Mints 1 and 2 (Fig. 4c,f). Notably, nuclei were not labeled. Staining throughout the neuropil was detected that was weaker than the cell body staining, indicating that low levels of Mints may be present at synapses as suggested by Okamoto et al. (2000).
Fig. 4.
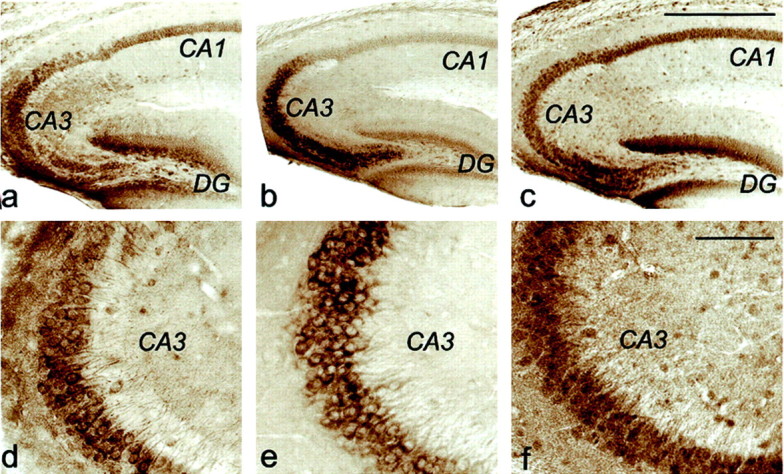
Immunoreactivity of Mint 1, Mint 2, and APP in mouse hippocampus. Vibratome sections (35 μm) from adult mouse hippocampus were stained for Mint 1 (a,d), Mint 2 (b, e), and APP (c, f) using the avidin–biotin peroxidase method. d, e, andf are enlarged images of CA3 regions froma, b, and c and show that the immunostaining is clearly present in both cell soma and proximal apical dendrites. Antibodies against Mint 1 were polyclonal and monoclonal against Mint 2. The polyclonal APP antibody that was used is directed against a C-terminal epitope in the cytoplasmic APP tail that is liberated after γ-cleavage. Scale bars (shown in cfor a–c): 0.5 mm; (shown inf for d–f): 0.1 mm.
To examine the subcellular distribution of Mints, we performed immunofluorescence staining of cultured hippocampal neurons (Figs. 5,6). Mints 1 and 2 were predominantly localized in a perinuclear compartment where they overlapped almost completely (Fig. 5a–c). Double labeling with antibodies to synapsins as a presynaptic marker failed to detect high levels of Mints in synapses (Fig. 5d–f) (and data not shown). APP was mostly colocalized with Mints (Fig. 5g–l). Compared with Mints, more APP appeared to be present in neurites, suggesting that at steady state, Mints are more concentrated in the perinuclear compartments than APP.
Fig. 5.

Immunofluorescence localization of Mints 1 and 2 in cultured hippocampal neurons. The left(red) and center(green) pictures show the separate fluorescence channels from double-immunofluorescence labeling experiments, whereas the right pictures show the merged images. Neurons were labeled with the following antibodies:a–c, antibodies to Mint 1 (a) and Mint 2 (b). Note that the pictures show a low-magnification overview with a high-magnification inset.d–f, antibodies to synapsins (d) and Mint 2 (e).g–i, Antibodies to APP (g) and Mint 1 (h).j–l, Antibodies to APP (j) and Mint 2 (k). Scale bars (shown in a for low-magnification views ind–l): 50 μm; (shown in l forinsets in a–c and the full images in a–l): 20 μm.
Fig. 6.
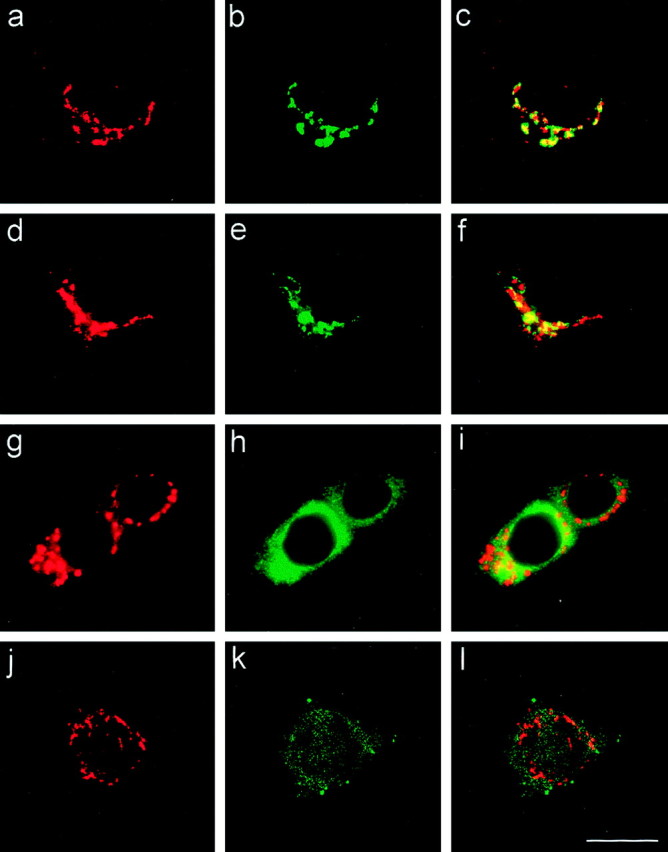
Mints 1 and 2 are concentrated in thetrans-Golgi complex. Subcellular distribution of Mints has been investigated further with a group of well characterized markers to Golgi apparatus, endoplasmic reticulum, and early endosome. Cultured hippocampal neurons were double labeled with antibodies to the following proteins: a–c, Mint 2 (a), TGN38 (b), and merged images (c); d–f, Mint 1 (d), the Golgi 58K protein (e), and merged images (f);g–i, Mint 2 (g), calnexin (h), and merged images (i); j–l, Mint 2 (j), EEA1 (k), and merged images (l). Scale bar (shown in lfor a–l): 15 μm.
In the next set of experiments, we double labeled cultured hippocampal neurons with antibodies to marker proteins to identify the perinuclear compartment containing Mints (Fig. 6). We found that Mints colocalize best with TGN38 (Fig. 6a–c) (and data not shown), a marker of the trans-Golgi complex (Luzio et al., 1990). A similar localization, but not quite as precise, was observed with the 58K Golgi protein (Fig. 6d–f) (and data not shown), a peripheral membrane protein that is enriched in, but also found outside of, the trans-Golgi complex (Bloom and Brashear, 1989). By contrast, double labeling of neurons with antibodies to Mint 1 or 2 and the endoplasmic reticulum protein calnexin (Wada et al., 1991) failed to detect an overlap in localization (Fig. 6g–i). Similarly, the early endosomal protein EEA1 (Mu et al., 1995) exhibited a different staining pattern (Fig. 6j–l). Together these data support the conclusion that in mature neurons in situ (Fig.4) and in culture (Figs. 5, 6), Mints 1 and 2 are colocalized in thetrans-Golgi complex, a localization consistent with a role in APP trafficking (Borg et al., 1998a; Sastre et al., 1998) and in directing proteins out of the trans-Golgi apparatus toward defined plasma membrane domains (Rongo et al., 1998; Whitfield et al., 1999). Thus at steady state, Mints exhibit a localization that is similar to that of APP but notably distinct from that of either CASK or Munc18–1.
A transactivation assay of APP cleavage
We recently described a function for the cytoplasmic tail of APP in activating transcription by forming a protein complex with Fe65 (Cao and Südhof, 2001). The transcriptional activation observed in these assays could potentially be used to measure APP cleavage but depends on Fe65, which binds to the same NPTY sequence of APP as Mints [(which, however, do not activate transcription (Cao and Südhof, 2001)]. To test the potential function of Mints in APP cleavage and signaling, we used a variant of our assay that allows monitoring of APP cleavage independent of Fe65 binding. For this purpose we introduced both Gal4 and VP16 into the cytoplasmic tail of APP695 at the cytoplasmic boundary of the TMR. This assay differs from the original assay (Cao and Südhof, 2001) in that the powerful viral transcriptional activator VP16 (Sadowski et al., 1988) is introduced together with the yeast DNA binding protein Gal4 into APP. Thus transactivation by APP-Gal4/VP16 is independent of the binding of cellular transcriptional activators but can occur only after APP is cleaved by γ-secretase and the released cytoplasmic tail fragment moves into the nucleus. We transfected the APP-Gal4/VP16 fusion protein into PC12, HEK293, COS, or HeLa cells and measured transactivation of transcription from a cotransfected Gal4-dependent reporter plasmid encoding luciferase. As a negative control, we used Gal4 alone without VP16 or APP (Cao and Südhof, 2001), and as a positive control, we used Gal4/VP16 without APP. In all experiments, cells were cotransfected with a constitutive β-galactosidase expression vector to control for transfection efficiency and to rule out direct effects of transfected proteins on transcription. Furthermore, in all cases, expression of transfected proteins was verified by immunoblotting.
In all cell types tested, full-length APP-Gal4/VP16 (APP-GV) transactivated Gal4-dependent transcription almost as strongly as Gal4/VP16 alone (∼500- to 2000-fold activation over Gal4 alone, depending on cell type), suggesting that cleavage of APP to release the intracellular fragment was not the major rate-limiting step (Fig.7, columns 1-3). A chimeric protein in which Gal4/VP16 was fused to the isolated cytoplasmic tail of APP (APPICF-GV) was an even more potent transactivator than Gal4/VP16 alone or full-length APP-Gal4/VP16 (∼4000 vs ∼500- to 2000-fold activation) (Fig. 7,column 5). Addition of the 12 hydrophobic residues from the TMR that are present in the initial γ-cleavage product had no inhibitory effect on transactivation but induced a moderate stimulation (Fig. 7, column 6). In contrast, the cytoplasmic APP tail containing only Gal4 without VP16 (APPICF-Gal4) was inactive when Fe65 was not cotransfected (less than fivefold activation) (Fig. 7, column 7) (Cao and Südhof, 2001). Cleavage of APP does not appear to require binding of endogenous cellular proteins to the NPTY tail sequence (residues 684–687 of APP695) because mutation of NPTY to NATA had no effect on transactivation. Specifically, no effect of this mutation was observed with either full-length APP or the cytoplasmic APP tail fused to Gal4/VP16 (Fig. 7,columns 4 and 7).
Fig. 7.
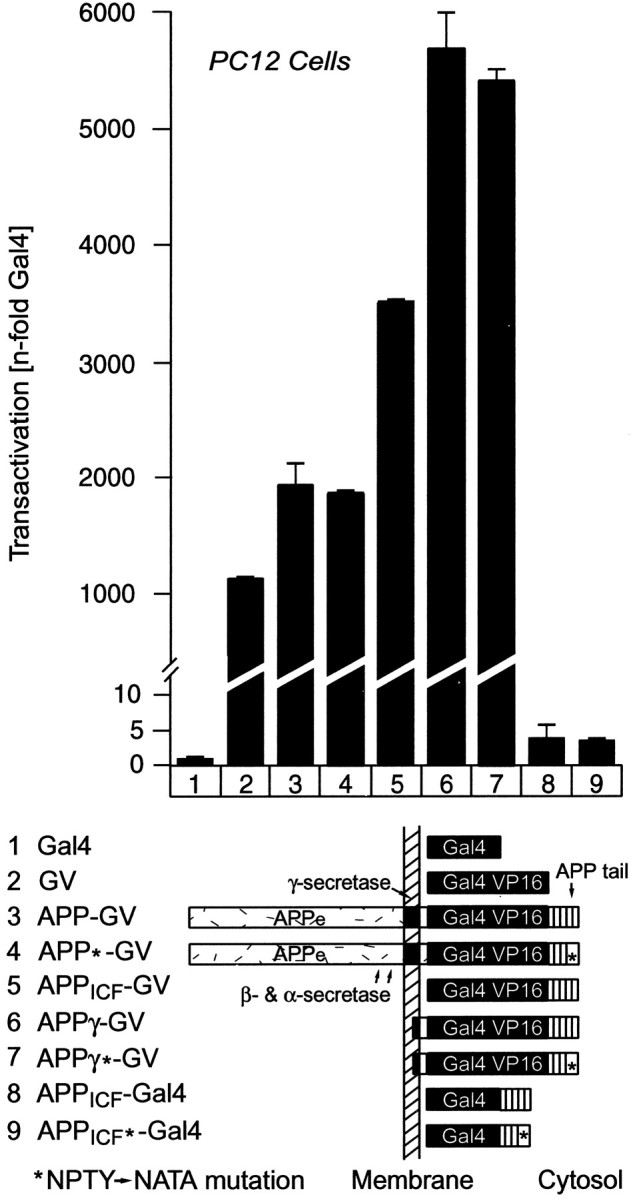
Use of APP-Gal4/VP16 fusion proteins to measure γ-cleavage of APP. The bar diagram ontop shows results from Gal4-transactivation assays in PC12 cells that were cotransfected with a Gal4-dependent luciferase reporter plasmid (to measure transactivation using luciferase expression), a β-galactosidase control plasmid (to normalize for transfection efficiency), and the test plasmids identified by numbersbelow the bars. The domain structures of the proteins encoded by the test plasmids are shown schematically below thebar diagram (APPe, extracellular sequences of APP; APPICF, cytoplasmic sequences of APP). Constructs marked with anasterisk (APP*-GV,APPICF*-GV, andAPPICF*-Gal4) contain a point mutation in which the NPTY sequence in the cytoplasmic tail of APP is replaced by NATA. Transfected cells were harvested 2 d after transfection, luciferase, and β-galactosidase activities were determined, and the luciferase activity was normalized for the β-galactosidase activity to control for transfection efficiency as described in Materials and Methods. The β-galactosidase-normalized luciferase activity is expressed in relation to the activity of cells cotransfected with Gal4 alone. Data shown are from a representative experiment repeated multiple times in PC12 cells and in COS, HEK293, and HeLa cells with similar results. GV, Gal4/VP16 module;APPICF, intracellular fragment of APP;APPγ, γ-secretase cleavage product of APP.
APP sequences required for cleavage
The assay described above uses transfected cells that overexpress the respective test proteins, raising the concern that transactivation by APP-Gal4/VP16 may be caused by nonspecific proteolysis of the APP-Gal4/VP16 fusion protein instead of specific α-/β- and γ-cleavage. To address this concern, we tested whether specific sequences of APP were required for transactivation by the embedded Gal4/VP16 module (Fig. 8).
Fig. 8.

Sequence requirements of APP γ-cleavage measured by Gal4/VP16-dependent transactivation. The bar diagrams on top show results of Gal4/VP16 transactivation assays obtained with the constructs that are schematically displayed and identified by numbers below the diagrams.A, Requirements of extracellular and intracellular APP sequences for transactivation. APP-Gal4/VP16 proteins that contain all APP sequences (construct 3) or lack the intracellular (construct 4) or extracellular and transmembrane sequences (construct 5) were analyzed as described in the legend to Figure 7. Gal4 (construct 1) and Gal4/VP16 (construct 2) were used as standardization controls to establish the background and maximal response, respectively.B, Test of the specific functions of extracellular versus intracellular APP sequences in γ-cleavage and of the effect of a dominant negative mutant of presenilin 2 (PS2DA) (Steiner et al., 1999). Transactivation was measured by constructs in which Gal4/VP16 is placed in all possible combinations into the context of the extracellular and intracellular sequences of APP (APPeandAPPICF= extracellular and cytoplasmic sequences of APP, respectively) or neurexin 1β (NRXeandNRXICF= extracellular and cytoplasmic sequences of neurexin, respectively). Gal4/VP16 constructs were transfected without (−) and with (+) the dominant negative presenilin 2 expression vector. All bar diagrams exhibit representative experiments in the cell types identified on thetop. Experiments were performed with test plasmids cotransfected with a Gal4 luciferase reporter plasmid and a β-galactosidase control plasmid as described in the legend to Figure7. In A, transactivation as measured by β-galactosidase-normalized luciferase activity is expressed relative to the activity of Gal4 alone, whereas in B, transactivation as measured by luciferase activity is shown in arbitrary units only normalized to β-galactosidase activities.
We first removed the cytoplasmic tail of APP and generated a “tailless” APP-Gal4/VP16 fusion protein in which the extracellular sequences and the TMR of APP were linked to intracellular Gal4/VP16 followed by a stop codon (APPe-GV). The tailless APP-Gal4/VP16 protein was almost completely inactive in transactivation assays compared with Gal4/VP16 alone or APP-Gal4/VP16 or APPγ-GV, which represents the initial γ-secretase cleavage product (Fig.8A, columns 2–5). This suggests that Gal4/VP16 is not released from APP-Gal4/VP16 by nonspecific degradation and that the tail of APP is required either for recognition by the APP cleavage enzymes or for trafficking of APP to the cleavage compartments.
To differentiate between these two possibilities, we inserted Gal4/VP16 into the cytoplasmic tail of neurexin 1β (NRX-GV). Neurexin 1β is expressed on the neuronal cell surface similar to APP but is not known to be processed by proteolytic cleavage (Ushkaryov et al., 1992). Neurexin 1β-Gal4/VP16 was nearly inactive in transactivation assays in contrast to APP-Gal4/VP16 (Fig. 8B, column 3 vs 6), suggesting that Gal4/VP16-mediated transactivation requires specific sequences in the APP molecule. To identify these sequences, we constructed chimeric Gal4/VP16-fusion proteins containing either extracellular APP sequences with the intracellular neurexin 1β region, or extracellular neurexin 1β sequences with the intracellular APP region. A fusion protein composed of the extracellular sequences and TMR of APP coupled to intracellular Gal4/VP16 and the cytoplasmic tail of neurexin 1 (APPe-GV-NRXICF) strongly transactivated transcription (Fig. 8B, column 8). In contrast, the reverse fusion protein of the extracellular neurexin sequences and the neurexin TMR with the cytoplasmic APP sequences (NRXe-GV-APPICF) was inactive (Fig. 8B, column 7). These experiments demonstrate that the extracellular sequences of APP are essential for proper cleavage, in agreement with results of Struhl and Adachi (2000). Although the intracellular sequences of APP are also essential for its processing (see tailless mutantAPPeGV, Fig. 8), they can be functionally replaced by the intracellular fragment of a plasma membrane protein like neurexin that exhibits no sequence similarity with APP, and in particular does not contain an NPxY sequence (Ushkaryov et al., 1992). This may indicate a role of the APP tail in trafficking to a cleavage compartment at the plasma membrane or derived from the plasma membrane. However, it should be noted that because APP is fused to a DNA-binding domain (Gal4) and a transactivation domain (VP16) in the present assay, this assay does not allow conclusions about the physiological function of the NPxY sequence in the transactivation process mediated by APP as described previously (Cao and Südhof, 2001).
Finally, to test whether presenilins are involved in transactivation by APP-Gal4/VP16, we cotransfected a dominant negative mutant of presenilin 2 (Steiner et al., 1999) with APP-Gal4/VP16 (Fig.8B). Transactivation of Gal4-dependent transcription by full-length APP-Gal4/VP16 was inhibited by the presenilin 2 mutant, whereas the small amount of residual transactivation observed with neurexin 1β-Gal4/VP16 was insensitive to presenilin 2 (Fig.8B, column 3 vs 6). As expected, mutant presenilin 2 also potently inhibited transactivation by the fusion protein of the extracellular domain of APP with intracellular neurexin sequences (Fig. 8B,column 8) but had no effect on the residual transactivation observed with the reverse fusion protein containing the extracellular domain of neurexin 1β coupled to the cytoplasmic tail of APP (Fig.8B, column 7).
Mint 1 inhibits transactivation mediated by APP-Gal4/VP16
We used the transactivation assay to study the effects of Mints on APP. Cotransfection of Mint 1 strongly inhibited transactivation mediated by APP-Gal4/VP16 (Fig.9A). This inhibition was abolished by mutation of the NPTY sequence in the cytoplasmic tail of APP-Gal4/VP16, consistent with the notion that direct binding of Mint 1 to APP is required. When we compared the activity of the three Mint isoforms in the transactivation assay, Mints 1 and 2 potently inhibited transactivation by APP-Gal4/VP16, whereas Mint 3 had no significant effect (Fig. 9B). Quantitative immunoblotting confirmed that all three Mints were expressed at high levels in the cotransfected cells (Fig. 9C).
Fig. 9.

Mints 1 and 2 but not Mint 3 inhibit transactivation by APP-Gal4/VP16 fusion proteins. A, Dose-dependent inhibition by Mint 1 of wild-type APP-Gal4/VP16 transactivation but not of APP-Gal4/VP16 carrying a point mutation in the cytoplasmic NPTY binding sequence for Mints (APP*-GV). A constant amount of wild-type or mutant APP-Gal4/VP16 plasmid (100 ng DNA per well) was cotransfected with the indicated amounts of Mint 1 expression vector into HEK293 cells. Transactivation and Mints levels in the cells were quantified in the same samples as described in Materials and Methods.B, Effects of Mints 1, 2, and 3 on APP-Gal4/VP16-dependent transactivation. Mint amounts in transactivation assay samples were quantified on immunoblots and are expressed as a fraction of the amount observed with the maximal amount of DNA transfected to control for the nonlinearity of the relation between transfected DNA and expressed protein. C, Quantitated immunoblot analysis of the Mints expressed in the experiment shown in B.
Because all three Mints bind to APP in vitro and in transfected cells (Figs. 1, 2), the selective inability of Mint 3 to inhibit transactivation was surprising. To gain insight into the mechanism by which Mint 1 inhibits APP-Gal4/VP16-mediated transactivation and to understand why Mint 3 has no effect, we examined a series of Mint mutants. We found that the inactivating point mutation in the first PDZ domain of Mint 1 or deletion of both PDZ domains that were studied above in the presenilin-binding experiments (Fig. 3) dramatically increased the inhibition of transactivation by Mint 1 (Fig. 10A). In contrast, the second PDZ domain point mutation did not alter the inhibitory effect of Mint 1. Because the various Mint 1 mutants exhibited different expression levels, the relative amounts of expressed protein were quantified using125I-labeled secondary antibodies. These quantitations showed that the PDZ domain deletion mutant was ∼10 times more potent than wild-type Mint 1 (Fig. 10A). We also tested a Mint 3 mutant with a PDZ domain deletion, which was completely inactive in the assay, similar to wild-type Mint 3 (Fig.10B,C). Control transfections showed that the various Mint 1 proteins did not inhibit general transcription but specifically impaired transactivation by APP-Gal4/VP16 (data not shown).
Fig. 10.

Structure–function analysis of Mint 1: role of PDZ domains in inhibiting APP-Gal4/VP16-dependent transactivation.A, HEK293 cells were cotransfected with a constant amount of APP-Gal4/VP16 expression vector and reporter plasmids and increasing amounts of expression vectors expressing wild-type Mint 1 or mutants of Mint 1 carrying point mutations in the first or second PDZ domains (Mint 1 PDZ1* or PDZ2*, respectively) or lacking both C-terminal PDZ domains (Mint 1 ΔPDZ). Transactivation and Mint 1 amounts in the cells were then quantified in the same samples as described in Materials and Methods. B, Transactivation observed in HEK293 cells cotransfected with APP-Gal4/VP16 and a control plasmid, or wild-type Mint 1, a Mint 1 mutant lacking the PDZ domains, wild-type Mint 3, or a Mint 3 mutant lacking both PDZ domains. All transactivation levels in A and B are normalized for the amount of transactivation observed under control conditions. C, Immunoblot analysis of the Mint mutants analyzed in B to control expression of the constructs in the experiments shown.
The results of Figure 10 suggest that Mint 1 binding to APP may couple it to another protein that binds to the first PDZ domain, implying that the two PDZ domains of Mint 1 are not equivalent. To examine this further, we measured the effect of mutating the PTB domain of Mint 1 on its inhibitory activity. On the basis of available structural information (Zhang et al., 1997), a mutant Mint 1 PTB* was designed in which critical residues involved in binding to the NPTY sequence of APP were altered. The PTB domain mutation decreased inhibition of transactivation but did not abolish it (Fig.11A), possibly because the mutant PTB domain retained residual binding activity for APP. Only the combination of the PTB domain mutation with the point mutation in the first PDZ domain of Mint 1 or with the deletion of both PDZ domains abolished its inhibitory effect on transactivation (Fig.11A) (and data not shown). Conversely, after deletion of the isoform-specific N-terminal residues of Mint 1 (the sequences that are N terminal to the PTB- and PDZ domains and account for 451 of the 839 residues of Mint 1) (Okamoto et al., 1997), the specific inhibitory activity of Mint 1 was also increased significantly (Fig.11B). The expression level of the N-terminal Mint 1 deletion mutant was low, suggesting that it may be partially cytotoxic (Fig. 11C).
Fig. 11.

The PTB and PDZ domains of Mint 1 cooperate in inhibiting transactivation by APP-Gal4/VP16. A, Effect of cotransfecting Mint 1 and various Mint 1 point mutants in the PTB domain and the first PDZ domain with APP-Gal4/VP16. The same DNA amount of control vector or Mint 1 vector or the vectors encoding the indicated Mint 1 mutants was cotransfected into HEK293 cells with the APP-Gal4/VP16 and reporter plasmids, and transactivation was determined as described in Materials and Methods. B, Effect of deleting the N-terminal isoform-specific Mint 1 sequences on inhibition of transactivation. Same amounts of myc-tagged full-length Mint 1 or an N-terminally truncated Mint 1 mutant containing only the PTB and PDZ domains were cotransfected with APP-Gal4/VP16 and reporter constructs into HEK293 cells, and their specific inhibitory activity on transactivation was determined. To exclude an effect of the myc-epitope, the specific activity of wild-type and myc-tagged Mint 1 was compared. C, Quantitated immunoblot analysis of Mint expression in the experiment shown in B. Expressed proteins were detected using antibodies against the myc epitope.
Effect of Mints on transactivation by the intracellular fragment of APP
A possible explanation for the effects of Mints on transactivation by APP-Gal4/VP16, on the basis of the binding of Mints to APP via their PTB domain and to presenilins via their PDZ domains (Fig. 1-3), would be that Mints 1 and 2 interfere with γ-cleavage of APP. However, the observations that Mint 3 also binds to APP and presenilins better than Mint 2 but does not inhibit transactivation, and that both Mint 1 PDZ domains bind to presenilin 1 in vitro but only the first PDZ domain is involved in the inhibition of transactivation, argue against this hypothesis. An alternative explanation for the transactivational inhibition is that Mints 1 and 2 act on the APP cytoplasmic tail after it has been released by γ-cleavage. To differentiate between these explanations, we measured the effect of Mints on transactivation by the “precleaved” cytoplasmic tail of APP fused to Gal4/VP16 (APPICF-GV). To distinguish specific, i.e., binding-dependent effects from nonspecific effects, we also analyzed in the same experiments the mutant cytoplasmic tail of APP that is unable to bind to Mints (APPICF*-GV). In addition, Mints were compared with Fe65 as another APP-binding protein and with CASK as an unrelated control (Fig. 12).
Fig. 12.

Binding-dependent effect of Mints on transactivation mediated by a fusion protein of the intracellular fragment of APP with Gal4/VP16. Constant amounts of plasmids encoding APPICF-GV or the NPTY (684–687) to NATA (684–687) mutant APPICF*-GV were cotransfected into HEK293 cells together with reporter plasmids and expression vectors for the indicated proteins. Note that Fe65 actually further enhances transcription even when Gal4 is fused to the cytoplasmic tail of APP together with the potent transcriptional activator VP16.
Mint 1 expression significantly inhibited transactivation mediated by the cytoplasmic tail of APP fused to Gal4/VP16; this inhibition was observed only when the APP tail contained a normal Mint binding sequence (Fig. 12). A similar inhibition was detected with Mint 2 (data not shown). Mint 3, by contrast, had no significant effect, in agreement with the results obtained with full-length APP-Gal4/VP16 (Figs. 9B, 10B). Fe65 enhanced transactivation, again only when the NPTY sequence in the cytoplasmic tail was intact (Fig. 12), consistent with the overall function of Fe65 in stimulating transcription (Cao and Südhof, 2001). CASK used as a negative control had no effect on transactivation. Identical effects of both Mints 1 and 3 and Fe65 were observed when transactivation was assayed by a construct that mimicked the initial γ-cleavage product of APP, i.e., that contained 12 hydrophobic residues from the TMR preceding the cytoplasmic tail of APP (data not shown). These results demonstrate that Mint 1 inhibits transactivation downstream of APP cleavage and that this effect is highly specific for neuronal versus ubiquitous Mints.
DISCUSSION
Mints, also known as X11 and mLin-10 proteins, are a family of adaptor proteins that have been associated with several localizations and implicated in many potential functions. In adult mammals, Mints 1 and 2 are specific for brain, whereas Mint 3 is expressed ubiquitously (Okamoto and Südhof, 1997, 1998). Mints are composed of N-terminal sequences that lack easily identifiable domains and differ between isoforms and C-terminal sequences that encode similar PTB and PDZ domains in all Mints. The combination of distinct and shared domains suggests that the isoform-specific N-terminal sequences of Mints may confer distinct functions onto the activities of their common C-terminal domains. However, what do Mints actually do and how similar or dissimilar are their biological roles? As a first step toward addressing these questions, we have focused on the best characterized interaction of Mints, their binding to APP (Borg et al., 1996;McLoughlin and Miller, 1996; Zhang et al., 1997), and related the biochemical properties and localizations of Mints to their functional effects on APP in a transcriptional transactivation assay.
We first showed that all Mints bind to APP and presenilins in vitro and increase the steady-state levels of APP in transfected cells (Figs. 1-3). These observations agree with and extend previous data (Borg et al., 1996, 1998; McLoughlin and Miller, 1996; Zhang et al., 1997; Sastre et al., 1998; Lau et al., 2000). Although the strength of APP binding by Mints and the stabilization of APP by Mints in transfected cells suggest that the APP–Mint interaction is physiologically important, the presenilin binding is questionable. The differences among Mint isoforms, the fact that both PDZ domains of Mint 1 bind equally, and the promiscuous binding properties of the PDZ domains involved give rise to caution. Not surprisingly, a large number of interactions mediated by these domains have been reported, the biological significance of which has been difficult to assess.
As one approach to evaluate the significance of the interaction of Mints with APP, we next examined the localization of endogenous Mints in neurons (Figs. 4-6). The observed predominant localization of Mints in the trans-Golgi network rules out the possibility that the majority of Mint 1 is part of a permanent presynaptic PDZ domain complex that could provide a scaffold for synaptic molecules as originally envisioned (Okamoto et al., 1997; Butz et al., 1998). Instead, in conjunction with the C. elegans data, these results indicate that the PDZ domain complexes formed by Mints may be transient, highlighting a potential role for Mints as escort proteins that mediate one particular phase in the life of other proteins, for example protein targeting to a specific part of the cell (Rongo et al., 1998; Whitfield et al., 1999). However, these results do not resolve the question of whether the in vitro interactions of Mints with CASK (Mint 1 only) or Munc18–1 (Mints 1 and 2) are physiologically relevant. In fact, one possibility is that Mints participate in trafficking proteins such as Munc18–1 and CASK to synapses, because we observed a steady-state localization and not a dynamic view.
Using a transactivation assay that depends on APP cleavage but is independent of transcriptional activators (Figs. 7, 8), we demonstrated that Mints 1 and 2 but not Mint 3 dramatically inhibit transactivation by APP-Gal4/VP16 (Figs. 9-12). Three principal findings were made. First, compared with the rather moderate effects of Mints on Aβ production and secretion (Borg et al., 1998a; Sastre et al., 1998), the inhibition that we observed was nearly complete at higher expression levels of Mints 1 and 2. This inhibition was specific because two other proteins that bind to the cytoplasmic tail of APP, Mint 3 and Fe65, were without inhibitory effect. Given the similarity between the biochemical properties of Mints 1 and 3 in binding to APP and presenilin (Figs. 1, 3) and in raising the steady-state levels of APP in transfected cells (Fig. 2), the lack of inhibition by Mint 3 is surprising. This observation suggests a functional differentiation of neuronal versus ubiquitous Mints. It also indicates that the inhibition cannot be explained simply by binding of Mints to APP and presenilin, because the inhibitory Mints 1 and 2 and the noninhibitory Mint 3 bind to APP comparably well (Figs. 1, 2), and the two inhibitory Mints 1 and 2 interact with presenilin with different strength. Nevertheless, inhibition appeared to involve direct binding of Mints 1 and 2 to APP because it required the Mint-binding site in the cytoplasmic tail of APP.
Second, we found that deletion of the two PDZ domains or of the isoform-specific N-terminal sequences of Mint 1 greatly potentiated its inhibitory activity, although the expression level of the N-terminally truncated mutant dropped precipitously, presumably because it is cytotoxic. This indicated that the inhibition may be caused by interference with a normal adaptor function of an endogenous protein and that this interference is enhanced by deletion of one of the domains. Interestingly, the PDZ domain mutations showed that the two PDZ domains are not equivalent because only the mutation in the first but not the second PDZ domain increased inhibition (Fig. 10). Both mutations were effective in disrupting PDZ domain function; both abolished ∼50% of presenilin binding, which was eliminated by the double PDZ domain mutation (Fig. 3C) (data not shown). Thus the inhibitory effects of Mint 1 are probably not mediated by presenilin binding because otherwise both PDZ domain mutations should have had analogous effects on the inhibition of transactivation. Presenilin binding as an inhibitory mechanism is also ruled out by the fact that Mints 1 and 2 but not 3 inhibit transactivation, but Mints 1 and 3 bind presenilin much better than Mint 2. The target of the N-terminal sequences of Mints is also unclear because coexpression of Munc18–1 or CASK (which bind to these N-terminal sequences) had no effect on the specific inhibitory activity of Mint 1 on APP transactivation (data not shown).
Third, we observed that the inhibitory effect of Mint 1 also operated on the “precleaved” cytoplasmic tail fragment (Fig. 12). Again, Mint 3 was ineffective. Although this finding does not rule out a role for Mints in regulating APP cleavage, it demonstrates that the effects of Mint 1 are unrelated at least in part to cleavage. As argued above, the inhibitory effect was specific because other binding proteins of the cytoplasmic tail of APP did not cause it. Thus simple masking of the APP tail, which might prevent nuclear translocation, is unlikely because one would expect that any protein that binds to the cytoplasmic tail of APP, such as Mint 3 and Fe65, would also cause steric hindrance and inhibit transactivation. Several possibilities could explain the inhibitory effect of Mint 1 on the precleaved cytoplasmic tail fragment, for example, destabilization of the fragment by targeting it for proteolysis or tethering it to other proteins that do not bind to Mint 3. Future experiments will have to address these hypotheses.
Viewed together, our results suggest that Mints are true adaptor proteins that function by connecting different proteins into a protein network via various protein–protein interaction domains. Our data do not reveal the nature of this function but support the notion that Mints are involved in regulating APP after APP cleavage. A point of action after APP cleavage is also supported by the lack of colocalization of Mints with the endosomal marker EEA1, because APP is primarily believed to be cleaved by γ-secretase after endocytosis (Selkoe, 1998; Sisodia and St. George-Hyslop, 2002). The observations described here raise a number of important questions. For example, what are the physiological interacting partners for the various N- and C-terminal sequences of Mints, and how does the activity of Mints on transactivation by APP-Gal4/VP16 relate to their in vivofunction in an intact animal? Future experiments using different types of approaches, ranging from knock-out mice to structural studies, will be required to address these questions.
Footnotes
This study was supported by a Pilot Project Grant from the Alzheimer's Disease Center at University of Texas Southwestern Medical Center (X.L.), Grant R37-MH52804-06 from National Institutes of Health (T.C.S.), and a fellowship from the Human Frontiers Science Program (T.B.). We thank I. Leznicki and E. Borowicz for excellent technical assistance.
Correspondence should be addressed to T. C. Südhof, The Center for Basic Neuroscience, Department of Molecular Genetics, and Howard Hughes Medical Institute, The University of Texas Southwestern Medical Center, 6000 Harry Hines Boulevard, NA4.118, Dallas TX 75390-9111. E-mail:Thomas.Sudhof@UTSouthwestern.edu.
REFERENCES
- 1.Bayer TA, Cappai R, Masters CL, Beyreuther K, Multhaup G. It all sticks together—the APP-related family of proteins and Alzheimer's disease. Mol Psychiatry. 1999;4:524–528. doi: 10.1038/sj.mp.4000552. [DOI] [PubMed] [Google Scholar]
- 2.Biederer T, Südhof TC. Mints as adaptors: direct binding to neurexins and recruitment of munc18. J Biol Chem. 2000;275:39803–39806. doi: 10.1074/jbc.C000656200. [DOI] [PubMed] [Google Scholar]
- 3.Biederer T, Südhof TC. CASK and protein 4.1 support F-actin nucleation on neurexins. J Biol Chem. 2001;276:47869–47876. doi: 10.1074/jbc.M105287200. [DOI] [PubMed] [Google Scholar]
- 4.Bloom GS, Brashear TA. A novel 58-kDa protein associates with the Golgi apparatus and microtubules. J Biol Chem. 1989;264:16083–16092. [PubMed] [Google Scholar]
- 5.Borg JP, Ooi J, Levy E, Margolis B. The phosphotyrosine interaction domains of X11 and FE65 bind to distinct sites on the YENPTY motif of amyloid precursor protein. Mol Cell Biol. 1996;16:6229–6241. doi: 10.1128/mcb.16.11.6229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borg JP, Yang Y, De Taddeo-Borg M, Margolis B, Turner RS. The X11alpha protein slows cellular amyloid precursor protein processing and reduces Abeta40 and Abeta42 secretion. J Biol Chem. 1998a;273:14761–14766. doi: 10.1074/jbc.273.24.14761. [DOI] [PubMed] [Google Scholar]
- 7.Borg JP, Straight SW, Kaech SM, de Taddeo-Borg M, Kroon DE, Karnak D, Turner RS, Kim SK, Margolis B. Identification of an evolutionarily conserved heterotrimeric protein complex involved in protein targeting. J Biol Chem. 1998b;273:31633–31636. doi: 10.1074/jbc.273.48.31633. [DOI] [PubMed] [Google Scholar]
- 8.Borg JP, Lopez-Figueroa MO, de Taddeo-Borg M, Kroon DE, Turner RS, Watson SJ, Margolis B. Molecular analysis of the X11-mLin-2/CASK complex in brain. J Neurosci. 1999;19:1307–1316. doi: 10.1523/JNEUROSCI.19-04-01307.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Butz S, Okamoto M, Südhof TC. A tripartite protein complex with the potential to couple synaptic vesicle exocytosis to cell adhesion in brain. Cell. 1998;94:773–782. doi: 10.1016/s0092-8674(00)81736-5. [DOI] [PubMed] [Google Scholar]
- 10.Cao X, Südhof TC. A transcriptionally active complex of APP with Fe65 and histone acetyltransferase tip60. Science. 2001;293:115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- 11.Cho KO, Hunt CA, Kennedy MB. The rat brain postsynaptic density fraction contains a homolog of the Drosophila discs-large tumor suppressor protein. Neuron. 1992;9:929–942. doi: 10.1016/0896-6273(92)90245-9. [DOI] [PubMed] [Google Scholar]
- 12.Cupers P, Orlans I, Craessaerts K, Annaert W, De Strooper B. The amyloid precursor protein (APP)-cytoplasmic fragment generated by gamma-secretase is rapidly degraded but distributes partially in a nuclear fraction of neurones in culture. J Neurochem. 2001;78:1168–1178. doi: 10.1046/j.1471-4159.2001.00516.x. [DOI] [PubMed] [Google Scholar]
- 13.Duclos F, Koenig M. Comparison of primary structure of a neuron-specific protein, X11, between human and mouse. Mamm Genome. 1995;6:57–58. doi: 10.1007/BF00350899. [DOI] [PubMed] [Google Scholar]
- 14.Fiore F, Zambrano N, Minopoli G, Donini V, Duilio A, Russo T. The regions of the Fe65 protein homologous to the phosphotyrosine interaction/phosphotyrosine binding domain of Shc bind the intracellular domain of the Alzheimer's amyloid precursor protein. J Biol Chem. 1995;270:30853–30856. doi: 10.1074/jbc.270.52.30853. [DOI] [PubMed] [Google Scholar]
- 15.Gao Y, Pimplikar SW. The γ-secretase-cleaved C-terminal fragment of amyloid precursor protein mediates signaling to the nucleus. Proc Natl Acad Sci USA. 2001;98:14979–14984. doi: 10.1073/pnas.261463298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Glenner GG, Wong CW. Alzheimer's disease and Down's syndrome: sharing of a unique cerebrovascular amyloid fibril protein. Biochem Biophys Res Commun. 1984;122:1131–1135. doi: 10.1016/0006-291x(84)91209-9. [DOI] [PubMed] [Google Scholar]
- 17.Guenette SY, Chen J, Jondro PD, Tanzi RE. Association of a novel human FE65-like protein with the cytoplasmic domain of the beta-amyloid precursor protein. Proc Natl Acad Sci USA. 1996;93:10832–10837. doi: 10.1073/pnas.93.20.10832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haass C, De Strooper B. The presenilins in Alzheimer's disease—proteolysis holds the key. Science. 1999;286:916–919. doi: 10.1126/science.286.5441.916. [DOI] [PubMed] [Google Scholar]
- 19.Hata Y, Butz S, Südhof TC. CASK: a novel dlg/PSD-95 homolog with an N-terminal CaM kinase domain identified by interaction with neurexins. J Neurosci. 1996;16:2488–2494. doi: 10.1523/JNEUROSCI.16-08-02488.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaech SM, Whitfield CW, Kim SK. The LIN-2/LIN-7/LIN-10 complex mediates basolateral membrane localization of the C. elegans EGF receptor LET-23 in vulval epithelial cells. Cell. 1998;94:761–771. doi: 10.1016/s0092-8674(00)81735-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Müller-Hill B. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 22.Kimberly WT, Zheng JB, Guenette SY, Selkoe DJ. The intracellular domain of the beta-amyloid precursor protein is stabilized by Fe65 and translocates to the nucleus in a notch-like manner. J Biol Chem. 2001;276:40288–40292. doi: 10.1074/jbc.C100447200. [DOI] [PubMed] [Google Scholar]
- 23.Kistner U, Wenzel BM, Veh RW, Cases-Langhoff C, Garner AM, Appeltauer U, Voss B, Gundelfinger ED, Garner CC. SAP90, a rat presynaptic protein related to the product of the Drosophila tumor suppressor gene dlg-A. J Biol Chem. 1993;268:4580–4583. [PubMed] [Google Scholar]
- 24.Kitaguchi N, Takahashi Y, Tokushima Y, Shiojiri S, Ito H. Novel precursor of Alzheimer's disease amyloid protein shows protease inhibitory activity. Nature. 1988;331:530–532. doi: 10.1038/331530a0. [DOI] [PubMed] [Google Scholar]
- 25.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;277:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 26.Lau KF, McLoughlin DM, Standen C, Miller CC. X11 alpha and x11 beta interact with presenilin-1 via their PDZ domains. Mol Cell Neurosci. 2000;16:557–565. doi: 10.1006/mcne.2000.0898. [DOI] [PubMed] [Google Scholar]
- 27.Luzio JP, Brake B, Banting G, Howell KE, Braghetta P, Stanley KK. Identification, sequencing and expression of an integral membrane protein of the trans-Golgi network (TGN38). Biochem J. 1990;270:97–102. doi: 10.1042/bj2700097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masters CL, Multhaup G, Simms G, Pottgiesser J, Martins RN, Beyreuther K. Neuronal origin of a cerebral amyloid: neurofibrillary tangles of Alzheimer's disease contain the same protein as the amyloid of plaque cores and blood vessels. EMBO J. 1985;4:2757–2763. doi: 10.1002/j.1460-2075.1985.tb04000.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McLoughlin DM, Miller CC. The intracellular cytoplasmic domain of the Alzheimer's disease amyloid precursor protein interacts with phosphotyrosine-binding domain proteins in the yeast two-hybrid system. FEBS Lett. 1996;397:197–200. doi: 10.1016/s0014-5793(96)01128-3. [DOI] [PubMed] [Google Scholar]
- 30.McLoughlin DM, Irving NG, Brownlees J, Brion JP, Leroy K, Miller CC. Mint2/X11-like colocalizes with the Alzheimer's disease amyloid precursor protein and is associated with neuritic plaques in Alzheimer's disease. Eur J Neurosci. 1999;11:1988–1994. doi: 10.1046/j.1460-9568.1999.00610.x. [DOI] [PubMed] [Google Scholar]
- 31.Mu FT, Callaghan JM, Steele-Mortimer O, Stenmark H, Parton RG, Campbell PL, McCluskey J, Yeo JP, Tock EP, Toh BH. EEA1, an early endosome-associated protein. EEA1 is a conserved alpha-helical peripheral membrane protein flanked by cysteine “fingers” and contains a calmodulin-binding IQ motif. J Biol Chem. 1995;270:13503–13511. doi: 10.1074/jbc.270.22.13503. [DOI] [PubMed] [Google Scholar]
- 32.Mueller HT, Borg JP, Margolis B, Turner RS. Modulation of amyloid precursor protein metabolism by X11alpha/Mint-1. A deletion analysis of protein-protein interaction domains. J Biol Chem. 2000;275:39302–39306. doi: 10.1074/jbc.M008453200. [DOI] [PubMed] [Google Scholar]
- 33.Okamoto M, Südhof TC. Mints, Munc18-interacting proteins in synaptic vesicle exocytosis. J Biol Chem. 1997;272:31459–31464. doi: 10.1074/jbc.272.50.31459. [DOI] [PubMed] [Google Scholar]
- 34.Okamoto M, Südhof TC. Mint 3: a ubiquitous mint isoform that does not bind to munc18–1 or 2. Eur J Cell Biol. 1998;77:161–165. doi: 10.1016/S0171-9335(98)80103-9. [DOI] [PubMed] [Google Scholar]
- 35.Okamoto M, Matsuyama T, Sugita M. Ultrastructural localization of mint1 at synapses in mouse hippocampus. Eur J Neurosci. 2000;12:3067–3072. doi: 10.1046/j.1460-9568.2000.00200.x. [DOI] [PubMed] [Google Scholar]
- 36.Okamoto M, Nakajima Y, Matsuyama T, Sugita M. Amyloid precursor protein associates independently and collaboratively with PTB and PDZ domains of mint on vesicles and at cell membrane. Neuroscience. 2001;104:653–656. doi: 10.1016/s0306-4522(01)00124-5. [DOI] [PubMed] [Google Scholar]
- 37.Rongo C, Whitfield CW, Rodal A, Kim SK, Kaplan JM. LIN-10 is a shared component of the polarized protein localization pathways in neurons and epithelia. Cell. 1998;94:751–759. doi: 10.1016/s0092-8674(00)81734-1. [DOI] [PubMed] [Google Scholar]
- 38.Sadowski I, Ma J, Triezenberg S, Ptashne M. GAL4-VP16 is an unusually potent transcriptional activator. Nature. 1988;335:563–564. doi: 10.1038/335563a0. [DOI] [PubMed] [Google Scholar]
- 39.Sastre M, Turner RS, Levy E. X11 interaction with beta-amyloid precursor protein modulates its cellular stabilization and reduces amyloid beta-protein secretion. J Biol Chem. 1998;273:22351–22357. doi: 10.1074/jbc.273.35.22351. [DOI] [PubMed] [Google Scholar]
- 40.Sastre M, Steiner H, Fuchs K, Capell A, Multhaup G, Condron MM, Teplow DB, Haass C. Presenilin-dependent gamma-secretase processing of beta-amyloid precursor protein at a site corresponding to the S3 cleavage of Notch. EMBO Rep. 2001;2:835–841. doi: 10.1093/embo-reports/kve180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Selkoe DJ. The cell biology of β-amyloid precursor protein and presenilin in Alzheimer's disease. Trends Cell Biol. 1998;8:447–453. doi: 10.1016/s0962-8924(98)01363-4. [DOI] [PubMed] [Google Scholar]
- 42.Setou M, Nakagawa T, Seog DH, Hirokawa N. Kinesin superfamily motor protein KIF17 and mLin-10 in NMDA receptor-containing vesicle transport. Science. 2000;288:1796–1802. doi: 10.1126/science.288.5472.1796. [DOI] [PubMed] [Google Scholar]
- 43.Sisodia SS, St. George-Hyslop PH. γ-Secretase, Notch, Abeta and Alzheimer's disease: where do the presenilins fit in? Nat Rev Neurosci. 2002;3:281–290. doi: 10.1038/nrn785. [DOI] [PubMed] [Google Scholar]
- 44.Steiner H, Duff K, Capell A, Romig H, Grim MG, Lincoln S, Hardy J, Yu X, Picciano M, Fechteler K, Citron M, Kopan R, Pesold B, Keck S, Baader M, Tomita T, Iwatsubo T, Baumeister R, Haass C. A loss of function mutation of presenilin-2 interferes with amyloid β-peptide production and notch signaling. J Biol Chem. 1999;274:28669–28673. doi: 10.1074/jbc.274.40.28669. [DOI] [PubMed] [Google Scholar]
- 45.Struhl G, Adachi A. Requirements for presenilin-dependent cleavage of notch and other transmembrane proteins. Mol Cell. 2000;6:625–636. doi: 10.1016/s1097-2765(00)00061-7. [DOI] [PubMed] [Google Scholar]
- 46.Tanzi RE, McClatchey AI, Lamperti ED, Villa-Komaroff L, Gusella JF, Neve RL. Protease inhibitor domain encoded by an amyloid protein precursor mRNA associated with Alzheimer's disease. Nature. 1988;331:528–530. doi: 10.1038/331528a0. [DOI] [PubMed] [Google Scholar]
- 47.Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ushkaryov YA, Petrenko AG, Geppert M, Südhof TC. Neurexins: synaptic cell surface proteins related to the α-latrotoxin receptor and laminin. Science. 1992;257:50–56. doi: 10.1126/science.1621094. [DOI] [PubMed] [Google Scholar]
- 49.Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer R, van den Berg TK, Missler M, Geuze H, Südhof TC. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science. 2000;287:864–869. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- 50.Wada I, Rindress D, Cameron PH, Ou WJ, Doherty JJ, II, Louvard D, Bell AW, Dignard D, Thomas DY, Bergeron JJ. SSR alpha and associated calnexin are major calcium binding proteins of the endoplasmic reticulum membrane. J Biol Chem. 1991;266:19599–19610. [PubMed] [Google Scholar]
- 51.Whitfield CW, Benard C, Barnes T, Hekimi S, Kim SK. Basolateral localization of the Caenorhabditis elegans epidermal growth factor receptor in epithelial cells by the PDZ protein LIN-10. Mol Biol Cell. 1999;10:2087–2100. doi: 10.1091/mbc.10.6.2087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wolfe MS, Haass C. The role of presenilins in gamma-secretase activity. J Biol Chem. 2001;276:5413–5416. doi: 10.1074/jbc.R000026200. [DOI] [PubMed] [Google Scholar]
- 53.Yu C, Kim SH, Ikeuchi T, Xu H, Gasparini L, Wang R, Sisodia SS. Characterization of a presenilin-mediated amyloid precursor protein carboxyl-terminal fragment gamma. Evidence for distinct mechanisms involved in gamma-secretase processing of the APP and Notch1 transmembrane domains. J Biol Chem. 2001;276:43756–43760. doi: 10.1074/jbc.C100410200. [DOI] [PubMed] [Google Scholar]
- 54.Zhang Z, Lee CH, Mandiyan V, Borg JP, Margolis B, Schlessinger J, Kuriyan J. Sequence-specific recognition of the internalization motif of the Alzheimer's amyloid precursor protein by the X11 PTB domain. EMBO J. 1997;16:6141–6150. doi: 10.1093/emboj/16.20.6141. [DOI] [PMC free article] [PubMed] [Google Scholar]