

Abstract
The identification of mutations in the Tau gene in frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) has made it possible to express human tau protein with pathogenic mutations in transgenic animals. Here we report on the production and characterization of a line of mice transgenic for the 383 aa isoform of human tau with the P301S mutation. At 5–6 months of age, homozygous animals from this line developed a neurological phenotype dominated by a severe paraparesis. According to light microscopy, many nerve cells in brain and spinal cord were strongly immunoreactive for hyperphosphorylated tau. According to electron microscopy, abundant filaments made of hyperphosphorylated tau protein were present. The majority of filaments resembled the half-twisted ribbons described previously in cases of FTDP-17, with a minority of filaments resembling the paired helical filaments of Alzheimer's disease. Sarkosyl-insoluble tau from brains and spinal cords of transgenic mice ran as a hyperphosphorylated 64 kDa band, the same apparent molecular mass as that of the 383 aa tau isoform in the human tauopathies. Perchloric acid-soluble tau was also phosphorylated at many sites, with the notable exception of serine 214. In the spinal cord, neurodegeneration was present, as indicated by a 49% reduction in the number of motor neurons. No evidence for apoptosis was obtained, despite the extensive colocalization of hyperphosphorylated tau protein with activated MAP kinase family members. The latter may be involved in the hyperphosphorylation of tau.
Keywords: hyperphosphorylation, MAP kinase family, neurodegeneration, tauopathy, tau filaments, Tau gene mutations
Abundant filaments made of hyperphosphorylated microtubule-associated protein tau constitute a defining characteristic of a number of neurodegenerative diseases, including Alzheimer's disease, Pick's disease, progressive supranuclear palsy, corticobasal degeneration, and frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17) (Lee et al., 2001). The discovery of coding region and intronic mutations in the Tau gene in FTDP-17 has firmly established that dysfunction of tau protein can cause neurodegeneration and dementia (Hutton et al., 1998; Poorkaj et al., 1998; Spillantini et al., 1998a).
In adult human brain, six tau isoforms are expressed from a single gene by alternative mRNA splicing (Goedert et al., 1988, 1989a,b; Andreadis et al., 1992; Poorkaj et al., 2001). They differ by the presence or absence of 29 and 58 aa inserts located in the N-terminal half and an additional 31 aa insert located in the C-terminal half. Inclusion of the latter, which is encoded by exon 10 of the Tau gene, gives rise to the three isoforms with four microtubule-binding repeats each. The other three isoforms have three repeats each. In normal brain, similar levels of three-repeat and four-repeat tau isoforms are expressed (Goedert and Jakes, 1990). Known mutations in theTau gene in FTDP-17 are either missense, deletion, and silent mutations in the coding region or intronic mutations located close to the splice donor site of the intron after exon 10. Functionally, they lead to a reduced ability of tau to interact with microtubules and/or a change in the ratio of three-repeat to four-repeat tau isoforms, resulting in the relative overproduction of four-repeat tau (Hasegawa et al., 1998; Hong et al., 1998; Hutton et al., 1998; Spillantini et al., 1998a; Yoshida et al., 2002). Some coding region mutations also promote the assembly of tau protein into filaments (Goedert et al., 1999; Nacharaju et al., 1999). In FTDP-17, the age of onset of disease varies, depending on the Taumutation. The same is true of the magnitude of the functional effects produced by individual mutations.
Mutation P301S in exon 10 of Tau causes an early onset of clinical signs and has strong functional effects, as evidenced by a reduced ability of mutant tau protein to promote microtubule assembly and a marked stimulatory effect on tau filament assembly (Bugiani et al., 1999; Goedert et al., 1999; Sperfeld et al., 1999; Yasuda et al., 2000; Morris et al., 2001). Here we describe the production and characterization of a line of transgenic mice that express, under the control of the murine thy1 promoter, the 383 aa isoform of human tau with the P301S mutation. At 5–6 months of age, homozygous animals developed motor symptoms characterized by a severe paraparesis. Brain and spinal cord contained insoluble hyperphosphorylated tau protein that migrated on gels at the same position as filamentous tau from human tauopathies. By electron microscopy, abundant tau filaments were found. In the spinal cord, neurodegeneration was present, as evidenced by a significant reduction in the number of motor neurons.
MATERIALS AND METHODS
Antibodies. A panel of anti-tau antibodies was used (Fig. 1). Antiserum BR133 recognizes the N terminus of tau; antiserum BR135 is directed against the microtubule-binding repeat region, and antiserum BR134 is directed against the C terminus of tau. All three antisera recognize the six adult human brain and three adult murine brain tau isoforms (Goedert et al., 1989b; Götz et al., 1995). T14 (a kind gift from Dr. V. M.-Y. Lee, University of Pennsylvania, Philadelphia, PA) recognizes residues 141–149 of human tau (in the numbering of the longest human brain tau isoform); it recognizes all human tau isoforms but fails to recognize murine tau (Kosik et al., 1988). Antiserum MT1 was raised in white Dutch rabbits using the synthetic peptide ARVASKDRTGNDEK (residues 114–127 of the longest isoform of mouse brain tau) as the immunogen. MT1 recognizes the three murine brain tau isoforms but fails to recognize human tau. Alz-50 (a kind gift from Dr. P. Davies, Albert Einstein College of Medicine, Bronx, NY) recognizes amino acids 5–15 and 312–322 of tau and is sensitive to the conformation of tau (Carmel et al., 1996). BR133, BR134, BR135, Tau14, MT1, and Alz-50 are phosphorylation-independent anti-tau antibodies. AT270 (Innogenetics, Gent, Belgium) recognizes human and murine tau phosphorylated at T181 (Goedert et al., 1994); AT8 (Innogenetics) recognizes human and murine tau phosphorylated at S202 and T205 (Goedert et al., 1995); AT100 (Innogenetics) recognizes human and murine tau phosphorylated at T212 and S214 (Zheng-Fischhöfer et al., 1998); pT212 (Biosource, Camarillo, CA) recognizes human and murine tau phosphorylated at T212 (Woods et al., 2001); CP3 (a kind gift from Dr. P. Davies) recognizes human and murine tau phosphorylated at S214 (Jicha et al., 1999); AT180 (Innogenetics) recognizes human and murine tau phosphorylated at T231 (Goedert et al., 1994); 12E8 (a kind gift from Dr. P. Seubert, Elan Pharmaceuticals, San Francisco, CA) recognizes human and murine tau phosphorylated at S262 and/or S356 (Seubert et al., 1995); AD2 (a kind gift from Dr. A. Delacourte, Institut National de la Santé et de la Recherche Médicale U422, Lille, France) and PHF1 (a kind gift from Dr. P. Davies) recognize human and murine tau phosphorylated at S396 and S404 (Otvos et al., 1994; Buée-Scherrer et al., 1996); PG5 (a kind gift from Dr. P. Davies) recognizes human and murine tau phosphorylated at S409 (Jicha et al., 1999); AP422 recognizes human and murine tau phosphorylated at S422 (Hasegawa et al., 1996). In addition to anti-tau antibodies, tissue sections were also stained with a number of other antibodies. They included antibodies to β-amyloid (a kind gift from Dr. V. M.-Y. Lee), parkin (Chemicon, Temecula, CA), ubiquitin (Dako, Glostrup, Denmark, and Chemicon), the 20S proteasome (Affiniti, Research Products, Exeter, UK), phospho-mitogen-activated protein (MAP) kinase kinase 3/6 (Cell Signaling Technology, Beverly, MA), phospho-MAP kinase (clone 12D4; Nanotools, Teningen, Germany), phospho-p38 MAP kinase (Cell Signaling Technology), phospho-c-Jun N-terminal protein kinase (JNK) (Cell Signaling Technology), phospho-glycogen synthase kinase-3 (GSK3) (specific for phosphorylated Y279 of GSK3α and phosphorylated Y216 of GSK3β, clone 5G-2F; Upstate Biotechnology, Lake Placid, NY), cyclin-dependent kinase 5 (cdk5) (Santa Cruz Biotechnology, Santa Cruz, CA), the p35 activator of cdk5 (Santa Cruz Biotechnology), Pin1 (N-19; Santa Cruz Biotechnology), active caspase-3 (PharMingen, San Diego, CA, and Cell Signaling Technology), cleaved α-fodrin (Cell Signaling Technology), heparan sulfate (Seikagaku Kogyo, Tokyo, Japan), heat shock protein 25 (Stressgen Biotechnologies, Victoria, Canada), heat shock protein 70 (Stressgen Biotechnologies), αB-crystallin (Stressgen Biotechnologies), and glial fibrillary acidic protein (GFAP; Dako).
Fig. 1.

Epitopes of anti-tau antibodies. A bar diagram of the 441 aa isoform of human tau is shown, with the microtubule-binding repeat region shown in black. Amino acids recognized by phosphorylation-dependent antibodies are indicated abovethe bar diagram (pS andpT). The epitopes of phosphorylation-independent antibodies are shown below the bar diagram.
Transgenic mice. Site-directed mutagenesis was used to introduce the P301S mutation (using the numbering of the longest human brain tau isoform) into the cDNA encoding the shortest human four-repeat tau isoform (383 aa isoform of human tau). The mutated cDNA was subcloned into a murine thy1.2 genomic expression vector (a kind gift from Dr. H. van der Putten, Novartis, Basel, Switzerland) (Lüthi et al., 1997). This tau expression construct was produced by subcloning P301S hTau43cDNA into a unique XhoI site of the expression vector. A Kozak consensus sequence had been introduced upstream of the initiation codon. Vector sequences were removed before microinjection. Transgenic mice were produced by pronuclear injection of (C57BL/6J × CBA/ca) F1 embryos. Founders were identified by PCR analysis of lysates from tail biopsies using the primer pair 5′-GGTTTTTGCTGGAATCCTGG-3′ and 5′-GGAGTTCGAAGTGATGGAAG-3′. Founder animals were intercrossed with C57BL/6J mice to establish lines. Homozygosity was determined by matings with nontransgenic animals.
Immunoblot analysis. Brains and spinal cords from transgenic mice were dounce homogenized in 2.5% (v/v) perchloric acid (0.5 ml/gm), allowed to stand on ice for 20 min, and centrifuged for 10 min at 10,000 × g. The supernatants were dialyzed against 50 mm Tris-HCl, pH 7.4, 1 mm dithiothreitol, and 0.1 mm phenylmethylsulfonyl fluoride (PMSF) and used for immunoblot analysis, as described previously (Goedert and Jakes, 1990). Comparison of the amount of expressed soluble human tau with endogenous mouse tau was determined by extracting brain and spinal cord samples from 5- to 6-month-old homozygous transgenic mice. Tissues were homogenized in 5 vol of extraction buffer (50 mmTris-HCl, pH 7.4, 0.5 m NaCl, 0.1 mm PMSF, 1 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mm sodium orthovanadate, 50 mm sodium fluoride, and 1 mm β-glycerophosphate). After a 15 min centrifugation at 543,000 × g, the supernatants were adjusted to 2% 2-mercaptoethanol, boiled for 10 min, chilled on ice for 10 min, and centrifuged, and the resulting supernatants were then brought to 50% ammonium sulfate. After centrifugation at 543,000 × g, the precipitates were resuspended in 50 mm Tris-HCl, pH 7.4, 0.1 mmPMSF, 1 μg/ml leupeptin, and 1 μg/ml pepstatin. Dephosphorylation of tau with Escherichia coli alkaline phosphatase (Sigma, St. Louis, MO) was performed as described previously (Goedert et al., 1992a). The relative tau levels were determined by densitometry using purified recombinant tau proteins as standards. Various amounts of the final fractions were run on 10% SDS-PAGE alongside recombinant human tau proteins and immunoblotted with anti-tau serum BR134. The blots were developed using enhanced chemiluminescence (Amersham Biosciences, Arlington Heights, IL), and the bands were scanned (Personal Densitometer SI; Molecular Dynamics, Sunnyvale, CA) and analyzed using the IQMac version 1.2 software (Molecular Dynamics). The readings given by the three mouse tau bands were added together and compared with the reading given by the single human tau band.
Histology and immunohistochemistry. Brain, spinal cord, and hindleg muscle from P301S tau transgenic mice were analyzed histologically. Animals were perfused transcardially with 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.2, and the tissues were postfixed for 2 hr. Histological staining and immunohistochemistry were performed on either 4 μm sections obtained from paraffin-embedded tissue blocks or 30 μm microtome sections. Thioflavin S staining and Bodian silver staining were done as described previously (Yamamoto and Hirano, 1986). For immunohistochemistry, sections were incubated overnight at 4°C with the primary antibodies. Immunostaining was visualized using the avidin–biotin system (Vectastain; Vector Laboratories, Burlingame, CA) and 3,3′-diaminobenzidine (Sigma) as the chromogen. The sections were counterstained with cresyl violet or hematoxylin and eosin. The colocalization of activated MAP kinase family members and hyperphosphorylated tau was investigated by double-labeling immunofluorescence. Nonspecific binding sites were blocked by incubating free-floating brain and spinal cord sections for 3 hr at room temperature with 3% bovine serum albumin (BSA) and 0.1% saponin in PBS. The tissue sections were then incubated overnight at room temperature with the following antibodies in the presence of either the polyclonal anti-tau antibody BR134 or the monoclonal antibody PHF1 (anti-phospho-MAP kinase kinase 3/6, anti-activated MAP kinase, anti-phospho-JNK, and anti-phospho-p38). The sections were washed five times in BSA/saponin/PBS and incubated with anti-mouse IgG conjugated to Alexa-488 (Molecular Probes, Eugene, OR) and anti-rabbit IgG conjugated to CY3 (Jackson ImmunoResearch, West Grove, PA) for 3 hr at room temperature. For the colocalization of Pin1 and hyperphosphorylated tau, tissue sections were subjected to microwave irradiation for antigen retrieval, followed by the addition of the polyclonal anti-Pin1 antibody and monoclonal antibody AT100. Donkey anti-goat CY3 and donkey anti-mouse CY2 (Jackson ImmunoResearch) were used as secondary antibodies. After washing, sections were mounted in Vectashield (Vector Laboratories) and imaged with a Radiance 2100 confocal microscope (Bio-Rad, Hercules, CA).
Electron microscopy of tissue sections. Tissues were fixed in 4% paraformaldehyde, postfixed in 1% osmium tetroxide, and embedded in Epon. One-micrometer-thick sections were stained with toluidine blue. Ultrathin sections were stained with uranyl acetate and lead citrate and viewed with a Philips 300 electron microscope. For immunoelectron microscopy, paraformaldehyde-fixed specimens (1 mm3) were washed in distilled water and placed in Tris-buffered saline, pH 7.6. Fifty-micrometer-thick sections were cut using a vibratome. Immunogold labeling with anti-tau antibody AT8 (1:5) was performed using 10 nm gold particles (Goldmark Biologicals, Phillipsburg, NJ) conjugated to goat anti-mouse IgG at a dilution of 1:10. Sections were postfixed in 2.5% glutaraldehyde, followed by 1% osmium tetroxide, dehydrated through a graded alcohol series, and embedded in epoxy resin. Semithin sections were stained with toluidine blue. Ultrathin sections were contrasted with lead citrate and uranyl acetate and scanned with the electron microscope.
Extraction and characterization of sarkosyl-insoluble tau.Sarkosyl-insoluble tau was extracted from whole brains and spinal cords of transgenic mice, as described for cerebral cortex from Alzheimer's disease brains. The sarkosyl-insoluble material was analyzed by immunoblotting and electron microscopy. For immunoblotting, SDS-PAGE and transfers were done, as described previously (Goedert and Jakes, 1990). Dephosphorylation using E. coli alkaline phosphatase was performed as described previously (Goedert et al., 1992a). For electron microscopy, aliquots of the sarkosyl-insoluble material were placed on carbon-coated 400 mesh grids and stained with 1% lithium phosphotungstate. Micrographs were recorded at a nominal magnification of 40,000× on a Philips EM208S microscope, as described previously (Crowther, 1991). Procedures for immunoelectron microscopy were as described previously (Crowther, 1991).
Nerve cell counts. The numbers of nerve cells in lamina II (substantia gelatinosa) and lamina IX (somatic motor neurons) of the lumbar spinal cord were counted in three 6-month-old transgenic mice and three age-matched controls. We used the atlas of Sidman et al. (1971) for the macroscopic identification of lumbar segments L2–L3 and the microscopic identification of laminas II and IX in cross sections. The widest part of the lumbar enlargement (level L2–L3) was serially sectioned (8 μm) and stained using hematoxylin and eosin, and laminas II and IX were identified. A field of each section was digitally captured using a Spot RT digital camera (Diagnostic Instruments, Sterling Heights, MI) attached to a Leica (Wetzlar, Germany) FMLB microscope with a 10× objective. The areas of laminas II and IX were measured using supplementary software of the digital camera. In each of the five serial sections from each mouse, the number of nerve cells was counted manually using a 20× or 40× objective. To avoid counting the same neuron in consecutive sections, only neurons with a nucleolus were included. The results were expressed as the mean neuronal density (number of neurons per millimeter square). Statistical analyses compared mean neuronal densities of control and transgenic mice. Hierarchical models were used which made it possible to correlate the density measurements of the five sections from each mouse (Goldstein, 1995). Individual mice were coded as random effect and the type of mouse (control or transgenic) as fixed effect.
Detection of apoptosis. The presence of DNA fragmentation in single cells was investigated by in situ end-labeling (ISEL) of sections obtained from paraffin-embedded tissue blocks, as described previously (Migheli et al., 1997). Briefly, sections were pretreated with 0.5–5 μg/ml proteinase K for 15 min at room temperature. They were then incubated with 20 U/ml terminal deoxynucleotidyl transferase (Roche Diagnostics, Indianapolis, IN) and 10 nmol/ml fluorescein-11-deoxyUTP (Roche Diagnostics) for 2 hr at 37°C. The reaction product was revealed with peroxidase-conjugated anti-fluorescein antibody (Roche Diagnostics) using diaminobenzidine as the chromogen. For immunohistochemistry with antibodies directed against activated caspase-3 and cleaved α-fodrin, tissue sections were subjected to microwave irradiation for antigen retrieval. They were then incubated with 5% ovalbumin for 15 min, followed by a 15 min incubation with 2% biotin. Immunohistochemistry was performed using streptavidin–biotin and cobalt chloride-intensified diaminobenzidine or 3-amino-9-ethylcarbazole as substrate.
RESULTS
Production of transgenic mouse lines and neurological phenotype
The neuron-specific elements of the mouse thy1 promoter were used to produce transgenic mice expressing the shortest human four-repeat tau isoform with the P301S mutation. Six founders were obtained, five of which transmitted the transgene in a Mendelian manner. Three of the five lines expressed high levels of human tau protein in the brain and spinal cord and were used to establish homozygous lines. The present report concentrates on line 2541. At 5–6 months of age, homozygous animals from this line developed a neurological phenotype that was characterized by general muscle weakness, tremor, and a severe paraparesis. When lifted by the tail, the mice were unable to extend their hindlimbs. In most animals, an eye inflammation was also present. Heterozygous animals developed a similar phenotype at 12–14 months of age.
Expression, phosphorylation, and solubility of mutant human tau protein
By immunoblotting, a broad transgenic tau band of 52–58 kDa apparent molecular mass was present in brains and spinal cords from 5- to 6-month-old homozygous transgenic mice (Fig.2A). It was reactive with anti-tau serum BR134, which recognizes both human and murine tau, and with antibody T14, which only recognizes human tau. The transgenic tau band was not immunoreactive with antibody MT1, which recognizes murine but not human tau (data not shown). Dephosphorylation with alkaline phosphatase was used to assess the expression level of transgenic human tau relative to endogenous mouse tau (Fig.2A). As shown previously (Götz et al., 1995;Kampers et al., 1999), dephosphorylated mouse brain tau runs as three bands that correspond to the three tau isoforms with four repeats. In transgenic mice, an additional human tau band of 52 kDa was observed that aligned with the 383 aa isoform of recombinant human brain tau. In brains and spinal cords from transgenic mice, human tau levels were approximately twofold higher than the levels of total mouse tau. The phosphorylation state of perchloric acid-soluble human tau protein from mouse brain and spinal cord was investigated by immunoblotting using 10 different phosphorylation-dependent anti-tau antibodies (Fig.2C). The human tau band was strongly immunoreactive with antibodies AT270, AT8, pT212, AT180, 12E8, AD2, PG5, and AP422. No reactivity was observed with antibodies AT100 or CP3.
Fig. 2.
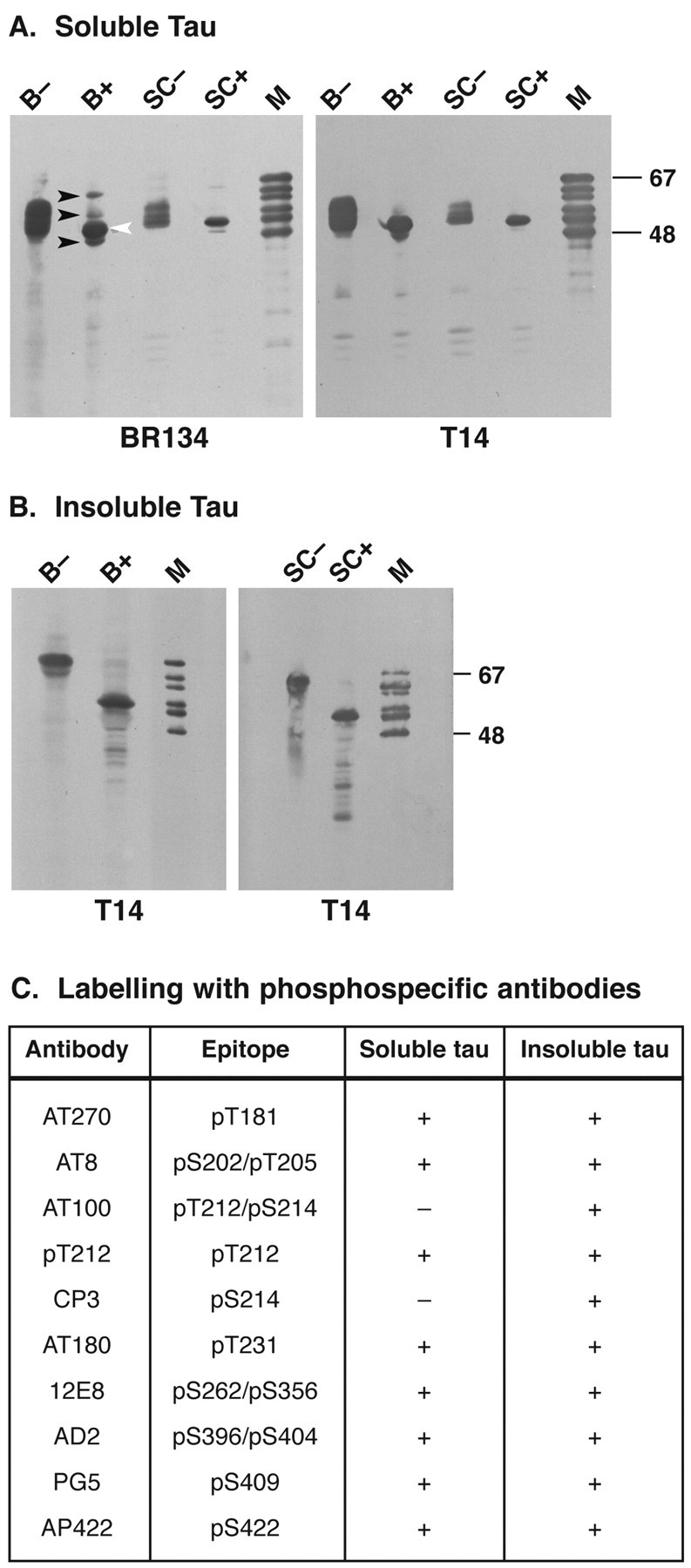
Immunoblot analysis of tau protein in brains and spinal cords from mice of the human P301S tau line. A, Perchloric acid-soluble tau was extracted from the brain (B) and spinal cord (SC) of 5- to 6-month-old mice and immunoblotted with anti-tau antibodies BR134 and T14 before (−) and after (+) alkaline phosphatase treatment.Black arrowheads point to the three mouse tau isoforms, with the white arrowhead pointing to the single human tau isoform. B, Sarkosyl-insoluble tau was extracted from brains (B) and spinal cords (SC) of 5- to 6-month-old transgenic mice and immunoblotted with T14 before (−) and after (+) alkaline phosphatase treatment. M, Mixture of the six recombinant human brain tau isoforms. C, Reactivities of perchloric acid-soluble and sarkosyl-insoluble tau with a panel of 10 different phosphorylation-dependent anti-tau antibodies.
Sarkosyl-insoluble tau protein was extracted from transgenic mouse brain and spinal cord. A strong band of 64 kDa apparent molecular mass was detected by immunoblotting with antibody T14 (Fig.2B). After dephosphorylation, sarkosyl-insoluble tau ran at 52 kDa and aligned with the recombinant 383 aa isoform of human tau, as did dephosphorylated perchloric acid-soluble tau (Fig.2A,B). Sarkosyl-insoluble human tau protein was strongly immunoreactive with all of the phosphorylation-dependent anti-tau antibodies, including AT100 and CP3 (Fig. 2C).
Immunohistochemical and histochemical staining of mutant human tau protein
The cellular distribution of tau protein was investigated in brains and spinal cords from 5- to 6-month-old transgenic mice by using phosphorylation-dependent and phosphorylation-independent anti-tau antibodies (Figs. 3,4; see Fig. 1 for antibody epitopes). Strong labeling of nerve cell bodies and processes was observed in most brain regions. For instance, in the cerebral cortex and the hippocampal formation, numerous nerve cells were immunopositive (Figs.3A,B, 4A). In frontal and temporal cortices, they were concentrated in layers 2, 4, and 5. The largest number of tau-immunoreactive nerve cells was present in the brainstem and spinal cord. Figure 3 shows the staining in a number of regions using the phosphorylation-dependent anti-tau antibody AT8. Overall, the tau staining was either homogenous or had the appearance of circumscribed inclusions. In some cells, particularly in the cerebral cortex, it was distinctly granular.
Fig. 3.
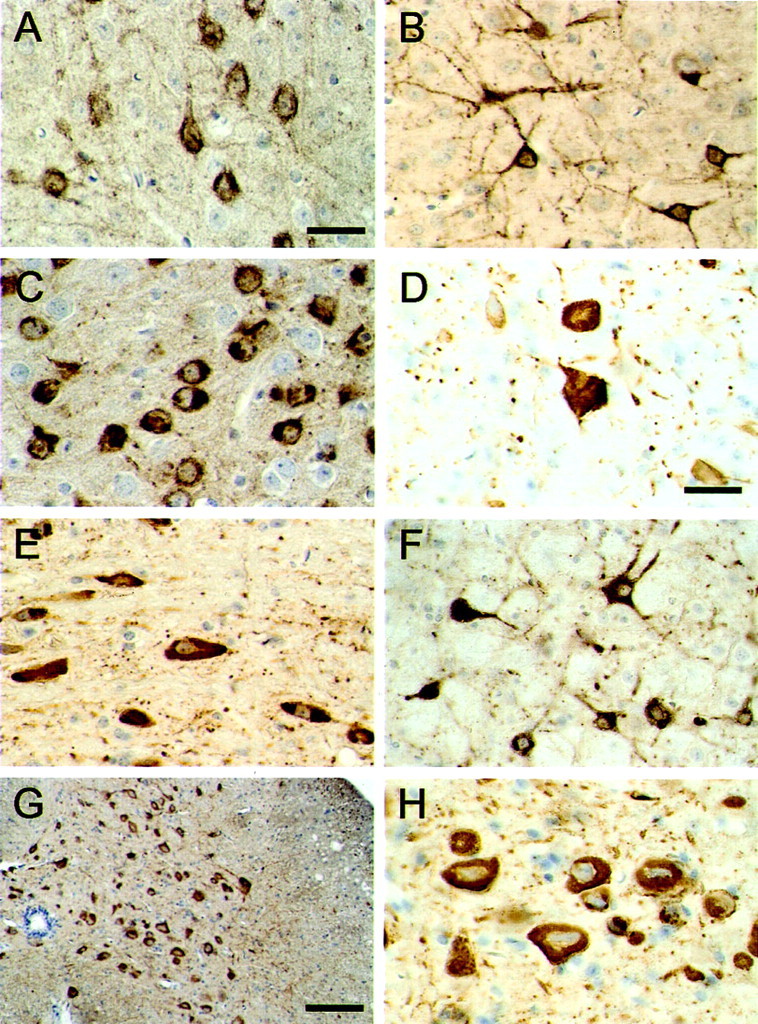
Tau protein immunoreactivity in brains and spinal cords from mice of the human P301S tau line. The phosphorylation-dependent anti-tau antibody AT8 was used to stain the cerebral cortex (A, B), amygdala (C), dentate nucleus of the cerebellum (D), brainstem (E,F), and spinal cord (G,H). The transgenic mice used were 5 months old. Scale bars: A, 40 μm (for A–C,E, F); D, 60 μm (for D, H); G, 250 μm.
Fig. 4.
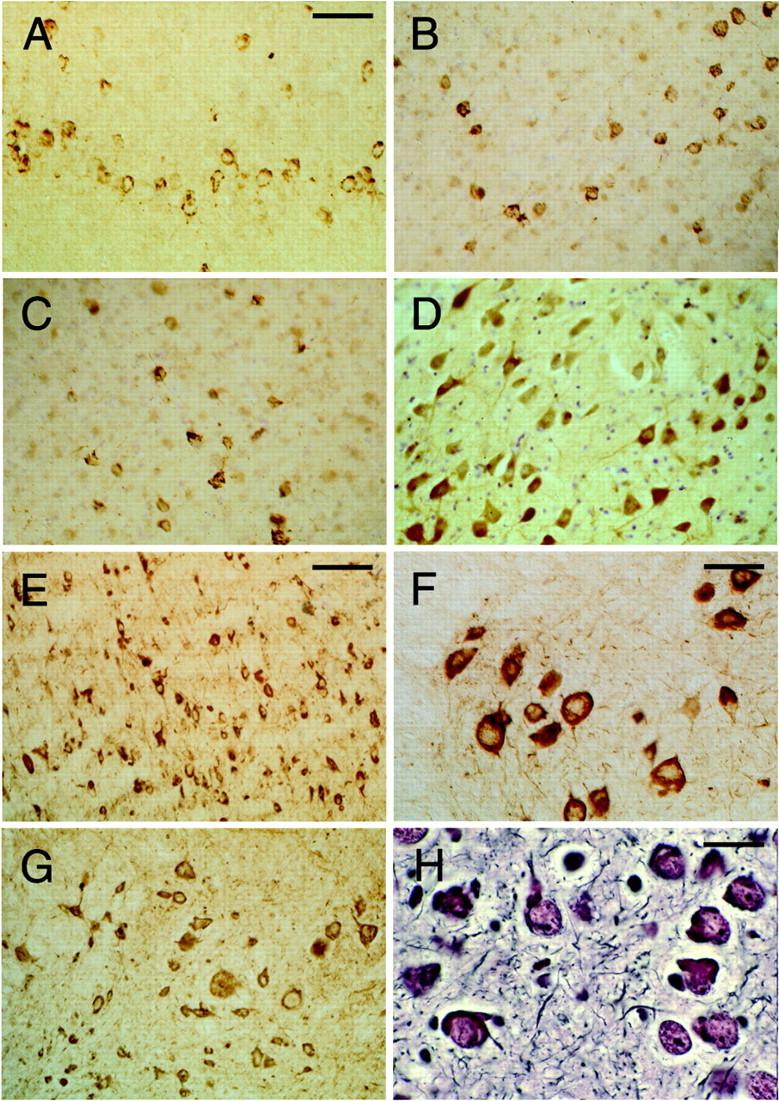
Tau protein immunoreactivity and silver staining in brains and spinal cords from mice of the human P301S tau line. The human-specific, phosphorylation-independent anti-tau antibody T14 was used to stain the cerebral cortex (A) and spinal cord (F). The phosphorylation-dependent anti-tau antibodies AP422 (B) and CP3 (C) were used to stain the cerebral cortex. The phosphorylation-dependent anti-tau antibodies 12E8 (D) and AT180 (E) were used to stain the brainstem. The phosphorylation-dependent anti-tau antibody AT100 was used to stain the spinal cord (G). Bodian silver staining of the amygdala is shown in H. The transgenic mice used were 5 months old. Scale bars: A, 125 μm (forA–D, G); E, 250 μm;F, 100 μm; H, 40 μm.
Immunoreactive nerve cells were observed with the phosphorylation-dependent anti-tau antibodies AT8, AT100, CP3, AT180, 12E8, PHF1, PG5, and AP422. Overall, the largest number of stained cells was observed with AT8, followed by AP422, AT180, and PG5. Fewer cells were labeled by AT100 and CP3, with the smallest number of cells being stained by 12E8 and PHF1. Antibody AD2 gave a staining pattern similar to that seen for PHF1. Regional differences were observed. For instance, strong and widespread staining for 12E8 was present in the spinal cord but not in the cerebral cortex. Staining with antibodies AP422, CP3, 12E8, AT180, and AT100 is illustrated in Figure 4. The phosphorylation-independent, human-specific anti-tau antibody T14 labeled numerous nerve cells (Fig.4A,F), as did Alz-50, BR133, and BR134. In contrast, antibody MT1, which is specific for murine tau, gave only diffuse neuropil staining. Antibodies directed against ubiquitin, parkin, the 20S proteasome, heparan sulfate, heat shock protein 25, heat shock protein 70, αB-crystallin, cdk5, and the p35 activator of cdk5 failed to stain the tau-positive cells. Most nerve cells were stained by the anti-phospho-GSK3 antibody, regardless of whether they were immunoreactive for human tau. No staining for β-amyloid was observed. Sections of brain and spinal cord from transgenic mice were also stained with the Bodian silver stain and thioflavin S. Numerous nerve cells were silver positive (Fig.4H) and fluorescent with thioflavin S (Fig.5). The thioflavin S fluorescence was weak (Fig. 5A–C) and resembled that given by the Pick bodies of sporadic Pick's disease (Fig. 5D).
Fig. 5.

Thioflavin S fluorescence in brains and spinal cords from mice of the human P301S tau line. Amygdala (A), brainstem (B), and spinal cord (C) from 6-month-old transgenic mice.D, Entorhinal cortex from a case of Pick's disease. Note the weak green fluorescence against ayellowish–green background in A–C and the similarly weak fluorescence intensity of Pick bodies against agreen background in D. Some of the positive cells in A–C are indicated byarrows, as are two positive Pick bodies inD. Scale bar, 80 μm.
Electron microscopy of tau filaments
Large numbers of abnormal filaments were observed in the cytoplasm and processes of nerve cells from 5- to 6-month-old transgenic mice. Representative sections of cerebral cortex, brainstem, and spinal cord are shown in Figure 6. The filaments were oriented randomly and were not membrane bound. Most filaments appeared ribbon like, with approximate diameters of 5–15 nm. According to immunoelectron microscopy on tissue sections, the filaments were strongly labeled by the phosphorylation-dependent anti-tau antibody AT8 (Fig. 6C,D).
Fig. 6.
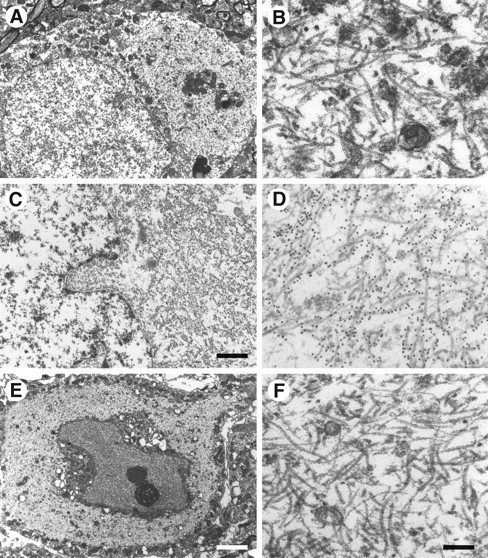
Electron microscopy and immunoelectron microscopy of nerve cells in brains and spinal cords from mice of the human P301S tau line. A, B, Cerebral cortex;C, D, brainstem; E,F, spinal cord. B, D,F, Higher magnifications of parts of the cytoplasmic regions from A, C, and E. Note the large numbers of abnormal filaments in the cytoplasm and apical dendrite. The electron micrographs in C andD show immunogold labeling of filaments using the phosphorylation-dependent anti-tau antibody AT8. Scale bars:C, 1.5 μm; E, 5.5 μm (forA, E); F, 300 nm (forB, D, F).
Isolated filaments were also characterized using sarkosyl-insoluble preparations extracted from brains and spinal cords of transgenic mice (Fig. 7). Labeling with antibodies against tau (see Fig. 1 for antibody epitopes) revealed reasonably abundant tau-containing filaments of rather irregular appearance but predominantly of two morphologies (Fig. 7). The majority resembled “half-twisted” ribbons, with an apparent width varying between 5 and 10 nm and with a very variable spacing between cross-overs. A minority of filaments (Fig. 7I,L) were more like Alzheimer-paired helical filaments with a cross-over spacing of ∼70–80 nm. The filaments were strongly labeled by phosphorylation-independent antibodies BR133 and BR134, raised against the N- and C-terminal regions of tau, but not by BR135, whose epitope lies in the repeat region. They were also labeled by the human tau-specific antibody T14. In contrast, only a small number of filaments were labeled by the mouse tau-specific antibody MT1. The filaments were not labeled by an anti-ubiquitin antibody. The filaments were labeled by a range of phosphorylation-dependent anti-tau antibodies but not 12E8, whose epitope lies in the repeat region. Similar labeling patterns were found, where tested, for filaments from brain and spinal cord.
Fig. 7.
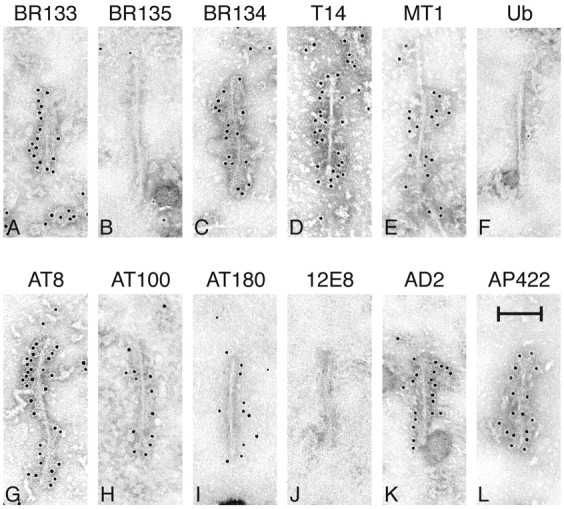
Immunoelectron microscopy of isolated filaments prepared by sarkosyl extraction of brains and spinal cords from mice of the human P301S tau line. A–E, Phosphorylation-independent anti-tau antibodies; F, anti-ubiquitin antibody (Ub); G–L, phosphorylation-dependent anti-tau antibodies. In each case, the antibody is named above the panel. The filaments in C and G came from spinal cord, the rest from brain. Antibodies BR135, Ub, and 12E8 did not label the filaments. Most filaments resembled half-twisted ribbons (A–H, J, K), whereas some filaments were reminiscent of paired helical filaments (I, L). The transgenic mice used were 5–6 months old. Scale bar, 100 nm.
Nerve cell numbers in the spinal cord
In ventral gray matter from 6-month-old homozygous human P301S tau mice, a reduction in the number of motor neurons and an increase in the number of glial cells were observed (Fig.8A,B). Neuronal cell bodies were often swollen or severely atrophic. In some instances, pyknotic neurons were surrounded by glial cells, suggestive of neuronophagia. Nerve cell counts showed a 49% reduction in the number of motor neurons in the anterior horn of the spinal cord from transgenic mice compared with age-matched controls (Fig.8C). Occasional severely atrophic neurons may not have been counted, because they lacked a visible nucleolus. The estimated mean was 8.14/mm2 in control mice and 4.02/mm2 in transgenic mice (p < 0.0001), with an estimated difference of 4.12/mm2 (95% confidence interval of 2.8–5.4). In contrast, the neuronal density in the substantia gelatinosa was not significantly different between control and transgenic mice (p > 0.7). The respective estimated means were 502.6 and 494.3, with an estimated difference of 8.3/mm2 (95% confidence interval of −38.3 to −54.9). The reduction in the number of motor neurons was accompanied by a reactive astrocytosis, as detected by a marked increase in GFAP staining (Fig.9A,B). In hindlimb skeletal muscle from transgenic mice, groups of atrophic, angulated muscle fibers were frequently observed amid normal-looking fibers, indicative of denervation atrophy (Fig. 9C).
Fig. 8.

Nerve cell loss in spinal cords from mice of the human P301S tau line. A, B, Hematoxylin and eosin-stained sections of the ventral gray matter of the spinal cord (level L2–L3) from a 6-month-old control mouse (A) and a transgenic mouse (B) of the same age. Swollen, abnormal material-containing motor neurons are indicated byarrows. Arrowheads point to atrophic motor neurons, with dashed arrows pointing to pyknotic cells that are surrounded by glial cells, suggestive of neuronophagia.C, Graph showing the density of motor neurons (expressed as number of neurons per millimeter square) in the anterior horn of the lumbar spinal cord from 6-month-old control and human P301S tau transgenic mice. Nerve cell numbers were determined in five consecutive sections from each animal, with the density of motor neurons from each section being represented by a circle. The results are expressed as the means ± SEM (n = 3); *p < 0.0001. Scale bar: A, 60 μm (for A, B).
Fig. 9.
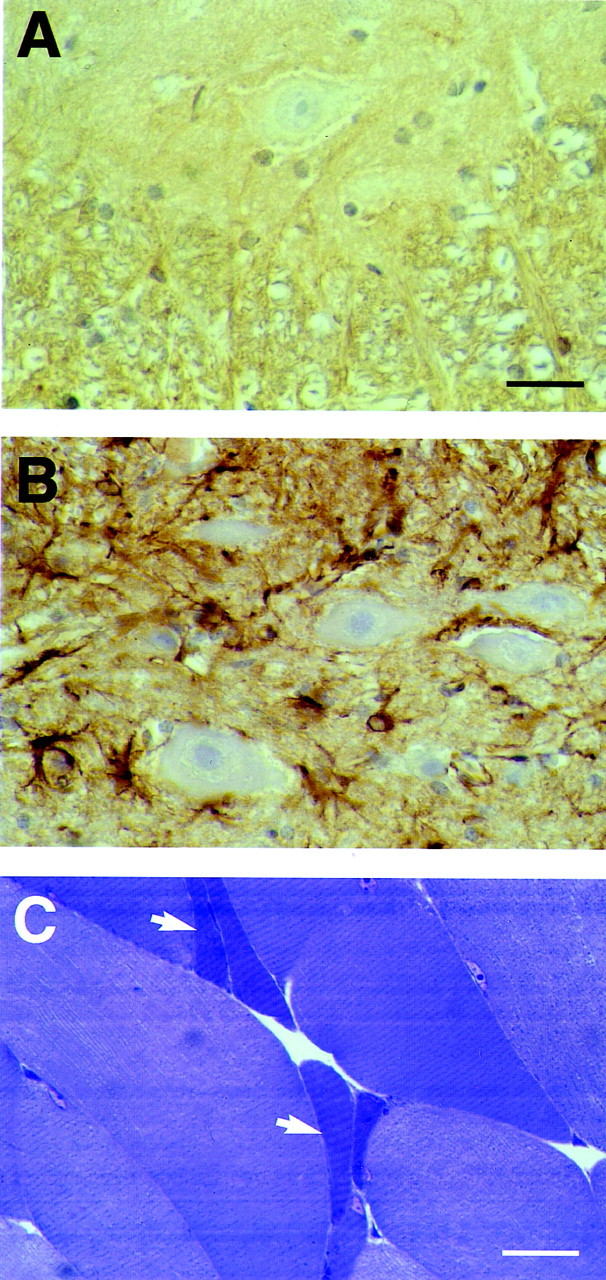
Astrocytosis in the spinal cord and denervation atrophy in skeletal muscle from mice of the human P301S tau line.A, B, The ventral gray matter of the spinal cord (level L2–L3) from a 6-month-old control mouse (A) and a transgenic mouse (B) of the same age was stained with an anti-glial fibrillary acidic protein antibody. Note the weak immunoreactivity in A and the much stronger staining inB, as reflected in a large number of immunopositive astrocytic cell bodies and coarse processes. C, Hindlimb skeletal muscle from a transgenic mouse was stained with toluidine blue. Groups of atrophic, angulated muscle fibers are indicated byarrows. Scale bars: A, 50 μm (forA, B); C, 60 μm.
Staining with apoptosis markers
Apoptotic profiles, as identified by ISEL and immunoreactivity of activated caspase-3 and cleaved α-fodrin, were extremely rare or absent in brains and spinal cords from 5- to 6-month-old transgenic mice. (Fig.10A,C,E). The number of caspase-3-positive cells never exceeded one or two in the entire brain. These cells, which were found at the same frequency in brains from control mice, were located primarily in the cerebral cortex and hippocampus. Brain sections from 8-d-old citron kinase−/− mice (Di Cunto et al., 2000) and 1-d-old control mice were used as positive controls. They showed apoptotic cells in the external granular layer of the cerebellum (Fig.10B,D,F).
Fig. 10.
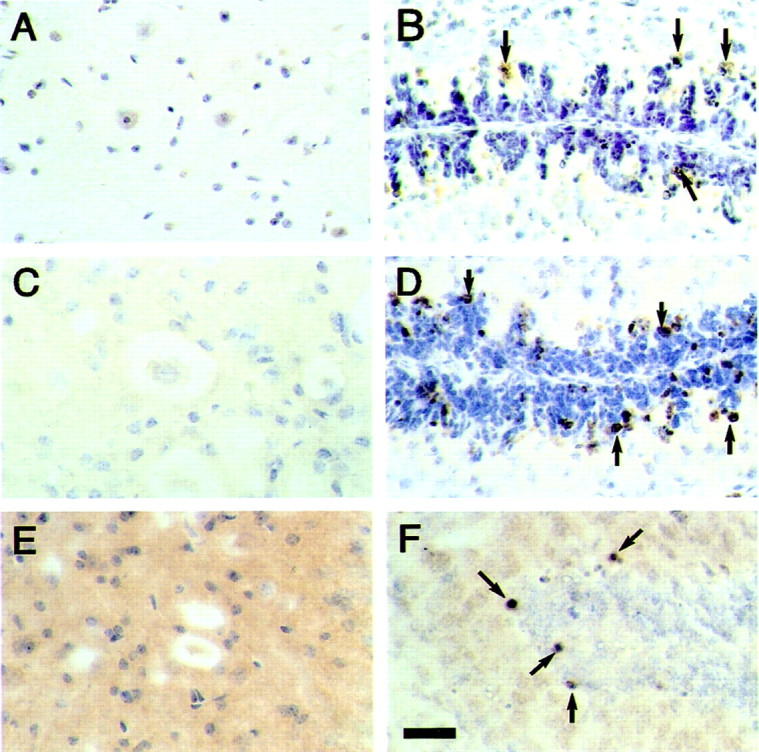
ISEL, staining of activated caspase-3, and staining of cleaved α-fodrin in spinal cords from mice of the human P301S tau line. A, B, ISEL;C, D, staining of activated caspase-3;E, F, staining of cleaved α-fodrin. The sections in A, C, and Eare from spinal cords of 6-month-old human P301S tau transgenic mice. The sections in B and D are from the cerebellum of 8-d-old citron kinase−/− mice, and the section in F is from the cerebellum of a 1-d-old control mouse. No specific reaction product is seen inA, C, and E. Some of the immunopositive cells in B, D, andF are indicated by arrows. Scale bar:F, 50 μm (for A–F).
Staining of activated MAP kinase family members and the prolyl isomerase Pin1
The colocalization of activated MAP kinase family members and hyperphosphorylated tau was investigated by double labeling immunofluorescence in brains and spinal cords from 5- to 6-month-old transgenic mice. Antibodies specific for activated MAP kinase, phospho-JNK, and phospho-p38 labeled the nerve cells that were tau positive (Fig.11A–F). The same was true of an antibody specific for phospho-MAP kinase kinase 3/6, the upstream activator of the p38 MAP kinase family (data not shown). The observed staining did not result from a cross-reaction with hyperphosphorylated tau, because the phosphorylation-dependent protein kinase antibodies failed to recognize the sarkosyl-insoluble 64 kDa tau band from brains and spinal cords of transgenic mice (data not shown). By immunoblotting, the antibody specific for phospho-p38 MAP kinase did not distinguish between p38α/stress-activated protein kinase (SAPK)2a, p38β/SAPK2b, p38γ/SAPK3, and p38δ/SAPK4 (data not shown). Staining of the prolyl isomerase Pin1 was only observed in a small minority (<1%) of AT100-positive cells in the cerebral cortex, in which some cytoplasmic granules were immunoreactive (Fig.11G,H). Pin1 staining was not seen in other brain regions or in the spinal cord.
Fig. 11.

Staining of activated MAP kinase family members (in red) and the prolyl isomerase Pin1 (inred) in cerebral cortex from mice of the human P301S tau line. Colocalization with hyperphosphorylated tau protein (ingreen) is shown. A, Anti-activated MAP kinase; C, anti-phospho-JNK; E, anti-phospho-p38; G, anti-Pin1. B, Staining with anti-tau antibody BR134 of the tissue section shown inA. D, F, Staining with anti-tau antibody PHF1 of the tissue sections shown in Cand E. H, Staining with anti-tau antibody AT100 of the tissue section shown in G. Scale bars:A, 15 μm (for A–F);G, 15 μm (for G,H).
DISCUSSION
We describe a transgenic mouse line that expresses in nerve cells four-repeat human tau protein with the P301S mutation. At 5–6 months of age, homozygous animals developed a neurological phenotype dominated by a severe paraparesis. By light microscopy, nerve cells in the brain and spinal cord were strongly immunoreactive for hyperphosphorylated human tau but not for mouse tau. Many nerve cells were also strongly silver positive and weakly fluorescent for thioflavin S. The largest number of tau-positive nerve cells was present in the brainstem and spinal cord. Numerous positive cells were also observed in other brain regions, including the cerebral cortex and hippocampus.
By electron microscopy on tissue sections, abundant filaments made of hyperphosphorylated tau protein were detected throughout the brain and in the spinal cord. The majority of filaments resembled the half-twisted ribbons described previously in some cases of FTDP-17 (Spillantini et al., 1998b; Mirra et al., 1999; Rizzini et al., 2000;Neumann et al., 2001). A minority of filaments were reminiscent of the paired helical filaments of Alzheimer's disease. Isolated filaments were decorated by antibodies directed against the N and C termini of tau but not by BR135, an antibody specific for the microtubule-binding repeat region, indicating that the repeat region of tau forms part of the core of the filaments. Identical findings have been reported previously for synthetic tau filaments (Goedert et al., 1996) and for filaments isolated from Alzheimer's disease and FTDP-17 brains (Goedert et al., 1992a, 1994; Hasegawa et al., 1996; Spillantini et al., 1997). Isolated filaments from transgenic mouse brain and spinal cord were made of mutant human tau and decorated by a large number of phosphorylation-dependent anti-tau antibodies. The one exception was antibody 12E8, whose epitope lies in the repeat region (Seubert et al., 1995) and was therefore inaccessible in the intact filaments. Isolated tau filaments were not decorated by anti-ubiquitin antibodies, which also failed to stain tissue sections from transgenic mice. This is consistent with previous findings which demonstrated that the ubiquitination of tau in Alzheimer's disease occurs after filament assembly (Goedert et al., 1992a; Morishima-Kawashima et al., 1993).
The morphological findings were mirrored at the biochemical level. Sarkosyl-insoluble tau from transgenic mouse brain and spinal cord ran as a hyperphosphorylated 64 kDa band that was labeled by all anti-human tau antibodies tested, including BR135 and 12E8. However, this band was not labeled by antibody MT1. In human tauopathies, the 383 aa tau isoform also runs as a hyperphosphorylated 64 kDa band (Goedert et al., 1992a; Mulot et al., 1994). The biochemical and morphological characteristics of sarkosyl-insoluble tau from the transgenic mice were therefore identical to those observed in human tauopathies. Abundant tau filaments have also been described in sections of brainstems and spinal cords from mice transgenic for human P301L tau (Lewis et al., 2000) but not in other mouse lines transgenic for human tau mutants (Götz et al., 2001a,b; Lim et al., 2001; Tanemura et al., 2001,2002). Unlike the present findings, tau inclusions were ubiquitin positive by light microscopy. The characteristics of isolated tau filaments were not reported (Lewis et al., 2000).
Perchloric acid-soluble tau from transgenic mouse brain and spinal cord ran as a broad band of 52–58 kDa that was immunoreactive with all phosphorylation-dependent anti-tau antibodies, except AT100 and CP3. In conjunction with our previous work (Probst et al., 2000), this provides the first demonstration of the existence of a pool of soluble tau protein phosphorylated at many but not all sites before filament assembly. It also indicates that immunoreactivity for phospho-S214 closely mirrors the presence of filaments, suggesting that phosphorylation of this site occurs after filament assembly. In transgenic mice, AT100 stained only a subset of the nerve cells that were immunoreactive with other antibodies, such as T14 or AT8, indicating that only this subset of nerve cells contained tau filaments.
Neurodegeneration was present in the spinal cord of 6-month-old transgenic mice, as indicated by a 49% reduction in the number of motor neurons. Both abnormally enlarged and severely atrophic motor neurons were observed. In some instances, the latter were surrounded by glial cells, suggestive of neuronophagia. Moreover, astrocytosis in spinal cord and denervation atrophy in skeletal muscle were in evidence. It remains to be determined whether nerve cells were also lost in the brain. In the spinal cord, tau filament formation thus correlated with nerve cell death. The same is true of nerve cells in the human tauopathies (Lee et al., 2001). A similar loss in the number of motor neurons was also reported in transgenic mice from the human P301L tau line with filaments (Lewis et al., 2000). In mice transgenic for wild-type human tau, no tau filaments or nerve cell loss was observed (Ishihara et al., 1999; Spittaels et al., 1999; Probst et al., 2000). In contrast, in Drosophila melanogaster expressing either wild-type or mutant human tau, nerve cell death by apoptosis was found in the apparent absence of tau filaments (Wittmann et al., 2001;Jackson et al., 2002).
In the human P301S tau line, no evidence for apoptosis was obtained when DNA fragmentation and staining for activated caspase-3 and cleaved α-fodrin were studied. This is in contrast to mice transgenic for human P301L tau, where the presence of terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling-positive nerve cells was reported (Götz et al., 2001a; Ho et al., 2001). However, the present findings are in line with work on the human tauopathies, where DNA fragmentation and staining for activated caspase-3 were not detected (Migheli et al., 1994; Atzori et al., 2001;Ferrer et al., 2001a).
In human diseases, tau inclusions are known to colocalize with activated MAP kinase family members, in particular, SAPKs (Hensley et al., 1999; Perry et al., 1999; Zhu et al., 2000; Atzori et al., 2001;Ferrer et al., 2001a,b). Similar results were obtained in the human P301S tau line, where activated MAP kinase, JNK, and p38 colocalized with hyperphosphorylated tau. The same was not true of other protein kinases, such as GSK3 and cdk5. MAP kinase and SAPKs phosphorylate tau at many sites in vitro (Drewes et al., 1992; Goedert et al., 1992b, 1997; Reynolds et al., 1997), suggesting that they may be involved in the hyperphosphorylation of tau and that specific inhibitors of these enzymes (Cohen, 2002) might be therapeutically beneficial. It remains to be determined whether activation of MAP kinase and SAPKs precedes the assembly of tau into filaments. Staining for activated MAP kinase family members was not observed in the transgenic mouse line ALZ17 (Probst et al., 2000), which expresses wild-type human tau protein but does not develop abundant tau filaments (our unpublished observations).
Activation of SAPKs is likely to have additional effects. Both JNK and p38 have been linked to the induction of apoptosis (Xia et al., 1995), although their effects are strongly cell and context dependent (Chang and Karin, 2001). The present findings, together with recent work on the human tauopathies (Atzori et al., 2001), indicate that in diseases with a filamentous tau pathology, SAPKs do not activate or regulate an apoptotic cascade. Like MAP kinase family members, the prolyl isomerase Pin1 has been reported to colocalize with filamentous tau deposits in Alzheimer's disease brain (Lu et al., 1999). It was proposed that this may contribute to nerve cell death through a reduction in the level of functional Pin1. However, in the mouse line expressing human P301S tau, Pin1 was only present in a small minority of tau-positive nerve cells in the cerebral cortex, where it localized to cytoplasmic granules.
In conclusion, the transgenic mouse line described here exhibits the essential features of a human tauopathy, including the formation of abundant filaments made of hyperphosphorylated tau protein and nerve cell degeneration. Although in human cases with the P301S mutation inTau, both nerve and glial cells in the brain are affected (Bugiani et al., 1999), in the mouse line, tau deposits are seen only in nerve cells. The transgenic mice do not exhibit the selective pattern of nerve cell degeneration characteristic of the human tauopathy, because they show spinal cord as well as widespread brain pathology. These differences in selectivity between mouse model and human disease may have resulted from our use of the murinethy1 promoter to drive expression of mutant human tau, the expression of a single isoform of mutant human tau protein on a wild-type mouse tau background, or a different neuronal vulnerability to mutant human tau in mice and humans. This notwithstanding, the mouse line expressing human P301S tau will be of great value for a better understanding of the molecular mechanisms by which mutant tau protein causes the dysfunction and death of nerve cells. It may for instance help to establish whether hyperphosphorylation of tau leads to filament assembly and whether the accumulation of tau filaments results in the demise of nerve cells. This may in turn lead to the design of new therapeutic strategies aimed at preventing tau dysfunction.
B.A., E.I., and M.T. contributed equally to this work.
This work was supported by the United Kingdom Alzheimer's Research Trust, the United Kingdom Medical Research Council, and United States Public Health Service Grants P30 AG10133 and R01 NS14426. We thank C. Alyea, R. Richardson, and B. Dupree for technical help and Drs. S. Hiu and A. Perkins for statistical assistance.
Correspondence should be addressed to Dr. Michel Goedert, Medical Research Council Laboratory of Molecular Biology, Hills Road, Cambridge CB2 2QH, United Kingdom. E-mail: mg@mrc-lmb.cam.ac.uk.
REFERENCES
- 1.Andreadis A, Brown MW, Kosik K. Structure and novel exons of the human tau gene. Biochemistry. 1992;31:10626–10633. doi: 10.1021/bi00158a027. [DOI] [PubMed] [Google Scholar]
- 2.Atzori C, Ghetti B, Piva R, Srinivasan AN, Zolo P, Delisle MB, Mirra SS, Migheli A. Activation of the JNK/p38 pathway occurs in diseases characterized by tau protein pathology and is related to tau phosphorylation but not to apoptosis. J Neuropathol Exp Neurol. 2001;60:1190–1197. doi: 10.1093/jnen/60.12.1190. [DOI] [PubMed] [Google Scholar]
- 3.Buée-Scherrer V, Condamines O, Mourton-Gilles C, Jakes R, Goedert M, Pau B, Delacourte A. AD2, a phosphorylation-dependent monoclonal antibody directed against tau proteins found in Alzheimer's disease. Brain Res Mol Brain Res. 1996;39:79–88. doi: 10.1016/0169-328x(96)00003-4. [DOI] [PubMed] [Google Scholar]
- 4.Bugiani O, Murrell JR, Giaccone G, Hasegawa M, Ghigo G, Tabaton M, Morbin M, Primavera A, Carella F, Solaro C, Grisoli M, Savoiardo M, Spillantini MG, Tagliavini F, Goedert M, Ghetti B. Frontotemporal dementia and corticobasal degeneration in a family with a P301S mutation in tau. J Neuropathol Exp Neurol. 1999;58:667–677. doi: 10.1097/00005072-199906000-00011. [DOI] [PubMed] [Google Scholar]
- 5.Carmel G, Mager EM, Binder LI, Kuret J. The structural basis of monoclonal antibody Alz-50's selectivity for Alzheimer's disease pathology. J Biol Chem. 1996;271:32789–32795. doi: 10.1074/jbc.271.51.32789. [DOI] [PubMed] [Google Scholar]
- 6.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 7.Cohen P. Protein kinases–the major drug targets of the twenty-first century? Nat Rev Drug Discov. 2002;1:309–315. doi: 10.1038/nrd773. [DOI] [PubMed] [Google Scholar]
- 8.Crowther RA. Straight and paired helical filaments in Alzheimer disease have a common structural unit. Proc Natl Acad Sci USA. 1991;88:2288–2292. doi: 10.1073/pnas.88.6.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Di Cunto F, Imarisio S, Hirsch E, Broccoli V, Bulfone A, Migheli A, Atzori C, Turco E, Triolo R, Dotto GP, Silengo L, Altruda F. Defective neurogenesis in citron kinase knockout mice by altered cytokinesis and massive apoptosis. Neuron. 2000;28:115–127. doi: 10.1016/s0896-6273(00)00090-8. [DOI] [PubMed] [Google Scholar]
- 10.Drewes G, Lichtenberg-Kraag B, Döring F, Mandelkow EM, Biernat J, Dorée M, Mandelkow E. Mitogen activated protein (MAP) kinase transforms tau protein into an Alzheimer-like state. EMBO J. 1992;11:2131–2138. doi: 10.1002/j.1460-2075.1992.tb05272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ferrer I, Blanco R, Carmona M, Ribera R, Goutan E, Puig B, Rey MJ, Cardozo A, Vinals F, Ribalta T. Phosphorylated MAP kinase (ERK1, ERK2) expression is associated with early tau deposition in neurones and glial cells, but not with increased nuclear DNA vulnerability and cell death, in Alzheimer disease, Pick's disease, progressive supranuclear palsy and corticobasal degeneration. Brain Pathol. 2001a;11:144–158. doi: 10.1111/j.1750-3639.2001.tb00387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferrer I, Blanco R, Carmona M, Puig B. Phosphorylated mitogen-activated protein kinase (MAPK/ERK-P), protein kinase of 38 kDa (p38-P), stress-activated protein kinase (SAPK/JNK-P), and calcium/calmodulin-dependent kinase II (CaM kinase II) are differentially expressed in tau deposits in neurons and glial cells in tauopathies. J Neural Transm. 2001b;108:1397–1415. doi: 10.1007/s007020100016. [DOI] [PubMed] [Google Scholar]
- 13.Goedert M, Jakes R. Expression of separate isoforms of human tau protein: correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990;9:4225–4230. doi: 10.1002/j.1460-2075.1990.tb07870.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: identification as the microtubule-associated protein tau. Proc Natl Acad Sci USA. 1988;85:4051–4055. doi: 10.1073/pnas.85.11.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in brain. EMBO J. 1989a;8:393–399. doi: 10.1002/j.1460-2075.1989.tb03390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: sequences and localization in neurofibrillary tangles of Alzheimer's disease. Neuron. 1989b;3:519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 17.Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of Alzheimer paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. 1992a;8:159–168. doi: 10.1016/0896-6273(92)90117-v. [DOI] [PubMed] [Google Scholar]
- 18.Goedert M, Cohen ES, Jakes R, Cohen P. p42 MAP kinase phosphorylation sites in microtubule-associated protein tau are dephosphorylated by protein phosphatase 2A1. Implications for Alzheimer's disease. FEBS Lett. 1992b;312:95–99. doi: 10.1016/0014-5793(92)81418-l. [DOI] [PubMed] [Google Scholar]
- 19.Goedert M, Jakes R, Crowther RA, Cohen P, Vanmechelen E, Vandermeeren M, Cras P. Epitope mapping of monoclonal antibodies to the paired helical filaments of Alzheimer's disease: identification of phosphorylation sites in tau protein. Biochem J. 1994;301:871–877. doi: 10.1042/bj3010871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goedert M, Jakes R, Vanmechelen E. Monoclonal antibody AT8 recognises tau protein phosphorylated at both serine 202 and threonine 205. Neurosci Lett. 1995;189:167–170. doi: 10.1016/0304-3940(95)11484-e. [DOI] [PubMed] [Google Scholar]
- 21.Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA. Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature. 1996;383:550–553. doi: 10.1038/383550a0. [DOI] [PubMed] [Google Scholar]
- 22.Goedert M, Hasegawa M, Jakes R, Lawler S, Cuenda A, Cohen P. Phosphorylation of microtubule-associated protein tau by stress-activated protein kinases. FEBS Lett. 1997;409:57–62. doi: 10.1016/s0014-5793(97)00483-3. [DOI] [PubMed] [Google Scholar]
- 23.Goedert M, Jakes R, Crowther RA. Effects of frontotemporal dementia FTDP-17 mutations on heparin-induced assembly of tau filaments. FEBS Lett. 1999;450:306–311. doi: 10.1016/s0014-5793(99)00508-6. [DOI] [PubMed] [Google Scholar]
- 24.Goldstein H. Multilevel statistical models. Edward Arnold Publishers; London: 1995. [Google Scholar]
- 25.Götz J, Probst A, Spillantini MG, Schäfer T, Jakes R, Bürki K, Goedert M. Somatodendritic localisation and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. EMBO J. 1995;14:1304–1313. doi: 10.1002/j.1460-2075.1995.tb07116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Götz J, Chen F, Barmettler R, Nitsch RM. Tau filament formation in transgenic mice expressing P301L tau. J Biol Chem. 2001a;276:529–534. doi: 10.1074/jbc.M006531200. [DOI] [PubMed] [Google Scholar]
- 27.Götz J, Tolnay M, Barmettler R, Chen F, Probst A, Nitsch RM. Oligodendroglial tau filament formation in transgenic mice expressing G272V tau. Eur J Neurosci. 2001b;13:2131–2140. doi: 10.1046/j.0953-816x.2001.01604.x. [DOI] [PubMed] [Google Scholar]
- 28.Hasegawa M, Jakes R, Crowther RA, Lee VMY, Ihara Y, Goedert M. Characterization of mAb AP422, a novel phosphorylation-dependent monoclonal antibody against tau protein. FEBS Lett. 1996;384:25–30. doi: 10.1016/0014-5793(96)00271-2. [DOI] [PubMed] [Google Scholar]
- 29.Hasegawa M, Smith MJ, Goedert M. Tau proteins with FTDP-17 mutations have a reduced ability to promote microtubule assembly. FEBS Lett. 1998;437:207–210. doi: 10.1016/s0014-5793(98)01217-4. [DOI] [PubMed] [Google Scholar]
- 30.Hensley L, Floyd RA, Zheng NY, Nael R, Robinson KA, Nguyen X, Pye QN, Stewart CA, Geddes J, Markesbery WR, Patel E, Johnson GVW, Bing G. p38 Kinase is activated in the Alzheimer's disease brain. J Neurochem. 1999;72:2053–2058. doi: 10.1046/j.1471-4159.1999.0722053.x. [DOI] [PubMed] [Google Scholar]
- 31.Ho L, Xiang Z, Mukherjee P, Zhang W, De Jesus N, Mirjani M, Yemul S, Pasinetti GM. Gene expression profiling of the tau mutant (P301L) transgenic mouse brain. Neurosci Lett. 2001;310:1–4. doi: 10.1016/s0304-3940(01)02044-4. [DOI] [PubMed] [Google Scholar]
- 32.Hong M, Zhukareva V, Vogelsberg-Ragaglia V, Wszolek Z, Reed L, Miller BL, Geschwind DH, Bird TD, McKeel D, Goate A, Morris JC, Wilhelmsen KC, Schellenberg GD, Trojanowski JQ, Lee VMY. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–1917. doi: 10.1126/science.282.5395.1914. [DOI] [PubMed] [Google Scholar]
- 33.Hutton M, Lendon CL, Rizzu P, Baker M, Froelich S, Houlden H, Pickering-Brown S, Chakraverty S, Isaacs A, Grover A, Hackett J, Adamson J, Lincoln S, Dickson D, Davies P, Petersen RC, Stevens M, de Graaff E, Wauters E, van Baren J. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393:702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 34.Ishihara T, Hong M, Zhang B, Nakagawa Y, Lee MK, Trojanowski JQ, Lee VMY. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron. 1999;24:751–762. doi: 10.1016/s0896-6273(00)81127-7. [DOI] [PubMed] [Google Scholar]
- 35.Jackson GR, Wiedau-Pazos M, Sang T-K, Wagle N, Brown CA, Massachi S, Geschwind DH. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathology in Drosophila. Neuron. 2002;34:509–519. doi: 10.1016/s0896-6273(02)00706-7. [DOI] [PubMed] [Google Scholar]
- 36.Jicha GA, Weaver C, Lane E, Vianna C, Kress Y, Rockwood J, Davies P. cAMP-dependent protein kinase phosphorylations on tau in Alzheimer's disease. J Neurosci. 1999;19:7486–7494. doi: 10.1523/JNEUROSCI.19-17-07486.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kampers T, Pangalos M, Geerts H, Wiech H, Mandelkow EM, Mandelkow E. Assembly of paired helical filaments from mouse tau: implications for the neurofibrillary pathology in transgenic mouse models of Alzheimer's disease. FEBS Lett. 1999;451:39–44. doi: 10.1016/s0014-5793(99)00522-0. [DOI] [PubMed] [Google Scholar]
- 38.Kosik KS, Orecchio LD, Binder LI, Trojanowski JQ, Lee VMY, Lee G. Epitopes that span the tau molecule are shared with paired helical filaments. Neuron. 1988;1:817–825. doi: 10.1016/0896-6273(88)90129-8. [DOI] [PubMed] [Google Scholar]
- 39.Lee VMY, Goedert M, Trojanowski JQ. Neurodegenerative Tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 40.Lewis J, McGowan E, Rockwood J, Melrose H, Nacharaju P, van Slegtenhorst M, Gwinn-Hardy K, Murphy MP, Baker M, Yu X, Duff K, Hardy J, Corral A, Lin WL, Yen SH, Dickson DW, Davies P, Hutton M. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- 41.Lim F, Hernandez F, Lucas JJ, Gomez-Ramos P, Moran MA, Avila J. FTDP-17 mutations in tau transgenic mice provoke lysosomal abnormalities and tau filaments in forebrain. Mol Cell Neurosci. 2001;18:702–714. doi: 10.1006/mcne.2001.1051. [DOI] [PubMed] [Google Scholar]
- 42.Lu PJ, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399:784–788. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- 43.Lüthi A, van der Putten H, Botteri FM, Mansuy IM, Meins M, Frey U, Sansig G, Portet C, Schmutz M, Schröder M, Nitsch C, Laurent JP, Monard D. Endogenous serine protease inhibitor modulates epileptic activity and hippocampal long-term potentiation. J Neurosci. 1997;17:4688–4698. doi: 10.1523/JNEUROSCI.17-12-04688.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Migheli A, Cavalla P, Marino S, Schiffer D. A study of apoptosis in normal and pathologic nervous tissue after in situ end-labeling of DNA strand breaks. J Neuropathol Exp Neurol. 1994;53:606–616. doi: 10.1097/00005072-199411000-00008. [DOI] [PubMed] [Google Scholar]
- 45.Migheli A, Piva R, Wei J, Attanasio A, Casolino S, Hodes ME, Dlouhy SR, Bayer SA, Ghetti B. Diverse cell death pathways result from a single missense mutation in weaver mouse. Am J Pathol. 1997;151:1629–1638. [PMC free article] [PubMed] [Google Scholar]
- 46.Mirra SS, Murrell JR, Gearing M, Spillantini MG, Goedert M, Crowther RA, Levey AI, Jones R, Green J, Shoffner JM, Wainer BH, Schmidt ML, Trojanowski JQ, Ghetti B. Tau pathology in a family with dementia and a P301L mutation in tau. J Neuropathol Exp Neurol. 1999;58:335–345. doi: 10.1097/00005072-199904000-00004. [DOI] [PubMed] [Google Scholar]
- 47.Morishima-Kawashima M, Hasegawa M, Takio K, Suzuki M, Yoshida H, Titani K, Ihara Y. Ubiquitin is conjugated with amino-terminally processed tau in paired helical filaments. Neuron. 1993;10:1151–1160. doi: 10.1016/0896-6273(93)90063-w. [DOI] [PubMed] [Google Scholar]
- 48.Morris HR, Khan MN, Janssen JC, Brown JR, Perez-Tur J, Baker M, Ozansoy M, Hardy J, Hutton M, Wood NW, Lees AJ, Revesz T, Lantos P, Rossor MN. The genetic and pathological classification of familial frontotemporal dementia. Arch Neurol. 2001;58:1813–1816. doi: 10.1001/archneur.58.11.1813. [DOI] [PubMed] [Google Scholar]
- 49.Mulot SFC, Hughes K, Woodgett JR, Anderton BH, Hanger DP. PHF-tau from Alzheimer's brain comprises four species on SDS-PAGE which can be mimicked by in vitro phosphorylation of human brain tau by glycogen synthase kinase-3β. FEBS Lett. 1994;349:359–364. doi: 10.1016/0014-5793(94)00702-0. [DOI] [PubMed] [Google Scholar]
- 50.Nacharaju P, Lewis J, Easson C, Yen S, Hackett J, Hutton M, Yen SH. Accelerated filament formation from tau protein with specific FTDP-17 missense mutations. FEBS Lett. 1999;447:195–199. doi: 10.1016/s0014-5793(99)00294-x. [DOI] [PubMed] [Google Scholar]
- 51.Neumann M, Schulz-Schaeffer W, Crowther RA, Smith MJ, Spillantini MG, Goedert M, Kretzschmar HA. Pick's disease associated with the novel tau gene mutation K369I. Ann Neurol. 2001;50:503–513. doi: 10.1002/ana.1223. [DOI] [PubMed] [Google Scholar]
- 52.Otvos L, Feiner L, Lang E, Szendrei GI, Goedert M, Lee VMY. Monoclonal antibody PHF-1 recognizes tau protein phosphorylated at serines 396 and 404. J Neurosci Res. 1994;39:669–673. doi: 10.1002/jnr.490390607. [DOI] [PubMed] [Google Scholar]
- 53.Perry G, Roder H, Nunomura A, Takeda A, Friedlich AL, Zhu X, Raina AL, Holbrook N, Siedlak SL, Harris PLR, Smith MA. Activation of neuronal extracellular receptor kinase (ERK) in Alzheimer disease links oxidative stress to abnormal phosphorylation. NeuroReport. 1999;10:2411–2415. doi: 10.1097/00001756-199908020-00035. [DOI] [PubMed] [Google Scholar]
- 54.Poorkaj P, Bird TD, Wijsman E, Nemens E, Garruto RM, Anderson L, Andreadis A, Wiederholt WC, Raskind M, Schellenberg GD. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43:815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- 55.Poorkaj P, Kas A, D'Souza I, Zhou Y, Pham Q, Stone M, Olson MV, Schellenberg GD. A genomic sequence analysis of the mouse and human microtubule-associated protein tau. Mamm Genome. 2001;12:700–712. doi: 10.1007/s00335-001-2044-8. [DOI] [PubMed] [Google Scholar]
- 56.Probst A, Götz J, Wiederhold KH, Tolnay M, Mistl C, Jaton AL, Hong M, Ishihara T, Lee VMY, Trojanowski JQ, Jakes R, Crowther RA, Spillantini MG, Bürki K, Goedert M. Axonopathy and amyotrophy in mice transgenic for human four-repeat tau protein. Acta Neuropathol. 2000;99:469–481. doi: 10.1007/s004010051148. [DOI] [PubMed] [Google Scholar]
- 57.Reynolds CH, Utton MA, Gibb GM, Yale A, Anderton BH. Stress-activated protein kinase/c-jun N-terminal kinase phosphorylates tau protein. J Neurochem. 1997;68:1736–1744. doi: 10.1046/j.1471-4159.1997.68041736.x. [DOI] [PubMed] [Google Scholar]
- 58.Rizzini C, Goedert M, Hodges JR, Smith MJ, Jakes R, Hills R, Xuereb JH, Crowther RA, Spillantini MG. Tau gene mutation K257T causes a tauopathy similar to Pick's disease. J Neuropathol Exp Neurol. 2000;59:990–1001. doi: 10.1093/jnen/59.11.990. [DOI] [PubMed] [Google Scholar]
- 59.Seubert P, Mawal-Dewan M, Barbour R, Jakes R, Goedert M, Johnson GVW, Litersky JM, Schenk D, Lieberburg I, Trojanowski JQ, Lee VMY. Detection of phosphorylated Ser262 in fetal tau, adult tau and paired helical filament tau. J Biol Chem. 1995;270:18917–18922. doi: 10.1074/jbc.270.32.18917. [DOI] [PubMed] [Google Scholar]
- 60.Sidman RL, Angevine JB, Pierce ET. Atlas of the mouse brain and spinal cord. Harvard UP; Cambridge, MA: 1971. [Google Scholar]
- 61.Sperfeld AD, Collatz MB, Baier H, Palmbach M, Storch A, Schwarz J, Tatsch K, Reske S, Joosse M, Heutink P, Ludolph AC. FTDP-17: an early-onset phenotype with parkinsonism and epileptic seizures caused by a novel mutation. Ann Neurol. 1999;46:708–715. doi: 10.1002/1531-8249(199911)46:5<708::aid-ana5>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 62.Spillantini MG, Goedert M, Crowther RA, Murrell JR, Farlow MR, Ghetti B. Familial multiple system tauopathy with presenile dementia: a disease with abundant neuronal and glial tau filaments. Proc Natl Acad Sci USA. 1997;94:4113–4118. doi: 10.1073/pnas.94.8.4113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA. 1998a;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Spillantini MG, Crowther RA, Kamphorst W, Heutink P, Van Swieten JC. Tau pathology in two Dutch families with mutations in the microtubule-binding region of tau. Am J Pathol. 1998b;153:1359–1363. doi: 10.1016/S0002-9440(10)65721-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Spittaels K, Van den Haute C, Van Dorpe J, Bruynseels K, Vandezande K, Laenen I, Geerts H, Mercken M, Sciot R, Van Lommel A, Loos R, Van Leuven F. Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. Am J Pathol. 1999;155:2153–2165. doi: 10.1016/S0002-9440(10)65533-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tanemura K, Akagi T, Murayama M, Kikuchi N, Murayama O, Hashikawa T, Yoshiike Y, Park JM, Matsuda K, Nakao S, Sun X, Sato S, Yamaguchi H, Takashima A. Formation of filamentous tau aggregations in transgenic mice expressing V337M human tau. Neurobiol Dis. 2001;8:1036–1045. doi: 10.1006/nbdi.2001.0439. [DOI] [PubMed] [Google Scholar]
- 67.Tanemura K, Murayama M, Akagi T, Hashikawa T, Tominaga T, Ishikawa M, Yamaguchi H, Takashima A. Neurodegeneration with tau accumulation in a transgenic mouse expressing V337M human tau. J Neurosci. 2002;22:133–141. doi: 10.1523/JNEUROSCI.22-01-00133.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wittmann CW, Wszolek MF, Shulman JM, Salvaterra PM, Lewis J, Hutton M, Feany MB. Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science. 2001;293:711–714. doi: 10.1126/science.1062382. [DOI] [PubMed] [Google Scholar]
- 69.Woods YL, Cohen P, Becker W, Jakes R, Goedert M, Wang X, Proud CG. The kinase DYRK phosphorylates protein-synthesis initiation factor eIF2Bε at Ser539 and the microtubule-associated protein tau at Thr212: potential role of DYRK as a glycogen synthase kinase 3-priming kinase. Biochem J. 2001;355:609–615. doi: 10.1042/bj3550609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 71.Yamamoto T, Hirano A. A comparative study of modified Bielschowsky, Bodian and thioflavin S stains on Alzheimer's neurofibrillary tangles. Neuropathol Appl Neurobiol. 1986;12:3–9. doi: 10.1111/j.1365-2990.1986.tb00677.x. [DOI] [PubMed] [Google Scholar]
- 72.Yasuda M, Yokoyama K, Nakayasu T, Nishimura Y, Matsui M, Yokohama T, Miyoshi K, Tanaka C. A Japanese patient with frontotemporal dementia and parkinsonism by a tau P301S mutation. Neurology. 2000;55:1224–1227. doi: 10.1212/wnl.55.8.1224. [DOI] [PubMed] [Google Scholar]
- 73.Yoshida H, Crowther RA, Goedert M. Functional effects of tau gene mutations ΔN296 and N296H. J Neurochem. 2002;80:548–551. doi: 10.1046/j.0022-3042.2001.00729.x. [DOI] [PubMed] [Google Scholar]
- 74.Zheng-Fischhöfer Q, Biernat J, Mandelkow EM, Illenberger S, Godemann R, Mandelkow E. Sequential phosphorylation of tau by glycogen synthase kinase-3β and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody AT100 and requires a paired helical filament-like conformation. Eur J Biochem. 1998;252:542–552. doi: 10.1046/j.1432-1327.1998.2520542.x. [DOI] [PubMed] [Google Scholar]
- 75.Zhu X, Rottkamp CA, Boux H, Takeda A, Perry G, Smith MA. Activation of p38 kinase links tau phosphorylation, oxidative stress, and cell cycle-related events in Alzheimer disease. J Neuropathol Exp Neurol. 2000;59:880–888. doi: 10.1093/jnen/59.10.880. [DOI] [PubMed] [Google Scholar]