

Abstract
Barbiturates are widely used as anesthetics, anticonvulsants, and neuroprotective agents. However, barbiturates may also inhibit mitochondrial respiration, and mitochondrial inhibitors are known to potentiate NMDA receptor-mediated neurotoxicity. Here we used rat cortical cultures to examine the effect of barbiturates on neuronal mitochondria and responses to NMDA receptor stimulation. The barbiturates tested, secobarbital, amobarbital, and thiamylal, each potentiated NMDA-induced neuron death at barbiturate concentrations relevant to clinical and experimental use (100–300 μm). By using rhodamine-123 under quenching conditions, barbiturates in this concentration range were shown to depolarize neuronal mitochondria and greatly amplify NMDA-induced mitochondrial depolarization. Barbiturate-induced mitochondrial depolarization was increased by the ATP synthase inhibitor oligomycin, indicating that barbiturates act by inhibiting electron transport sufficiently to cause ATP synthase reversal. Barbiturates similarly amplified the effects of NMDA on cytoplasmic free calcium concentrations. The cell-impermeant barbiturate N-glucoside amobarbital did not influence mitochondrial potential or potentiate NMDA neurotoxicity or calcium responses. However, all of the barbiturates attenuated NMDA-induced calcium elevations and cell death when present at millimolar concentrations. Whole-cell patch-clamp studies showed that these effects may be attributable to actions at the cell membrane, resulting in a block of NMDA-induced current flux at millimolar barbiturate concentrations. Together, these findings reconcile previous reports of opposing effects on barbiturates on NMDA neurotoxicity and show that barbiturate effects on neuronal mitochondria can be functionally significant. Effects of barbiturates on neuronal mitochondria should be considered in experimental and clinical application of these drugs.
Keywords: amobarbital, N-glucoside amobarbital, secobarbital, calcium, glutamate, NMDA, rhodamine-123, fura-2
Barbiturates reduce glutamate-induced neuronal depolarization (Barker and Ransom, 1978), block voltage-dependent Ca2+ channels (Werz and Macdonald, 1985), attenuate the Ca2+ influx of NMDA-induced currents (Daniell, 1994; Charlesworth et al., 1995), and depress neuronal activity and metabolism (Sokoloff et al., 1977; Steen and Michenfelder, 1980). These actions suggest that barbiturates should effectively reduce excitotoxic neuronal injury, particularly under conditions of energy deprivation. Therefore, it is surprising that barbiturates do not attenuate neuronal death in cell-culture models of glucose or combined oxygen–glucose deprivation (Giffard et al., 1993) that produce NMDA receptor-dependent toxicity.
The present study examines whether the potential neuroprotective effects of barbiturates may be limited or outweighed by inhibitory effects on mitochondrial function. Barbiturates have long been known to depress mitochondrial respiration (Aldrich and Parker, 1960; Chance and Hollunger, 1963). In isolated mitochondria, compounds of the oxybarbiturate class (including secobarbital, amobarbital, and amytal) inhibit respiration by interfering with complex 1 of the electron transport chain (Aldrich and Parker, 1960). Thiobarbiturates (including thiamylal and thiopental) may additionally have an “uncoupling” effect on oxidative phosphorylation (Aldrich and Parker, 1960; Chance and Hollunger, 1963). These effects are incomplete and do not appreciably depress cell ATP levels under basal metabolic conditions; however, barbiturate effects on cellular energy metabolism may become physiologically significant during periods of reduced substrate delivery or high energy demand (Swanson and Seid, 1998).
Barbiturate actions on mitochondria could be particularly important during NMDA receptor activation. Mitochondrial inhibitors have been shown to amplify NMDA receptor-mediated intracellular calcium ([Ca2+]i) elevations, by both impairment of intracellular calcium homeostasis and plasma membrane depolarization and resultant increased Ca2+ flux through NMDA-gated cation channels (White and Reynolds, 1995; Greene et al., 1998). Several mitochondrial inhibitors can potentiate NMDA receptor activation and excitotoxic neuronal death (Novelli et al., 1988; Beal, 1992; Greene and Greenamyre, 1996; Greene et al., 1998), suggesting that barbiturates might have similar effects. In this study, we used cortical neuron cultures to examine the effects of barbiturates on NMDA-mediated mitochondrial depolarization, intracellular Ca2+ elevations, and cell death. The findings demonstrate that barbiturates have direct effects on neuronal mitochondria and thereby potentiate responses to NMDA.
MATERIALS AND METHODS
Materials.5(R/S)-5-ethyl-1-(1-β-d-glucopyranosyl)-5-(3-methylbutyl)-2,3,6-(1H,3H,5H)-pyrimidinetrione (N-glucoside amobarbital) was synthesized as described previously (Soine et al., 1986). All other reagents were purchased from Sigma (St. Louis, MO) except where otherwise stated.
Astrocyte–neuron cocultures. Cortical cultures were prepared from newborn and neonatal rats using protocols approved by the San Francisco Veterans Affairs Medical Center animal use review committee and following National Institutes of Health guidelines. Cocultures were prepared by seeding neurons onto a pre-existing astrocyte layer. Astrocyte cultures were prepared from cortices harvested from 1-d-old Sprague Dawley rats (Simonsen, Gilroy, CA). After removal of meninges, the cells were dissociated by incubation in papain/DNase, followed by trituration. The dissociated cells were washed, suspended in Eagle's minimum essential medium (MEM) with 10% fetal bovine serum (FBS; Hyclone, Ogden, UT) and 2 mm glutamine, and plated in Falcon 24 well tissue culture plates at an approximate density of 5 × 104cells/cm2. For some studies, the astrocytes were plated onto glass coverslips placed on the bottoms of the culture wells. The cultures were maintained in a humidified, 5% CO2, 37°C incubator and received a medium exchange every 7 d. Neurons prepared from fetal (embryonic day 16) rats were plated onto the astrocytes after 14–20 d, when the astrocytes form a confluent monolayer. The fetal rat forebrain cortices were removed, and cells were dissociated by the same method used for the astrocyte preparations. These cells were plated at an approximate density of 1 × 105cells/cm2, and the resulting cocultures were maintained in a 5% CO2 atmosphere. Proliferation of other cell types was inhibited by the addition of 10 μm cytosine arabinoside 3 d after plating. This medium was replaced after 48 hr with glial-conditioned medium prepared by placing MEM with 2 mm glutamine and 5% FBS into a flask of confluent cortical astrocytes for 72 hr. The coculture medium was subsequently exchanged with fresh glial-conditioned media every 7 d and on the evening before use of the cells. Experiments were conducted when the neurons were 18–22 din vitro.
Experimental media. Experimental incubations were performed at 37°C in a balanced salt solution (BSS) containing (in mm): 135 NaCl, 3.1 KCl, 1.2 CaCl2, 1.2 MgSO4, 0.5 KH2PO4, 5 1,4-piperazinediethanesulfonic acid, and 2 glucose, pH 7.2. All drugs were added from concentrated iso-osmolar-buffered stock solutions, pH 7.2.
Neuron death. Cells were washed by three partial (85%) exchanges into prewarmed BSS containing 0.1% bovine serum albumin (BSA). Barbiturates were added 5 min before additions of NMDA or H2O2. Incubations were terminated after 5 min with NMDA or 10 min with H2O2 by washing back into MEM containing 0.1% BSA. Neuronal death was determined 18–24 hr after NMDA exposures by measurement of lactate dehydrogenase (LDH) in the medium (Koh and Choi, 1987). LDH values corresponding to 100% neuronal death were established for each 24 well culture plate by treating four of the wells with 10 mm NMDA to kill all neurons. The LDH values from the other 20 wells were then normalized to these values to express results as a percentage of neuronal death. Subtraction of background LDH values, representing neuronal death under control conditions, was performed only where noted. Propidium iodide (PI) staining (Edidin, 1970) was used to assess neuron death 18–24 hr after H2O2 exposure and in selected NMDA-treated cultures to corroborate the LDH results and confirm that LDH release reflected only neuronal death. PI (0.01 mg/ml) was added to each well, and both live and dead (PI stained) cells were counted in random optical fields in each of four quadrants, totaling >500 cells per well. Neurons were distinguished from the underlying glial layer by a phase-bright, process-bearing appearance under phase-contrast optics (Ying et al., 1999).
Intracellular calcium. Intracellular cytoplasmic Ca2+ measurements were performed by fura-2 ratiometric imaging. Cultures on coverslips were loaded with 8 μm fura-2 acetoxymethyl ester (Molecular Probes, Eugene, OR) for 15 min at room temperature, washed, and then treated with barbiturates and NMDA as indicated. Coverslips were placed in a chamber on an Olympus inverted microscope (Scientific Instruments, Sunnyvale, CA) with a 40× objective for fluorescence imaging. Data were recorded from 5 to 20 cells per coverslip and were averaged from three or more coverslips. Cells were excited alternately at 340 and 380 nm, and emission was recorded at 510 nm. Calcium measurements were determined as the ratio of emission intensities at 340 and 380 nm excitation wavelengths by using standard imaging techniques and analysis with Metafluor software from Universal Imaging (West Chester, PA). The NMDA-induced change in fluorescence ratio was integrated over time for each imaged neuron using an in-house macro program for Microsoft Excel (Redmond, WA) spreadsheets.
Mitochondrial membrane potential. Changes in mitochondrial potential in individual neurons were monitored using the cationic fluorescent dye rhodamine-123 (Molecular Probes), as described previously (Ward et al., 2000). Cells plated on 12-mm-diameter coverslips were incubated at 37°C for 25 min with 2.6 μm rhodamine-123 to achieve a quenching concentration of dye within the mitochondrial matrix. Coverslips were washed and placed in a BSS-containing open-bath imaging chamber (Warner Instruments, Hamden, CT) maintained at 37°C. Single-cell imaging was performed using an Olympus IX70 inverted epifluorescence microscope with a 40× oil immersion objective (Olympus America, Melville, NY). The dye was excited at 485 nm using a Spectramaster monochromator (PerkinElmer, Cambridge, UK), and fluorescence was measured using a fura-2/rhodamine dichroic mirror and emission filter set (Chroma Technology, Brattleboro, VT). Images were collected and analyzed using the Imaging Suite software (Olympus America). Data were recorded for 5–10 individual neurons per coverslip and from multiple coverslips. The extent of the barbiturate-induced mitochondrial membrane depolarization was estimated using a mathematical simulation of single-cell fluorescent responses, as described previously (Ward et al., 2000). The following parameters were assumed for the simulation: resting plasma membrane potential of −60 mV; initial mitochondrial membrane potential (Δψm) of −150 mV; cell volume occupied by matrix, 1%; permeability constant for plasma membrane equilibration, 0.0003 sec−1; quench limit for probe in the matrix, 20 μm; and estimated extracellular probe concentration, 80 nm.
Whole-cell patch-clamp recording. Current recordings were obtained using the whole-cell patch-clamp technique at room temperature. The pipette solution contained (in mm): 135 KCl, 3 Mg-ATP, 5 EGTA, and 10 HEPES, pH 7.2, with KOH. The external solution was Mg2+-free BSS containing 1 μm glycine to potentiate NMDA-induced currents. For both pipette and external solutions, osmolarity was adjusted to 300 mOsm with sucrose. Pipettes had resistance between 4 and 6 MΩ. Series resistance was <10 mΩ and was compensated to the maximum extent possible (usually >60%) without causing oscillation or overshoot. Connection of the perfusion chamber to ground was established via a 3m KCl agarose bridge. The U-tube method (Duan and Cooke, 1999) was used for rapid solution change.
Statistics. Statistical differences between experimental groups were determined by performing one-way ANOVA followed, where applicable, by Dunnett's test for multiple comparisons against a control group or Student Newman–Keuls test for comparisons between multiple experimental groups.
RESULTS
Barbiturates potentiate NMDA neurotoxicity through actions at an intracellular site
Previous studies have shown that secobarbital at high concentrations (1–3 mm) can reduce NMDA-mediated neuronal death (Giffard et al., 1993). To more fully define the effects of barbiturates on NMDA toxicity, we measured NMDA-induced neuronal death over a range of NMDA and secobarbital concentrations. Using NMDA concentrations that produced a moderate degree of neuron death (<50%) (Fig. 1A), we found that secobarbital concentrations in the 100–300 μm range amplified NMDA toxicity, with this effect becoming less pronounced at higher secobarbital concentrations (Fig. 1B,C). The more lipid-soluble thiobarbiturate, thiamylal, also displayed a biphasic effect on NMDA toxicity, with NMDA-induced neuron death potentiated by 30 μm thiamylal but significantly reduced by 1 mm thiamylal (Fig. 1C). Propidium iodide staining performed in cultures 24 hr after NMDA and barbiturate exposures confirmed that only neuronal death contributed to the LDH signal (data not shown). Figure 1C also shows that the increase in NMDA toxicity produced by secobarbital and thiamylal was eliminated by the addition of 10 μm(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate (MK-801), a noncompetitive NMDA receptor antagonist. Together with the lack of toxicity of the barbiturates alone, these results suggest that the barbiturates may act by potentiating NMDA toxicity specifically. This was supported by an additional set of studies that showed that secobarbital had a negligible effect on H2O2-induced neuronal death. The LD50 for H2O2 was ∼50 μm in both the presence and absence of 100 μm secobarbital (data not shown).
Fig. 1.
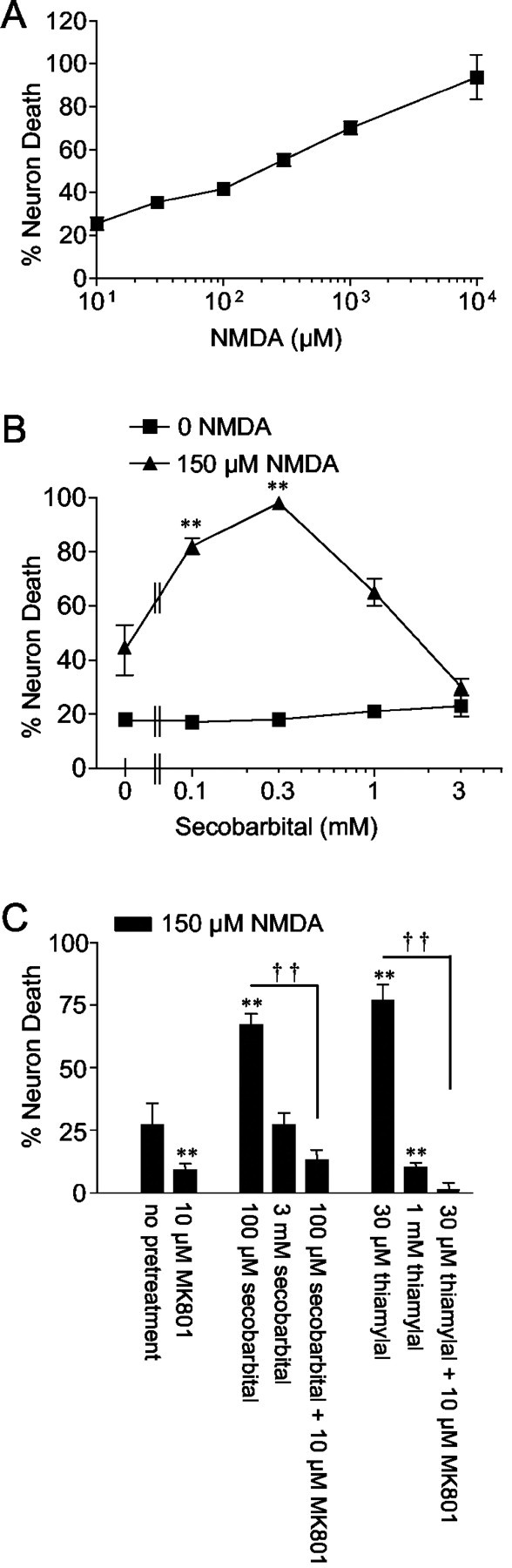
Effects of barbiturates on NMDA neurotoxicity.A, Exposure to NMDA for 5 min resulted in neuronal death with an EC50 of ∼200 μm. B, Secobarbital at 100 and 300 μm potentiated NMDA neurotoxicity, but this effect was reversed at higher secobarbital concentrations. Vertical hatches mark the beginning of the log scale. ∗∗p < 0.01 versus the 0 secobarbital control in each of the two treatment groups;n ≥ 7. C, The thiobarbiturate thiamylal produced a similar biphasic effect. For both thiamylal and secobarbital, MK-801 completely blocked the toxicity produced by NMDA plus barbiturate. ∗∗p < 0.01 versus the 0 barbiturate group; ††p < 0.01 between indicated groups; n ≥ 7. All data are means ± SE. C, Data are shown after subtraction of 12% neuronal death observed in the control (wash only) condition.
To determine whether barbiturates amplify NMDA neurotoxicity through actions at the plasma membrane or at intracellular sites, experiments were also performed using N-glucoside amobarbital, a hydrophilic N-glucoside derivative of amobarbital with limited plasma membrane permeability (Tang, 1990).N-glucoside amobarbital did not potentiate NMDA toxicity but instead exhibited only neuroprotective effects (Fig.2A). In contrast, the parent compound amobarbital had a biphasic effect on NMDA-induced cell death over the same concentration range (Fig. 2A), similar to that observed with secobarbital and thiamylal. The failure of N-glucoside amobarbital to amplify NMDA toxicity suggests that barbiturates potentiate NMDA toxicity through actions at an intracellular site. Additionally, the marked reduction in NMDA toxicity observed with N-glucoside amobarbital suggests that the reduction in NMDA toxicity observed with high concentrations (>300 μm) of the standard, cell-permeant barbiturates may be mediated by actions at the neuronal cell membrane. These possibilities were also investigated by measuring the effects of barbiturates on NMDA-induced currents in whole-cell patch-clamped neurons. Secobarbital reduced peak NMDA-induced current in a monophasic, concentration-dependent manner, achieving a 61 ± 9% reduction at 1 mm (Fig.2B,C). N-glucoside amobarbital produced a similar inhibition of NMDA-induced currents (Fig. 2B,C).
Fig. 2.
Comparisons between a cell-permeant and a cell-impermeant barbiturate. A, The cell-impermeant barbiturate N-glucoside (N-glu) amobarbital reduced NMDA toxicity at all concentrations tested, whereas the parent compound had a biphasic effect on NMDA toxicity. Vertical hatches mark the beginning of the log scale. B, Representative patch-clamp recordings from different neurons exposed to NMDA in the presence of sequentially elevated concentrations of secobarbital orN-glucoside amobarbital. C, Pooled data showing a concentration-dependent inhibition of the NMDA current with both barbiturates. Currents from each recorded neuron are normalized to the peak current recorded in the absence of barbiturate. Data are means ± SE; ∗p < 0.05; ∗∗p < 0.01 versus control;n = 3–6.
Barbiturates depolarize mitochondria by inhibiting mitochondrial ATP synthesis
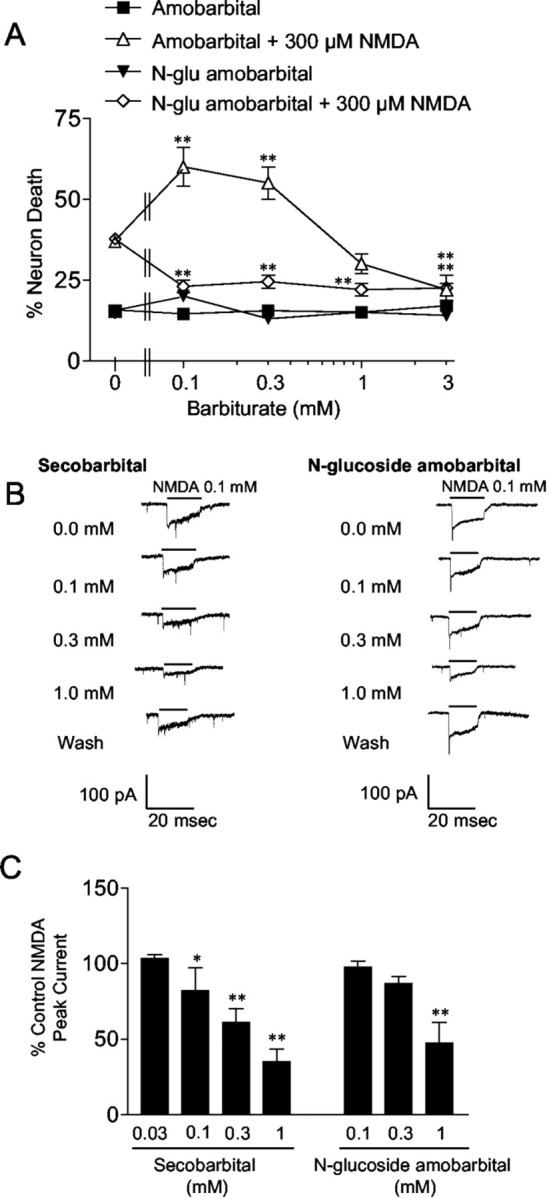
Previous studies have suggested that mitochondria are an intracellular site of barbiturate action (Aldrich and Parker, 1960). Barbiturate effects on mitochondrial membrane potential (Δψm) were assessed in individual neurons using the fluorescent cationic dye rhodamine-123. In 25 of 29 neurons analyzed, secobarbital (100 μm) caused significant mitochondrial depolarization (Fig.3A), as reflected by an increase in total cell fluorescence caused by release of rhodamine-123 from the quenched environment of the matrix (for review, see Nicholls and Ward, 2000). The dye quench limit was surpassed because carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP) was able to increase whole-cell fluorescence (data not shown). Subsequent application of the ATP synthase inhibitor oligomycin (2.9 μg/ml) produced additional mitochondrial depolarization (Fig.3A), indicating that Δψm had been sustained by “reverse” operation of ATP synthase. Oligomycin added to cells with normal Δψm produced either no change in Δψm or a slight hyperpolarization consistent with slowed proton influx through the mitochondrial ATP synthase complex (Fig. 3B). As expected, the cell-impermeant barbiturate N-glucoside amobarbital had no effect on Δψm (Fig. 3B), whereas the parent compound amobarbital produced significant depolarization (data not shown).
Fig. 3.
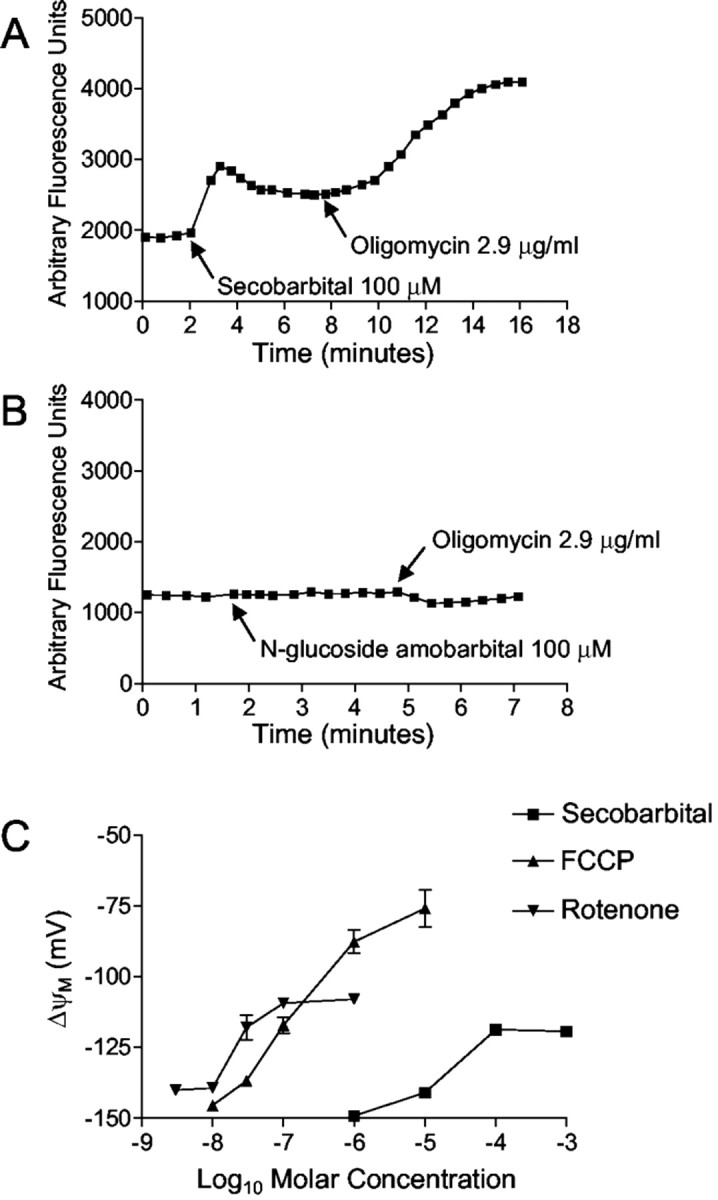
Barbiturates cause mitochondrial depolarizationin situ. A, Secobarbital induced an increase in total cell rhodamine-123 fluorescence, indicative of mitochondrial depolarization. The addition of oligomycin caused additional depolarization, indicating that secobarbital induces ATP synthase reversal. B, The cell-impermeable barbiturateN-glucoside amobarbital had no effect on ΔΨm. Oligomycin caused a slight membrane hyperpolarization. C, Secobarbital and the complex 1 inhibitor rotenone increased peak mitochondrial depolarization to a plateau at approximately −110 mV, whereas the uncoupler FCCP increased peak depolarization dose dependently. The EC50 for secobarbital is ∼30 μm. Data in A andB are representative traces. Data inC are means ± SE of ≥10 neurons from two different coverslips.
Mitochondrial depolarization with reduced ATP synthesis can result from impaired electron transport (or substrate supply) or enhanced inner membrane proton permeability. In cells with active glycolysis, the former will result in only a partial depolarization, because ATP synthase reversal can maintain a suboptimal membrane potential even in the face of total respiratory inhibition by consuming ATP generated by glycolysis. In contrast, increasing concentrations of a protonophore would decrease Δψm asymptotically toward zero. In an attempt to distinguish between these alternatives, the relationship between mitochondrial depolarization and secobarbital concentration was compared with that of a known mitochondrial complex 1 inhibitor (rotenone) and an established uncoupler, FCCP. Figure3C shows that both rotenone and secobarbital produced a concentration-dependent mitochondrial depolarization. This plateaued at a residual mitochondrial membrane potential of approximately −110 mV, as estimated by a simulation that predicts Δψm from whole-cell fluorescence intensity (Ward et al., 2000). In contrast, increasing concentrations of FCCP produced a near linear depolarization with no observable plateau. Because maximal inhibition of respiration produces only partial mitochondrial depolarization, secobarbital displayed characteristics of a respiration inhibitor.
Because barbiturates were found to potentiate NMDA neurotoxicity, we also assessed the effect of barbiturates on NMDA-induced changes in Δψm. NMDA alone caused a small mitochondrial depolarization (Fig.4A), indicative of increased mitochondrial Ca2+ uptake (Ward et al., 2000). In the presence of 100 μmsecobarbital, the NMDA-induced mitochondrial depolarization was greatly increased (Fig. 4B). This result is congruent with the increased neurotoxicity of NMDA in the presence of 100 μm secobarbital (Fig. 1).
Fig. 4.

Secobarbital potentiates NMDA-induced mitochondrial depolarization. A, Bath-applied NMDA (40 μm) produced an increase in total cell rhodamine-123 fluorescence, indicative of a slight mitochondrial depolarization.B, Pretreatment with 100 μm secobarbital enhanced NMDA-induced depolarization. Data are representativetraces of 12 individual neurons from two separate coverslips for each treatment regimen.
Barbiturates enhance NMDA-induced intracellular calcium elevations
Mitochondrial dysfunction can lead to neuron death by disrupting Ca2+ homeostasis (Stout et al., 1998;Murphy et al., 1999; Nicholls and Ward, 2000; Sattler and Tymianski, 2000). NMDA-induced Ca2+ elevations were monitored in the presence or absence of secobarbital using ratiometric fura-2 imaging. Barbiturates had biphasic, concentration-dependent effects on free Ca2+ that paralleled their effects on NMDA-induced cell death. As shown in Figure5A, NMDA applied at a concentration that normally induced a negligible increase in intracellular Ca2+ (5 μm) was found to induce a large Ca2+ increase when applied in the presence of 100 μm secobarbital but not 1 mm secobarbital. When NMDA was applied at a concentration sufficient to induce a substantial Ca2+ elevation (20 μm), this increase was attenuated in the presence of 1 mm amobarbital, 1 mmN-glucoside amobarbital (Fig.5B), or 1 mm secobarbital (data not shown).
Fig. 5.

Barbiturate effects on NMDA-induced cytoplasmic Ca2+ elevations. A, A 5 μm concentration of NMDA alone produced a small rise in intracellular Ca2+. This increase was significantly increased in the presence of 100 μm secobarbital but nearly eliminated in the presence of 1 mm secobarbital.Secobarb, Secobarbital. B, The larger increase in intracellular Ca2+ induced by 20 μm NMDA was reduced by both 1 mm amobarbital and 1 mmN-glucoside amobarbital. Error bars are omitted for clarity. Values adjacent to the tracings are the mean ± SE of the integrated NMDA-induced changes in the fura-2 fluorescence ratio integrated over the 4 min interval beginning 10 sec after the addition of NMDA. ∗∗p < 0.01 versus NMDA alone; n = 35–49 in A;n = 12–19 in B. Barb, Barbiturate.
DISCUSSION
These findings suggest that barbiturates potentiate NMDA neurotoxicity by inhibiting mitochondrial respiration and thereby amplifying NMDA-induced mitochondrial depolarization and intracellular Ca2+ dysregulation. NMDA neurotoxicity was increased by all three barbiturates tested: secobarbital, amobarbital, and thiobarbital. Thiamylal was the most potent of these, but all three agents showed maximal effects in the 30–100 μmconcentration range. Because these agents represent members of both the oxybarbiturate and thiobarbiturate classes, the findings suggest that potentiation of NMDA toxicity is likely a general property common to all cell-permeant barbiturates.
Importantly, the poorly permeant congener N-glucoside amobarbital did not potentiate NMDA toxicity, and this provides one line of evidence that barbiturate influences on NMDA toxicity occur at an intracellular site. N-glucoside amobarbital was originally synthesized for studies of barbiturate metabolism (Soine et al., 1986, 1991). The pharmacology of this compound has not been extensively characterized, and it remains possible that its failure to amplify the effects of NMDA is attributable to some property other than poor cell entry. However, the studies presented here show thatN-glucoside amobarbital is not inert. N-glucoside amobarbital was found to attenuate NMDA toxicity over the same concentration range that other barbiturates, including amobarbital, had the opposite effect. Moreover, N-glucoside amobarbital also attenuated NMDA-induced intracellular Ca2+elevations and blocked NMDA-induced currents in whole-cell patch-clamped neurons. These results suggest that the neuroprotective effects of N-glucoside amobarbital occur at an extracellular site, whereas the NMDA-potentiating effects of permeable barbiturates are intracellular.
Previous studies have shown that barbiturates exacerbate neuronal cell death induced by glucose deprivation but not by oxygen deprivation (Giffard et al., 1993). These studies also showed that 100–300 μm secobarbital amplified ATP depletion during glucose deprivation but not oxygen deprivation. One explanation for these results is that barbiturate inhibition of mitochondrial respiration (Aldrich and Parker, 1960) is sufficient to limit oxidative ATP production. Barbiturate effects on mitochondria could likewise mediate exacerbation of NMDA neurotoxicity, in accord with effects of other known mitochondrial inhibitors (White and Reynolds, 1995; Greene et al., 1998). In support of this idea, results obtained in this study provide the first direct evidence that barbiturates produce mitochondrial membrane depolarization in neurons. For secobarbital, a maximal depolarization was achieved at 100 μm, and the EC50 was ∼30 μm, which corresponds to the concentrations at which secobarbital potentiated NMDA neurotoxicity.
Mitochondrial depolarization can result from increased ATP demand or increased Ca2+ influx (Nicholls and Ward, 2000). In both cases, mitochondrial ATP synthase continues to function and support the cytosolic ATP:ADP ratio. Alternatively, mitochondrial depolarization can result from enhanced inner membrane proton permeability (classical uncoupling) or from impaired respiration. In these conditions, cytosolic ATP is consumed by ATP synthase as it functions in reverse mode to extrude matrix protons and maintain Δψm. In the present studies, the ATP synthase inhibitor oligomycin increased mitochondrial depolarization in neurons treated with secobarbital, indicating that ATP synthase was functioning in reverse mode and that Δψm was being supported by cytoplasmic ATP.
The mechanism for barbiturate-induced disruption of ATP synthesis was probed by comparing secobarbital to a known protonophoric uncoupler (FCCP) and a known respiration inhibitor (rotenone). High protonophore concentrations can totally collapse Δψm. In contrast, respiratory chain inhibitors, even after 100% blockade of electron transport, only partially depolarize Δψm caused by ATP synthase reversal. Thus, the plot of mitochondrial depolarization versus concentration for rotenone reaches a submaximal plateau, as depicted in Figure3C. An analogous plot for secobarbital revealed a maximal depolarizing capability similar to rotenone, between 30 and 100 μm. This suggests that secobarbital is an inhibitor of mitochondrial respiration, in agreement with classical studies using isolated mitochondria (Aldrich and Parker, 1960). The single-cell fluorescence simulation (Ward et al., 2000) suggests a maximal secobarbital-induced depolarization of ∼25 mV.
The effects of secobarbital on NMDA-induced mitochondrial depolarization were paralleled by its effects on intracellular free Ca2+ concentrations. Secobarbital alone caused little or no elevation in intracellular Ca2+, but when present during NMDA exposure, it increased the cytoplasmic Ca2+ concentrations by several-fold. This result is consistent with previous studies showing that disrupted intracellular Ca2+ homeostasis correlates with the extent of glutamate-induced mitochondrial depolarization (Vergun et al., 1999; Ward et al., 2000). The NMDA concentrations used for the studies of intracellular free Ca2+changes were lower than those used for the cell death studies because of the concern that higher NMDA concentrations would saturate the high-affinity fura-2 calcium indicator (Hyrc et al., 1997). It is possible that Ca2+ elevations produced by higher NMDA concentrations might not exhibit the same degree of amplification in the presence of barbiturates, but this possibility seems unlikely given that the barbiturate influences on NMDA-induced cell death, mitochondrial membrane depolarization, and calcium elevations were all consistent.
Barbiturates have numerous effects on cell metabolism, and consequently, there are several ways, both direct and indirect, that barbiturates could influence mitochondrial function and NMDA neurotoxicity. The results provided here and in previous studies indicate that barbiturates act by a direct effect on mitochondrial electron transport. It has been known since the 1960s that barbiturates can block complex 1 activity in isolated liver mitochondria at submillimolar concentrations (Aldrich and Parker, 1960; Chance and Hollunger, 1963). The present studies show that secobarbital has the same pattern of effects on the mitochondrial membrane potential of intact neurons as the complex 1 inhibitor rotenone (although rotenone is more potent). A causative link between the effects of barbiturates on neuronal mitochondria and their potentiating effect on NMDA toxicity is supported by the fact that these effects, as well as the amplification of NMDA-induced calcium elevations, are all observed over the 30–100 μm range. Together, these observations support the idea that barbiturates act in a manner similar to other mitochondrial inhibitors (Novelli et al., 1988; Beal, 1992; Greene and Greenamyre, 1996; Greene et al., 1998) to potentiate NMDA-induced calcium elevations and neurotoxicity. It remains possible, however, that there are additional effects of barbiturates that also contribute to these processes or that the increased calcium elevations are not directly related to increased neuronal death.
Our findings also identify a second mechanism by which barbiturates influence excitotoxicity. At concentrations of ≥1 mm, all of the barbiturates tested were found to attenuate NMDA-induced inward currents. These observations are consistent with previous reports (Daniell, 1994; Charlesworth et al., 1995) and may reconcile the opposing effects of micromolar and millimolar barbiturate concentrations on excitotoxicity; barbiturates in the 30–100 μm range exacerbate excitotoxicity by impairing mitochondrial respiration, but this effect becomes moot at millimolar barbiturate concentrations, because flux through the NMDA receptor complex is then effectively blocked. An additional contributory factor may be that the effect of barbiturates on mitochondrial membrane potential plateaus at ∼100 μm and does not increase further at the higher barbiturate concentrations (Fig. 3).
Although effects of barbiturates on mitochondrial energy metabolism are well established, they have not been recognized previously as physiologically significant in neurons. Studies with cardiac muscle provide a precedent for functionally significant effects resulting from barbiturate-induced mitochondrial inhibition. Bhayana et al. (1980)showed in perfused rat hearts that pentobarbital and thiopental reduce both state 3 respiration and myocardial contractile force at 0.1–0.5 mm. Although other mechanisms could not be excluded, it is likely that the impaired respiration contributed to the impaired contractility, because cardiac muscle relies almost exclusively on oxidative ATP production. Similarly, ATP levels and glutamate uptake were shown to be reduced in astrocytes exposed to barbiturates at submillimolar concentrations (Swanson et al., 1997). These effects were strikingly potentiated by glucose deprivation, consistent with a greater reliance of astrocytes on glycolytic rather than oxidative metabolism (Swanson, 1992; Silver et al., 1997). Studies of brain and kidney in situ have demonstrated elevated cellular β-nicotinamide adenine dinucleotide, reduced form (NADH) levels during barbiturate infusions, consistent with respiratory chain inhibition, but did not examine the effects of this impairment on cell function (Chance et al., 1962). The present study provides the first demonstration that barbiturate inhibition of neuronal mitochondrial function can become functionally significant during NMDA receptor stimulation.
Barbiturates are widely used in studies of neuronal function. The present findings suggest that interpretation of experiments using barbiturates should include consideration of mitochondrial function and cellular energetics in addition to effects on excitable membranes. Barbiturates are also widely used as neuroprotective agents (Cheng et al., 1997). The barbiturate dose range shown to potentiate NMDA-mediated neuronal death in this study is within the range of barbiturate concentrations achieved in clinical use. Barbiturate treatment of refractory status epilepticus targets EEG burst suppression as a treatment goal (Lowenstein et al., 1988; Van Ness, 1990). Plasma thiopental concentrations of 80–200 μmproduce a burst suppression EEG pattern in human subjects (Airey et al., 1982; Stanski et al., 1984), which is considered by many authors to signal the optimal anesthesia depth for neuroprotection (Zaidan et al., 1991; Frawley et al., 1994). Barbiturate concentrations in CSF are typically 15–40% of serum levels, and a clinical study found that the CSF concentration of thiopental required to produce an anticonvulsant effect in a patient with status epilepticus was 15–90 μm(Airey et al., 1982). In addition, brain concentrations of barbiturates increase with extended use because of tolerance, requiring dose escalation, and because of accumulation in lipids (Bolander et al., 1984; Koskela and Wahlström, 1989). Whether barbiturate potentiation of excitotoxicity is sufficient to produce a net negative effect on neuronal survival in vivo is not clear; however, the present studies suggest that barbiturates could exacerbate excitotoxic injury, particularly if glucose supply is impaired.
Footnotes
This work was supported by American Heart Association Grant-in-Aid 9750537N, National Institutes of Health Grant RO1 NS31914, the Department of Veterans Affairs, the Canadian Institutes of Health Research, and the Heart and Stroke Foundation of Canada. We thank Dr. Conrad Alano and Elizabeth Gum for advice and technical assistance.
Correspondence should be addressed to Dr. Raymond A. Swanson, Neurology Service (127), Veterans Affairs Medical Center, 4150 Clement Street, San Francisco, CA 94121. E-mail: ray@itsa.ucsf.edu.
S. Duan's present address: Institute of Neuroscience, Chinese Academy of Sciences, Shanghai 200031, China.
REFERENCES
- 1.Airey IL, Smith PA, Stoddart JC. Plasma and cerebrospinal fluid barbiturate levels during prolonged continuous thiopentone infusion. Anaesthesia. 1982;37:328–331. doi: 10.1111/j.1365-2044.1982.tb01110.x. [DOI] [PubMed] [Google Scholar]
- 2.Aldrich WN, Parker VH. Barbiturates and oxidative phosphorylation. Biochem J. 1960;76:47–56. doi: 10.1042/bj0760047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barker JL, Ransom BR. Pentobarbitone pharmacology of mammalian central neurones grown in tissue culture. J Physiol (Lond) 1978;280:355–372. doi: 10.1113/jphysiol.1978.sp012388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Beal MF. Does impairment of energy metabolism result in excitotoxic neuronal death in neurodegenerative illnesses? Ann Neurol. 1992;31:119–130. doi: 10.1002/ana.410310202. [DOI] [PubMed] [Google Scholar]
- 5.Bhayana V, Alto LE, Dhalla NS. Effects of pentobarbital and pentothal on rat heart contractile force and oxidative phosphorylation activities. Gen Pharmacol. 1980;11:375–377. doi: 10.1016/0306-3623(80)90102-0. [DOI] [PubMed] [Google Scholar]
- 6.Bolander HG, Wahlström G, Norberg L. Reevaluation of potency and pharmacokinetic properties of some lipid-soluble barbiturates with an EEG-threshold method. Acta Pharmacol Toxicol (Copenh) 1984;54:33–40. doi: 10.1111/j.1600-0773.1984.tb01892.x. [DOI] [PubMed] [Google Scholar]
- 7.Chance B, Hollunger G. Inhibition of electron and energy transfer in mitochondria. J Biol Chem. 1963;278:418–431. [PubMed] [Google Scholar]
- 8.Chance B, Cohen P, Jobsis F, Schoener B. Intracellular oxidation-reduction states in vivo. Science. 1962;137:499–508. doi: 10.1126/science.137.3529.499. [DOI] [PubMed] [Google Scholar]
- 9.Charlesworth P, Jacobson I, Richards CD. Pentobarbitone modulation of NMDA receptors in neurones isolated from the rat olfactory brain. Br J Pharmacol. 1995;116:3005–3013. doi: 10.1111/j.1476-5381.1995.tb15956.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng MA, Theard MA, Tempelhoff R. Intravenous agents and intraoperative neuroprotection. Beyond barbiturates. Crit Care Clin. 1997;13:185–199. doi: 10.1016/s0749-0704(05)70301-8. [DOI] [PubMed] [Google Scholar]
- 11.Daniell LC. Effect of anesthetic and convulsant barbiturates on N-methyl-d-aspartate receptor-mediated calcium flux in brain membrane vesicles. Pharmacology. 1994;49:296–307. doi: 10.1159/000139246. [DOI] [PubMed] [Google Scholar]
- 12.Duan S, Cooke IM. Selective inhibition of transient K+ current by La3+ in crab peptide-secretory neurons. J Neurophysiol. 1999;81:1848–1855. doi: 10.1152/jn.1999.81.4.1848. [DOI] [PubMed] [Google Scholar]
- 13.Edidin M. A rapid, quantitative fluorescence assay for cell damage by cytotoxic antibodies. J Immunol. 1970;104:1303–1306. [PubMed] [Google Scholar]
- 14.Frawley JE, Hicks RG, Horton DA, Gray LJ, Niesche JW, Matheson JM. Thiopental sodium cerebral protection during carotid endarterectomy: perioperative disease and death. J Vasc Surg. 1994;19:732–738. doi: 10.1016/s0741-5214(94)70049-4. [DOI] [PubMed] [Google Scholar]
- 15.Giffard RG, Weiss JH, Swanson RA, Choi DW. Secobarbital attenuates excitotoxicity but potentiates oxygen-glucose deprivation neuronal injury in cortical cell culture. J Cereb Blood Flow Metab. 1993;13:803–810. doi: 10.1038/jcbfm.1993.102. [DOI] [PubMed] [Google Scholar]
- 16.Greene JG, Greenamyre JT. Bioenergetics and glutamate excitotoxicity. Prog Neurobiol. 1996;48:613–634. doi: 10.1016/0301-0082(96)00006-8. [DOI] [PubMed] [Google Scholar]
- 17.Greene JG, Sheu SS, Gross RA, Greenamyre JT. 3-Nitropropionic acid exacerbates N-methyl-d-aspartate toxicity in striatal culture by multiple mechanisms. Neuroscience. 1998;84:503–510. doi: 10.1016/s0306-4522(97)00389-8. [DOI] [PubMed] [Google Scholar]
- 18.Hyrc K, Handran SD, Rothman SM, Goldberg MP. Ionized intracellular calcium concentration predicts excitotoxic neuronal death: observations with low-affinity fluorescent calcium indicators. J Neurosci. 1997;17:6669–6677. doi: 10.1523/JNEUROSCI.17-17-06669.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koh JY, Choi DW. Quantitative determination of glutamate mediated cortical neuronal injury in cell culture by lactate dehydrogenase efflux assay. J Neurosci Methods. 1987;20:83–90. doi: 10.1016/0165-0270(87)90041-0. [DOI] [PubMed] [Google Scholar]
- 20.Koskela T, Wahlström G. Comparison of anaesthetic and kinetic properties of thiobutabarbital, butabarbital and hexobarbital after intravenous threshold doses in the male rat. Pharmacol Toxicol. 1989;64:308–313. doi: 10.1111/j.1600-0773.1989.tb00653.x. [DOI] [PubMed] [Google Scholar]
- 21.Lowenstein DH, Aminoff MJ, Simon RP. Barbiturate anesthesia in the treatment of status epilepticus: clinical experience with 14 patients. Neurology. 1988;38:395–400. doi: 10.1212/wnl.38.3.395. [DOI] [PubMed] [Google Scholar]
- 22.Murphy AN, Fiskum G, Beal MF. Mitochondria in neurodegeneration: bioenergetic function in cell life and death. J Cereb Blood Flow Metab. 1999;19:231–245. doi: 10.1097/00004647-199903000-00001. [DOI] [PubMed] [Google Scholar]
- 23.Nicholls DG, Ward MW. Mitochondrial membrane potential and neuronal glutamate excitotoxicity: mortality and millivolts. Trends Neurosci. 2000;23:166–174. doi: 10.1016/s0166-2236(99)01534-9. [DOI] [PubMed] [Google Scholar]
- 24.Novelli A, Reilly JA, Lysko PG, Henneberry RC. Glutamate becomes neurotoxic via the N-methyl-d-aspartate receptor when intracellular energy levels are reduced. Brain Res. 1988;451:205–212. doi: 10.1016/0006-8993(88)90765-2. [DOI] [PubMed] [Google Scholar]
- 25.Sattler R, Tymianski M. Molecular mechanisms of calcium-dependent excitotoxicity. J Mol Med. 2000;78:3–13. doi: 10.1007/s001090000077. [DOI] [PubMed] [Google Scholar]
- 26.Silver IA, Deas J, Erecinska M. Ion homeostasis in brain cells: differences in intracellular ion responses to energy limitation between cultured neurons and glial cells. Neuroscience. 1997;78:589–601. doi: 10.1016/s0306-4522(96)00600-8. [DOI] [PubMed] [Google Scholar]
- 27.Soine WH, Soine PJ, Overton BW, Garrettson LK. Product enantioselectivity in the N-glucosylation of amobarbital. Drug Metab Dispos. 1986;14:619–621. [PubMed] [Google Scholar]
- 28.Soine WH, Soine PJ, England TM. Barbital N-glucoside is not detected as a urinary excretion product of barbital in humans. J Pharm Biomed Anal. 1991;9:747–752. doi: 10.1016/0731-7085(91)80216-v. [DOI] [PubMed] [Google Scholar]
- 29.Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD, Sakurada O, Shinohara M. The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem. 1977;28:897–916. doi: 10.1111/j.1471-4159.1977.tb10649.x. [DOI] [PubMed] [Google Scholar]
- 30.Stanski DR, Hudson RJ, Homer TD, Saidman LJ, Meathe E. Pharmacodynamic modeling of thiopental anesthesia. J Pharmacokinet Biopharm. 1984;12:223–240. doi: 10.1007/BF01059279. [DOI] [PubMed] [Google Scholar]
- 31.Steen PA, Michenfelder JD. Mechanisms of barbiturate protection. Anesthesiology. 1980;53:183–185. [PubMed] [Google Scholar]
- 32.Stout AK, Raphael HM, Kanterewicz BI, Klann E, Reynolds IJ. Glutamate-induced neuron death requires mitochondrial calcium uptake. Nat Neurosci. 1998;1:366–373. doi: 10.1038/1577. [DOI] [PubMed] [Google Scholar]
- 33.Swanson RA. Astrocyte glutamate uptake during chemical hypoxia in vitro. Neurosci Lett. 1992;147:143–146. doi: 10.1016/0304-3940(92)90580-z. [DOI] [PubMed] [Google Scholar]
- 34.Swanson RA, Seid LL. Barbiturates impair astrocyte glutamate uptake. Glia. 1998;24:365–371. doi: 10.1002/(sici)1098-1136(199812)24:4<365::aid-glia1>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 35.Swanson RA, Farrell K, Stein BA. Astrocyte energetics, function, and death under conditions of incomplete ischemia: a mechanism of glial death in the penumbra. Glia. 1997;21:142–153. doi: 10.1002/(sici)1098-1136(199709)21:1<142::aid-glia16>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 36.Tang BK. Drug glucosidation. Pharmacol Ther. 1990;46:53–56. doi: 10.1016/0163-7258(90)90034-y. [DOI] [PubMed] [Google Scholar]
- 37.Van Ness PC. Pentobarbital and EEG burst suppression in treatment of status epilepticus refractory to benzodiazepines and phenytoin. Epilepsia. 1990;31:61–67. doi: 10.1111/j.1528-1157.1990.tb05361.x. [DOI] [PubMed] [Google Scholar]
- 38.Vergun O, Keelan J, Khodorov BI, Duchen MR. Glutamate-induced mitochondrial depolarisation and perturbation of calcium homeostasis in cultured rat hippocampal neurones. J Physiol (Lond) 1999;519:451–466. doi: 10.1111/j.1469-7793.1999.0451m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ward MW, Rego AC, Frenguelli BG, Nicholls DG. Mitochondrial membrane potential and glutamate excitotoxicity in cultured cerebellar granule cells. J Neurosci. 2000;20:7208–7219. doi: 10.1523/JNEUROSCI.20-19-07208.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Werz MA, Macdonald RL. Barbiturates decrease voltage-dependent calcium conductance of mouse neurons in dissociated cell culture. Mol Pharmacol. 1985;28:269–277. [PubMed] [Google Scholar]
- 41.White RJ, Reynolds IJ. Mitochondria and Na+/Ca2+ exchange buffer glutamate-induced calcium loads in cultured cortical neurons. J Neurosci. 1995;15:1318–1328. doi: 10.1523/JNEUROSCI.15-02-01318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ying W, Han SK, Miller JW, Swanson RA. Acidosis potentiates oxidative neuronal death by multiple mechanisms. J Neurochem. 1999;73:1549–1556. doi: 10.1046/j.1471-4159.1999.0731549.x. [DOI] [PubMed] [Google Scholar]
- 43.Zaidan JR, Klochany A, Martin WM, Ziegler JS, Harless DM, Andrews RB. Effect of thiopental on neurologic outcome following coronary artery bypass grafting. Anesthesiology. 1991;74:406–411. doi: 10.1097/00000542-199103000-00003. [DOI] [PubMed] [Google Scholar]