

Abstract
Aggregates of green fluorescent protein (GFP)-fused truncated N-terminal huntingtin containing abnormally long polyglutamine tracts (150 repeats of glutamine residue) were purified from an ecdysone-inducible mutant neuro2A cell line (HD150Q-28) by using a fluorescence-activated cell sorter. To analyze the aggregate-interacting proteins, we subjected the purified aggregates to SDS-PAGE; prominent protein bands in the gel were digested with Achromobactor lysyl endopeptidase, followed by a HPLC-mass spectrometry (MS) analysis. The resulting data of tandem MS analysis revealed that, in addition to ubiquitin and widely reported chaperone proteins such as heat shock cognate 70 (HSC70), human DNA J-1 (HDJ-1), and HDJ-2, the translational elongation factor-1α (EF-1α) and heat shock protein 84 (HSP84) also were recognized as aggregate-interacting proteins. Sequestration of these proteins to aggregates was confirmed further by several immunochemical methods. We confirmed that, in addition to the other known proteins, EF-1α and HSP84 also colocalized with the intracellular aggregates. An assay of the transient expression of EF-1α and HSP84 in HD150Q-28 cells revealed that both proteins improved cell viability. Moreover, the rate of aggregate formation decreased in both transfectants. Our study suggests that both EF-1α and HSP84 are involved in the neurodegenerative process of polyglutamine diseases.
Keywords: huntingtin, polyglutamine aggregate, GFP, cell sorter, mass spectrometry, HSP84, EF-1α
Intracellular inclusions are a common pathological feature in the group of neurodegenerative disorders known as CAG repeat diseases or polyglutamine (polyQ) diseases. At least nine members are involved in this disease group, including Huntington disease (HD) (Zoghbi and Orr, 2000; Nakamura et al., 2001). They are caused by CAG repeat expansion in the coding region of their responsible genes being translated to mutant proteins containing an abnormal length of polyQ tracts. Such an expansion of polyQ repeats in the mutant proteins leads to the formation of intracellular aggregates (Scherzinger et al., 1997). These intracellular aggregates have been observed in the nucleus (intranuclear inclusions) of neurons in the brains of transgenic mice (Davies et al., 1997) and of HD patients (DiFiglia et al., 1997). Lines of evidence have demonstrated that the accumulation of aggregates in brain neurons closely correlates with the progression of polyQ diseases (Davies et al., 1997; Ona et al., 1999;Paulson, 1999; Yamamoto et al., 2000). It is hypothesized that the expanded polyQ tract causes a toxic gain of function in the form of abnormal protein–protein interactions (Trottier et al., 1995; Davies et al., 1997). In this regard, the identification of aggregate-interacting proteins (AIPs) will provide a precise history of the aggregate-forming process, thereby helping to reveal each toxic function of the mutant protein, which in turn may help in developing better therapies for polyQ diseases. Some chaperones such as human DNA J-1 (HDJ-1) and HDJ-2 have been shown to colocalize with aggregates in several polyQ diseases, including HD (Cummings et al., 1998; Stenoien et al., 1999; Jana et al., 2000; Waelter et al., 2001). Also, aggregates have been shown immunohistochemically to be highly ubiquitinated in the brains of transgenic mice and patients of HD (Davies et al., 1997; DiFiglia et al., 1997), and the 20S proteasome protein has been found to colocalize with aggregates (Chai et al., 1999). In the nucleus, transcription factors such as cAMP-responsive element-binding protein (CREB)-binding protein, TATA-binding protein (TBP), and TBP-associated factor [TAF(II)130] have been demonstrated to be colocalized with aggregates (Perez et al., 1998; Kazantsev et al., 1999; Shimohata et al., 2000; Steffan et al., 2000; Nucifora et al., 2001).
The above findings elucidate the cellular dysfunction linked to the distortion of the protein-folding process, ubiquitin–proteasome system, and transcription machinery. Recent reports (Sanchez et al., 1999; U et al., 2001) demonstrating the sequestration of caspase-8 within the aggregates suggests that the aggregates are involved more directly in apoptosis. In addition to these candidates, more AIPs may remain to be unveiled. For surveying such candidates, it is necessary to isolate intact and highly purified aggregates. We previously established ecdysone-inducible mutant neuro2A (N2A) cell lines that were designed to express extended polyQ-containing (150 repeats) truncated N-terminal huntingtin (tNhtt) fused with green fluorescence protein (GFP; tNhtt-150Q-GFP) (Wang et al., 1999) for studying HD. In the present study we purified tNhtt-150Q-GFP aggregates from the mutant N2A cells with a fluorescence-activated cell sorter (FACS) and analyzed AIPs by a proteomics procedure that used a HPLC-mass spectrometry (MS) system.
MATERIALS AND METHODS
Mice. Heterozygous HD exon 1 transgenic male mice of the R6/2 (145 CAG repeats) strain [Jackson code, B6CBA-TgN (HD exon1) 62] were obtained from Jackson Laboratory (Bar Harbor, ME) and were maintained by crossing carrier males with CBA females. The genotype was determined by a PCR assay, and the CAG repeat length was estimated by Genescan, as described previously (Mangiarini et al., 1996). R6/2 mice and their age-matched controls at 4, 8, and 12 weeks of age were killed with ether anesthesia, and their brains were collected. The brain samples were embedded in Tissue-Tek compound, frozen by using powdered solid CO2, and stored at −80°C.
Antibodies. Antibodies were purchased from the following sources. Mouse monoclonal 1C2 (MAB1574), anti-ubiquitin (MAB1510), anti-tubulin β-III isoform (MAB1637), anti-α-actin (MAB1501), and rabbit polyclonal anti-histone H2A (AB3052) were from Chemicon(Temecula, CA). Rabbit polyclonal anti-ubiquitin (Z-0458) was from Dako Japan (Kyoto, Japan). Rabbit polyclonal anti-HSP84 (PA3-012) and anti-HSP86 (PA3-013) were from Affinity Bioreagents (Golden, CO). Mouse monoclonal anti-HDJ-2 (clone KA2A5.6) was from NeoMarkers (Fremont, CA). Mouse monoclonal anti-elongation factor (EF)-1α (clone CBP-KK1) was from Upstate Biotechnology (Lake Placid, NY). Rabbit polyclonal anti-HDJ-1 (SPA-400) was from StressGen Biotechnologies (Victoria, British Columbia, Canada). Goat polyclonal anti-huntingtin N-terminal fragment (N-18; SC-8767), anti-HSC70 (SC-1059), and rabbit polyclonal anti-N-terminal TFIID (TBP; SC-204) were from Santa Cruz Biotechnology (Santa Cruz, CA). Mouse monoclonal anti-GFP (clone 7.1&13.1) was from Roche Diagnostics (Tokyo, Japan). Antibody against v5-epitope was from Invitrogen (Carlsbad, CA).
Alexa Fluor 488-labeled and Alexa Fluor 546-labeled goat anti-mouse IgG and goat anti-rabbit IgG (A-11029, A-11030, A-11034, and A-11035) were obtained from Molecular Probes (Eugene, OR) as secondary antibodies for indirect immunofluorescence. Horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG and goat anti-rabbit IgG were from Amersham Biosciences (Piscataway, NJ). HRP-conjugated anti-goat IgG (SC-2020) was from Santa Cruz Biotechnology.
Induction of expression of tNhtt-polyQ-GFP in N2A cells. The mutant N2A cell lines were established as described previously (Wang et al., 1999) by using an ecdysone-inducible mammalian expression system purchased from Invitrogen. The mutant cell lines bearing tNhtt-150Q-GFP (containing 150 repeats of glutamine residue) and tNhtt-16Q-GFP (containing 16 repeats of glutamine residue) were named as HD150Q-28 and HD16Q-23, respectively. They were stimulated with 1 μm ponasterone A (Invitrogen), a synthetic analog of ecdysone, to induce the expression of tNhtt-polyQ-GFP.
Isolation of tNhtt-150Q-GFP aggregates. HD150Q-28 cells (5 × 107) were plated in 10 of the 100 mm tissue culture dishes (5 × 106 cells/dish) and cultured overnight in DMEM containing 10% fetal bovine serum with penicillin/streptomycin at 37°C under 5% CO2. db-cAMP (5 mm; Nacalai Tesque, Kyoto, Japan) and 1 μm ponasterone A were added to the medium, and the cells were cultured further. After 2 d of the drug treatment the cells in 10 dishes were collected in an ice-cooled homogenization glass pot with 3 ml of PBS containing 10 mmMgCl2 plus 1500 U of DNase I (Nacalai Tesque) and 3 U of RNase A (Nacalai Tesque). The homogenization was performed with a Potter-Elvehjem-type homogenizer with 30 strokes of up-and-down motion of the glass pot under the spinning Teflon pestle (at 3000 rpm) jointed to a Digital Homogenizer (As One, Osaka, Japan). The temperature of the glass pot was kept below 4°C by an occasional dipping in an ice bath. The homogenate was applied directly to an argon laser-loaded fluorescence-activated cell sorter (Epics Elite ESP, Beckman Coulter, Fullerton, CA) with an outlet nozzle of 100 μm in diameter. The flow rate was adjusted to ∼500 events/min, and GFP fluorescence was monitored for sorting. The sorted aggregates were collected in 16 × 100 mm culture tubes (code 9834-1610, Iwaki Glass, Chiba, Japan) and spun down by centrifugation at 1500 ×g for 30 min. The precipitated aggregates were dissolved in 4% Sarcosyl containing PBS and combined and then transferred into a 1.5 ml Eppendorf tube. The aggregates in the Eppendorf tube were washed twice more with 4% Sarcosyl containing PBS to remove nonspecifically bound contaminants from the aggregates; then the aggregate pellet was stored at −80°C.
Normalization of the amount of purified aggregates. Because the protein content of the aggregate cannot be estimated by normal protein assay methods because of its insolubility to buffers, the number of the fluorescent aggregate particles laid on a hemocytometer was measured with a fluorescent microscope, and the amount of the aggregate was normalized by this number.
HPLC-MS analysis. Purified aggregates were boiled in the sample buffer for SDS-PAGE and electrophoresed in 5–20% polyacrylamide gradient gel. The separated protein bands in the gel were stained with Coomassie brilliant blue. After intensive washing of the gel with Millipore water, gel pieces of prominent bands were excised. Each gel piece was immersed fully in 40 μl of peptidase solution containing 0.1 m Tris-HCl, pH 9.0, 1 mm EDTA, 0.1% SDS, and 0.4 mU ofAchromobacter lyticus M497–1 lysyl endopeptidase (LysC; EC3.4.21.50) (Wako Pure Chemical Industries, Osaka, Japan) and then incubated overnight at 37°C. The eluate from the gel piece containing the LysC-digested peptides was transferred to a 96-well plate, and the plate was placed in an auto sampler (HTC-PAL, CTC Analytics, Zwingen, Switzerland) linked to a HPLC (Agilent 1100 system, Agilent Technologies, Palo Alto, CA)-MS (LCQ, Thermo Finnigan, San Jose, CA) system. The ionization of the enzymatically digested peptides was induced by the nano-electrospray method immediately after passing a reverse-phase HPLC column (Mightysil RP-18, Kanto Chemical, Tokyo, Japan). The MS/MS spectrum data were analyzed by searching mouse proteins in the NCBInr database (National Center for Biotechnology Information, Bethesda, MD) by using the mass spectrometry analysis software Turbo Sequest (Thermo Finnigan).
Western blot analysis. Cell lysate or purified aggregate was subjected to SDS-PAGE by using a 9-cm-square slab gel, with aggregates applied at ∼14,000 particles/well. The proteins separated in the gel were transferred electrically to polyvinylidene fluoride (PVDF) membrane and were probed with the indicated primary antibodies. The dilutions of the antibodies included 1:2000 in 0.05% Tween 20 containing Tris-buffered saline (TBST) for 1C2, 1:5000 for anti-v5 antibody, and 1:500 for all other primary antibodies. After treatment with HRP-conjugated secondary antibodies at 1:2000 dilutions, the signal was detected via enhanced chemiluminescence (ECL) reagents (Amersham Biosciences).
Immunofluorescence methods. For immunocytochemistry the cells were cultured in chamber slides and were differentiated by adding db-cAMP in the medium together with ponasterone A to induce tNhtt-150Q-GFP expression. At 2 d after the drug treatment the cells were washed twice with Tris-buffered saline (TBS) and fixed for 10 min with 4% paraformaldehyde (PFA) in PBS. Also for immunohistochemistry the frozen brain samples were sectioned at 10 μm thickness in a cryostat (Coldtome CM-502, Sakura Instrument, Tokyo, Japan). Sections were air dried for 30 min, rehydrated with TBS for 5 min, and then fixed for 10 min with 4% PFA in PBS. The plasma membrane of cells in chamber slides or in tissue sections was permeabilized for 5 min with 0.5% Triton X-100 in TBS and then washed and immersed for 1 hr in 5% nonfat dried milk in TBST for blocking. Primary antibody (1:200 dilutions in TBST) was applied, and then the cells or tissue sections were incubated for 12–36 hr at 4°C. After being washed with TBST, the cells or tissue sections were incubated for 40 min with the secondary antibody (1:2000 dilutions), washed several times, and mounted by layering 50% glycerol solution with a cover glass. The specimens were observed under a confocal laser fluorescence microscope (Fluoview FV300, Olympus, Tokyo, Japan).
Electron microscopy and immunogold labeling of purified polyQ aggregates. For morphological observation the aggregates (∼500 particles) were adsorbed onto 300-mesh grids coated by a glow-charged supporting membrane. These grids were fixed by floating with 2% PFA/2.5% glutaraldehyde in 0.1 m phosphate buffer for 5 min, negatively stained with neutralized 2% sodium phosphotungstic acid, and observed by an electron microscope (LEO-912AB; LEO, Oberkochen, Germany). For immunoelectron microscopy the adsorbed aggregates on grids were incubated in a primary antibody (anti-actin, -tubulin, -GFP, -HDJ2, -EF-1α, or -HSP84; 1:500 dilutions in 0.1m TBS) at 4°C overnight. After being washed, the aggregates were incubated with colloidal gold-conjugated antibody (5 nm in diameter, diluted 1:20; British BioCell, Cardiff, UK) for 2 hr, fixed with 2% glutaraldehyde, and processed by the negative-staining method in preparation for electron microscopy.
DNA transfection. cDNA for murine EF-1α and HSP84 was obtained by reverse transcription (RT)-PCR from the total RNA fraction of R6/2 mouse brain. Each cDNA was cloned in TOPO-pcDNA3.1-v5-His mammalian expression vector (Invitrogen). HD16Q-23 or HD150Q-28 cells were plated in six-well plates at a cell density of 1 × 106 cells/well in 1 ml of DMEM containing 10% fetal bovine serum with penicillin and streptomycin. Cells were cultured overnight at 37°C under 5% CO2. Then 1 μg of the vector containing EF-1α, HSP84, LacZ (negative control), or HDJ-1 (positive control) dissolved in 50 μl of OPTI-MEM (Invitrogen) and 2 μl of Lipofectamine 2000 (Invitrogen) dissolved in 50 μl of OPTI-MEM were prepared separately. The vector solution and Lipofectamine 2000 solution were mixed 5 min later. After standing for 20 min, 100 μl of the DNA–Lipofectamine complex was added to the cells in each well. The cells were cultured for 12 hr and then transferred to the wells of 48-well plates at a density of 5 × 104 cells per 0.25 ml of medium/well. The protein expressions of the transfected genes were confirmed by SDS-PAGE of the cell lysate, followed by Western blot analysis with the anti-v5 antibody as a probe.
Measuring cell viability and aggregate formation. Cells plated in 48-well plates and transfected with four different DNAs as described above were cultured for 24 hr. Each transfectant was divided into two blocks; one of the blocks was treated with 5 mmdb-cAMP, whereas the other was treated with 5 mm db-cAMP plus 1 μm ponasterone A. Cells were cultured further for 1, 2, 3, or 4 d and then subjected to 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay to measure the cell viability. The MTT assay was performed as described previously (Wang et al., 1999).
For measuring the effect of EF-1α or HSP84 on aggregate formation, we treated each transfectant simultaneously with 5 mm db-cAMP and 0.1 μm ponasterone A. After 24 hr of culture the cells were fixed with 4% PFA in PBS; then each overexpressing protein was stained with anti-v5 antibody, followed by detection with Alexa Fluor 546-labeled secondary antibody. The numbers of the cells expressing transfected DNA and the fluorescent aggregates were counted under a confocal laser fluorescent microscope. The frequency of aggregates in each transfectant was estimated as a percentage of the numbers of aggregate-positive cells in the cells expressing transfected DNA.
Data were analyzed statistically by using the statistical data analysis software StatView (SAS Institute, Cary, NC).
Filter retardation assay. The filter retardation assay for evaluating the amount of the aggregated form of tNhtt was performed as reported previously (Sittler et al., 1998; Nagao et al., 2000). LacZ-, EF-1α-, or HSP84-transfected cells were homogenized in PBS containing 10 mm MgCl2 plus 1500 U of DNase I and 3 U of RNase A. After 1 hr of incubation at 37°C, 25 μg of protein of each homogenate was diluted with 1% SDS and 8% β-mercaptoethanol in PBS and filtered through a cellulose acetate membrane (0.2 μm pore size; Schleicher & Schuell, Dassel, Germany). Then the membrane was subjected to the procedure of general Western blot analysis by using anti-N-terminal huntingtin antibody as a probe, and the immunoreactive spots were detected by ECL. The density of the spots was analyzed by Scion Image software (Scion, Frederick, MD) to quantify the amount of the immunoreactivity.
RESULTS
Conditional expression of tNhtt-150Q-GFP in mutant N2A cells
To confirm that treatment of the mutant N2A cell lines with db-cAMP and ponasterone A could induce differentiation and expression of transfected genes, we observed morphological change and GFP fluorescence under a fluorescent microscope. Cells exposed to db-cAMP and ponasterone A began to show neuronal differentiation within 12 hr after treatment (Fig.1a,c), and this differentiation continued for 3 or 4 d. The expression of tNhtt-16Q-GFP and tNhtt-150Q-GFP began to be seen within 6 hr of treatment and continued to increase over the subsequent days. In HD16Q-23 cells the entire cytosol appeared diffusely luminous under soluble tNhtt-16Q-GFP fluorescence (Fig. 1b). In HD150Q-28 cells, on the other hand, tNhtt-150Q-GFP gradually formed aggregates that appeared as bright fluorescent particles mostly in the perikaryotic area of the cytosol (Fig. 1d). The aggregates were seen rarely in the nucleus, possibly because of the rapid aggregation in the cytosol. At 3 d after the drug treatment the HD150Q-28 cells abruptly entered the apoptotic phase, as observed in our previous work (Wang et al., 1999). For further study we collected the cells after 2 d of treatment with ponasterone A, which seemed to be the best time point for obtaining a high number of aggregates before cell death.
Fig. 1.

Expression of tNhtt-16Q-GFP and tNhtt-150Q-GFP in differentiated N2A cells. HD16Q-23 (a, b) and HD150Q-28 (c, d) were treated with db-cAMP and ponasterone A. After 2 d of culture their morphology and tNhtt-GFP expressions were observed in phase contrast (a, c) and corresponding fluorescence (b, d) images. Scale bars, 20 μm.
Isolation of tNhtt-150Q-GFP aggregates by the use of a cell sorter
The lysate of the collected cells was prepared by homogenization; then the aggregates were isolated from the lysate by using a FACS. For effective separation of aggregates from other cellular components, intensive homogenization of the cell suspension was necessary. On the other hand, a mild denaturing procedure was preferable so that the possible interacting proteins could remain associated with the aggregates. The lysate basically was prepared in nondetergent buffer by using a mechanical homogenizer. The effect of detergent treatment with or without brief sonication was tested; however, no significant improvement in the extracting efficiency as estimated by counting aggregates was observed (data not shown). In the scattergram (Fig.2a) of the flow cytometric analysis of the lysates, particles with the highest fluorescence of GFP were observed mostly in the region with minimum forward and side scattering (region A), which consisted of 64% of the whole particles. Among the particles in region A, 18% showed high fluorescence for GFP (Fig. 2a). The bright particles were sorted and subjected to reanalysis. As shown in Figure 2b, the sorted particles were highly homogeneous and showed high fluorescence for GFP. The average size of a sorted aggregate was estimated as 5 μm in diameter under laser confocal microscopic observation (Fig. 2c). The results shown in Figure 2 were highly reproducible in the indicated number of independent experiments.
Fig. 2.
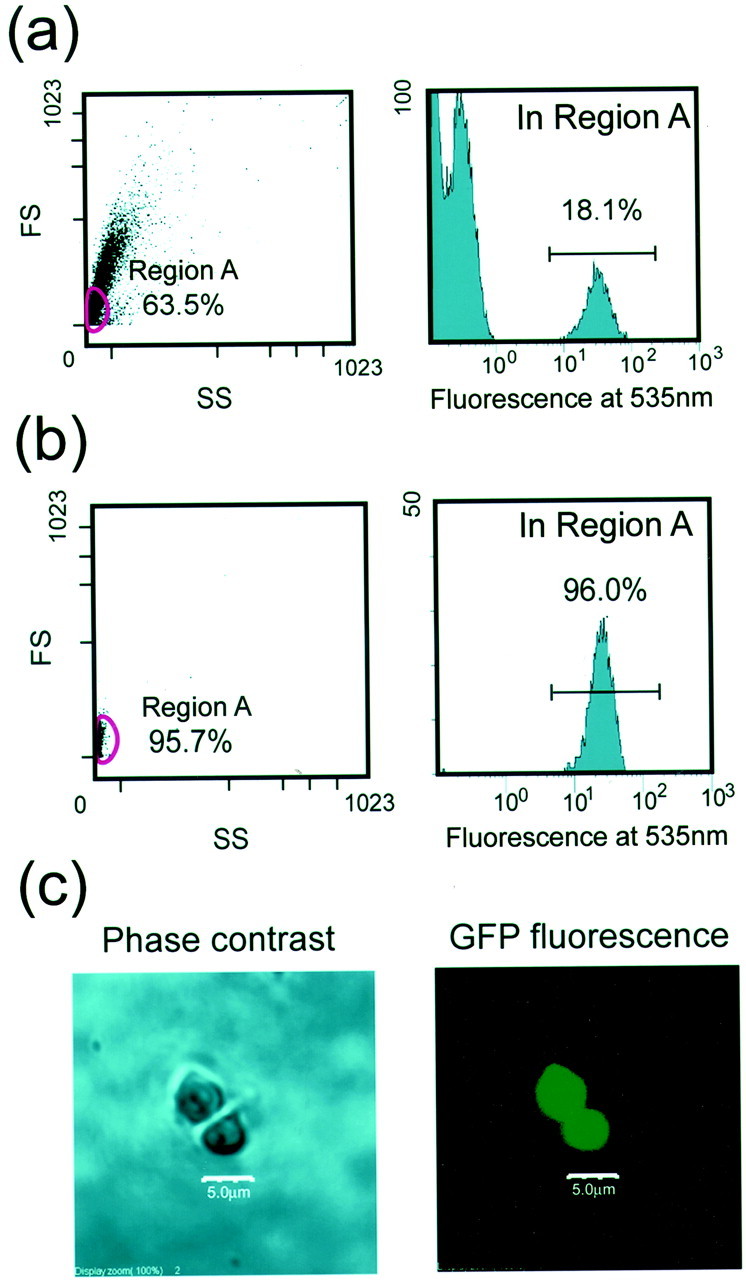
Purification of tNhtt-150Q-GFP aggregates with a cell sorter. a, Sorting profiles of aggregates from HD150Q-28 lysate are shown in a scattergram [left; forward scattering (FS) vs side scattering (SS)] and in a histogram for the fluorescence at 535 nm (right). Aggregates with bright fluorescence are observed mostly in the particles within Region A of theleft panel. Numbers in theleft and right panels are the relative frequency of particles in Region A against the total particles and the frequency of bright particles in Region A, respectively. b, Results of the reanalysis of the sorted aggregates. Note that sorted particles are homogeneous with respect to the forward scattering, side scattering, and the fluorescence at 535 nm. c, Microscopic observation of purified aggregates presented by phase contrast image (left) and corresponding fluorescence image (right). Scale bars, 5 μm.
SDS-PAGE of isolated tNhtt-150Q-GFP aggregates and protein assignment by MS/MS analysis
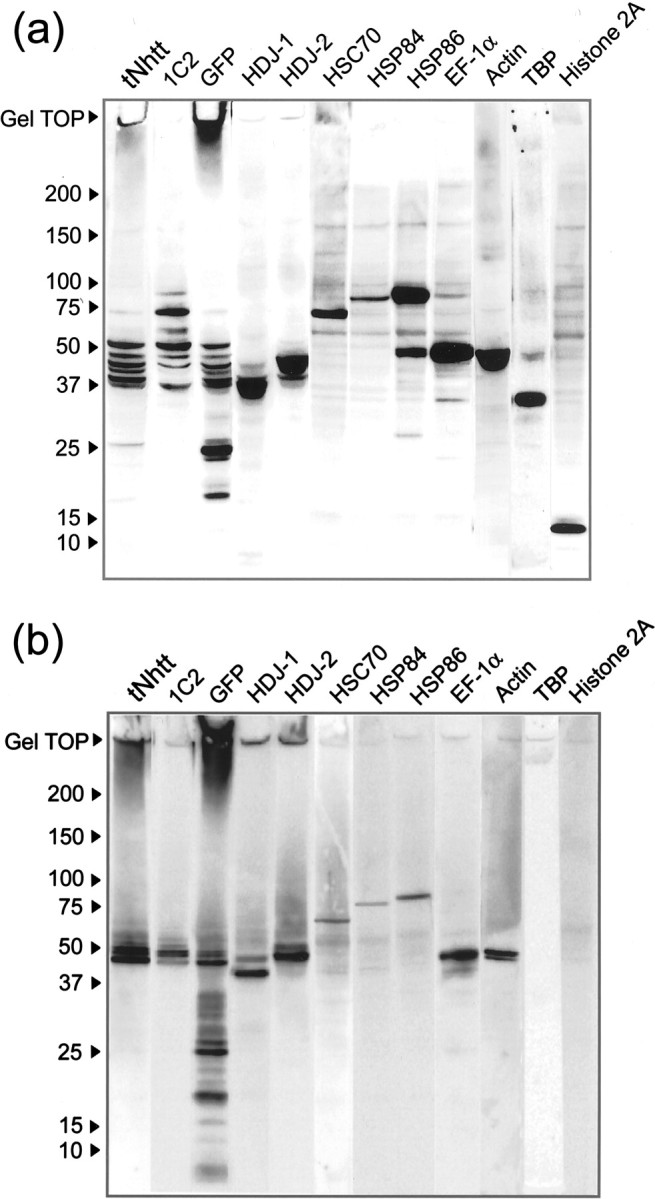
To detect proteins that coexisted, or were recruited to polyQ aggregates, we subjected the isolated aggregates to SDS-PAGE, followed by Coomassie brilliant blue (CBB) staining. The lane on which the aggregate fraction was charged showed smearing of the CBB staining in the range from ∼100 to ∼50 kDa; however, ∼15 bands were recognized clearly. The main bands were excised and subjected to tandem MS analysis after in-gel digestion with LysC. The results of the protein assignment for the PAGE bands are shown in Figure3. Bands overlapped with the smear contained a great abundance of ubiquitin. Because such a high level of ubiquitin interfered with the effective detection of other fragment ions, the masses expected as ubiquitin fragments were set to be rejected from the ion selection for MS/MS analysis. After that setting, the fragments of well studied AIPs such as HSC70, HDJ-1, and HDJ-2 (Fig. 3) were detected clearly as well as the fragments of tNhtt-150Q-GFP itself from their corresponding gel bands. In addition to those expected proteins we detected two more proteins, HSP84 and EF-1α, as candidates of AIPs in this analysis. Actin and tubulin were detected also. The detection of the band for the GFP monomer indicates that a GFP fragment was generated from the tNhtt-150Q-GFP fusion protein by a partial enzymatic cleavage. The strong staining with CBB at the gel top indicates that there existed a highly insoluble portion of the tNhtt-150Q-GFP even after boiling in SDS sample buffer. There were several unknown protein bands that could not be assigned by the current system. So that these bands can be analyzed in the future, it will be necessary to pool more bands of the same molecular weight and apply a different method, such as Edman digestion. The HPLC-MS/MS analysis was applied for three independent experiments, and the results were reproducible.
Fig. 3.

SDS-PAGE of purified aggregates and protein assignment to the CBB-stained bands. Approximately 17,000 purified aggregates were subjected to SDS-PAGE. Coomassie blue-stained bands in the gel were excised, followed by in-gel digestion with LysC. HPLC-MS/MS analysis of the enzyme-digested gel extracts assigned the indicated proteins to the corresponding bands. The white arrowheads with question marks indicate the protein bands that were applied to the HPLC-MS analysis but that could not be identified in this study.
Confirming candidates for AIPs by Western blot analysis
The potential AIPs detected in Figure 3 were confirmed further by Western blot analysis (Fig. 4). For detecting main components of the aggregate (i.e., tNhtt-150Q-GFP), we used the antibody 1C2 and antibodies against tNhtt and GFP. 1C2 is a mouse monoclonal antibody that specifically reacts with abnormally expanded polyQ tracts (Trottier et al., 1995). In lanes 1–3of both blots from lysate (Fig. 4a) and from aggregates (Fig. 4b), several bands in the molecular weight range of 20–70 kDa were detected clearly. Each band was thought to contain GFP-fused tNhtt with different lengths of polyQ tracts. As recognized in Figure 3, a GFP fragment was detected in the blot from aggregates as well as in the blot from lysate. Interestingly, the gel top in which the insoluble aggregates were retained was immunoreactive with anti-tNhtt and anti-GFP antibodies, but not with 1C2. The polyQ structure in the gel top may be different from that migrated in the gel. Because the well known AIPs such as HDJ-1, HDJ-2, and HSC70 are detected clearly in blots from both aggregates and lysate, the purified aggregates in this study must have retained the in vivointeraction with AIPs. HSP84, EF-1α, and actin also were detected specifically in the blots from aggregates and lysate. Because these proteins are quite abundant in cells, it should be evaluated carefully whether they are associated inevitably to aggregates in vivo. However, at this stage, the results of Figure 4 can be considered consistent with those in Figure 3, where HSP84 and EF-1α were taken as possible AIPs. Because some sequences of the fragments of HSP84 that were presented by the tandem MS analysis overlapped with that of HSP86, we checked the immunoreactivity for anti-HSP86 antibody in the blots and detected clear immunoreactive bands for HSP86 in blots from both aggregates and lysate. Suhr et al. (2001) demonstrated recently that intranuclear aggregates formed from the nuclear localization signal-fused tNhtt-96Q sequestrated TBP in human embryonic kidney (HEK) 293 cells. Because, in contrast to their system, the aggregates from tNhtt-150Q-GFP in HD150Q-28 were formed mostly in perikaryotic area in the cytosol but little in the nucleus, immunoreactivity for anti-TBP antibody was not expected as well as that for the anti-histone H2A antibody used as a negative control in the blot from aggregates; this was confirmed.
Fig. 4.
Western blot analysis of AIP candidates. HD150Q-28 cell lysate (a) and purified aggregates (b) were subjected to SDS-PAGE, followed by Western blotting probed with antibodies against the proteins that are indicated on the top of each panel. HRP-conjugated secondary antibodies were used, and the signal was detected by ECL.
Colocalization of EF-1α and HSP84 with tNhtt-150Q-GFP aggregates in N2A cells
Whether the candidate AIPs colocalize with aggregates was examined by fluorescent immunocytochemistry. Differentiated and tNhtt-150Q-GFP-induced HD150Q-28 cells on a culture slide were subjected to the study. The top panel of Figure5a shows a typical image of colocalization of HDJ-2 with the aggregates; this image was taken as an appropriate positive control. EF-1α and HSP84 exist abundantly and are stained widely throughout the whole cytosol so that the colocalization is not as clear as in the case of HDJ-2. However, a clear circular accumulation of Alexa Fluor 546 fluorescence is observed along the outskirts where the GFP aggregates are located. This observation was supported further by the images obtained from the induced and differentiated HD150Q-28 cells transiently transfected with expression constructs containing v5-tagged EF-1α and HSP84 (Fig.5a). This suggests that at least some of the EF-1α and HSP84 molecules localize on the surface area of the aggregates. Immunostaining for actin and tubulin also was performed under the same conditions (Fig. 5b). Actin and tubulin were stained widely throughout the whole cytosol, with holes at the positions where aggregates existed. In contrast to the above cases, no clear sign of accumulation of fluorescence was seen in the vicinity of the holes. On the basis of this observation it was hard to conclude that actin and tubulin colocalized with aggregates.
Fig. 5.
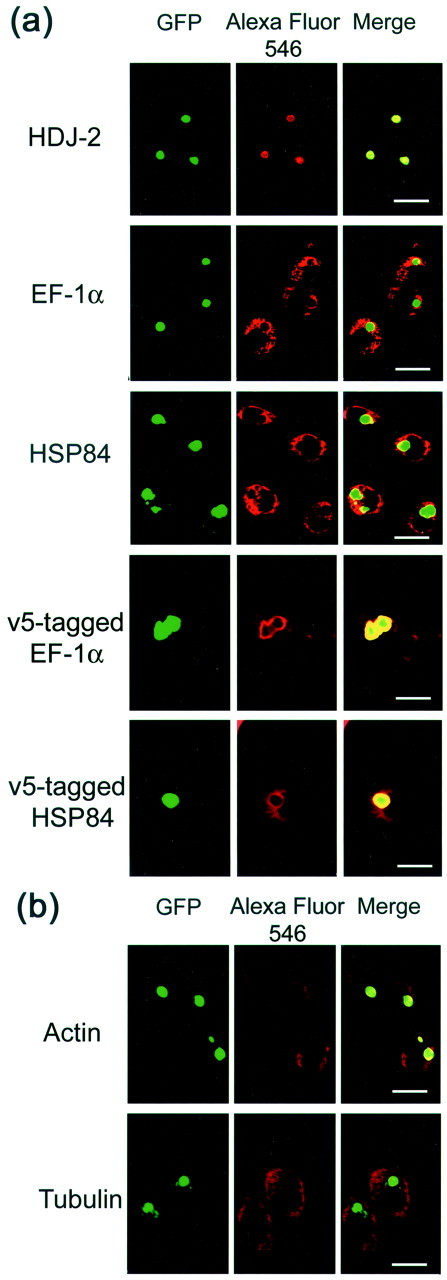
Immunocytochemical analysis of AIP candidates in HD150Q-28 cells. Cells of 2 d in culture after differentiation and tNhtt-150Q-GFP induction were used. Fixed cells were incubated with antibodies against the proteins that are indicated at theleft and then were labeled with Alexa Fluor 546-labeled anti-rabbit or anti-mouse secondary antibodies. Colocalization (a) and uncolocalization (b) of AIP candidates with aggregates are presented. Left column, Fluorescence of aggregates of tNhtt-150Q-GFP. Middle column, Alexa Fluor 546-labeled proteins. Right column, Merged images of the two signals. Scale bars, 20 μm.
Immunoelectron microscopic analysis of association of EF-1α and HSP84 with purified tNhtt-150Q-GFP aggregates
To confirm the association of EF-1α and HSP84 with polyQ aggregates more precisely at the ultrastructural level, we subjected purified aggregates to immunoelectron microscopic analysis. The electron microscopic image of the aggregate of tNhtt-150Q-GFP shows fibrous structures crossing each other in random directions (Fig.6a). Immunogold labeling of HDJ-2 is shown as the positive control (Fig. 6b). Immunogold also specifically labeled EF-1α (Fig. 6c) and HSP84 (Fig.6d) on the aggregate fibers. The gold particles are seen mainly at the perimeters of the fiber tangle but are almost completely absent from the core area. No immunogold labeling is seen in the images of actin (Fig. 6e) and tubulin (Fig. 6f). These results are consistent with the images in the immunocytochemistry (Fig. 5) and further confirm that EF-1α and HSP84 associate with aggregates, but actin and tubulin do not. As abundant cytosolic proteins, actin and tubulin may have been involved nonspecifically in aggregates during the process of their formation and probably washed out in the procedure for immunoelectron microscopy.
Fig. 6.
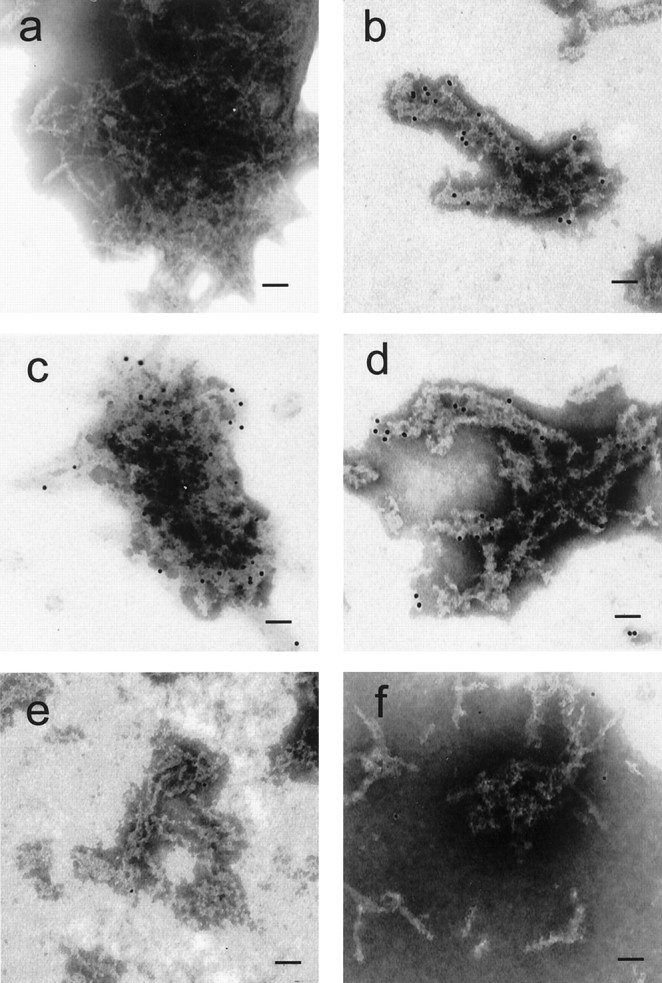
Immunogold labeling of AIP candidates on purified aggregates. The ultrastructure of purified aggregates was observed by electron microscope (a). For immunogold labeling the aggregates were treated with antibodies against HDJ-2 (b), EF-1α (c), HSP84 (d), actin (e), and tubulin (f), followed by probing with immunogold-conjugated anti-IgG antibodies. Black dots on the aggregate fibers represent the labeled gold particles. Scale bars, 30 nm.
Colocalization of EF-1α and HSP84 with tNhtt-150Q-GFP aggregates in an R6/2 transgenic mouse brain
Next we examined whether EF-1α and HSP84 colocalized with aggregates in HD mutant mice brains. Frozen brain sections of an R6/2 mouse were subjected to double-fluorescent immunohistochemical staining. Because aggregates in R6/2 mice brains exist as highly ubiquitinated nuclear inclusions (NIs) in neurons, dual fluorescent labeling of NIs with anti-ubiquitin antibody and anti-EF-1α or anti-HSP84 antibody was performed. The top panel of Figure 7 shows a typical image of the colocalization of HDJ-2 with ubiquitin; this image was taken as an appropriate positive control. Consistent with the immunocytochemical results, the expression of both EF-1α and HSP84 was observed widely in the cytosol in contrast to the appearance of HDJ-2. Here again, however, a clear accumulation of the molecules was observed at the location at which ubiquitin was stained.
Fig. 7.
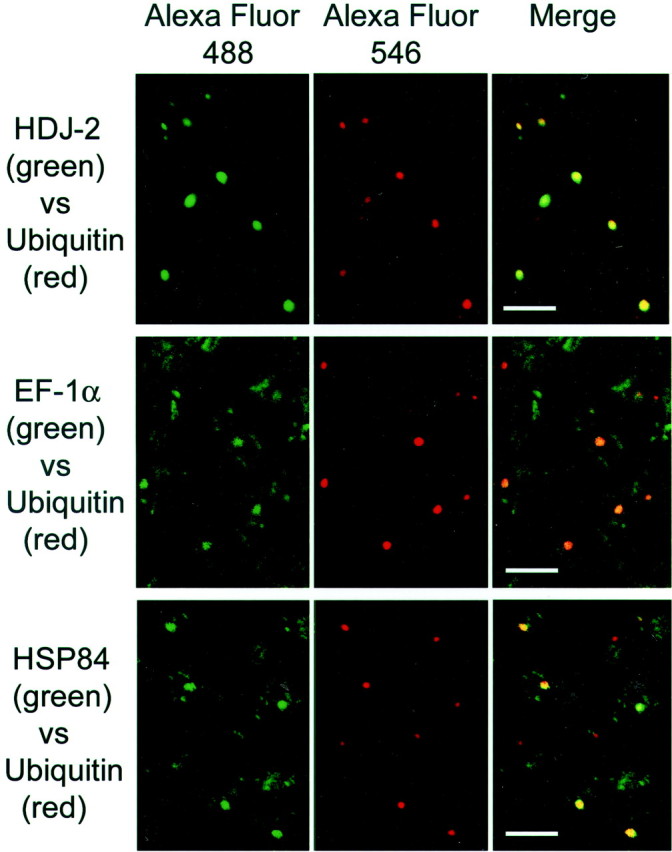
Immunohistochemical analysis of aggregate-interacting candidates in an R6/2 transgenic mouse brain. Frozen brain sections of an R6/2 transgenic mouse were double labeled with anti-ubiquitin antibody, together with the antibodies against the indicated proteins at the left. Anti-ubiquitin antibody was probed with Alexa Fluor 546-labeled secondary antibody, and the antibodies against the indicated proteins were probed with Alexa Fluor 488-labeled secondary antibody. Left column, The Alexa Fluor 488-labeled proteins. Middle column, The localization of Alexa Fluor 546-labeled ubiquitin. Right column, The merged images. Scale bars, 20 μm.
Partial restoration of cell viability of aggregate-expressing N2A cells by overexpression of EF-1α or HSP84
Having demonstrated the colocalization of EF-1α and HSP84 with aggregates by several different immunochemical methods as described above, we next examined whether sequestration of these molecules into aggregates would affect cell viability. For examining this proposition, we transiently transfected HD150Q-28 cells with mammalian expression vectors containing EF-1α and HSP84 genes. At 4 d after the treatment with db-cAMP and ponasterone A to induce differentiation and tNhtt-150Q-GFP expression, we performed an MTT assay to estimate the cell viability. As presented in Figure8a, the control transfectant that expresses LacZ showed a significant decrease in viability to 44.1 ± 2.72% (mean ± SEM; n = 12) when depositing aggregates compared with that of the ponasterone A-untreated control. HDJ-1 transfectant reversed the viability to 69.0 ± 2.84% (mean ± SEM; n = 12) compared with the control. This is consistent with our previous result (Jana et al., 2000). EF-1α transfectant and HSP84 transfectant also reversed the viability to 57.7 ± 2.29 and 61.3 ± 1.61% (mean ± SEM; n = 12), respectively, compared with the control. These results suggest that the sequestration of EF-1α and HSP84 by aggregates is related partially to polyQ-mediated cellular toxicity.
Fig. 8.
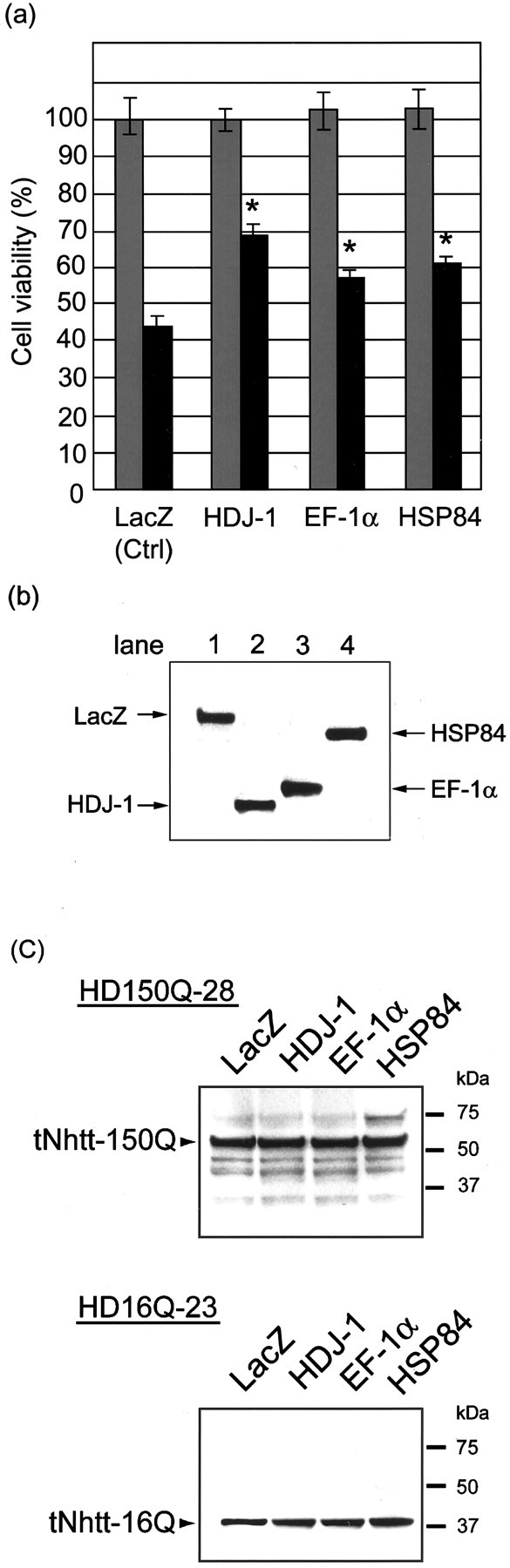
Reduction of polyQ-mediated cellular toxicity in HSP84- and EF-1α-overexpressing mutant N2A cells. a, HD150Q-28 cells were transfected with expression plasmids encoding LacZ (control), HDJ-1, EF-1α, or HSP84. After 24 hr the cells were replated to the 48-well plates at a density of 5 × 104/well and then differentiated by the addition of 5 mm db-cAMP (light gray bars), or the cells were differentiated and induced tNhtt-150Q-GFP by 5 mmdb-cAMP plus 1 μm ponasterone A (dark gray bars). Cell viability on the fourth day of induction was measured by MTT assay and presented as a percentage of differentiated-only LacZ-overexpressing control (indicated asCtrl). Values are the means ± SEM;n = 12. *p < 0.01, compared by Student's t test with a ponasterone A-treated LacZ-overexpressing experiment. b, Expressions of LacZ (lane 1), HDJ-1 (lane 2), EF-1α (lane 3), and HSP84 (lane 4) in the transfected HD150Q-28 cells were confirmed by Western blotting at 2 d after the treatment with db-cAMP and ponasterone A. Bands were detected with anti-v5 antibody. c, Western blot analysis to check the effect of transient overexpression of HDJ-1, EF-1α, and HSP84 on the expression of tNhtt-polyQ-GFP from differentiated and induced HD150Q-28 and HD16Q-23 cell lines. The expression level of tNhtt-polyQ-GFP was detected by anti-N-terminal huntingtin antibody, showing no differences among those transfected cells.
The expression of transfected genes in the cells was confirmed by Western blotting by using anti-v5 antibody as a probe (Fig.8b). Because EF-1α is one of the essential components of translation machinery, we checked whether the transient overexpression of EF-1α changed the expression of stably transfected tNhtt-polyQ-GFP that would affect the cell viability. The Western blot analysis that used anti-N-terminal huntingtin antibody revealed no significant difference of the expression level of tNhtt among HDJ-1-, EF-1α-, HSP84-, and LacZ-overexpressing HD150Q-28 and HD16Q-23 cells in the differentiated and induced state (Fig. 8c).
EF-1α and HSP84 affect the rate of aggregate formation
In addition to the viability, we also examined the frequency of aggregate formation in EF-1α- and HSP84-overexpressing cells. The transfection of LacZ, HDJ-1, EF-1α, and HSP84 vectors to HD150Q-28 cells revealed that ∼60% of total cells were positive for these vectors (Fig. 9a). Among the LacZ transfection-positive cells, 28.4 ± 3.61% (mean ± SEM; n = 10) deposited aggregates. As reported previously (Jana et al., 2000), HDJ-1-overexpressing cells show a reduction of aggregate formation to 9.0 ± 1.77% (mean ± SEM; n = 10) compared with that in LacZ-expressing control cells (Fig. 9b). In the same system the frequencies of aggregate formation in EF-1α- and HSP84-overexpressing cells showed a significant decrease to 11.7 ± 1.74% (mean ± SEM;n = 10) and 13.1 ± 1.98% (mean ± SEM;n = 10), respectively, in comparison with that in the control cells (Fig. 9b).
Fig. 9.
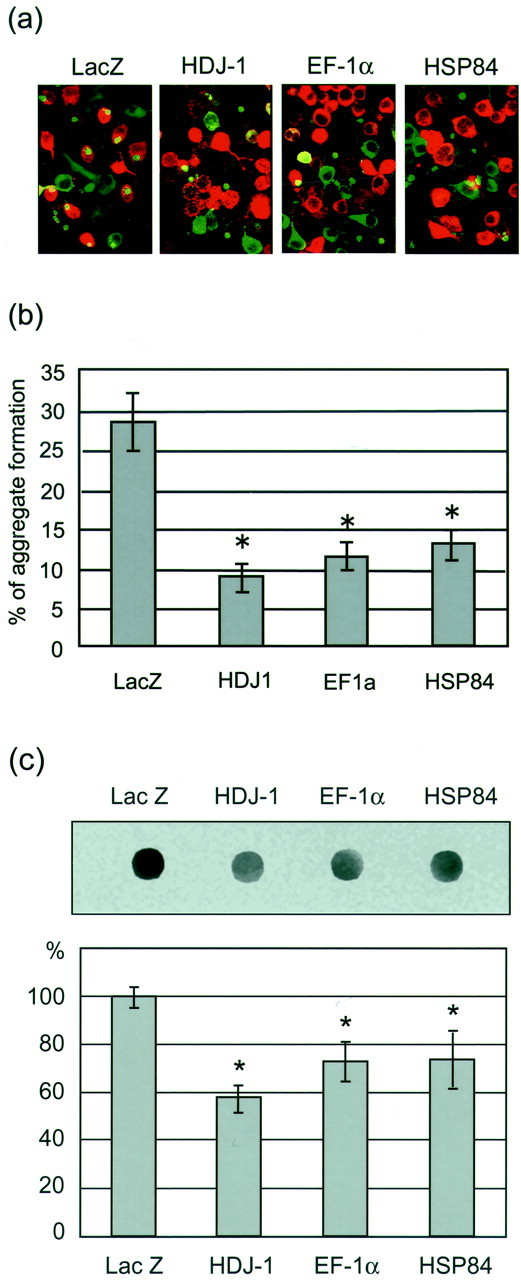
Effect of EF-1α and HSP84 on aggregate formation. a, HD150Q-28 cells overexpressing LacZ, HDJ-1, EF-1α, and HSP84, followed by differentiation and induction with 5 mm db-cAMP plus 0.1 μm ponasterone A, were probed with anti-v5 antibody and stained with Alexa Fluor 546-labeled secondary antibody. The panels show the typical immunofluorescent images. Red, green, andyellow indicate v5, tNhtt-150Q-GFP, and the coexistence of both molecules, respectively. b, The frequency of aggregate formation in the v5-positive cells was measured by counting the numbers of aggregate-positive cells. The data are presented as the percentage of aggregate and v5 double-positive cells in total v5-positive cells. Values are the means ± SEM;n = 10. *p < 0.01, compared by Student's t test with a control experiment that used LacZ-expressing cells. c, Filter retardation assay for comparing aggregate formation in control (LacZ-), HDJ-1-, EF-1α-, and HSP84-overexpressing cells. The aggregated form of tNhtt-150Q-GFP in the cells was trapped on the cellulose acetate membrane; the tNhtt-immunoreactive spots were detected (top), and the density was quantified (bottom) as described in Materials and Methods. The significant reduction of SDS insoluble aggregates was observed in HDJ-1-, EF-1α-, and HSP84-overexpressing cells. Values are the means ± SEM; n = 4. *p < 0.01, compared by Student's ttest with a control experiment that used LacZ-expressing cells.
The reduction of aggregate formation in HDJ-1-, EF-1α-, and HSP84-overexpressing cells compared with that in control cells was confirmed further by the filter retardation assay (Fig. 9c). The densities of the spots from the HDJ-1, EF-1α, and HSP84 samples were observed as significantly lower than that from the control sample. The densitometric analysis showed a decreased amount of aggregate formation to 58.6 ± 0.19, 73.4 ± 0.30, and 74.4 ± 0.44% (mean ± SEM; n = 4 in each case) in HDJ-1-, EF-1α-, and HSP84-overexpressing cells, respectively, in comparison with that in the control cells.
The results suggest that the rescue effect of the overexpression of EF-1α and HSP84 shown in Figure 8 partly involves the attenuation of aggregate formation.
DISCUSSION
Generally, isolation of aggregates is performed by using the detergent insolubility of aggregates or density gradient fractionation (Suhr et al., 2001). In this report we used a cell sorter to purify aggregates of GFP-fused huntingtin mutant protein. We successfully obtained a quality and quantity of purified aggregates that was sufficient for surveying AIPs. Because each aggregate appears as a fairly homogeneous spherical shape with a strong intensity of GFP fluorescence, the whole aggregates were well localized in a small area of the scattergram such that the procedure could be performed with good reproducibility to collect highly pure, homogeneous aggregates. Although we have not compared the different methods of purifying aggregates, the notable advantage of our cell sorter method is that the aggregates are treated as mildly as possible during the whole procedure so that the putative candidates of AIPs can be kept in an associated state. Further, this method can be applied to other GFP fusion-containing aggregates from different disease models.
In previous studies EF-1α was never described as a candidate for an AIP. As for HSP90, its role as an AIP has not yet been well studied, although several reports demonstrated that it colocalized with aggregates (Stenoien et al., 1999; Sittler et al., 2001). In the present study we have shown that both EF-1α and HSP84 are candidates for AIPs. Because they are abundant and ubiquitous cytosolic proteins as well as actin and tubulin, we were concerned that such abundant cytosolic proteins just might diffuse nonspecifically into aggregates rather than interact positively with polyQ-extended tNhtt protein. The electron microscopic study clarified that actin and tubulin were nonspecific contaminants, whereas EF-1α and HSP84 were the actual AIPs. EF-1α is responsible for the GTP-dependent recruitment of aminoacyl-tRNAs to the ribosome during the elongation cycle of protein translation. In addition, regardless of its name, EF-1α is now known to have several different functions aside from protein synthesis. Such functions include actin filament bundling (Yang et al., 1990), oncogenic transformation (Tatsuka et al., 1992), microtubule severing (Shiina et al., 1994), ubiquitin-dependent proteolysis of N terminus-blocked proteins (Gonen et al., 1994), and activation of serum starvation-induced or p53-induced apoptosis with its upregulation or overexpression (Kato et al., 1997; Duttaroy et al., 1998). These functions were demonstrated by using cells of non-neuronal origin, such as erythroleukemia cells (Kato et al., 1997) and mouse 3T3 fibroblasts (Duttaroy et al., 1998). In all of these cases the increase of the intracellular level of EF-1α occurred in parallel with the induction of apoptosis. In contrast, we have demonstrated that overexpression of EF-1α in HD150Q-28 cells results in a partial restoration of the cell viability from the significant reduction in control cells after 3–4 d of differentiation with aggregate deposition. Recently, Khalyfa et al. (2001) reported a correlation between EF-1α deficiency and neurodegeneration. EF-1α consists of two isoforms, EF1A-1 and EF1A-2/S1. EF1A-1 is expressed in most of the tissues, but in the brain, heart, and muscle it is replaced with EF1A-2/S1 during development. Khalyfa et al. (2001) demonstrated that the replacement did not proceed properly in the heterozygous wasted mice mutant, resulting in the deficiency of EF-1α in the brain, heart, and muscle. This caused severe muscle wasting and motor neuron degeneration. Together with our present findings, these results suggest that the change of EF-1α level may induce a susceptibility to apoptosis. The toxicity induced by overexpression or underexpression of EF-1α may depend on the cell types and/or apoptotic pathways.
HSP90 belongs to a highly conserved chaperone family, the members of which include HSP90α, HSP90β, and Grp94 (Csermely et al., 1998). HSP86 and HSP84 are the mouse homolog of HSP90α and HSP90β, respectively. They are expressed rather constitutively in the cytosol and account for as much as 1–2% of all cytosolic proteins. Stress further stimulates their expression. HSP90 forms heterocomplexes and works on the refolding of denatured proteins as HSP70 and HSP60 do with the help of a conformational change with ATP binding. HSP90 generally does not act in nascent protein folding (Nathan et al., 1997) but, rather, associates with a number of signaling molecules such as steroid hormone receptors and signaling protein kinases (Picard et al., 1990;Xu and Lindquist, 1993; Nathan and Lindquist, 1995). In terms of correlation with apoptosis, it first was described that overexpression of HSP90 increased TNF- and cycloheximide-induced apoptosis in U937 cells (Galea-Lauri et al., 1996). Later, however, HSP90 was shown to inhibit staurosporine-induced cell death in the same cell system (Pandey et al., 2000). Its role in apoptosis has been proposed to consist of interaction with components in the apoptotic signal cascade, predominantly for the purpose of blocking cell death.
Jana et al. (2001) suggested recently that disruption of the ubiquitin–proteasome system in aggregate-bearing cells was the primary cause of cell death in a polyQ disease cellular model. They demonstrated that both normal and polyQ-extended tNhtt proteins were degraded by proteasome, but the rate of degradation was reverse-proportional to the polyQ length. The undegraded polyQ-expanded tNhtt forms aggregate, and thus the proteasomal components are sequestered into aggregates. This causes a decrease in the availability of the proteasome for degrading other target proteins, including p53, and this eventually leads to disruption of the mitochondrial membrane potential to initiate the mitochondria-related apoptosis pathway.
We have not yet examined whether EF-1α and HSP84 are connected with the above pathway. However, some lines of evidence suggest that these molecules are involved in the course of the linkage between proteasome function and apoptosis. Gonen et al. (1994) have pointed out that EF-1α may play a role as either a ubiquitin C-terminal hydrolase or a chaperone; in either case EF-1α would help proteins that are the target for proteasome, such as Nα-acetylated proteins, be more accessible to the 26S proteasome complex (Gonen et al., 1994). This suggestion leads us to take EF-1α as a participant in the proteasome disruption hypothesis.
As for HSP90, it recently has been closed up in connection with a mechanism of protein triage decision that regulates the process of recognizing unfolded or misfolded proteins. As stated previously, HSP90 can recognize misfolded proteins and assist their conversion to a functional conformation. However, binding of the C terminus of the HSC70-interacting protein (CHIP) with a tetratricopeptide acceptor site in the HSP90 molecule modulates this process. Such binding causes disruption of the HSP90 heterocomplexes and induces ubiquitylation of the misfolded proteins to put them in a ubiquitin–proteasome degradation pathway rather than continuing the reconformation process.Connell et al. (2001) confirmed the existence of such a protein triage decision derived from CHIP–HSP90 interaction by showing increased ubiquitylation of a glucocorticoid receptor, a well characterized HSP90 substrate, in CHIP-overexpressed COS-7 cells. Murata et al. (2001) also have advocated that in such a function CHIP should be regarded as “a quality-control E3 ligase.” These reports suggest that HSP84 might be one of the key molecules that work to insert the misfolded mutant huntingtin protein into the ubiquitin–proteasome pathway to save the viability of the cells, and in this regard our data may support this prospect in our cell system.
In contrast to the above positive view regarding the role of HSP90 in neurodegeneration, some evidence suggests that the participation of HSP90 in polyQ diseases is negative. Sittler et al. (2001) used Gelnadamycin, a drug that bound to HSP90 to activate the heat shock response in COS-1 cells transiently expressing EGFP-HD79Q fusion protein, to see which chaperones would be connected directly to the aggregate function. Their data showed that expression of HSP70, HSP40, and HSP90 was increased by Gelnadamycin, possibly triggering dissociation of the heat shock factor (HSF)–HSP90 complex; then HSP70 and HSP40 were recruited to aggregates, whereas few HSP90 molecules were colocalized with the aggregates. The overexpression of HSP70 and HSP40 prevented the formation of aggregates, whereas the overexpressed HSP90 did not affect the aggregate formation. However, in our experiment, overexpression of HSP84 in HD150Q-28 cells also resulted in a recovery of the reduced viability after 3–4 d of differentiation with reduced aggregate deposition. Again, these contradictory observations may be attributable to the difference of cell types and/or apoptotic pathways.
The present results at least suggest that EF-1α and HSP84 have the ability to reduce the rate of aggregate formation in the manner of such chaperones as HSP70, HDJ-1, and HDJ-2 and that in this way they may protect cell viability, although there is another possibility that their association with expanded polyQ-containing huntingtin may sterically hinder the intermolecular interaction of polyQ stretches, resulting in the reduction of aggregate formation.
It also should be noted that we could observe the colocalization of both EF-1α and HSP84 with NIs in the brain tissue of HD transgenic mice. This suggests that the sequestration of these molecules into aggregates is not an event limited to our cell culture system but that it also occurs in brains of HD transgenic animals. Our method used in this report can purify relatively large aggregates. The development of a purification method for much smaller aggregates would reveal other specific AIPs. Further intensive studies for AIPs are needed to clarify the precise pathological cascade of polyglutamine diseases.
Footnotes
This study was supported in part by grants-in-aid from the Ministry of Health, Labor, and Welfare and the Ministry of Education, Culture, Sports, Science, and Technology of Japan. We thank Professor Hiroyuki Nishimura at Toin University of Yokohama for his expert advice on the FACS analysis. We also thank Dr. Atsushi Iwata and Hitoshi Doi for valuable help with immunohistochemical studies.
Correspondence should be addressed to Nobuyuki Nukina, Laboratory for CAG Repeat Diseases, RIKEN Brain Science Institute, 2-1 Hirosawa, Wako-shi, Saitama 351-0198, Japan. E-mail: nukina@brain.riken.go.jp.
REFERENCES
- 1.Chai Y, Koppenhafer SL, Shoesmith SJ, Perez MK, Paulson HL. Evidence for proteasome involvement in polyglutamine disease: localization to nuclear inclusions in SCA3/MJD and suppression of polyglutamine aggregation in vitro. Hum Mol Genet. 1999;8:673–682. doi: 10.1093/hmg/8.4.673. [DOI] [PubMed] [Google Scholar]
- 2.Connell P, Ballinger CA, Jiang J, Wu Y, Thompson LJ, Hohfeld J, Patterson C. The co-chaperone CHIP regulates protein triage decisions mediated by heat shock proteins. Nat Cell Biol. 2001;3:93–96. doi: 10.1038/35050618. [DOI] [PubMed] [Google Scholar]
- 3.Csermely P, Schnaider T, Soti C, Prohaszka Z, Nardai G. The 90 kDa molecular chaperone family: structure, function, and clinical applications. A comprehensive review. Pharmacol Ther. 1998;79:129–168. doi: 10.1016/s0163-7258(98)00013-8. [DOI] [PubMed] [Google Scholar]
- 4.Cummings CJ, Mancini MA, Antalffy B, DeFranco DB, Orr HT, Zoghbi HY. Chaperone suppression of aggregation and altered subcellular proteasome localization imply protein misfolding in SCA1. Nat Genet. 1998;19:148–154. doi: 10.1038/502. [DOI] [PubMed] [Google Scholar]
- 5.Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 6.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 7.Duttaroy A, Bourbeau D, Wang XL, Wang E. Apoptosis rate can be accelerated or decelerated by overexpression or reduction of the level of elongation factor-1α. Exp Cell Res. 1998;238:168–176. doi: 10.1006/excr.1997.3819. [DOI] [PubMed] [Google Scholar]
- 8.Galea-Lauri J, Richardson AJ, Latchman DS, Katz DR. Increased heat shock protein 90 (Hsp90) expression leads to increased apoptosis in the monoblastoid cell line U937 following induction with TNF-α and cycloheximide: a possible role in immunopathology. J Immunol. 1996;157:4109–4118. [PubMed] [Google Scholar]
- 9.Gonen H, Smith CE, Siegel NR, Kahana C, Merrick WC, Chakraburtty K, Schwartz AL, Ciechanover A. Protein synthesis elongation factor EF-1α is essential for ubiquitin-dependent degradation of certain Nα-acetylated proteins and may be substituted for by the bacterial elongation factor EF-Tu. Proc Natl Acad Sci USA. 1994;91:7648–7652. doi: 10.1073/pnas.91.16.7648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jana NR, Tanaka M, Wang G, Nukina N. Polyglutamine length-dependent interaction of Hsp40 and Hsp70 family chaperones with truncated N-terminal huntingtin: their role in suppression of aggregation and cellular toxicity. Hum Mol Genet. 2000;9:2009–2018. doi: 10.1093/hmg/9.13.2009. [DOI] [PubMed] [Google Scholar]
- 11.Jana NR, Zemskov EA, Wang G, Nukina N. Altered proteasomal function due to the expression of polyglutamine-expanded truncated N-terminal huntingtin induces apoptosis by caspase activation through mitochondrial cytochrome c release. Hum Mol Genet. 2001;10:1049–1059. doi: 10.1093/hmg/10.10.1049. [DOI] [PubMed] [Google Scholar]
- 12.Kato MV, Sato H, Nagayoshi M, Ikawa Y. Upregulation of the elongation factor-1α gene by p53 in association with death of an erythroleukemic cell line. Blood. 1997;90:1373–1378. [PubMed] [Google Scholar]
- 13.Kazantsev A, Preisinger E, Dranovsky A, Goldgaber D, Housman D. Insoluble detergent-resistant aggregates form between pathological and nonpathological lengths of polyglutamine in mammalian cells. Proc Natl Acad Sci USA. 1999;96:11404–11409. doi: 10.1073/pnas.96.20.11404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khalyfa A, Bourbeau D, Chen E, Petroulakis E, Pan J, Xu S, Wang E. Characterization of elongation factor-1A (eEF1A-1) and eEF1A-2/S1 protein expression in normal and wasted mice. J Biol Chem. 2001;276:22915–22922. doi: 10.1074/jbc.M101011200. [DOI] [PubMed] [Google Scholar]
- 15.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 16.Murata S, Minami Y, Minami M, Chiba T, Tanaka K. CHIP is a chaperone-dependent E3 ligase that ubiquitylates unfolded protein. EMBO Rep. 2001;2:1133–1138. doi: 10.1093/embo-reports/kve246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagao Y, Ishiguro H, Nukina N. DMSO and glycerol reduce bacterial death induced by expression of truncated N-terminal huntingtin with expanded polyglutamine tracts. Biochim Biophys Acta. 2000;1502:247–256. doi: 10.1016/s0925-4439(00)00047-8. [DOI] [PubMed] [Google Scholar]
- 18.Nakamura K, Jeong SY, Uchihara T, Anno M, Nagashima K, Nagashima T, Ikeda S, Tsuji S, Kanazawa I. SCA17, a novel autosomal dominant cerebellar ataxia caused by an expanded polyglutamine in TATA-binding protein. Hum Mol Genet. 2001;10:1441–1448. doi: 10.1093/hmg/10.14.1441. [DOI] [PubMed] [Google Scholar]
- 19.Nathan DF, Lindquist S. Mutational analysis of Hsp90 function: interactions with a steroid receptor and a protein kinase. Mol Cell Biol. 1995;15:3917–3925. doi: 10.1128/mcb.15.7.3917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nathan DF, Vos MH, Lindquist S. In vivo functions of the Saccharomyces cerevisiae Hsp90 chaperone. Proc Natl Acad Sci USA. 1997;94:12949–12956. doi: 10.1073/pnas.94.24.12949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nucifora FC, Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA. Interference by huntingtin and atrophin-1 with CBP-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- 22.Ona VO, Li M, Vonsattel JP, Andrews LJ, Khan SQ, Chung WM, Frey AS, Menon AS, Li XJ, Stieg PE, Yuan J, Penney JB, Young AB, Cha JH, Friedlander RM. Inhibition of caspase-1 slows disease progression in a mouse model of Huntington's disease. Nature. 1999;399:263–267. doi: 10.1038/20446. [DOI] [PubMed] [Google Scholar]
- 23.Pandey P, Saleh A, Nakazawa A, Kumar S, Srinivasula SM, Kumar V, Weichselbaum R, Nalin C, Alnemri ES, Kufe D, Kharbanda S. Negative regulation of cytochrome c-mediated oligomerization of Apaf-1 and activation of procaspase-9 by heat shock protein 90. EMBO J. 2000;19:4310–4322. doi: 10.1093/emboj/19.16.4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paulson HL. Protein fate in neurodegenerative proteinopathies: polyglutamine diseases join the (mis)fold. Am J Hum Genet. 1999;64:339–345. doi: 10.1086/302269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perez MK, Paulson HL, Pendse SJ, Saionz SJ, Bonini NM, Pittman RN. Recruitment and the role of nuclear localization in polyglutamine-mediated aggregation. J Cell Biol. 1998;143:1457–1470. doi: 10.1083/jcb.143.6.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Picard D, Khursheed B, Garabedian MJ, Fortin MG, Lindquist S, Yamamoto KR. Reduced levels of Hsp90 compromise steroid receptor action in vivo. Nature. 1990;348:166–168. doi: 10.1038/348166a0. [DOI] [PubMed] [Google Scholar]
- 27.Sanchez I, Xu CJ, Juo P, Kakizaka A, Blenis J, Yuan J. Caspase-8 is required for cell death induced by expanded polyglutamine repeats. Neuron. 1999;22:623–633. doi: 10.1016/s0896-6273(00)80716-3. [DOI] [PubMed] [Google Scholar]
- 28.Scherzinger E, Lurz R, Turmaine M, Mangiarini L, Hollenbach B, Hasenbank R, Bates GP, Davies SW, Lehrach H, Wanker EE. Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell. 1997;90:549–558. doi: 10.1016/s0092-8674(00)80514-0. [DOI] [PubMed] [Google Scholar]
- 29.Shiina N, Gotoh Y, Kubomura N, Iwamatsu A, Nishida E. Microtubule severing by elongation factor-1α. Science. 1994;266:282–285. doi: 10.1126/science.7939665. [DOI] [PubMed] [Google Scholar]
- 30.Shimohata T, Nakajima T, Yamada M, Uchida C, Onodera O, Naruse S, Kimura T, Koide R, Nozaki K, Sano Y, Ishiguro H, Sakoe K, Ooshima T, Sato A, Ikeuchi T, Oyake M, Sato T, Aoyagi Y, Hozumi I, Nagatsu T, Takiyama Y, Nishizawa M, Goto J, Kanazawa I, Davidson I, Tanese N, Takahashi H, Tsuji S. Expanded polyglutamine stretches interact with TAFII130, interfering with CREB-dependent transcription. Nat Genet. 2000;26:29–36. doi: 10.1038/79139. [DOI] [PubMed] [Google Scholar]
- 31.Sittler A, Walter S, Wedemeyer N, Hasenbank R, Scherzinger E, Eickhoff H, Bates GP, Lehrach H, Wanker EE. SH3GL3 associates with the Huntingtin exon 1 protein and promotes the formation of polygln-containing protein aggregates. Mol Cell. 1998;2:427–436. doi: 10.1016/s1097-2765(00)80142-2. [DOI] [PubMed] [Google Scholar]
- 32.Sittler A, Lurz R, Lueder G, Priller J, Hayer-Hartl MK, Hartl FU, Lehrach H, Wanker EE. Geldanamycin activates a heat shock response and inhibits huntingtin aggregation in a cell culture model of Huntington's disease. Hum Mol Genet. 2001;10:1307–1315. doi: 10.1093/hmg/10.12.1307. [DOI] [PubMed] [Google Scholar]
- 33.Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM. The Huntington's disease protein interacts with p53 and CREB-binding protein and represses transcription. Proc Natl Acad Sci USA. 2000;97:6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stenoien DL, Cummings CJ, Adams HP, Mancini MG, Patel K, DeMartino GN, Marcelli M, Weigel NL, Mancini MA. Polyglutamine- expanded androgen receptors form aggregates that sequester heat shock proteins, proteasome components, and SRC-1, and are suppressed by the HDJ-2 chaperone. Hum Mol Genet. 1999;8:731–741. doi: 10.1093/hmg/8.5.731. [DOI] [PubMed] [Google Scholar]
- 35.Suhr ST, Senut MC, Whitelegge JP, Faull KF, Cuizon DB, Gage FH. Identities of sequestered proteins in aggregates from cells with induced polyglutamine expression. J Cell Biol. 2001;153:283–294. doi: 10.1083/jcb.153.2.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tatsuka M, Mitsui H, Wada M, Nagata A, Nojima H, Okayama H. Elongation factor-1α gene determines susceptibility to transformation. Nature. 1992;359:333–336. doi: 10.1038/359333a0. [DOI] [PubMed] [Google Scholar]
- 37.Trottier Y, Lutz Y, Stevanin G, Imbert G, Devys D, Cancel G, Saudou F, Weber C, David G, Tora L. Polyglutamine expansion as a pathological epitope in Huntington's disease and four dominant cerebellar ataxias. Nature. 1995;378:403–406. doi: 10.1038/378403a0. [DOI] [PubMed] [Google Scholar]
- 38.U M, Miyashita T, Ohtsuka Y, Okamura-Oho Y, Shikama Y, Yamada M. Extended polyglutamine selectively interacts with caspase-8 and -10 in nuclear aggregates. Cell Death Differ. 2001;8:377–386. doi: 10.1038/sj.cdd.4400819. [DOI] [PubMed] [Google Scholar]
- 39.Waelter S, Boeddrich A, Lurz R, Scherzinger E, Lueder G, Lehrach H, Wanker EE. Accumulation of mutant huntingtin fragments in aggresome-like inclusion bodies as a result of insufficient protein degradation. Mol Biol Cell. 2001;12:1393–1407. doi: 10.1091/mbc.12.5.1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang GH, Mitsui K, Kotliarova S, Yamashita A, Nagao Y, Tokuhiro S, Iwatsubo T, Kanazawa I, Nukina N. Caspase activation during apoptotic cell death induced by expanded polyglutamine in N2a cells. NeuroReport. 1999;10:2435–2438. doi: 10.1097/00001756-199908200-00001. [DOI] [PubMed] [Google Scholar]
- 41.Xu Y, Lindquist S. Heat shock protein Hsp90 governs the activity of pp60v-src kinase. Proc Natl Acad Sci USA. 1993;90:7074–7078. doi: 10.1073/pnas.90.15.7074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- 43.Yang F, Demma M, Warren V, Dharmawardhane S, Condeelis J. Identification of an actin-binding protein from Dictyostelium as elongation factor-1a. Nature. 1990;347:494–496. doi: 10.1038/347494a0. [DOI] [PubMed] [Google Scholar]
- 44.Zoghbi HY, Orr HT. Glutamine repeats and neurodegeneration. Annu Rev Neurosci. 2000;23:217–247. doi: 10.1146/annurev.neuro.23.1.217. [DOI] [PubMed] [Google Scholar]