

Abstract
Activation of protein kinase C (PKC) constitutes a key event in the upregulation of secretory strength in neurons and neurosecretory cells during extensive stimulation, presumably by speeding up vesicle supply. However, the molecular targets and their mode of action remain elusive. We studied the only PKC-dependent phosphorylation site in the neuronal solubleN-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex, Ser187, in synaptosome-associated protein of 25 kDa (SNAP-25). This phosphorylation site is located within the negatively charged C-terminal end of SNAP-25, which has been shown to be of critical importance in calcium-triggered exocytosis. We combined mutational studies that used overexpression in chromaffin cells with capacitance measurements and flash photolysis of caged calcium, allowing for high time resolution during both the stimulation and measurement of exocytosis. Overexpression of mutants simulating the phosphorylated form of Ser187 accelerated vesicle recruitment after the emptying of the releasable vesicle pools. Overexpression of mutants simulating the nonphosphorylated form, or block of PKC, impaired the refilling of the vesicle pools to similar extents. Biochemical studies verified the phosphorylation of a subpopulation of SNAP-25 after elevation of intracellular calcium concentrations. Some of the mutations led to a moderately decreased fast exocytotic burst component, which did not seem to be associated with the phosphorylation state of SNAP-25. Thus the C terminus of SNAP-25 plays a role for both fast exocytosis triggering and vesicle recruitment, and the latter process is regulated by PKC-dependent phosphorylation.
Keywords: chromaffin cell, exocytosis, membrane capacitance, protein kinase C, SNARE proteins, SNAP-25
During extensive stimulation neurosecretory cells and neurons undergo use-dependent changes in exocytotic strength. An initial phase of secretory depression caused by a depletion of the release-ready vesicle pool is followed by an upregulation of vesicle recruitment (Zucker, 1999). This process is widely believed to rely on protein phosphorylation by calcium-activated protein kinases, among which protein kinase C (PKC) is the one most studied (Majewski and Iannazzo, 1998; Turner et al., 1999). For instance, in chromaffin cells a PKC inhibitor can block activity-dependent increases in secretory strength (Smith, 1999), and phorbol esters increase both the size and the rate of replenishment of the readily releasable vesicle pool (Gillis et al., 1996; Misonou et al., 1998; Smith et al., 1998).
Although several targets for PKC phosphorylation have been identified, the link to vesicle recruitment is not clear. Among the candidate targets are the plasma membrane solubleN-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins syntaxin and SNAP-25 (synaptosome-associated protein of 25 kDa) and the vesicle-associated SNARE synaptobrevin 2 (VAMP2). These proteins form a stable ternary complex consisting of a twisted four-helix bundle linking the vesicle to the plasma membrane (Sutton et al., 1998; Jahn and Südhof, 1999). The formation of the SNARE complex is required for vesicular release competence (“priming”; Jahn and Südhof, 1999; Xu et al., 1999b), and the final fusion event may be driven by the zippering up of the complex toward the C-terminal ends of syntaxin and synaptobrevin (Hanson et al., 1997).
Of the three neuronal SNAREs only SNAP-25 is phosphorylated by protein kinase C (Shimazaki et al., 1996). Phosphorylation was induced in PC12 cells by treatment with phorbol ester or neuronal growth factor (Shimazaki et al., 1996; Kataoka et al., 2000) and in hippocampal neurons by long-term potentiation (LTP) induction (Genoud et al., 1999). The phosphorylation site is a serine (Ser187) in the C-terminal end of SNAP-25 between the cleavage sites for botulinum toxin A and E (BoNT/A and E; see Fig. 7A). The importance of this region in SNAP-25 has been documented extensively: BoNT/A treatment inhibits secretion, whereas BoNT/E treatment abolishes secretion (Xu et al., 1998; Gerona et al., 2000). Neutralization of negative amino acids within this end of SNAP-25 decreases secretion (Sørensen et al., 2002) and interferes with synaptotagmin I binding (Zhang et al., 2002).
Fig. 7.
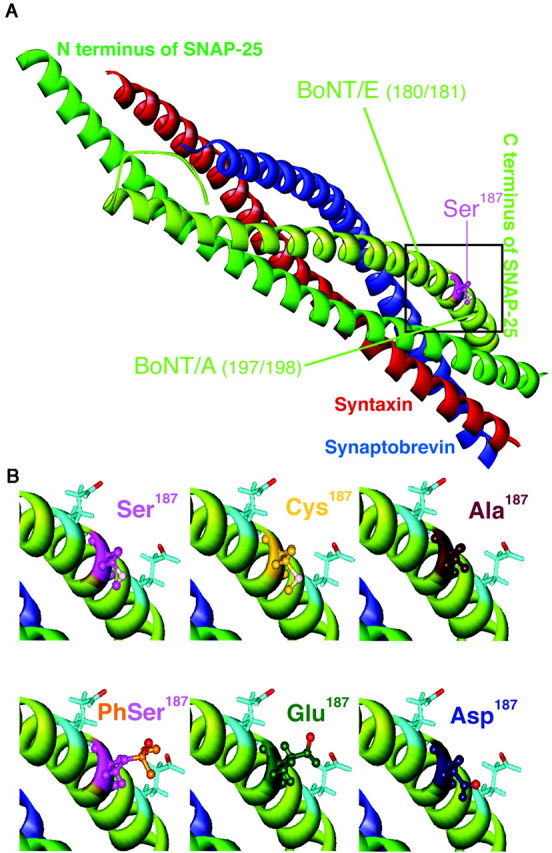
Structural arrangement of in vivoamino acids and mutants at position 187 in SNAP-25. A, An overall view of the core complex indicating the localization of Ser187 (modified after Sutton et al., 1998).B, The top row displays the structural configuration of Ser187 (top left) and its nonphosphorylated substitutes, cysteine (top middle) and alanine (top right), in higher magnification. The side chains of the neighboring negatively charged amino acids Glu183 and Glu194 in the C-terminal end also are indicated with light blue sticks in all panels. The polar oxygen and sulfur are displayed in light pink. The bottom row displays the in vivo phosphoserine (bottom left) and the two amino acids that were used to simulate it, glutamate (bottommiddle) and aspartate (bottom right). The phosphoserine can adopt just one possible rotamer conformation in the complex. Although both glutamate and aspartate have more possible rotamers, glutamate can simulate phosphoserine satisfactorily, whereas aspartate most likely cannot (the negative oxygen is emphasized with red).
Our goal was to investigate whether the phosphorylation of SNAP-25 at Ser187 plays a modulatory role for secretion in chromaffin cells. The conclusion of studies that have used phorbol esters is hampered by the presence of phorbol ester receptors other than PKC with extensive effects on secretion (Munc13; Betz et al., 1998; Rhee et al., 2002) and the simultaneous phosphorylation of a wide range of proteins. To study the effect of phosphorylation of SNAP-25 in isolation, we therefore used a mutagenesis approach in combination with high time resolution capacitance measurements. We found that phosphomimetic mutants of SNAP-25 accelerate vesicle recruitment after an emptying stimulus, whereas nonphosphomimetic mutants, or inhibition of PKC, inhibit vesicle pool refilling. Thus phosphorylation of SNAP-25 may constitute a link between secretory activity and vesicle pool refilling.
MATERIALS AND METHODS
Generation of plasmids and Semliki Forest viruses.The viral vector pSFV1 (Invitrogen, San Diego, CA) was modified by the introduction of an oligonucleotide cassette into itsXmaI site to generate singular ClaI andBssHII restriction sites. Green fluorescent protein (GFP)-SNAP-25A was cloned into the modified pSFV1 as described previously (Wei et al., 2000). SNAP-25 mutants were generated by site-directed mutagenesis. The sequence of all constructs was verified by DNA sequencing. Virus production and transfection were performed as described previously (Ashery et al., 1999).
Chromaffin cell preparation. Cell preparation was modified compared with the previous description (Ashery et al., 1999) by the omission of the Percoll gradient purification step. Bovine adrenal glands were collected, injected with collagenase, and opened; the medulla was dissected out as described previously (Ashery et al., 1999). Pieces of the medulla were collected in a plastic tube with ∼10 ml of Locke's solution and centrifuged at 1000 rpm (190 ×g) for 2 min at 20°C. After the supernatant was removed, the tissue was minced gently for ∼4 min through a 50 μm nylon mesh and washed with Locke's solution. The cell suspension was centrifuged at 600 rpm (70 × g) for 8 min at 20°C. The supernatant was removed; then the cells were resuspended in ∼10 ml of Locke's solution and centrifuged three more times or until the cell preparation was clean. After resuspension in ∼5 ml of enriched DMEM, the cells were plated on glass coverslips. Approximately 2 ml of enriched DMEM was added to each dish, and the cells were incubated at 37°C in 8% CO2. Enriched DMEM (Linaris, Wertheim-Bettingen, Germany) contained 2.2 gm/l NaHCO3, 4.5 gm/l d-glucose, 1.028 gm/l l-glutamine, 10 ml/l insulin-transferrin-selenium-X (Invitrogen), and 1:250 penicillin and streptomycin (10,000 U/ml; Invitrogen).
Solutions. The external solution contained (in mm): 145 NaCl, 2.8 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES plus 1 mg/mld-glucose, pH 7.2 (osmolarity was adjusted to 310 mOsm). Equimolar NaCl was replaced by 37.2 mm KCl and 8 mm CaCl2 in the high potassium/high calcium stimulation for biochemical experiments. The pipette solution contained (in mm): 100 Cs-glutamate, 8 NaCl, 4 CaCl2, 32 HEPES, 2 Mg-ATP, 0.3 GTP, 5 nitrophenyl-EGTA (supplied by G. Ellis-Davies, MCP Hahnemann University, Philadelphia, PA), 0.2 fura-2 (Molecular Probes, Eugene, OR), 0.3 furaptra (Molecular Probes), pH 7.2 (osmolarity was adjusted to 300 mOsm). The PKC inhibitor PKC 19–31 (Calbiochem, Bad Soden, Germany) was diluted in 5% acetic acid as a stock solution at 500 μm. Stock solutions of phorbol 12-myristate 13-acetate (PMA; 200 μm), bisindolylmaleimide I (BIS; 1 or 10 mm; Calbiochem), Gö 6976 (2 mm;Calbiochem), and fura-2 AM (1 mm; Calbiochem) were prepared in DMSO. Calyculin A (Calbiochem) and cypermethrin (Calbiochem) were diluted in DMSO as 10 mm and 10 μm stock solutions, respectively. Cyclosporine A (Calbiochem) was diluted in 70% ethanol as a 1 mm stock solution. The final concentration of DMSO was 0.15% in all biochemical experiments.
All chemicals were from Sigma-Aldrich (Steinheim, Germany) unless otherwise noted.
Electrophysiological and electrochemical measurements.Conventional whole-cell recordings were performed at 30°C with Sylgard-coated 3–5 MΩ pipettes (Kimax-51; Kimble/Kontes, Vineland, NJ) 12–30 hr after transfection. An EPC-9 patch-clamp amplifier was used together with the Pulse software package (HEKA Electronics, Lambrecht, Germany). Capacitance measurements were performed by using the Lindau–Neher technique implemented as the “sine + dc” mode of the software lock-in extension of Pulse, which allowed long-duration capacitance measurements in a single sweep (Gillis, 1995). A 1000 Hz, 70 mV peak-to-peak sinusoid voltage stimulus was superimposed onto a DC holding potential of –70 mV. Currents were filtered at 3 kHz and sampled at 12 kHz.
Carbon fiber electrodes were prepared as described previously (Xu et al., 1999a). A constant voltage of 780 mV versus an Ag/AgCl reference was applied to the electrode. The tip of the carbon fiber was pressed gently against the cell surface. The amperometric current was recorded with an EPC-7 amplifier (HEKA Electronics). Signals were filtered at 3 kHz and sampled at 12 kHz.
The capacitance and amperometric traces were imported to IgorPro (WaveMetrics, Lake Oswego, OR) for analysis. Displayed traces are averages for each condition, with the number of cells (n) given in the figure legends. To control for variation between preparations, we always compared transfected cells with control cells obtained from the same preparation. Kinetic data were obtained by fitting individual capacitance recordings with a sum of three exponential functions. Data are given as the mean ± SEM; the Mann–Whitney test was used for statistical analysis.
Photolysis of caged Ca2+ and measurements of [Ca2+]i. Flashes of UV light were generated by a flash lamp (Rapp Optoelektronik, Hamburg, Germany), and fluorescence excitation light was generated by a monochromator (TILL Photonics, Planegg, Germany) as described previously (Gillis et al., 1996; Xu et al., 1998); these were coupled into the epifluorescence port of an inverted Axiovert 100 microscope with a 40× Fluor objective (Zeiss, Oberkochen, Germany). The fluorescent dyes were excited at 350/380 nm, and the illumination area was reduced to cover only the diameter of the cell. Emitted light was detected with a photomultiplier, filtered at 3 kHz, and sampled at 12 kHz by Pulse software (Ashery et al., 2000). [Ca2+]i was calculated from the fluorescence ratio after calibration as described by Voets (2000). Fluorescent excitation light was used not only to measure [Ca2+]ibut also to adjust [Ca2+]i before and after the flash (Voets, 2000). The calcium concentration before the flash was 200–500 nm.
Western blot. The cells were incubated with PMA or high K+/high Ca2+solution, put on ice, and washed once with ice-cold PBS. Equal amounts of proteins were separated on a 12% SDS-polyacrylamide gel and were blotted onto nitrocellulose membranes. Primary antibodies were mouse anti-SNAP-25, 1:10,000 (Cl 71.2) (Bruns et al., 1997), a kind gift from R. Jahn (Max-Planck-Institute for Biophysical Chemistry, Göttingen, Germany), and rabbit anti-Pi-SNAP-25, 1:250, a kind gift from M. Takahashi (Mitsubishi Korei Institute of Life Sciences, Tokyo, Japan) (Iwasaki et al., 2000). After incubation with secondary antibodies (goat anti-rabbit/anti-mouse horseradish peroxidase-conjugated IgG, 1:10,000; Jackson ImmunoResearch, West Grove, PA), the membranes were washed three times and incubated in ECL Western blotting detection reagent (Amersham Biosciences, Piscataway, NJ). Chemiluminescence-emitting signals were detected by Hyperfilm ECL.
Computer simulations. Molecular simulations were performed by using the InsightII software package (Molecular Simulations, San Diego, CA). Minimization of chains a–d of the crystal structure file1SFC (Protein Data Bank, Brookhaven National Laboratory, Upton, NY) was performed with the Discover program by using the consistent valence forcefield (CVFF) to reach a root mean square of 0.1. The side chain of SNAP-25 Ser187 was modified with the Biopolymer module, and possible rotamers were minimized by using the CVFF. Iterations were stopped at a root mean square of 0.1. For the simulation of phosphorylated Ser187 the hydroxyl group was exchanged against a phosphate group by using the Builder module; rotamers were checked and minimized as described above.
RESULTS
Mutants mimicking the phosphorylated state of Ser187 increase the sustained component of secretion
To study the effect of the phosphorylation of Ser187 in SNAP-25, we replaced Ser187 with either aspartate (S187D) or glutamate (S187E), both of which mimic the constitutively phosphorylated state because of their negative charge. We overexpressed the mutants by using the Semliki Forest virus system in bovine chromaffin cells (Ashery et al., 1999). GFP was attached to the N terminus of SNAP-25 as an indicator of expression. We have shown previously that overexpression of wild-type GFP-linked SNAP-25 leads to ∼25-fold overexpression over the native SNAP-25 and that it assembles into SNARE complexes in vivo without changing significantly the secretion of the transfected cells (Wei et al., 2000). Thus a basic assumption of this method is that the expressed GFP-linked SNAP-25 substitutes functionally for the native SNAP-25 in the cell. The fluorescence of expressing cells was measured, and cells were selected for experiments on the basis of similar fluorescence levels.
Cells were held in whole-cell patch-clamp configuration and dialyzed through the pipette with the calcium-cage nitrophenyl-EGTA and a mixture of two calcium-sensitive fluorescent dyes, fura-2 and furaptra, which allow a precise measurement of [Ca2+]i over a large concentration range (Voets, 2000). At 2–3 min after the establishment of the whole-cell configuration, a strong UV flash was applied to the cells, causing the photolysis of nitrophenyl-EGTA and leading to a step-like homogenous increase of [Ca2+]i. Flash-induced exocytosis was evaluated by high time resolution capacitance measurements. The membrane capacitance increase typically consists of a rapid burst phase, corresponding to the complete emptying of the releasable vesicle pools, followed by a slower sustained phase representing the recruitment of vesicles and consecutive exocytosis. To verify that the capacitance increase resulted from secretion of large dense-core vesicles, we simultaneously monitored secretion by amperometry.
Overexpression of the two phosphomimetic mutants led to an increased exocytotic response (Fig.1A,C) because of a larger sustained component of secretion. We applied a second UV flash ∼80 sec after the first one, which allows enough time for the releasable vesicle pools to refill in control cells. Thus this second flash stimulation evoked a similar response in control cells as the first flash did. Figure 1, B and D, demonstrates that the sustained component of the secretion also was increased after the second flash with the two phosphomimetic mutants.
Fig. 1.

Mutants mimicking the phosphorylated state of Ser187 (aspartate and glutamate substitution) lead to an increased sustained component of secretion. A, Averaged calcium concentration (top), capacitance (middle), and amperometric responses (bottom) after a step-like elevation of [Ca2+]i induced by flash photolysis of a calcium cage (flash at arrow). The capacitance trace displays a burst-like increase within the first 1 sec after the flash, which is followed by a slower sustained phase of secretion, representing vesicle recruitment (priming) and consecutive fusion. Overexpression of S187D mutant (gray trace;n = 22) led to an increased sustained phase of the secretion compared with the nontransfected control cells (black trace; n = 18). B, The corresponding response evoked by the second flash stimulation 80 sec later shows a similar increase in the sustained component of secretion.C, Overexpression of S187E (gray trace; n = 21) also led to an increase in the sustained phase of exocytosis caused by the first flash compared with control cells (black trace; n = 27). D, The corresponding response evoked by the second flash stimulation 80 sec later shows a similar increase in the sustained component of secretion.
These data indicate that SNAP-25 mutants simulating the PKC-phosphorylated state accelerate the sustained component of release, which assays the reaction that refills the releasable vesicle pools.
Mutants mimicking the nonphosphorylated state of Ser187 impair vesicle pool refilling
Alanine substitution is used widely to mimic the nonphosphorylated form of serine. However, the side chain of alanine is nonpolar, whereas the side chain of serine is polar. Cysteine appears to be a better simulation of the nonphosphorylated state of serine because of its similar side chain, although there is the risk of creating sulfur bridges between neighboring cysteines. Therefore, we constructed both S187A and S187C mutants to simulate the nonphosphorylated serine.
These mutants had hardly any effect on the overall response to the first flash (Fig.2A,C), although there was a slight depression with the alanine mutant (discussed below). Both mutants exhibited a marked decrease in the exocytotic burst component caused by the second flash, indicating that the releasable pools were not refilled completely during the ∼80 sec pause between the stimuli (Fig. 2B,D). The decrease in amperometric current during the burst phase of the second flash-evoked event is in agreement with the capacitance measurements (Fig. 2B,D,bottom traces).
Fig. 2.

Mutants mimicking the nonphosphorylated state of Ser187 (alanine and cysteine substitution) reduce the responses evoked by the second flash (flash atarrow). A, Overexpression of S187A mutant (gray trace; n = 18) slightly decreased the overall secretory response to the first flash (flash atarrow) compared with the control group (black trace; n = 18). B, A substantial reduction in the second flash-evoked secretion was observed for the S187A mutant, mainly because of a decrease in the exocytotic burst phase. C, Overexpression of S187C (gray trace; n = 19) did not affect the response to the first flash compared with the control cells (black trace; n = 19).D, The response to the second flash-evoked secretion was diminished in the S187C mutant-expressing cells because of a decrease in the exocytotic burst phase.
Because in this case the mutations that were studied (S187A, S187C) led to a loss-of-function phenotype, we considered whether this may be caused by subtle dominant-negative effects of overexpressing SNAP-25 rather than because of the mutations that have been introduced. We therefore repeated our previous experiment (Wei et al., 2000) and overexpressed wild-type SNAP-25-GFP. The results confirmed that SNAP-25-GFP overexpression has no effect on secretion after the first or the second flash stimulation (Fig.3A,B).
Fig. 3.

Protein kinase C inhibitor peptide, but not SNAP-25 overexpression, reduces the response to the second flash.A, B, Overexpression of wild-type SNAP-25-GFP (gray trace; n = 22) did not affect the response to the first or second flash stimulation (flash atarrow) compared with control cells (black trace; n = 16). C, Inclusion of 10 μm PKC inhibitor peptide (PKC 19–31; gray trace; n = 16) in the pipette had no effect on the secretory response evoked by the first flash compared with control cells (black trace; n = 14).D, PKC 19–31 inhibited the response evoked by the second flash because of a reduced burst phase of exocytosis.
Inhibition of PKC also impairs vesicle pool refilling
The reduction in the response to the second flash that was observed with the two nonphosphorylated mutants implies that PKC-dependent phosphorylation of SNAP-25 on Ser187 facilitates the refilling of the releasable vesicle pools after an emptying stimulus. On the other hand, because the response to the first flash is normal in the S187C mutant, these data also indicate that PKC-dependent phosphorylation is not involved in setting the size of the releasable vesicle pools before stimulation. To test these two interpretations directly, we introduced the pseudosubstrate protein kinase C inhibitor peptide (PKC 19–31;House and Kemp, 1987) into control cells through the patch pipette. We used 10 μm PKC 19–31 in the pipette solution and waited for 3 min before the first flash stimulation. We calculated conservatively that, with an access resistance of 20 MΩ (in reality, the access resistance varied from 5 to 20 MΩ), the concentration of the peptide in the cell would be 7 μm at 3 min after establishing the whole-cell configuration (Pusch and Neher, 1988). This is far above the concentration yielding maximal inhibition of PKC without inhibiting other kinases (House and Kemp, 1987).
Figure 3, C and D, presents the results with PKC 19–31. Although the inhibitor left the response to the first flash, given after 3 min, completely unaffected (Fig. 3C), there was a substantial reduction in the secretion caused by the second flash (Fig. 3D). Thereby, the effect of PKC 19–31 was almost identical to the effect of overexpression of the S187C mutant of SNAP-25 (Fig. 2), which supports the hypothesis that PKC-dependent phosphorylation of SNAP-25 is necessary for rapid refilling of the vesicle pools after, but not before the first flash.
Kinetic analysis: S187D and S187A have a detrimental effect on the fast burst component
In the above discussion of results, we distinguished between only the burst phase (0–1 sec after the flash) and the sustained component (>1 sec after the flash). However, it is evident from Figures 1-3that S187D and S187A decreased the size of the exocytotic burst, whereas for S187E and S187C mutants or with PKC 19–31 treatment the burst phase was almost normal.
Detailed kinetic analysis revealed that the capacitance response evoked by flash photolysis of a Ca2+ cage follows a triple exponential time course (Xu et al., 1998; Voets et al., 1999;Voets, 2000). The fast burst component (time constant, τ = 20–30 msec) and the slow burst component (τ = 200–300 msec) represent the parallel fusion of two release-competent pools, the rapidly and the slowly releasable pools, respectively. The sustained component (τ > 1 sec) represents the refilling of the releasable vesicle pools from a larger pool of docked but unprimed vesicles (Parsons et al., 1995; Ashery et al., 2000). To address the question of whether S187D and S187A mutants have a selective detrimental effect on one of the two burst components, we fit a triple exponential function to the responses from individual cells (Fig.4A, inset) The amplitudes of the first two exponential components provide estimates of the fast and slow burst components, respectively. The size of the sustained component was found by measuring the capacitance increase 5 sec after the flash and subtracting the size of the two releasable pools. To guard against preparation-specific variations, we normalized pool sizes to the control values (Fig. 4). The kinetic analysis revealed that the smaller burst phase of both the S187D and the S187A mutants was attributable to a statistically significant decrease (by ∼50%) in the fast burst component, whereas the size of the slow burst component was unaffected by these mutations. Note that the fast burst elicited by the first flash in the cells expressing either S187E or S187C mutants was almost normal (no statistical significant difference) (Fig. 4A). Moreover, the sustained components were increased almost twofold for both phosphomimetic mutants. The nonphosphomimetic mutants as well as PKC 19–31 did not change the sustained phase but led to a dramatic decrease in both the fast and the slow burst components of the secretion caused by the second flash (Fig. 4B). The time constants for both the fast and slow burst components were unchanged for all mutants and conditions (data not shown).
Fig. 4.
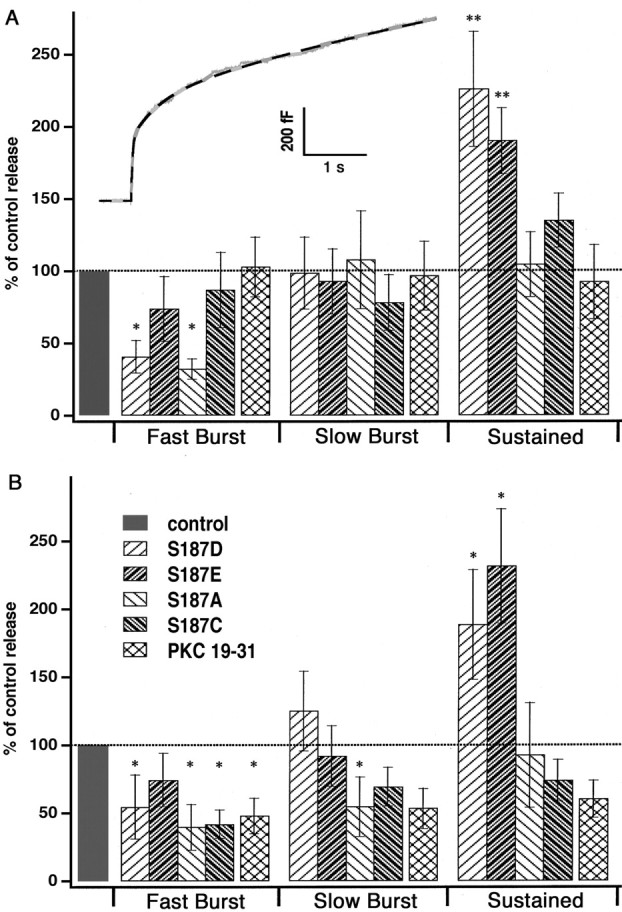
Analysis of the three kinetic components of secretion. A, The inset shows a typical secretory response to the flash in bovine chromaffin cells (gray trace) and, superimposed on it, a triple exponential fit that was used to determine the amplitudes and time constants of the kinetic components (black dotted line). The bar diagram shows the amplitude of each kinetic component of secretion, normalized to control values obtained from the same cell preparations. The time constants did not vary significantly between mutants and conditions and therefore are not displayed. Both of the phosphomimetic mutants (S187D, S187E) caused a statistically significant increase in the amplitude of the sustained component of the secretion evoked by the first flash. The substitution of Ser187 with aspartate (S187D) or alanine (S187A) significantly reduced the fast burst component of secretion. S187C as well as the inhibition of PKC (PKC 19–31) had no significant effects on the different kinetic components of secretion evoked by the first flash stimulation. B, Overexpression of nonphosphomimetic mutants (S187A, S187C) and inhibition of PKC (PKC 19–31) led to a depression of both the fast and the slow component of the exocytotic burst evoked by the second flash. The increase in the sustained component was still present with the two phosphomimetic mutants (S187D, S187E) as well as the depression of the fast burst component in the case of S187D. *p < 0.05; **p < 0.01. Data are displayed as mean ± SEM.
We conclude that two of the mutants decrease the size of the rapidly releasable vesicle pool. Because this was the case for one mutant mimicking the phosphorylated state of Ser187 (S187D) and one mutant mimicking the unphosphorylated state (S187A), this phenomenon apparently is not related to phosphorylation (see Discussion).
The phosphorylation of SNAP-25 alone is not sufficient to allow refilling of the vesicle pools
Inhibition of PKC revealed that the activation of PKC after the first flash is essential for refilling the depleted releasable vesicle pools. Moreover, we observed a similar depression of secretion caused by the second flash with the nonphosphomimetic mutants of SNAP-25, but not with the phosphomimetic mutants. From these experiments we cannot tell whether PKC-dependent phosphorylation of SNAP-25 is the only event required to maintain vesicle supply under our experimental conditions.
To address this question, we inhibited PKC with PKC 19–31 in cells overexpressing S187E and compared them with nontransfected cells in which PKC also was inhibited. The response to the first flash was very similar to results obtained without the inhibition of PKC in both transfected and control cells (compare Figs.5A and 1C,3C). However, S187E could not prevent the reduction in the burst phase of the second flash-evoked secretion caused by PKC inhibition (compare Figs. 5B and 1D,3D). Note that the increase in the sustained component with the S187E mutant persisted even under these circumstances. We therefore conclude that SNAP-25 is not the only substrate for PKC phosphorylation to maintain secretion.
Fig. 5.

Overexpression of the SNAP-25 S187E mutant cannot prevent the reduction of the response to the second flash induced by PKC inhibition. A, Averaged calcium concentration (top), capacitance (middle), and amperometric responses (bottom) measured after the introduction of 10 μm PKC inhibitor (PKC 19–31) into the cells through the pipette. Overexpression of S187E in the presence of PKC inhibition still led to an increase in the sustained phase of exocytosis evoked by the first flash (gray traces; n = 18) compared with the nontransfected cells (black traces;n = 15). The amplitudes of the three kinetic components are indicated at the bottom(black, nontransfected; gray, S187E). When the two conditions are compared, only the difference between the two sustained phases is statistically significant; *p < 0.05. B, In the secretory response caused by the second flash stimulation in the transfected cells (gray traces) and the nontransfected cells (black traces), both are reduced compared with the results without PKC inhibitor (compare with Fig.1D). Dashed capacitance traces indicate the expected secretion without PKC inhibitor (gray for mutants and black for control cells). At the bottom, a triple exponential fit reveals a tendency toward an increase in the sustained phase of the transfected cells even under these conditions; ∼p< 0.1. Data are displayed as mean ± SEM.
Ser187 of SNAP-25 is phosphorylated by PKCα at stimulation
We next tested whether SNAP-25 actually is phosphorylated at Ser187 when the cells are stimulated. For this we used an antibody directed against phosphorylated Ser187 of SNAP-25 (Iwasaki et al., 2000). As a control for the total amount of SNAP-25 we also immunoblotted with an antibody directed against another epitope of SNAP-25 (Bruns et al., 1997). As demonstrated in Figure6A (top panel), although the phosphorylated SNAP-25 was hardly detectable under control conditions, phorbol ester treatment led to massive phosphorylation (Fig. 6A, comparefirst and last lanes). Stimulation by high potassium/high calcium conditions led to the phosphorylation of a smaller fraction of the available SNAP-25 (Fig. 6A,third lane). The high potassium/high calcium-evoked phosphorylation could be blocked completely with the PKC inhibitors BIS and Gö 6976, which is an inhibitor of conventional PKC isoforms (α, β, and γ). We also tested whether parallel phosphatase activity might affect the amount of phosphorylated SNAP-25 under resting conditions and during stimulation. We blocked all major phosphatase types with a mixture of calyculin A (type 1 and 2A), cypermethrin, and cyclosporine A (type 2B) (Hunter, 1995). Application of the phosphatase inhibitors did not increase the level of phosphorylated SNAP-25 under both control and stimulated conditions (Fig. 6A). To quantify the amount of SNAP-25 that was phosphorylated after calcium influx, we performed quantitative densitometric Western blot analysis and displayed the amount of phosphorylated SNAP-25 as a fraction of the amount that was phosphorylated in separate experiments by PMA. The result (Fig.6B) showed, despite large variability between preparations, that only 5% of the SNAP-25 was phosphorylated under basal conditions. After exposure to a solution causing calcium influx, the amount approximately doubled within 30 sec to 10%, with little change over the next couple of minutes. After longer-lasting stimulation (10 min) the amount of phosphorylated SNAP-25 increased to 25% of that obtained in the presence of PMA (Fig. 6B).
Fig. 6.
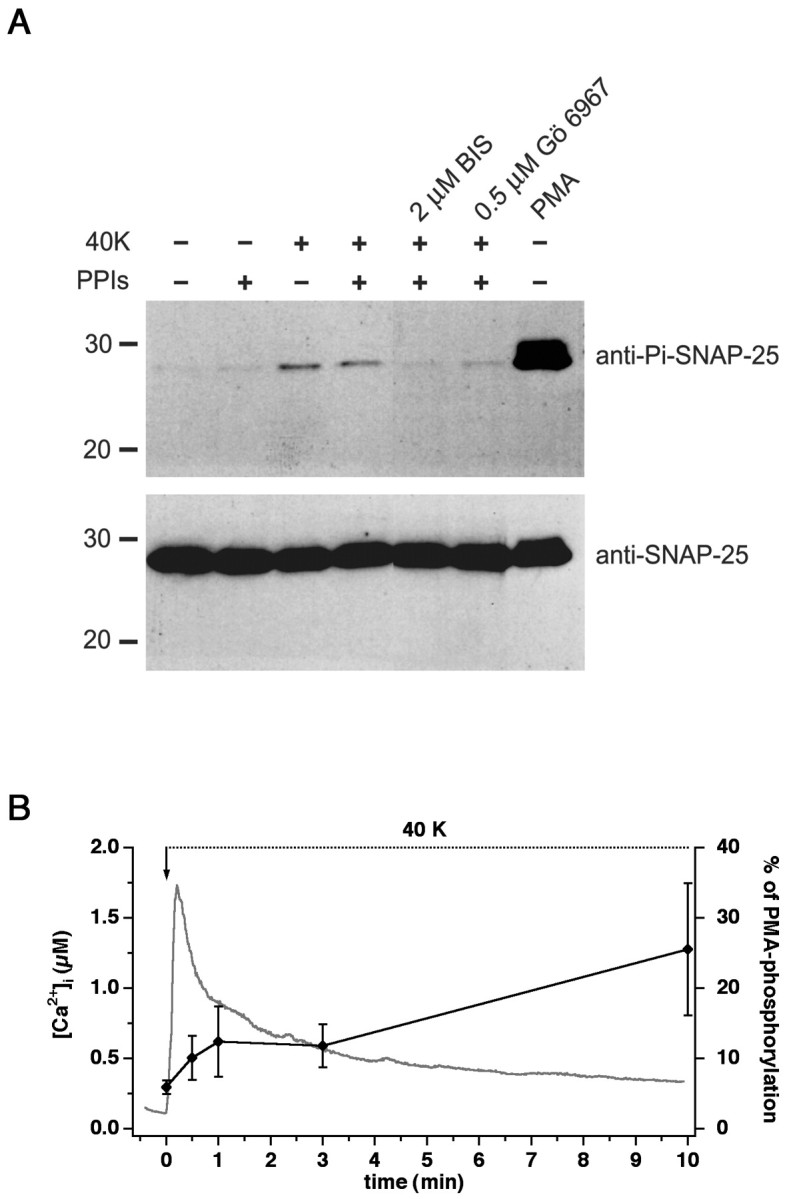
Phosphorylation of SNAP-25 at Ser187. A, Western blot analysis with an antibody against the Ser187-phosphorylated form of SNAP-25 (top). Control cells had very low amounts of phosphorylated SNAP-25. The extracellular application of 40 mm K+ together with 10 mmCa2+ (40K) for 10 min led to the phosphorylation of a subpopulation of SNAP-25. Phosphorylation could be blocked by preincubation with 2 μm BIS or 500 nm Gö 6976, indicating that phosphorylation was mediated by PKC. Adding 10 μm calyculin A, 1 nm cypermethrin, and 1 μm cyclosporine A (protein phosphatase inhibitors, PPIs) did not alter the amount of phosphorylated SNAP-25. Stimulation of the cells with 100 nm PMA for 20 min led to massive phosphorylation (last lane). The bottom panels show the detection of SNAP-25 with a monoclonal antibody against the N terminus of the protein to demonstrate loading of similar amounts of protein into each lane. Marker lines are shown in kilodaltons.B, The time course of the phosphorylation caused by the 40K solution was studied by quantitative Western blot analysis and displayed as a fraction of the PMA-induced phosphorylation in separate experiments (black symbols, right axis; error bars indicate mean ± SEM; n = 5–7). Depolarization caused an increase in phosphorylation of SNAP-25 within 30 sec and a further increase after 10 min. Also plotted is the time course of [Ca2+]i change in fura-2 AM-loaded cells (n = 8) exposed to the depolarizing solution (continuous gray curve, left axis).
In our previous experiments we stimulated the cells by using flash photolysis of caged calcium loaded through the patch pipette, a method that obviously cannot be used when cells are stimulated in the scale required for biochemical experiments. To compare the intensity of flash stimulation with the high potassium/high calcium stimulation used for Western blot analysis, we loaded cells with fura-2 AM, exposed them to the depolarizing solution, and measured the [Ca2+]. The calcium transient that followed depolarization (Fig. 6B) was biphasic, with a peak phase in which the [Ca2+] increased to >1 μm and with a plateau phase of several hundred nanomolars. Separate experiments (data not shown) showed that most of the PMA-induced phosphorylation was blocked by 500 nm BIS or 1 μm Gö6976, whereas 5 μm BIS was required for a complete block. Because PKCα is the only conventional PKC isoform present in chromaffin cells (Sena et al., 2001), we conclude that SNAP-25 is phosphorylated mainly by PKCα in chromaffin cells.
DISCUSSION
Calcium, PKC, and vesicle recruitment
In this report we demonstrated that phosphomimetic mutants of SNAP-25 Ser187 increase the sustained phase of secretion, which reflects the rate of priming of vesicles, almost twofold. Conversely, blocking this phosphorylation by overexpression of mutants simulating the nonphosphorylated Ser187 or by PKC inhibition leads to severe depression of exocytosis after the emptying of the pools. However, not S187C, S187A (except for the effect on the fast burst component discussed later), nor PKC 19–31 affected the secretion caused by the first flash. Biochemical experiments showed that the level of phosphorylated SNAP-25 in resting cells is low but that a partial phosphorylation is seen when [Ca2+]iis increased. This makes it likely that the phosphorylation of SNAP-25 required for fast vesicle recruitment takes place between the first and second stimulation. However, it cannot be ruled out that the basal phosphorylation level of SNAP-25 suffices for fast refilling but that this becomes noticeable only after the emptying of the releasable vesicle pools.
These findings are in agreement with previous studies, which showed that PKC activation plays a modulatory role for the release process in chromaffin cells and PC12 cells (Terbush and Holz, 1990;Iwasaki et al., 2000). An increase in [Ca2+]i was found to cause translocation of PKC to the plasma membrane in chromaffin cells (Terbush et al., 1988). The apparent role of SNAP-25 phosphorylation in vesicle recruitment fits well with the observation that the recruitment of vesicles is PKC-dependent in chromaffin cells (Tsuboi et al., 2001; Shoji-Kasai et al., 2002). A large body of evidence indicates that activity-dependent PKC activation is associated with increased noradrenaline release in CNS and peripheral sympathetic nerves (for review, see Majewski and Iannazzo, 1998). Therefore, the stimulation-induced PKC-dependent phosphorylation of SNAP-25 might be a general feature of adrenaline/noradrenaline-secreting cell types.
Overexpression of the phosphomimetic mutant S187E could not prevent the detrimental effect of PKC inhibitor on the burst size of the second stimulation; therefore, we conclude that phosphorylation of SNAP-25 at Ser187 is necessary, but not sufficient, for maintaining high levels of secretion under our experimental conditions.
Previous investigations in chromaffin cells have identified two pathways of Ca2+-dependent modulation of secretory strength: one is PKC-independent and relies on Ca2+-dependent priming processes of unknown origin (Smith, 1999; Voets, 2000). The other form is PKC-dependent because it is sensitive to PKC inhibitors (Smith, 1999). Most papers studying the latter pathway have emphasized the increase in the size of the rapidly releasable pool (RRP), which can be induced by phorbol esters (Gillis et al., 1996; Smith et al., 1998) or stimulation trains (Smith, 1999) and which results from increased vesicle priming (Smith et al., 1998). However, the interpretation of these results became more complicated by the discovery of the presynaptic phorbol ester receptor Munc13 (Betz et al., 1998), which acts as a priming factor independently of PKC activation (Augustin et al., 1999; Ashery et al., 2000; Rhee et al., 2002). For this reason we avoided phorbol esters in the present study and relied instead on the overexpression of phosphorylation mutants and the endogenous PKC activation after calcium increases.
Surprisingly, we observed that the potentiation of vesicle pool refilling in the SNAP-25 S187E mutant is not correlated with an increase in the size of the releasable vesicle pools; in fact, the size of the releasable vesicle pools remained constant (Fig. 1). Conversely, with nonphosphomimetic mutants or PKC inhibition we found no change in the sustained component of release after the first flash, but we still observed that the vesicle pools were smaller when the cells were stimulated 80 sec later (Figs. 2, 3). These data indicate that, at variance with previous interpretations, there is not a direct relationship between the sustained component of release and the size of releasable vesicle pools at equilibrium. In previous models of the chromaffin cell (Heinemann et al., 1993; Voets et al., 1999;Ashery et al., 2000) it was assumed that vesicles move through a series of linearly arranged pools:
where the depot pool is considered to be a large reserve pool of vesicles that can prime into the slowly releasable pool (SRP) and further mature into the RRP, which is characterized by faster release kinetics than the SRP (i.e., at typical postflash calcium concentrations γ2 ≫ γ1). Vesicles are released into the pool C by exocytosis. In such a scheme the priming rate,k1, is related intimately to the size of the SRP and the RRP at equilibrium. With our data this scheme seems too simple to account for chromaffin cell secretion. One possible explanation might be that priming involves (at least) a second-order process such as the association of the vesicle with a receptor for priming, the availability of which can limit the size of the releasable vesicle pools independently of the refilling rate.
Iwasaki and colleagues (2000) stimulated PC12 cells with phorbol esters and found no correlation between the increase in secretion of dopamine and acetylcholine and the phosphorylation of SNAP-25 at Ser187. They concluded that the effect of PMA was not caused by protein kinase C activation, but possibly by Munc13 activation. Based on the above considerations, the involvement of Munc13 is, indeed, indicated. It is an open question whether the acceleration of vesicle recruitment seen by SNAP-25 Ser187 phosphorylation would have been picked up in the release assay used by Iwasaki and colleagues (2000) or whether in the presence of phorbol ester the massive Munc13 recruitment would mask any phosphorylation effect. We also note that in our case phosphorylation of a small proportion of SNAP-25 apparently is sufficient to cause vesicle recruitment.
The molecular mechanism by which phosphorylation of SNAP-25 at Ser187 causes stimulation of vesicle recruitment remains an open question. So far the only biochemical study of this process demonstrated that phosphorylation of SNAP-25 accelerated the dissociation of SNAP-25 from syntaxin in vitro (Shimazaki et al., 1996). Recently, it was demonstrated that two molecules of syntaxin and one SNAP-25 can associate to form a binary SNARE complex. One syntaxin molecule then has to be replaced by synaptobrevin for productive assembly of the ternary SNARE complex (Margittai et al., 2001). It can be suggested tentatively that phosphorylation of SNAP-25 speeds up disassembly of nonproductive binary complexes, thereby favoring the formation of productive ternary complexes.
A dual role of the C-terminal end of SNAP-25 in exocytosis
A critical role of the C-terminal end of SNAP-25 in exocytosis was demonstrated by treatment with BoNT/E, which cleaves 26 amino acids off the C terminus (including Ser187) and abolishes catecholamine secretion in chromaffin cells (Xu et al., 1998). The exact role of the C terminus of SNAP-25 is, however, still not clear. Some reports indicate that the five acidic amino acids Asp179, Glu183, Asp186, Asp193, and Glu194, which confer a net negative charge to the C terminus, are necessary for exocytosis, maybe via the binding of synaptotagmin I (Gerona et al., 2000; Sørensen et al., 2002; Zhang et al., 2002). Conversely, in BoNT/E-treated cracked open PC12 cells C-terminal SNAP-25 peptides cate that the C-terminal end of SNAP-25 may have a dual role in exocytosis in chromaffin cells. Elimination of the last nine amino acids from the C terminus of SNAP-25 (by BoNT/A treatment or overexpression) led to a reduction in the fast burst component but also to a reduction in the sustained phase and in the response to the second stimulation, which is an indication of slowed recruitment (Xu et al., 1998; Wei et al., 2000).
In the present study phosphomimetic mutants increased vesicle recruitment, whereas nonphosphomimetic mutants decreased it. However, another effect was noted also: two of the mutations caused a significant decrease in fast burst amplitude (Fig.4A). Notably, this was the case for one phosphomimetic (S187D) and one nonphosphomimetic mutant (S187A). This indicates that the change in fast burst amplitude was not related to phosphorylation of Ser187, but to other structural changes induced by the mutations. To investigate this question, we simulated the three-dimensional structure of the neuronal core complex on the basis of the published crystal structure (Sutton et al., 1998), with the substituted mutations as well as the in vivo amino acids in position 187. No gross structural changes in the core complex were induced by the mutations. Not surprising is the prediction that cysteine is a good substitution for serine, whereas alanine as a nonpolar amino acid is not (Fig.7B). Phosphoserine turned out to have only one possible rotamer, which did not lead to clashes with neighboring groups. Glutamate, which is quite similar to phosphoserine, had several possible rotamers, one of which simulated the phosphoserine quite well (displayed in Fig. 7B). Aspartate, on the other hand, did not have any rotamer bringing the negative charge into a similar position (Fig. 7B). These studies are consistent with the view that a very delicate structural arrangement in the C-terminal end of SNAP-25 is necessary for the fast burst component of exocytosis. However, apparently, the fine structure of the C-terminal end is less important during vesicle recruitment, with aspartate being as effective as glutamate during this phase. Therefore, it is likely that sequential molecular interactions of the C-terminal end of SNAP-25 with other proteins are responsible for recruitment and fast triggering of exocytosis.
Footnotes
This work was supported by grants from the Deutsche Forschungsgemeinschaft (Sonderforschungsbereich 530 to J.R., Sonderforschungsbereich 523 to E.N.) and the Human Frontier Science Program to T.B. G.N. is a PhD student of the International MD/PhD Program in the Neurosciences of the International Max Planck Research School and holds a fellowship by the Deutsche Forschungsgemeinschaft (Graduiertenkolleg 521). We thank Uri Ashery, Reinhard Jahn, Corey Smith, Kevin Gillis, and Thomas Voets for commenting on this manuscript. We thank Reinhard Jahn and Masami Takahashi for providing antibodies. We also thank Ina Herfort, Anke Bührmann, Dirk Reuter, and Stefanie Feldhege for expert technical assistance.
Correspondence should be addressed to Jakob B. Sørensen, Max Planck Institute for Biophysical Chemistry, Am Fassberg 11, 37077 Göttingen, Germany. E-mail: jsoeren@gwdg.de.
U. Matti's and J. Rettig's present address: Institute of Physiology, Saarland University, 66421 Homburg, Germany.
REFERENCES
- 1.Ashery U, Betz A, Xu T, Brose N, Rettig J. An efficient method for infection of adrenal chromaffin cells using the Semliki Forest virus gene expression system. Eur J Cell Biol. 1999;78:525–532. doi: 10.1016/s0171-9335(99)80017-x. [DOI] [PubMed] [Google Scholar]
- 2.Ashery U, Varoqueaux F, Voets T, Betz A, Thakur P, Koch H, Neher E, Brose N, Rettig J. Munc13-1 acts as a priming factor for large dense-core vesicles in bovine chromaffin cells. EMBO J. 2000;19:3586–3596. doi: 10.1093/emboj/19.14.3586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Augustin I, Rosenmund C, Südhof TC, Brose N. Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature. 1999;400:457–461. doi: 10.1038/22768. [DOI] [PubMed] [Google Scholar]
- 4.Betz A, Ashery U, Rickmann M, Augustin I, Neher E, Südhof TC, Rettig J, Brose N. Munc13-1 is a presynaptic phorbol ester receptor that enhances neurotransmitter release. Neuron. 1998;21:123–136. doi: 10.1016/s0896-6273(00)80520-6. [DOI] [PubMed] [Google Scholar]
- 5.Bruns D, Engers S, Yang C, Ossig R, Jeromin A, Jahn R. Inhibition of transmitter release correlates with the proteolytic activity of tetanus toxin and botullinus toxin A in individual cultured synapses of Hirudo medicinalis. J Neurosci. 1997;17:1898–1910. doi: 10.1523/JNEUROSCI.17-06-01898.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen YA, Scales SJ, Duvvuri V, Murthy M, Patel SM, Schulman H, Scheller RH. Calcium regulation of exocytosis in PC12 cells. J Biol Chem. 2001;276:26680–26687. doi: 10.1074/jbc.M103522200. [DOI] [PubMed] [Google Scholar]
- 7.Genoud S, Pralong W, Riederer BM, Eder L, Catsicas S, Muller D. Activity-dependent phosphorylation of SNAP-25 in hippocampal organotypic cultures. J Neurochem. 1999;72:1699–1706. doi: 10.1046/j.1471-4159.1999.721699.x. [DOI] [PubMed] [Google Scholar]
- 8.Gerona RRL, Larsen EC, Kowalchyk JA, Martin TFJ. The C terminus of SNAP-25 is essential for Ca2+-dependent binding of synaptotagmin to SNARE complexes. J Biol Chem. 2000;275:6328–6336. doi: 10.1074/jbc.275.9.6328. [DOI] [PubMed] [Google Scholar]
- 9.Gillis KD. Techniques for membrane capacitance measurements. In: Sakmann B, Neher E, editors. Single-channel recording, 2nd Ed. Plenum; New York: 1995. pp. 155–198. [Google Scholar]
- 10.Gillis KD, Mössner R, Neher E. Protein kinase C enhances exocytosis from the chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron. 1996;16:1209–1220. doi: 10.1016/s0896-6273(00)80147-6. [DOI] [PubMed] [Google Scholar]
- 11.Hanson PI, Heuser JE, Jahn R. Neurotransmitter release—four years of SNARE complexes. Curr Opin Neurobiol. 1997;7:310–315. doi: 10.1016/s0959-4388(97)80057-8. [DOI] [PubMed] [Google Scholar]
- 12.Heinemann C, von Rüden L, Chow RH, Neher E. A two-step model of secretion control in neuroendocrine cells. Pflügers Arch. 1993;424:105–112. doi: 10.1007/BF00374600. [DOI] [PubMed] [Google Scholar]
- 13.House C, Kemp BE. Protein kinase C contains a pseudosubstrate prototope in its regulatory domain. Science. 1987;238:1726–1728. doi: 10.1126/science.3686012. [DOI] [PubMed] [Google Scholar]
- 14.Hunter T. Protein kinases and phosphatases: the yin and yang of protein phosphorylation and signaling. Cell. 1995;80:225–236. doi: 10.1016/0092-8674(95)90405-0. [DOI] [PubMed] [Google Scholar]
- 15.Iwasaki S, Kataoka M, Sekiguchi M, Shimazaki Y, Sato K, Takahashi M. Two distinct mechanisms underline the stimulation of neurotransmitter release by phorbol esters in clonal rat pheochromocytoma PC12 cells. J Biochem (Tokyo) 2000;128:407–414. doi: 10.1093/oxfordjournals.jbchem.a022768. [DOI] [PubMed] [Google Scholar]
- 16.Jahn R, Südhof TC. Membrane fusion and exocytosis. Annu Rev Biochem. 1999;68:863–911. doi: 10.1146/annurev.biochem.68.1.863. [DOI] [PubMed] [Google Scholar]
- 17.Kataoka M, Kuwahara R, Iwasaki S, Shoji-Kasai Y, Takahashi M. Nerve growth factor-induced phosphorylation of SNAP-25 in PC12 cells: a possible involvement in the regulation of SNAP-25 localization. J Neurochem. 2000;74:2058–2066. doi: 10.1046/j.1471-4159.2000.0742058.x. [DOI] [PubMed] [Google Scholar]
- 18.Majewski H, Iannazzo L. Protein kinase C: a physiological mediator of enhanced transmitter output. Prog Neurobiol. 1998;55:463–475. doi: 10.1016/s0301-0082(98)00017-3. [DOI] [PubMed] [Google Scholar]
- 19.Margittai M, Fasshauer D, Pabst S, Jahn R, Langen R. Homo- and hetero-oligomeric SNARE complexes studied by site-directed spin labeling. J Biol Chem. 2001;276:13169–13177. doi: 10.1074/jbc.M010653200. [DOI] [PubMed] [Google Scholar]
- 20.Misonou H, Ohara-Imaizumi M, Murakami T, Kawasaki M, Ikeda K, Wakai T, Kumakura K. Protein kinase C controls the priming step of regulated exocytosis in adrenal chromaffin cells. Cell Mol Neurobiol. 1998;18:379–390. doi: 10.1023/A:1022593330685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parsons TD, Coorssen JR, Horstmann H, Almers W. Docked granules, the exocytotic burst, and the need for ATP hydrolysis in endocrine cells. Neuron. 1995;15:1085–1096. doi: 10.1016/0896-6273(95)90097-7. [DOI] [PubMed] [Google Scholar]
- 22.Pusch M, Neher E. Rates of diffusional exchange between small cells and a measuring patch pipette. Pflügers Arch. 1988;411:204–211. doi: 10.1007/BF00582316. [DOI] [PubMed] [Google Scholar]
- 23.Rhee J-S, Betz A, Pyott S, Reim K, Varoqueaux F, Augustin I, Hesse D, Südhof TC, Takahashi M, Rosenmund R, Brose N. β-Phorbol ester- and diacylglycerol-induced augmentation of transmitter release is mediated by Munc13s and not by PKC. Cell. 2002;108:121–133. doi: 10.1016/s0092-8674(01)00635-3. [DOI] [PubMed] [Google Scholar]
- 24.Sena CM, Santos RM, Standen NB, Boarder MR, Rosário L. Isoform-specific inhibition of voltage-sensitive Ca2+ channels by protein kinase C in adrenal chromaffin cells. FEBS Lett. 2001;492:146–150. doi: 10.1016/s0014-5793(01)02252-9. [DOI] [PubMed] [Google Scholar]
- 25.Shimazaki Y, Nishiki T-I, Omori A, Sekiguchi M, Kamata Y, Kozaki S, Takahashi M. Phosphorylation of 25 kDa synaptosome-associated protein. J Biol Chem. 1996;271:14548–14553. doi: 10.1074/jbc.271.24.14548. [DOI] [PubMed] [Google Scholar]
- 26.Shoji-Kasai Y, Itakura M, Kataoka M, Yamamori S, Takahashi M. Protein kinase C-mediated translocation of secretory vesicles to plasma membrane and enhancement of neurotransmitter release from PC12 cells. Eur J Neurosci. 2002;15:1390–1394. doi: 10.1046/j.1460-9568.2002.01972.x. [DOI] [PubMed] [Google Scholar]
- 27.Smith C. A persistent activity-dependent facilitation in chromaffin cells is caused by Ca2+ activation of protein kinase C. J Neurosci. 1999;19:589–598. doi: 10.1523/JNEUROSCI.19-02-00589.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smith C, Moser T, Xu T, Neher E. Cytosolic Ca2+ acts by two separate pathways to modulate the supply of release-competent vesicles in chromaffin cells. Neuron. 1998;20:1243–1253. doi: 10.1016/s0896-6273(00)80504-8. [DOI] [PubMed] [Google Scholar]
- 29.Sørensen JB, Matti U, Wei S, Nehring RB, Voets T, Ashery U, Binz T, Neher E, Rettig J. The SNARE protein SNAP-25 is linked to fast calcium triggering of exocytosis. Proc Natl Acad Sci USA. 2002;99:1627–1632. doi: 10.1073/pnas.251673298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sutton RB, Fasshauer D, Jahn R, Brünger AT. Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution. Nature. 1998;395:347–353. doi: 10.1038/26412. [DOI] [PubMed] [Google Scholar]
- 31.Terbush DR, Holz RW. Activation of protein kinase C is not required for exocytosis from bovine adrenal chromaffin cells. J Biol Chem. 1990;265:21179–21184. [PubMed] [Google Scholar]
- 32.Terbush DR, Bittner MA, Holz RW. Ca2+ influx causes rapid translocation of protein kinase C to membranes. J Biol Chem. 1988;263:18873–18879. [PubMed] [Google Scholar]
- 33.Tsuboi T, Kikuta T, Warashina A, Terakawa S. Protein kinase C-dependent supply of secretory granules to the plasma membrane. Biochem Biophys Res Commun. 2001;282:621–628. doi: 10.1006/bbrc.2001.4603. [DOI] [PubMed] [Google Scholar]
- 34.Turner KM, Burgoyne RD, Morgan A. Protein phosphorylation and the regulation of synaptic membrane traffic. Trends Neurosci. 1999;22:459–464. doi: 10.1016/s0166-2236(99)01436-8. [DOI] [PubMed] [Google Scholar]
- 35.Voets T. Dissection of the three Ca2+-dependent steps leading to secretion in chromaffin cells from mouse adrenal slices. Neuron. 2000;28:537–545. doi: 10.1016/s0896-6273(00)00131-8. [DOI] [PubMed] [Google Scholar]
- 36.Voets T, Neher E, Moser T. Mechanisms underlying phasic and sustained secretion in chromaffin cells from mouse adrenal slices. Neuron. 1999;23:607–615. doi: 10.1016/s0896-6273(00)80812-0. [DOI] [PubMed] [Google Scholar]
- 37.Wei S, Xu T, Ashery U, Kollewe A, Matti U, Antonin W, Rettig J, Neher E. Exocytotic mechanism studied by truncated and zero layer mutants of the C-terminus of SNAP-25. EMBO J. 2000;19:1279–1289. doi: 10.1093/emboj/19.6.1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu T, Binz T, Niemann H, Neher E. Multiple kinetic components of exocytosis distinguished by neurotoxin sensitivity. Nat Neurosci. 1998;1:192–200. doi: 10.1038/642. [DOI] [PubMed] [Google Scholar]
- 39.Xu T, Ashery U, Burgoyne RD, Neher E. Early requirement for α-SNAP and NSF in the secretory cascade in chromaffin cells. EMBO J. 1999a;18:3294–3304. doi: 10.1093/emboj/18.12.3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu T, Rammner B, Margittai M, Artalejo AR, Neher E, Jahn R. Inhibition of SNARE complex assembly differentially affects kinetic components of exocytosis. Cell. 1999b;99:713–722. doi: 10.1016/s0092-8674(00)81669-4. [DOI] [PubMed] [Google Scholar]
- 41.Zhang X, Kim-Miller MJ, Fukuda M, Kowalchyk JA, Martin TFJ. Ca2+-dependent synaptotagmin binding to SNAP-25 is essential for Ca2+-triggered exocytosis. Neuron. 2002;34:599–611. doi: 10.1016/s0896-6273(02)00671-2. [DOI] [PubMed] [Google Scholar]
- 42.Zucker RS. Calcium- and activity-dependent synaptic plasticity. Curr Opin Neurobiol. 1999;9:305–313. doi: 10.1016/s0959-4388(99)80045-2. [DOI] [PubMed] [Google Scholar]