

Abstract
During development, sympathetic neurons innervating rodent sweat glands undergo a target-induced change in neurotransmitter phenotype from noradrenergic to cholinergic. Although the sweat gland innervation in the adult mouse is cholinergic and catecholamines are absent, these neurons continue to express tyrosine hydroxylase (TH), the rate-limiting enzyme in catecholamine synthesis. The developmental suppression of noradrenergic function in these mouse sympathetic neurons is not well understood. We investigated whether the downregulation of the enzyme aromatic l-amino acid decarboxylase (AADC) or the TH cofactor tetrahydrobiopterin (BH4) could account for the loss of catecholamines in these neurons. AADC levels did not decrease during development, and adult cholinergic sympathetic neurons were strongly immunoreactive for AADC. In contrast, BH4 levels dropped significantly in murine sweat gland-containing footpads during the time period when the gland innervation was switching from making norepinephrine to acetylcholine. Immunoreactivity for the rate-limiting BH4 synthetic enzyme GTP cyclohydrolase (GCH) became undetectable in the sweat gland neurons during this phenotypic conversion, suggesting that sweat glands reduce BH4 levels by suppressing GCH expression during development. Furthermore, extracts from sweat gland-containing footpads suppressed BH4 in cultured mouse sympathetic neurons, and addition of the BH4 precursor sepiapterin rescued catecholamine production in neurons treated with footpad extracts. Together, these results suggest that the mouse sweat gland-derived cholinergic differentiation factor functionally suppresses the noradrenergic phenotype during development by inhibiting production of the TH cofactor, BH4. These data also indicate that GCH expression, which is often coordinately regulated with TH expression, can be controlled independently of TH during development.
Keywords: tetrahydrobiopterin, GTP cyclohydrolase, sympathetic neuron, development, noradrenergic, cholinergic differentiation factor
Neuronal transmitter and peptide phenotype can be developmentally regulated by postsynaptic environmental influences, including neurotrophins, and target-derived differentiation factors that induce a new transmitter phenotype while suppressing the initial transmitter phenotype. An example is the target-induced noradrenergic to cholinergic/peptidergic switch in the sympathetic neurons innervating rodent sweat glands (Landis, 1990;Schotzinger and Landis, 1990; Rao and Landis, 1993). Neurons innervating the sweat glands initially exhibit a noradrenergic phenotype, but in response to a sweat gland-derived cholinergic differentiation factor (CDF), catecholamine histofluorescence disappears, while cholinergic and peptidergic properties appear. In the rat, the acquisition of cholinergic properties is accompanied by decreased tyrosine hydroxylase (TH), the rate-limiting enzyme in catecholamine synthesis (Landis et al., 1988). In the mouse, however, this functional switch is not accompanied by the loss of TH (Rao et al., 1994; Guidry and Landis, 1995, 1998). Although the idea that TH was not rate limiting for catecholamine synthesis in these neurons was first proposed several years ago (Rao et al., 1994), the factor limiting catecholamine synthesis has remained unclear.
Several things could account for the loss of catecholamines in murine TH-containing sympathetic neurons. Tyrosine hydroxylase, a tightly regulated enzyme, could be inhibited so that nol-dihydroxyphenylalanine (l-DOPA) is produced. Alternatively, the enzyme aromatic l-amino acid decarboxylase (AADC) could be absent, thereby preventing the conversion of l-DOPA to dopamine. Finally, the vesicular monoamine transporter (VMAT2) could be missing, resulting in the breakdown of catecholamines by monoamine oxidase (MAO). Although we cannot rule out any of these or other explanations for the absence of catecholamines in murine TH-positive neurons, some are more likely than others.
Tyrosine hydroxylase activity is dependent on the cofactor tetrahydrobiopterin (BH4) (Kaufman, 1978; Zigmond et al., 1989). The sweat gland-derived CDF is part of a family of cytokines (Habecker et al., 1997) that decreases BH4 content in sympathetic neurons by suppressing the expression of GTP cyclohydrolase (GCH) (Stegenga et al., 1996), the rate-limiting enzyme in BH4 production (Thöny et al., 2000). Therefore, developmental downregulation of BH4 is a good candidate for catecholamine suppression in the sweat gland innervation.
Although AADC activity is not tightly regulated like TH activity, the absence of AADC activity would prevent the formation of dopamine in TH-containing neurons. Conditioned media containing CDFs decrease AADC expression in cultured sympathetic neurons (Swerts et al., 1983;Raynaud et al., 1987), suggesting that suppression of AADC is also a candidate for inhibiting catecholamine production during development. In contrast, VMAT2 is not decreased in sympathetic neurons by CDFs (Habecker et al., 2000), suggesting that the loss of VMAT2 is unlikely to account for the absence of catecholamines in the sweat gland innervation.
We investigated whether the developmental suppression of AADC or the TH cofactor BH4 was associated with the loss of catecholamines in the sweat gland innervation. We found that AADC levels did not decrease during development, but that BH4 levels dropped significantly during the switch from norepinephrine (NE) to ACh. Extracts from sweat gland-containing footpads (FPs) suppressed BH4 production in cultured sympathetic neurons, and the BH4 precursor sepiapterin restored catecholamine production in those neurons. These results suggest that the sweat gland-derived CDF suppresses the noradrenergic phenotype during development by inhibiting production of the TH cofactor BH4.
MATERIALS AND METHODS
Cell culture reagents were obtained from Invitrogen (Carlsbad, CA) and Centricon 10 kDa filters were provided by Amicon (Danvers, MA). Biochemicals and hormones were purchased from Sigma (St. Louis, MO) except as noted. Dispase was obtained from Boehringer Mannheim (Indianapolis, IN), collagenase type II from Worthington Biochemicals(Freehold, NJ), leukemia inhibitory factor (LIF) and ciliary neurotrophic factor (CNTF) from R & D Systems (Minneapolis, MN), and NGF from Austral Biologicals (San Ramon, CA). Rabbit anti-GCH was a kind gift from Dr. Gregory Kapatos (Wayne State University, Detroit, MI), guinea pig anti-VIP was a kind gift from Dr. Story Landis (National Institute of Neurological Disorders and Stroke, Bethesda, MD), rabbit anti-AADC was from Protos Biotech (New York, NY), rabbit anti-TH was from Chemicon (Temecula, CA), and goat anti-rabbit secondary antibodies were from Cappel (Durham, NC). Horseradish peroxidase-conjugated IgG was also from Cappel, and chemiluminescence reagents were from DuPont NEN (Wilmington, DE). Protein concentrations were determined using a protein assay kit from Pierce (Rockford, IL).
Preparation of tissue extracts. Extracts of soluble protein were prepared from the hindlimb footpads of adult mice by a procedure described previously (Rao et al., 1992; Habecker et al., 1995). The procedure was modified as follows: frozen footpad tissue was pulverized with a stainless steel mortar and pestle and chilled on dry ice, before homogenization with a Polytron. Soluble proteins were concentrated by centrifugation through Centricon 10 kDa filters. For tissue extracts to be used for Western blot analysis, frozen footpads were pulverized with a stainless steel mortar and pestle and homogenized 40 times with a ground-glass homogenizer in disruption buffer (7.5 mmNa2HPO4, 10 mm EDTA, and 1% SDS) supplemented with the protease inhibitors leupeptin (1 μg/ml), pepstatin (1 μg/ml), and phenylmethylsulfonylfluoride (100 μg/ml). Debris was removed by low-speed centrifugation, and supernatants were collected for protein assays and either stored at −20°C or diluted in loading buffer for immediate Western blot analysis.
Primary cell culture. Cultures of sympathetic neurons were prepared from the superior cervical ganglia of newborn mice, essentially as described previously (Hawrot and Patterson, 1979; Rao and Landis, 1990). Cells were grown in L15-CO2-complete, supplemented with NGF (50 ng/ml), penicillin G (100 U/ml), streptomycin sulfate (100 μg/ml), and either 10% fetal bovine serum or 10% rat serum. Neurons were preplated for ≥2 hr and grown in 96 well plates coated with poly-l-lysine and laminin or 96 well BioCoat plates. Cells were maintained in the antimitotic agent fluorodeoxyuridine/uridine (10 μm) for 2 d to reduce the number of non-neuronal cells. There were ∼1000–2000 neurons per well. Cytokines and tissue extracts were diluted in culture medium and filter sterilized before addition to the culture dishes. Sepiapterin was dissolved in DMSO and diluted in medium before addition to cultures. N-acetyl serotonin (NAS) was dissolved in ethanol and diluted in medium. The final volume of DMSO and/or ethanol in cultures was <1%.
Western blots. Neuron or footpad extracts were diluted in SDS loading buffer (2% SDS, 60 mm Tris, pH 6.8, 0.01% bromophenol blue, 5% β-mercaptoethanol, and 10% glycerol) and denatured at 60°C for 30 min. Equivalent amounts of each extract were separated on 8.5% SDS-polyacrylamide gels and then transferred to nitrocellulose blots for identification of AADC or TH. All incubations were performed at room temperature. Blots were incubated for 1 hr in blocking solution (5% nonfat dry milk in 10 mmTris, pH 8.0, 150 mm NaCl, and 0.1% Tween 20; TBS-T) and then incubated overnight in blocking solution with rabbit anti-AADC (1:1000) or rabbit anti-TH (1:1000). Bound antibodies were detected using horseradish peroxidase-conjugated IgG (1:10,000) and visualized by chemiluminescence. Unsaturated films were scanned, and the pixel density of the image was analyzed using LabWorks software (UVP, Upland, CA).
HPLC analysis of (6R)-5,6,7,8-BH4.Tetrahydrobiopterin was quantified by an HPLC procedure using electrochemical detection (ED) described by Hylands et al. (Howells et al., 1986; Howells and Hyland, 1987). Footpad tissue was frozen on dry ice and homogenized in acid-washed glass homogenizers at 4°C in the HPLC mobile phase consisting of 50 mm sodium acetate, 5 mm citric acid, pH 5.22, and 50 μm EDTA, supplemented with 1 mg/ml of both dithioerythritol (DTE) and diethylenetriaminepentaacetic acid (DTPA) (Howells and Hyland, 1987). Residual protein was removed by centrifugation at 4°C through 10 kDa Centricon filters (Howells et al., 1986). Sympathetic neuron cultures were homogenized at 4°C by trituration in the HPLC mobile phase, supplemented with DTE and DTPA, and spun down at 4°C to remove cell debris. The BH4 in the samples was stable under these conditions for ≥5 hr at 4°C and for 1 month at −70°C. Tetrahydrobiopterin was chromatographed by reversed-phase HPLC-ED on a C18 column (15 × 0.46 cm, 5 μm particle size; Rainin, Ridgefield, NJ) (Howells et al., 1986; Howells and Hyland, 1987). An ESA Coulochem multielectrode detector (Chelmsford, MA) was used to quantify the BH4 with the electrodes set as follows: electrode 1, +0.18 V; electrode 2, −0.07 V. Solutes in the column eluents were first oxidized by electrode 1, and then BH4 was reduced by electrode 2. The limits of detection for the BH4 reduction signal in electrode 2 were ∼60 fmol.
HPLC analysis of catecholamines. Catecholamines were measured by HPLC-ED (Felice et al., 1978; Woodward et al., 1987). Sympathetic neurons were triturated through a pipette tip in 75 μl of ice-cold 0.2 m perchloric acid containing 0.2 mm EDTA, 1 μm ascorbate, and 250 nm dihydroxyl-benzylamine (an internal standard) and were centrifuged. An aliquot of the supernatant was chromatographed by HPLC on a C18 reversed-phase column (15 × 0.46 cm, 5 μm; Rainin) using a mobile phase containing 50 mm sodium phosphate and 50 mm sodium acetate, pH 3.0, 360 mg/l sodium octane sulfonate, 100 μl/l triethylamine, and 30% (v/v) acetonitrile. Catecholamines were quantified using an ESA Coulochem detector with the electrode potential set at +0.18 V. The detection limit for dopamine was estimated to be <50 fmol. Although norepinephrine is the catecholamine produced by sympathetic neurons in vivo, dopamine is the predominant catecholamine produced by these neuronsin vitro and was quantified as a measure of noradrenergic function in the sympathetic neurons (Woodward et al., 1987).
Immunohistochemistry. Mice of the appropriate age were perfused through the heart with 4% paraformaldehyde in 0.1m phosphate buffer, pH 7.4, for 10 min. Footpads were postfixed in 4% paraformaldehyde for 1 hr and cryoprotected overnight in a solution of 30% sucrose in 0.1 mphosphate buffer, pH 7.4. Ten micrometer cryostat sections were thaw mounted onto charged slides, rinsed in PBS, preincubated in dilution buffer (2% BSA, 0.1% sodium azide, and 0.3% Triton X-100 in PBS) for 1 hr, and incubated overnight with primary antisera raised against AADC (rabbit anti-AADC diluted 1:300), VIP (guinea pig anti-VIP diluted 1:300), TH (rabbit anti-TH diluted 1:300), or GCH (rabbit anti-GCH diluted 1:300) (Hirayama and Kapatos, 1998). Sections were then rinsed in PBS, incubated for 1–2 hr with species-specific fluorescent secondary antibodies diluted 1:300 in dilution buffer containing 5% goat serum, and rinsed again with PBS before visualization by fluorescence microscopy.
Statistics. Statistical analyses were performed with Prism 3.0 Graphpad Software (San Diego, CA).
RESULTS
Although postganglionic sympathetic neurons innervate the sweat glands of adult mice, these neurons are cholinergic and peptidergic and do not contain catecholamines (Rao et al., 1994; Francis et al., 1997). In addition to the peptides VIP and calcitonin gene-related peptide (Rao et al., 1994; Guidry and Landis, 1995; Francis et al., 1997), the neurons innervating sweat glands synthesize ACh and express acetylcholinesterase (Rao et al., 1994; Francis et al., 1997) and the vesicular ACh transporter (Guidry and Landis, 1998). Despite the cholinergic and peptidergic phenotype and the lack of catecholamines, however, these neurons contain abundant tyrosine hydroxylase immunoreactivity (TH-IR) (Rao et al., 1994; Guidry and Landis, 1995,1998).
To determine whether the absence of catecholamines in the TH-containing mouse sweat gland innervation could be attributed to the loss of AADC, footpad tissue was examined for the presence of AADC by Western blot. Extracts from sweat gland-containing footpads were collected before [postnatal day 5 (P5)], during (P10), and after (P21, adult) the loss of catecholamines and blotted for AADC. Footpad AADC protein increased throughout development and appeared to be highest in the adult (Fig.1A). Because the footpad tissue used for Western blot analysis contains many cell types, it is possible that a loss of AADC in the sweat gland innervation is compensated for by increases in AADC in other cell types in the tissue. AADC immunohistochemistry, however, confirmed that the enzyme was present in the neurons innervating adult sweat glands (Fig.1B) along with the neuropeptide VIP (Fig.1C), which is induced during the switch from noradrenergic to cholinergic function. Factors related to the sweat gland-derived CDF decrease transcription of the human AADC promoter (Chireux et al., 1994) and inhibit expression of AADC protein (Swerts et al., 1983;Raynaud et al., 1987), but there is no evidence that cholinergic differentiation factors decrease AADC activity (Berry et al., 1996). Therefore, although we have not directly demonstrated that the AADC present in this tissue can decarboxylate l-DOPA to form dopamine, we think it unlikely that the AADC is inactive in the adult sweat gland innervation.
Fig. 1.

Developmental expression of AADC in the sweat gland innervation. A, Western blot analysis of AADC in mouse sweat gland-containing FPs. Aliquots of tissue extracts (5 μg of protein) from adult adrenal gland and from the hindlimb footpads of P5, P10, P21, and adult mice were separated on 8.5% SDS-polyacrylamide gels and blotted for AADC. Footpad extracts from all ages contained AADC. Data shown are representative of results from three animals at each age. Immunohistochemistry of AADC (B) and VIP (C) in adult mouse footpad is shown. Ten micrometer cryostat sections were stained with an antibody directed against AADC (diluted 1:300) or VIP (diluted 1:300). Cholinergic fibers innervating sweat glands are strongly immunoreactive for AADC (B, arrowheads) and the neuropeptide VIP (C, arrowheads). Similar results were obtained in four animals.
Tyrosine hydroxylase requires the cofactor BH4; therefore, the loss of BH4 could block catecholamine synthesis. To determine whether BH4 production was suppressed during the switch from noradrenergic to cholinergic phenotype in the gland innervation, BH4 levels were measured in sweat gland-containing footpads collected before (P4 and P7), during (P10 and P14), and after (P21 or P22) the change in phenotype. A significant decrease in footpad BH4 levels was observed in both the mouse (Fig.2A) and the rat (Fig.2B), and this decrease correlated with the disappearance of catecholaminergic properties and the appearance of cholinergic properties in the respective species. These data suggest that suppression of BH4 may be a common feature of cholinergic differentiation. Although BH4 did not disappear completely from footpad extracts (Fig. 2), and BH4 content was increased in adult footpads compared with P21 or P22 animals [BH4 levels in adult mouse, 282 ± 49 pmol/gm (n = 5); BH4 levels in adult rat, 246 ± 31 pmol/gm (n = 3); mean ± SEM], the developmental time course of BH4 suppression was nonetheless consistent with a role for BH4 in the downregulation of catecholamine production in cholinergic sympathetic neurons.
Fig. 2.

Developmental expression of BH4 in sweat gland-containing footpads. BH4 levels were quantified in hindlimb footpads from mice (A) or rats (B) collected before (P4 and P7), during (P10 and P14), and after (P21 and P22) the phenotypic switch in the gland innervation. Data shown are the mean ± SEM of three animals.A, In mouse footpad, BH4 decreased by ∼44% between P7 (396 ± 49 pmol/gm) and P21 (174 ± 8 pmol/gm) (p = 0.01; two-tailed unpairedt test). B, In rat footpad, BH4 decreased by 50% between P7 (266 ± 29 pmol/gm) and P14 (133 ± 19 pmol/gm) (p < 0.05; two-tailed unpairedt test).
Inasmuch as BH4 is a cofactor for several enzymes, including nitric oxide synthase (NOS), we expected that even if BH4 synthesis was suppressed completely in neurons innervating the adult sweat glands, BH4 production in other tissues within the developing footpad might be maintained or even increased, thereby accounting for the elevation of BH4 in adult footpad. Therefore, we examined the immunolocalization of GCH, the rate-limiting enzyme in the synthesis of BH4, to ascertain the sites of BH4 production within the footpad tissue. Mouse footpad sections were collected before (P5), during (P10), and after (adult) the change in phenotype and were analyzed by immunohistochemistry using an affinity-purified antibody directed against GCH (a generous gift from Dr. Gregory Kapatos, Wayne State University). GCH-IR was present in nerve fibers projecting to the developing glands at P5 and P10 (Fig.3A,B) but was undetectable in the sympathetic innervation of the adult sweat glands, despite its presence in other tissues within the adult footpad (Fig. 3C). The lack of GCH-IR is in contrast to the presence of TH-IR in neurons innervating adult sweat glands (Fig.3D). The loss of GCH-IR in sympathetic neurons innervating sweat glands and the decreased BH4 content in footpad extracts suggest that BH4 is absent from the sympathetic neurons innervating sweat glands in the adult. It is, however, still present in other cells in the adult footpad tissue, including the noradrenergic vascular innervation (Fig. 3C, arrowhead) and endothelial cells (Fig. 3C, inset), which require BH4 as a cofactor for NOS.
Fig. 3.

Developmental expression of GCH in the sweat gland innervation. Sections of mouse footpad from P5 (A), P10 (B), and adult (C, D) animals were stained with an antibody directed against GTP cyclohydrolase (A–C; diluted 1:300) or tyrosine hydroxylase (D; diluted 1:300). GCH immunoreactivity was present in the neurons innervating developing gland anlage at P5 and P10 (A,B, arrowheads). Although GCH was not detected in the innervation of the adult sweat glands (C, arrows), it was found in other tissues within the footpad (C, arrowhead) and in vascular endothelial cells (C,inset). The absence of GTP cyclohydrolase is in contrast to abundant TH immunoreactivity in the adult sweat gland innervation (D, arrowheads). Similar results were obtained with at least three animals of each age. Scale bar, 10 μm.
In vitro treatment of rat sympathetic neurons with extracts of sweat gland-containing footpads, LIF, or CNTF induces a switch in neurotransmitter phenotype from noradrenergic to cholinergic that is similar to the one observed in the sweat gland innervation in vivo (Fukada, 1985; Saadat et al., 1989; Rao and Landis, 1990; Rao et al., 1992; Rohrer, 1992). To determine whether the developmental loss of BH4 was caused by the action of a sweat gland-derived cholinergic differentiation factor, mouse sympathetic neurons were grown in vitro and treated with soluble extracts from murine sweat gland-containing footpads. Extracts of mouse footpads, LIF, and CNTF caused a switch from noradrenergic to cholinergic phenotype in mouse sympathetic neurons in vitro (Fig.4). LIF substantially decreased TH immunoreactivity (−63%) (Fig. 4A) and dopamine levels (−60%) (Fig. 4B) in mouse sympathetic neurons but had no significant effect on AADC levels (−8%) (Fig.4A). Footpad extracts also significantly decreased dopamine (−60%) (Fig. 4B) but only slightly decreased TH immunoreactivity and AADC immunoreactivity (−13 and −13%, respectively) (Fig. 4A). Footpad extracts and CNTF decreased BH4 content significantly (−70 and −80%, respectively; p < 0.01) (Fig. 4C), as did LIF (data not shown) (Stegenga et al., 1996). Thus, mouse footpad extracts caused a pattern of neurochemical changes in murine sympathetic neurons that was similar to that observed in the sweat gland innervation during development. These data are consistent with the notion that a sweat gland-derived CDF suppresses synthesis of catecholamines primarily by inhibiting production of BH4.
Fig. 4.
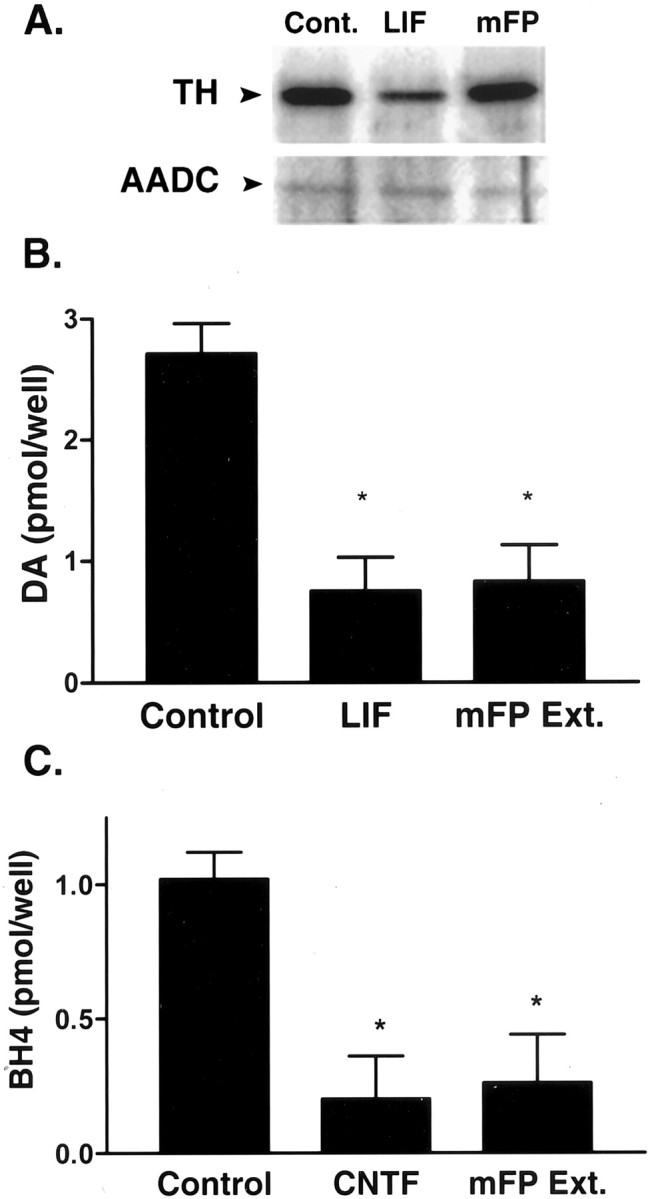
Mouse footpad extracts suppress BH4 in sympathetic neurons. Mouse sympathetic neurons were treated for 5–7 d with 10 ng/ml LIF or CNTF or 300 μg/ml soluble extracts from mouse footpad (mFP Ext.). Cells from each treatment were assayed for TH and AADC, dopamine content, or BH4 content. A, One microgram aliquots of neuron extracts were separated on SDS-polyacrylamide gels, transferred to nitrocellulose, and blotted for identification of TH and AADC. LIF decreased TH to a greater extent (−63%) than mouse FP (−13%), but neither treatment significantly altered AADC. The blots shown are representative of results obtained in three experiments. B, LIF and mouse FP extracts decreased dopamine, as measured by HPLC-ED, by ∼60% compared with control cells (*p < 0.01; ANOVA with Dunnett multiple comparison post hoc test). Data shown are the mean ± SEM of three experiments. C, CNTF and mouse FP extracts decreased BH4 content, as measured by HPLC-ED, by ∼80 and 70%, respectively, compared with control cells (*p < 0.01, ANOVA with Dunnett multiple comparison post hoc test). Data shown are from a single experiment (n = 4–6; mean ± SEM) and are representative of three separate experiments.
To establish that BH4 levels control the production of catecholamines in neurons innervating mouse sweat glands, sympathetic neurons were grown in the presence or absence of the BH4 precursor sepiapterin (Fig.5A). Sister cultures, also grown under these conditions, were treated with either CNTF or footpad extracts. In each case, the addition of sepiapterin caused a severalfold increase in dopamine levels when compared with neurons of the same treatment condition that did not receive sepiapterin (Fig.5B). In the case of CNTF or footpad extract-treated neurons, the increased dopamine levels in the presence of sepiapterin significantly exceeded the dopamine content of control neurons not grown in the presence of sepiapterin. This suggests that BH4 is rate limiting for catecholamine production in these sympathetic neurons.
Fig. 5.

Sepiapterin (SEP) rescue of dopamine and tetrahydrobiopterin production. A, BH4 synthesis: GTP is converted to 6-pyruvoyl-5,6,7,8-tetrahydropterin (PTP) by GCH. 6-Pyruvoyl-5,6,7,8-tetrahydropterin is converted to sepiapterin by the enzyme 6-pyruvoyl-tetrahydropterin synthase (PTPS). Sepiapterin is then reduced to BH4 by sepiapterin reductase (SR), which is inhibited by NAS.B, Sympathetic neurons were treated for 5–7 d with 10 ng/ml CNTF or 300 μg/ml mouse footpad extract (mFP Ext.), with or without 20 μm sepiapterin. CNTF and FP extracts decreased dopamine (DA) content by 50 and 90%, respectively, compared with control cells (p < 0.01; ANOVA; Dunnett multiple comparison of cells without SEP). The addition of sepiapterin increased dopamine compared with control cells without sepiapterin and within each treatment condition (p < 0.001; ANOVA; Tukey multiple comparison of all conditions). The fold increase in dopamine after sepiapterin addition is indicated inparentheses above each set of bars. The data shown are from a single experiment (n = 6–8; mean ± SEM) and are representative of four independent experiments. C, Cellular BH4 levels increased approximately threefold with sepiapterin addition (p < 0.001; ANOVA; Dunnett multiple comparison post hoc test). Administration of the sepiapterin reductase inhibitor NAS (2.5 mm) (Kapatos et al., 1992) decreased BH4 production, even in the presence of exogenous sepiapterin (p < 0.001; ANOVA; Dunnett multiple comparison post hoc test). Data shown are from a single experiment (n = 3–4; mean ± SEM) and are representative of three independent experiments.
To strengthen the linkage between BH4 and catecholamine production and to confirm that sepiapterin was not having an effect on catecholamine production that was independent of the BH4 synthetic pathway, we measured BH4 levels in sympathetic neurons grown in the presence and absence of sepiapterin. The presence of sepiapterin in the culture medium significantly increased (approximately threefold) the BH4 content of the sympathetic neurons (Fig. 5C). Moreover, the sepiapterin reductase inhibitor NAS (Fig. 5A) reduced BH4 levels, probably by blocking the endogenous synthesis of BH4, and the addition of sepiapterin along with the inhibitor did not result in any increase in BH4 levels (Fig. 5C). In sister cultures treated with NAS, the dopamine content was significantly reduced, regardless of whether the neurons were grown in the presence or absence of sepiapterin (−52 or −60%, respectively) (data not shown). Similar changes in BH4 and dopamine content were observed in neurons treated with FP extracts, although the levels of BH4 and dopamine were lower in these neurons, because of the extract treatment (data not shown). These results demonstrate that sepiapterin increases BH4 levels in sympathetic neurons by bypassing GCH, the rate-limiting step in BH4 synthesis. Furthermore, these results provide compelling evidence that BH4 regulates catecholamine production in these neurons.
DISCUSSION
The sympathetic neurons innervating rodent sweat glands undergo a switch from noradrenergic to cholinergic neurotransmission. In the mouse, this functional switch is not accompanied by a decrease in immunoreactivity for tyrosine hydroxylase (Rao et al., 1994; Guidry and Landis, 1998), the enzyme that is rate-limiting in catecholamine synthesis. This suggests that some other aspect of catecholamine synthesis is responsible for blocking catecholamine production during the developmental switch to acetylcholine, an idea that was first proposed by Rao et al. (1994).
We set out to determine what mediated the target-induced suppression of catecholamine accumulation in the TH-IR mouse sweat gland innervation. There are at least three possible explanations: (1) there is inhibition of TH activity caused by the loss of cofactor or post-translational modification of the enzyme, (2) there is inhibition at the second step in catecholamine synthesis (i.e., the conversion of l-DOPA to dopamine by AADC), or (3) there is a loss of vesicular uptake for catecholamines, resulting in rapid metabolism of catecholamines by MAO. Although we cannot ignore the latter two possibilities, they seem less attractive, inasmuch as blocking the first step in catecholamine biosynthesis, a branch point in metabolism, would be the most parsimonious use of the energy resources of the cell. We examined the regulation of both TH and AADC because in vitro studies indicated that cholinergic differentiation factors suppressed GCH, BH4, and AADC in sympathetic neurons (Swerts et al., 1983; Raynaud et al., 1987; Stegenga et al., 1996).
Our results are consistent with inhibition of catecholamine synthesis occurring at the TH-catalyzed step in the biosynthetic pathway. We found that as catecholamine production waned during cholinergic differentiation of sweat gland innervation, there was a concomitant loss of BH4 content in sweat gland-containing footpads. Moreover, immunohistochemical localization of GCH suggested that the loss of BH4 was confined to the sympathetic neurons innervating sweat glands and not to other tissues in the vicinity, such as blood vessels, which contain enzymes requiring BH4 as a cofactor. However, there was no detectable decrease in AADC during this developmental period.
Neuronal culture studies supported the conclusion that the loss of BH4 inhibited catecholamine synthesis in sympathetic neurons. Extracts of sweat gland-containing footpads reduced the levels of BH4 and catecholamines in cultured sympathetic neurons, but catecholamine production could be partially restored in these neurons by the addition of sepiapterin to the culture medium, which increased cellular BH4 content. The sepiapterin-induced restoration of catecholamine production in extract-treated neurons demonstrates that TH and AADC are both present and functional. Furthermore, the accumulation of cellular catecholamine stores in the absence of MAO inhibitors implies that vesicular uptake of catecholamines is not impaired in these cells. Together, our data strongly suggest that the primary action of the sweat gland-derived CDF is to suppress the noradrenergic phenotype by inhibiting the production of the TH cofactor BH4, and that the other cellular machinery for catecholamine synthesis and packaging is present and remains functional in these cholinergic sympathetic neurons.
This conclusion differs from the current thinking, based on the rat model, that cholinergic differentiation factors inhibit noradrenergic function by decreasing the expression of catecholamine synthetic enzymes, especially tyrosine hydroxylase. The latter model was based onin vivo and in vitro data from the rat indicating that cholinergic differentiation factors suppress TH expression and activity. The switch from noradrenergic to cholinergic phenotype during rat sweat gland innervation is characterized by decreased TH-IR (Landis et al., 1988). Likewise, culture studies indicate that CDFs, including LIF, CNTF, and footpad extracts, suppress TH mRNA and protein in cultured rat sympathetic neurons (Saadat et al., 1989; Rao and Landis, 1990; Nawa et al., 1991; Lewis et al., 1994). Although TH-IR does not disappear completely from the gland innervation in vivo or from sympathetic neurons in vitro, these data are consistent with a role for the suppression of TH in the regulation of catecholamine synthesis and have led to the model that decreased TH expression is the crucial step in the loss of noradrenergic function.
The observation that TH content in the sympathetic innervation of mouse sweat glands did not change suggested that an alternative model was needed to explain the developmental suppression of catecholamine production, at least in mice (Rao et al., 1994). Our data provide an explanation for the loss of catecholamines in TH-containing cholinergic neurons during development. BH4 is decreased in mouse and rat neurons because of the loss of GCH, and this loss of TH cofactor is likely to be the factor responsible for curtailing catecholamine production in both species. These results are consistent with studies of genetically engineered replacement of dopamine in rats and mice which indicate that the genes encoding GCH and TH are both required for behavioral rescue of dopamine deficits (Bencsics et al., 1996; Mandel et al., 1998;Szczypka et al., 1999). Although a decrease in BH4 levels does not necessarily result in a proportional decrease in TH activity (Kapatos et al., 1992), the absence of GCH and BH4 prevents catecholamine production in cells containing TH (Nagatsu et al., 1997).
Attempts to substitute for the loss of cofactor in sympathetic neuronsin vitro by adding BH4 rather than sepiapterin caused an unexpected decrease in dopamine production (data not shown). The addition of 20 μm sepiapterin to the culture medium resulted in a fivefold to 20-fold increase in catecholamines (Fig. 5), whereas BH4 concentrations in the range of 6–600 μm either had little effect or decreased catecholamine content. Concentrations of BH4 of >50 μm have been shown to inhibit TH activity in a cell-free system (Alterio et al., 1998); thus, it seems likely that the excess BH4 added to the culture medium caused inhibition of TH activity. It is, however, less clear why the lower levels of BH4 were not able to stimulate TH activity. A potential explanation comes from work by Choi et al. (2000), who reported that extracellular BH4 is toxic to catecholamine-producing cells in culture because of the generation of reactive oxygen species. In contrast, sepiapterin is able to elevate intracellular BH4 levels, increase TH activity, and is not toxic (Choi et al., 2000). Although our BH4-treated cultures did not exhibit morphological changes, the decreases in dopamine levels may have resulted from BH4 toxicity.
Attempts to regulate catecholamine production in vivo by replacing BH4 also produced mixed results. Injection of BH4 into adult mice (with or without MAO inhibitors) did not consistently stimulate accumulation of catecholamines in the cholinergic innervation of the sweat glands or the noradrenergic innervation of the salivary gland or heart (data not shown). Other investigators have used BH4 in vivo with varied results. Injections of BH4 into BH4-deficienthph mice increased central BH4 content but did not alter CNS dopamine levels (Hyland et al., 1996). Viral coexpression of GCH and TH in the CNS produced behavioral rescue of feeding in dopamine-deficient mice, but peripheral administration of BH4 did not complement expression of TH and rescue behavior (Szczypka et al., 1999). Intravitreal injection of BH4 increased l-DOPA in dark-adapted retinas but had no effect on l-DOPA levels in light-adapted retinas (Iuvone et al., 1985). Likewise, oral administration of BH4 increased nitric oxide production in insulin-resistant rats but not control rats (Shinozaki et al., 2000). Thus, the effectiveness of exogenous BH4 as a cofactor for TH or other enzymes in vivo seems to be influenced by complex environmental factors.
The differential regulation of TH and GCH during development of the mouse sweat gland innervation is somewhat unique. Many factors that control expression of these enzymes regulate both of them in a coordinate manner. For example, LIF and CNTF suppress GCH and BH4 together with TH in cultured rat sympathetic neurons (Stegenga et al., 1996), and our data indicate that LIF suppresses both TH and BH4 in cultured mouse sympathetic neurons. Additional studies indicate that nerve growth factor induces the expression of both GCH and TH in sympathetic neurons (Hirayama and Kapatos, 1995) and in PC12 cells (Anastasiadis et al., 1996). Likewise, the two enzymes are regulated similarly by cAMP (Abou-Donia et al., 1986; Anastasiadis et al., 1998) and by some disease states (Serova et al., 1999).
The differential expression of TH and GCH seen during development is particularly interesting given the coordinate regulation of these enzymes by LIF and CNTF. Several lines of evidence suggest that the mouse sweat gland-derived CDF uses the same receptor complex and intracellular signaling pathways that are activated by LIF and CNTF (Habecker et al., 1997). The observation that the mouse sweat gland-derived CDF has little effect on TH expression in mouse sympathetic neurons in vivo, whereas LIF suppresses TH in mouse sympathetic neurons both in vitro (Fig. 4) andin vivo (Bamber et al., 1994), suggests that there are unanticipated differences in downstream signal transduction. This is the first example of a functional difference between LIF, CNTF, and the sweat gland-derived CDF with regard to the suppression of NE or induction of ACh. Elucidation of the mechanisms involved will likely require cloning the gland-derived differentiation factor.
The studies described here identify a novel role for BH4 in regulating neurotransmitter phenotype during development. They also indicate that GCH expression and activity can be regulated quite differently than TH and other catecholamine synthetic enzymes. The revelation that catecholamine content is controlled at the level of a cofactor rather than an enzyme expands our understanding of the ways in which environmental factors regulate neuronal differentiation.
Footnotes
This work was supported by American Heart Association Grant 9750083N and National Institutes of Health Grant HL68231 (B.A.H.). We thank Dr. Gregory Kapatos of Wayne State University for the anti-GCH antibody and Dr. Un Jung Kang of Northwestern University for the suggestion to use sepiapterin in replacement experiments.
Correspondence should be addressed to Dr. Beth A. Habecker, Department of Physiology and Pharmacology, L334, Oregon Health and Sciences University, 3181 Southwest Sam Jackson Park Road, Portland, OR 97239. E-mail: habecker@ohsu.edu.
REFERENCES
- 1.Abou-Donia MM, Wilson SP, Zimmerman TP, Nichol CA, Viveros OH. Regulation of guanosine triphosphate cyclohydrolase and tetrahydrobiopterin levels and the role of the cofactor in tyrosine hydroxylation in primary cultures of adrenomedullary chromaffin cells. J Neurochem. 1986;46:1190–1199. doi: 10.1111/j.1471-4159.1986.tb00637.x. [DOI] [PubMed] [Google Scholar]
- 2.Alterio J, Ravassard P, Haavik J, Le CJ, Biguet NF, Waksman G, Mallet J. Human tyrosine hydroxylase isoforms. Inhibition by excess tetrahydropterin and unusual behavior of isoform 3 after camp-dependent protein kinase phosphorylation. J Biol Chem. 1998;273:10196–10201. doi: 10.1074/jbc.273.17.10196. [DOI] [PubMed] [Google Scholar]
- 3.Anastasiadis PZ, Kuhn DM, Blitz J, Imerman BA, Louie MC, Levine RA. Regulation of tyrosine hydroxylase and tetrahydrobiopterin biosynthetic enzymes in PC12 cells by NGF, EGF and IFN-gamma. Brain Res. 1996;713:125–133. doi: 10.1016/0006-8993(95)01494-2. [DOI] [PubMed] [Google Scholar]
- 4.Anastasiadis PZ, Bezin L, Gordon LJ, Imerman B, Blitz J, Levine RA. Vasoactive intestinal peptide induces both tyrosine hydroxylase activity and tetrahydrobiopterin biosynthesis in PC12 cells. Neuroscience. 1998;86:179–189. doi: 10.1016/s0306-4522(97)00611-8. [DOI] [PubMed] [Google Scholar]
- 5.Bamber BA, Masters BA, Hoyle GW, Brinster RL, Palmiter RD. Leukemia inhibitory factor induces neurotransmitter switching in transgenic mice. Proc Natl Acad Sci USA. 1994;91:7839–7843. doi: 10.1073/pnas.91.17.7839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bencsics C, Wachtel SR, Milstien S, Hatakeyama K, Becker JB, Kang UJ. Double transduction with GTP cyclohydrolase I and tyrosine hydroxylase is necessary for spontaneous synthesis of l-DOPA by primary fibroblasts. J Neurosci. 1996;16:4449–4456. doi: 10.1523/JNEUROSCI.16-14-04449.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berry MD, Juorio AV, Li XM, Boulton AA. Aromatic l-amino acid decarboxylase: a neglected and misunderstood enzyme. Neurochem Res. 1996;21:1075–1087. doi: 10.1007/BF02532418. [DOI] [PubMed] [Google Scholar]
- 8.Chireux M, Raynal JF, Le Van TA, Cadas H, Bernard C, Martinou I, Martinou JC, Weber MJ. Multiple promoters of human choline acetyltransferase and aromatic l-amino acid decarboxylase genes. J Physiol (Paris) 1994;88:215–227. doi: 10.1016/0928-4257(94)90084-1. [DOI] [PubMed] [Google Scholar]
- 9.Choi HJ, Jang YJ, Kim HJ, Hwang O. Tetrahydrobiopterin is released from and causes preferential death of catecholaminergic cells by oxidative stress. Mol Pharmacol. 2000;58:633–640. [PubMed] [Google Scholar]
- 10.Felice LJ, Felice JD, Kissinger PT. Determination of catecholamines in rat brain parts by reverse-phase ion-pair liquid chromatography. J Neurochem. 1978;31:1461–1465. doi: 10.1111/j.1471-4159.1978.tb06573.x. [DOI] [PubMed] [Google Scholar]
- 11.Francis NJ, Asmus SE, Landis SC. CNTF and LIF are not required for the target-directed acquisition of cholinergic and peptidergic properties by sympathetic neurons in vivo. Dev Biol. 1997;182:76–87. doi: 10.1006/dbio.1996.8464. [DOI] [PubMed] [Google Scholar]
- 12.Fukada K. Purification and partial characterization of a cholinergic differentiation factor. Proc Natl Acad Sci USA. 1985;82:8795–8799. doi: 10.1073/pnas.82.24.8795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guidry G, Landis SC. Sympathetic axons pathfind successfully in the absence of target. J Neurosci. 1995;15:7565–7574. doi: 10.1523/JNEUROSCI.15-11-07565.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guidry G, Landis SC. Target-dependent development of the vesicular acetylcholine transporter in rodent sweat gland innervation. Dev Biol. 1998;199:175–184. doi: 10.1006/dbio.1998.8929. [DOI] [PubMed] [Google Scholar]
- 15.Habecker BA, Tresser SJ, Rao MS, Landis SC. Production of sweat gland cholinergic differentiation factor depends on innervation. Dev Biol. 1995;167:307–316. doi: 10.1006/dbio.1995.1025. [DOI] [PubMed] [Google Scholar]
- 16.Habecker BA, Symes AJ, Stahl N, Francis NJ, Economides A, Fink JS, Yancopoulos GD, Landis SC. A sweat gland-derived differentiation activity acts through known cytokine signaling pathways. J Biol Chem. 1997;272:30421–30428. doi: 10.1074/jbc.272.48.30421. [DOI] [PubMed] [Google Scholar]
- 17.Habecker BA, Klein MG, Cox BC, Packard BA. Norepinephrine transporter expression in cholinergic sympathetic neurons: differential regulation of membrane and vesicular transporters. Dev Biol. 2000;220:85–96. doi: 10.1006/dbio.2000.9631. [DOI] [PubMed] [Google Scholar]
- 18.Hawrot E, Patterson PH. Long-term culture of dissociated sympathetic neurons. Methods Enzymol. 1979;58:574–584. doi: 10.1016/s0076-6879(79)58174-9. [DOI] [PubMed] [Google Scholar]
- 19.Hirayama K, Kapatos G. Regulation of GTP cyclohydrolase I gene expression and tetrahydrobiopterin content by nerve growth factor in cultures of superior cervical ganglia. Neurochem Int. 1995;27:157–161. doi: 10.1016/0197-0186(95)00008-v. [DOI] [PubMed] [Google Scholar]
- 20.Hirayama K, Kapatos G. Nigrostriatal dopamine neurons express low levels of GTP cyclohydrolase I protein. J Neurochem. 1998;70:164–170. doi: 10.1046/j.1471-4159.1998.70010164.x. [DOI] [PubMed] [Google Scholar]
- 21.Howells DW, Hyland K. Direct analysis of tetrahydrobiopterin in cerebrospinal fluid by high-performance liquid chromatography with redox electrochemistry: prevention of autoxidation during storage and analysis. Clin Chim Acta. 1987;167:23–30. doi: 10.1016/0009-8981(87)90081-7. [DOI] [PubMed] [Google Scholar]
- 22.Howells DW, Smith I, Hyland K. Estimation of tetrahydrobiopterin and other pterins in cerebrospinal fluid using reversed-phase high-performance liquid chromatography with electrochemical and fluorescence detection. J Chromatogr. 1986;381:285–294. doi: 10.1016/s0378-4347(00)83594-x. [DOI] [PubMed] [Google Scholar]
- 23.Hyland K, Gunasekera RS, Engle T, Arnold LA. Tetrahydrobiopterin and biogenic amine metabolism in the hph-1 mouse. J Neurochem. 1996;67:752–759. doi: 10.1046/j.1471-4159.1996.67020752.x. [DOI] [PubMed] [Google Scholar]
- 24.Iuvone PM, Reinhard JF, Jr, Abou-Donia MM, Viveros OH, Nichol CA. Stimulation of retinal dopamine biosynthesis in vivo by exogenous tetrahydrobiopterin: relationship to tyrosine hydroxylase activation. Brain Res. 1985;359:392–396. doi: 10.1016/0006-8993(85)91459-3. [DOI] [PubMed] [Google Scholar]
- 25.Kapatos G, Hirayama K, Hasegawa H. Tetrahydrobiopterin turnover in cultured rat sympathetic neurons: developmental profile, pharmacologic sensitivity, and relationship to norepinephrine synthesis. J Neurochem. 1992;59:2048–2055. doi: 10.1111/j.1471-4159.1992.tb10093.x. [DOI] [PubMed] [Google Scholar]
- 26.Kaufman S. Establishment of tetrahydrobiopterin as the hydroxylase cofactor and a review of some recent studies in man. Psychopharmacol Bull. 1978;14:38–40. [PubMed] [Google Scholar]
- 27.Landis SC. Target regulation of neurotransmitter phenotype. Trends Neurosci. 1990;13:344–350. doi: 10.1016/0166-2236(90)90147-3. [DOI] [PubMed] [Google Scholar]
- 28.Landis SC, Siegel RE, Schwab M. Evidence for neurotransmitter plasticity in vivo. II. Immunocytochemical studies of rat sweat gland innervation during development. Dev Biol. 1988;126:129–140. doi: 10.1016/0012-1606(88)90246-1. [DOI] [PubMed] [Google Scholar]
- 29.Lewis SE, Rao MS, Symes AJ, Dauer WT, Fink JS, Landis SC, Hyman SE. Coordinate regulation of choline acetyltransferase, tyrosine hydroxylase, and neuropeptide mRNAs by ciliary neurotrophic factor and leukemia inhibitory factor in cultured sympathetic neurons. J Neurochem. 1994;63:429–438. doi: 10.1046/j.1471-4159.1994.63020429.x. [DOI] [PubMed] [Google Scholar]
- 30.Mandel RJ, Rendahl KG, Spratt SK, Snyder RO, Cohen LK, Leff SE. Characterization of intrastriatal recombinant adeno-associated virus-mediated gene transfer of human tyrosine hydroxylase and human GTP-cyclohydrolase I in a rat model of Parkinson's disease. J Neurosci. 1998;18:4271–4284. doi: 10.1523/JNEUROSCI.18-11-04271.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nagatsu I, Sakai M, Takeuchi T, Arai R, Karasawa N, Yamada K, Nagatsu T. Tyrosine hydroxylase (TH)-only-immunoreactive non-catecholaminergic neurons in the brain of wild mice or the human TH transgenic mice do not contain GTP cyclohydrolase I. Neurosci Lett. 1997;228:55–57. doi: 10.1016/s0304-3940(97)00352-2. [DOI] [PubMed] [Google Scholar]
- 32.Nawa H, Nakanishi S, Patterson PH. Recombinant cholinergic differentiation factor (leukemia inhibitory factor) regulates sympathetic neuron phenotype by alterations in the size and amounts of neuropeptide mRNAs. J Neurochem. 1991;56:2147–2150. doi: 10.1111/j.1471-4159.1991.tb03479.x. [DOI] [PubMed] [Google Scholar]
- 33.Rao MS, Landis SC. Characterization of a target-derived neuronal cholinergic differentiation factor. Neuron. 1990;5:899–910. doi: 10.1016/0896-6273(90)90350-o. [DOI] [PubMed] [Google Scholar]
- 34.Rao MS, Landis SC. Cell interactions that determine sympathetic neuron transmitter phenotype and the neurokines that mediate them. J Neurobiol. 1993;24:215–232. doi: 10.1002/neu.480240208. [DOI] [PubMed] [Google Scholar]
- 35.Rao MS, Patterson PH, Landis SC. Multiple cholinergic differentiation factors are present in footpad extracts: comparison with known cholinergic factors. Development. 1992;116:731–744. doi: 10.1242/dev.116.3.731. [DOI] [PubMed] [Google Scholar]
- 36.Rao MS, Jaszczak E, Landis SC. Innervation of footpads of normal and mutant mice lacking sweat glands. J Comp Neurol. 1994;346:613–625. doi: 10.1002/cne.903460412. [DOI] [PubMed] [Google Scholar]
- 37.Raynaud B, Clarous D, Vidal S, Ferrand C, Weber MJ. Comparison of the effects of elevated K+ ions and muscle-conditioned medium on the neurotransmitter phenotype of cultured sympathetic neurons. Dev Biol. 1987;121:548–558. doi: 10.1016/0012-1606(87)90190-4. [DOI] [PubMed] [Google Scholar]
- 38.Rohrer H. Cholinergic neuronal differentiation factors: evidence for the presence of both CNTF-like and non-CNTF-like factors in developing rat footpad. Development. 1992;114:689–698. doi: 10.1242/dev.114.3.689. [DOI] [PubMed] [Google Scholar]
- 39.Saadat S, Sendtner M, Rohrer H. Ciliary neurotrophic factor induces cholinergic differentiation of rat sympathetic neurons in culture. J Cell Biol. 1989;108:1807–1816. doi: 10.1083/jcb.108.5.1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schotzinger RJ, Landis SC. Acquisition of cholinergic and peptidergic properties by sympathetic innervation of rat sweat glands requires interaction with normal target. Neuron. 1990;5:91–100. doi: 10.1016/0896-6273(90)90037-g. [DOI] [PubMed] [Google Scholar]
- 41.Serova LI, Nankova BB, Feng Z, Hong JS, Hutt M, Sabban EL. Heightened transcription for enzymes involved in norepinephrine biosynthesis in the rat locus coeruleus by immobilization stress. Biol Psychiatry. 1999;45:853–862. doi: 10.1016/s0006-3223(98)90360-2. [DOI] [PubMed] [Google Scholar]
- 42.Shinozaki K, Nishio Y, Okamura T, Yoshida Y, Maegawa H, Kojima H, Masada M, Toda N, Kikkawa R, Kashiwagi A. Oral administration of tetrahydrobiopterin prevents endothelial dysfunction and vascular oxidative stress in the aortas of insulin-resistant rats. Circ Res. 2000;87:566–573. doi: 10.1161/01.res.87.7.566. [DOI] [PubMed] [Google Scholar]
- 43.Stegenga SL, Hirayama K, Kapatos G. Regulation of GTP cyclohydrolase I gene expression and tetrahydrobiopterin content in cultured sympathetic neurons by leukemia inhibitory factor and ciliary neurotrophic factor. J Neurochem. 1996;66:2541–2545. doi: 10.1046/j.1471-4159.1996.66062541.x. [DOI] [PubMed] [Google Scholar]
- 44.Swerts JP, Le Van TA, Vigny A, Weber MJ. Regulation of enzymes responsible for neurotransmitter synthesis and degradation in cultured rat sympathetic neurons. I. Effects of muscle-conditioned medium. Dev Biol. 1983;100:1–11. doi: 10.1016/0012-1606(83)90195-1. [DOI] [PubMed] [Google Scholar]
- 45.Szczypka MS, Mandel RJ, Donahue BA, Snyder RO, Leff SE, Palmiter RD. Viral gene delivery selectively restores feeding and prevents lethality of dopamine-deficient mice. Neuron. 1999;22:167–178. doi: 10.1016/s0896-6273(00)80688-1. [DOI] [PubMed] [Google Scholar]
- 46.Thöny B, Auerbach G, Blau N. Tetrahydrobiopterin biosynthesis, regeneration and functions. Biochem J. 2000;347:1–16. [PMC free article] [PubMed] [Google Scholar]
- 47.Woodward WR, Seil FJ, Hammerstad JP. Cerebellum plus locus coeruleus in tissue culture. II. Development and metabolism of catecholamines. J Neurosci Res. 1987;17:184–188. doi: 10.1002/jnr.490170214. [DOI] [PubMed] [Google Scholar]
- 48.Zigmond RE, Schwarzschild MA, Rittenhouse AR. Acute regulation of tyrosine hydroxylase by nerve activity and by neurotransmitters via phosphorylation. Annu Rev Neurosci. 1989;12:415–461. doi: 10.1146/annurev.ne.12.030189.002215. [DOI] [PubMed] [Google Scholar]