

Abstract
A large body of evidence indicates important interactions between the adenosine and opioid systems in regulating pain at both the spinal and supraspinal level. Mice lacking the A2A receptor gene have been developed successfully, and these animals were shown to be hypoalgesic. To investigate whether there are any compensatory alterations in opioid systems in mutant animals, we have performed quantitative autoradiographic mapping of μ, δ, κ, and opioid receptor-like (ORL1) opioid receptors in the brains and spinal cords of wild-type and homozygous A2A receptor knock-out mice. In addition, μ-, δ-, and κ−mediated antinociception using the tail immersion test was tested in wild-type and homozygous A2A receptor knock-out mice. A significant reduction in [3H]deltorphin-I binding to δ receptors and a significant increase in [3H]CI-977 binding to κ receptors was detected in the spinal cords but not in the brains of the knock-out mice. μ and ORL1 receptor expression were not altered significantly. Moreover, a significant reduction in δ-mediated antinociception and a significant increase in κ-mediated antinociception were detected in mutant mice, whereas μ-mediated antinociception was unaffected. Comparison of basal nociceptive latencies showed a significant hypoalgesia in knock-out mice when tested at 55°C but not at 52°C. The results suggest a functional interaction between the spinal δ and κ opioid and the peripheral adenosine system in the control of pain pathways.
Keywords: A2A knock-out, μ opioid receptor, δ opioid receptor, κ opioid receptor, ORL1 receptor, autoradiography, opioid-mediated antinociception
There is evidence indicating that both acute and chronic effects of opioids are partly mediated by adenosine (Sawynok, 1998). Morphine enhances adenosine release from the spinal cord in vitro and in vivo(Sweeney et al., 1987, 1989; Cahill et al., 1995, 1996). Although neither δ- nor κ-selective opioid agonists enhance adenosine release from spinal cord synaptosomes (Cahill et al., 1995), a synergistic interaction between μ and δ receptors in mediating adenosine release has been suggested (Cahill et al., 1996). Adenosine in turn activates A1 receptors in the spinal cord, which suppresses the transmission of nociceptive information (Sawynok et al., 1989). A2A receptors (A2ARs) have been suggested to be involved in the expression of opioid-mediated antinociception. An additive interaction between μ-selective opioid receptor agonists and the A2A-selective adenosine agonist CGS21680 has been observed at the spinal level (DeLander and Keil, 1994). However, synergistic or multiplicative interactions between δ- or κ-selective opioid receptor agonists and CGS21680 suggest a more complex functional interaction between those receptors (DeLander and Keil, 1994).
In addition to the spinal cord, opioid–adenosine interactions have been reported to be present in the brain. Morphine enhances purine release in vivo and in vitro from the cortex of rodents (Fredholm and Vernet, 1978; Phillis et al., 1980; Stone, 1981). A2A receptors play a role in the development of opioid dependence, and chronic exposure to morphine has been demonstrated to downregulate adenosine A2Areceptors in the striatum of rats (De Montis, 1992). A specific interaction has been suggested between A2A and δ receptors, because a selective δ receptor agonist has been shown to inhibit the A2A receptor-mediated increase in DARPP-32 (dopamine- and cAMP-regulated phosphoprotein) occurring in striatopallidal neurons (Lindskog et al., 1999). Finally, the involvement of the opioid-related peptide nociceptin in stress and anxiety behaviors (Mogil et al., 1996; Jenck et al., 1997), combined with the modulatory role of A2A adenosine receptors in these behaviors (Svenningsson et al., 1999), has led to the suggestion of a possible interaction between opioid receptor-like (ORL1) and A2A receptors (Dassesse et al., 2000).
The generation of mice deficient in the A2AR gene has been reported (Ledent et al., 1997; Chen et al., 1999). The knock-out mice generated by Ledent et al. (1997) show increased aggression, are more anxious, and are hypoalgesic. Moreover, there are decreases in transcripts for proenkephalin and protachykinin in the striatum (Ledent et al., 1997). These results suggest the likelihood of alterations in pain processing in these mice and in modulatory effects of the opioid systems. To test the hypothesis that opioid systems are altered in the absence of the A2A receptor gene, we examined by quantitative autoradiography whether there are any changes in μ, δ, κ, and ORL1 opioid receptors in brains and spinal cords of A2A adenosine receptor knock-out mice. To investigate further the involvement of A2A adenosine receptors in modulating opioid effects, μ, δ, and κ receptor-mediated antinociception was also investigated in these animals.
MATERIALS AND METHODS
Generation of knock-out mice and experimental conditions. The experimental methodology for the generation of A2A adenosine receptor-deficient mice has been described in detail elsewhere (Ledent et al., 1997). Knock-out and wild-type mice from the same litters were bred from heterozygous mice and genotyped by PCR at weaning. Male mice aged between 10 and 12 weeks were used in all studies. Mice were housed in groups in a temperature-controlled room with a 12 hr light/dark schedule. Food and water were available ad libitum. For antinociception experiments, animals were tested in the same building in which they were housed between 2 and 5 P.M. and were allowed to acclimatize to the testing room for at least 1 hr before each experiment. All studies were performed in accordance with protocols approved by the Home Office (Animals Act 1986) UK.
Autoradiographic procedures and quantitative analysis. Autoradiography was performed as detailed previously (Kitchen et al., 1997; Clarke et al., 2001). Adjacent 20 μm coronal sections were cut at intervals of 300 μm from wild-type (+/+) and homozygous (−/−) brains and transverse sections from spinal cords for the determination of total and nonspecific binding of [3H]d-Ala2-MePhe4-Gly-ol5enkephalin (DAMGO), [3H]d-Ala2deltorphin-I (deltorphin-I), [3H] ((-)-N-methyl-N-[7-(1-pyrrodinyl)-1-oxospirol[4,5]dec-8-yl]-4-benzofuranacetamide (CI-977), and [3H] nociceptin at μ, δ, κ, and ORL1 opioid receptors, respectively. Ligand concentrations were ∼3–4× KD with [3H]DAMGO used at a concentration of 4 nm, [3H]deltorphin-I at 7 nm, [3H]CI-977 at 2.5 nm, and [3H]nociceptin at 0.4 nm. Nonspecific binding was defined in the presence of naloxone (1 μm for [3H]DAMGO and [3H]CI-977 and 10 μm for [3H]deltorphin-I) or unlabeled nociceptin (100 μm for [3H]nociceptin). An incubation time of 60 min was used for μ, δ, and κ ligands, whereas 180 min was used for ORL1. Sections from +/+ and −/− animals were processed together in a paired protocol. After binding, brain sections were apposed to [3H] Hyperfilm (Amersham) for a period of 3 weeks (μ, δ, and ORL1 receptors) or 6 weeks (κ receptors), whereas spinal cord sections were apposed for 11 weeks (μ receptors), 14 weeks (δ receptors), 18 weeks (k receptors), or 8 weeks (ORL1 receptors). Films were developed using 50% Kodak D19 developer. Quantitative analysis of brain receptors was performed as detailed previously (Kitchen et al., 1997; Clarke et al., 2001) using an MCID image analyzer (Imaging Research). Measurements for quantitative analysis of spinal cords were taken from both right and left sides for each region, therefore representing a duplicate determination apart from lamina X, where only one measurement was taken. All anatomical areas of the spinal cord were analyzed by freehand drawing. Brain structures were identified using the mouse brain atlas of Franklin and Paxinos (1997), and spinal cord structures were referenced to the rat atlas of Paxinos and Watson (1986). [3H] CGS21680 (10 nm) binding to spinal cord and brain sections taken at the level of the striatum of each mouse used in this study was determined as described by Bailey et al. (2002) to confirm the genotype.
Drug administration and assay for antinociception. A time course for antinociceptive effects was established. All drugs were administered intraperitoneally in a volume of 0.1 ml. Antinociceptive responses to the μ-selective agonist morphine, the δ-selective agonist [d-Ala2]deltorphin (deltorphin-I), and the κ-selective agonist CI-977 were studied using the tail immersion test as described by Janssen et al. (1963). The thermal stimulus for the tail immersion assay was warm water at 55°C in the studies with morphine and 52°C for deltorphin-I and CI-977. Control latencies were determined by measuring the time required for the mouse to flick or withdraw its tail from the water 20 min before drug treatment. A 10 sec cutoff time was used to prevent tissue damage. Each naive +/+ and −/− mouse was administered vehicle or a dose of an opioid agonist intraperitoneally. Each mouse was tested for antinociceptive responses 5, 15, 30, 45, and 60 min after the administration of morphine, 5, 10, 15, 30 and 45 min after the administration of deltorphin-I, and 5, 10, 20, 30, 45, 60, and 120 min after the administration of CI-977. Dose–response curves for each agonist were plotted from data at times when the analgesic response to the opioid agonists was maximum (45 min for morphine, 15 min for deltorphin-I, 10 min for CI-977). Basal nociceptive responses of wild-type and A2A receptor knock-out mice to mechanical stimuli were determined by the use of the tail pressure test as described by Kitchen (1984).
Statistics. Comparison of quantitative measures of autoradiographic binding for each ligand in brains and spinal cords from +/+ and −/− animals was performed using two-way ANOVA (for factors region or lamina and genotype). Comparison of antinociceptive dose–response curves of +/+ and −/− animals for each agonist was performed using repeated measures two-way ANOVA (for factors dose and genotype) followed by Scheffe's post hocanalysis. Comparison of basal control latencies from +/+ and −/− animals was performed using Student's t test. Comparison of tail immersion latencies of +/+ and −/− vehicle-treated animals was performed using repeated measures two-way ANOVA (for factors time and genotype).
Materials and drug preparation. [3H]DAMGO, 56.0 Ci/mmol, [3H]deltorphin-I, 50.0 Ci/mmol, [3H]CI-977, 41 Ci/mmol, and [3H]nociceptin, 164 Ci/mmol, were purchased from Amersham International (Buckinghamshire, UK). [3H]CGS21689, 42.5 Ci/mmol, was purchased from NEN Life Science Products (Hounslow, UK). Naloxone and morphine sulfate were purchased from Sigma-Aldrich (Dorset, UK), unlabeled nociceptin and deltorphin-I were from Bachem (Essex, UK), and unlabeled CI-977 was a gift from Parke-Davis (Cambridge, UK). Morphine sulfate and CI-977 were dissolved in saline, and deltorphin-I was dissolved in saline that was acidified with glacial acetic acid (1% v/v) and buffered with NaOH (75 mm) to pH 7.
RESULTS
Quantitative autoradiography of μ, δ, κ, and ORL1 opioid receptors in brains and spinal cords of A2A adenosine receptor knock-out mice
The qualitative and quantitative distribution of μ, δ, κ, and ORL1 opioid receptors labeled with [3H]DAMGO (4 nm), [3H]deltorphin-I (7 nm), [3H]CI-977 (2.5 nm), and [3H]nociceptin (0.4 nm) in coronal sections of brain of wild-type mice were similar to previous studies reported by our group (Kitchen et al., 1997; Slowe et al., 1999; Clarke et al., 2001) (Fig. 1). In spinal cord, the highest levels of [3H]DAMGO, [3H]deltorphin-I, and [3H]CI-977 binding were detected in the superficial layers. High levels of [3H]nociceptin binding were detected in the superficial layers and in lamina X of the spinal cord (Fig. 2). The pattern of distribution of all receptor types studied in homozygous A2A adenosine receptor knock-out mice brains and spinal cords was identical to wild type. Binding of A2A adenosine receptors with [3H]CGS21680 showed a complete absence of A2A sites in homozygous mice, which confirmed the genotype of the A2A adenosine receptor knock-out mice used in this study (Fig. 1). Finally no [3H]CGS21680 binding was detected in wild-type or knock-out spinal cords (Fig. 2).
Fig. 1.
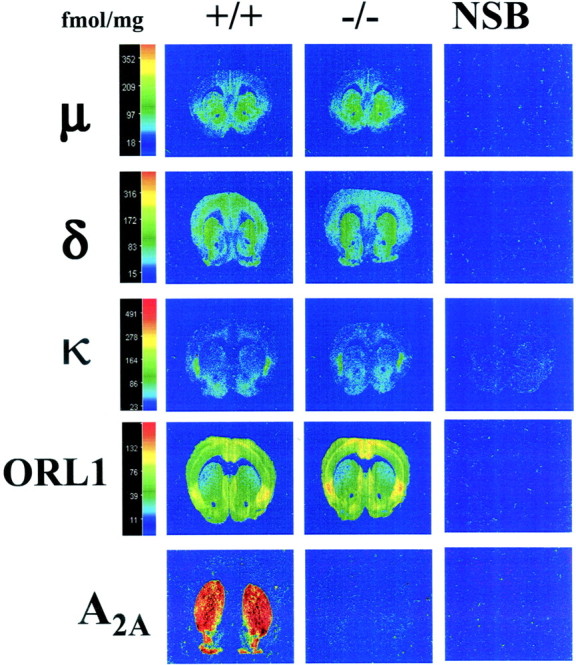
Computer-enhanced autoradiograms of coronal brain sections from wild-type (+/+) and homozygous (−/−) A2A adenosine receptor knock-out mice. All the sections shown are from the level of the caudate (bregma 1.34 mm, apart from A2A receptor-labeled sections, which were bregma 1.10 mm). μ receptors were labeled with [3H]DAMGO (4 nm), δ receptors with [3H]deltorphin-I (7 nm), κ receptors with [3H]CI-977 (2.5 nm), ORL1 receptors with [3H] nociceptin (0.4 nm), and A2A receptors with [3H]CGS216800 (10 nm). Nonspecific binding (NSB), shown in the far right column, was determined in the presence of naloxone (1 μm for μ and κ and 10 μm for δ), unlabeled nociceptin (100 μm for ORL1), or 5′-N-ethylcarboxamidoadenosine (20 μm for A2A). The color bar shows a pseudocolor interpretation of the relative density of the black and white film image calibrated in femtomoles per milligram of tissue. Sections from +/+ and −/− brains were processed in parallel.
Fig. 2.
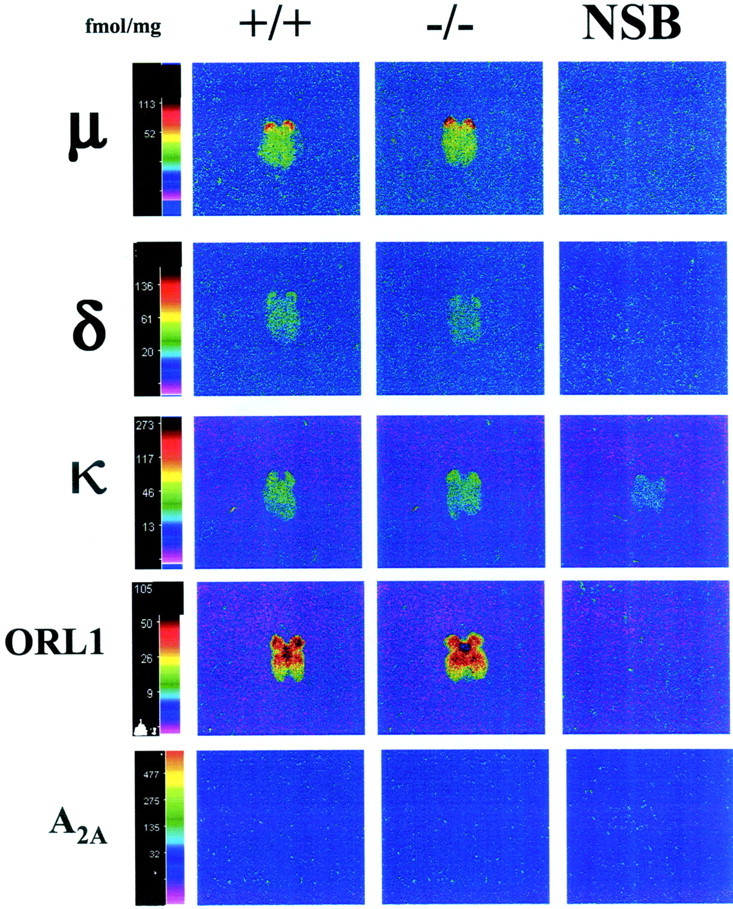
Computer-enhanced autoradiograms of coronal sacral spinal cord sections from wild-type (+/+) and homozygous (−/−) A2A knock-out mice. The sections were taken were the S4 segment according to the rat atlas of Paxinos and Watson (1986). μ receptors were labeled with [3H]DAMGO (4 nm), δ receptors with [3H]deltorphin-I (7 nm), κ receptors with [3H]CI-977 (2.5 nm), ORL1 receptors with [3H]nociceptin (0.4 nm), and A2A receptors with [3H]CGS21680 (10 nm). Nonspecific binding (NSB), shown in the far right column, was determined in the presence of naloxone (1 μm for μ and κ and 10 μm for δ), unlabeled nociceptin (100 μmfor ORL1), or 5′-N-ethylcarboxamidoadenosine (20 μm for A2A). The color bar shows a pseudocolor interpretation of the relative density of the black and white film image calibrated in femtomoles per milligram of tissue. Sections from +/+ and −/− spinal cords were processed in parallel.
In the brain, there were no significant differences in the levels of μ, δ, κ, and ORL1 opioid expression between genotypes for between 40 and 50 brain regions analyzed for each receptor (p > 0.05). For illustration, Table1 shows only noncortical regions that either exhibit high level of receptor expression or are known to play an important role in pain. Binding in all cortical structures was very similar across genotypes for each receptor (data not shown). In contrast, in the spinal cord a significant quantitative difference in the levels of [3H]deltorphin-I binding between genotypes was observed (p < 0.01), and three-quarters of the regions analyzed in A2Aknock-out mice showed a downregulation of δ receptor expression. The largest loss of δ sites (up to 19%) was found in the superficial layers of the spinal cord (Table 2). There was also a significant quantitative increase in the levels of [3H]CI-977 binding between genotypes (p < 0.05), and the greatest overall upregulation was detected in the superficial layers of the spinal cord (up to 27%) (Table 3). In contrast to δ and κ binding, there were no significant differences in the levels of μ or ORL1 receptor expression between genotypes in the spinal cord (p > 0.05) (Tables4,5).
Table 1.
Representative noncortical regions of quantitative autoradiography of μ, δ, κ, and ORL1 receptor binding in wild-type (+/+) and homozygous (−/−) A2A mutant mice brains
| Region | Bregma coordinates (mm) | [3H]ligand-specific binding (fmol/mg) | % Change in binding | |
|---|---|---|---|---|
| Wild-type (+/+) | Homozygous (−/−) | (−/−) | ||
| [3H]DAMGO binding | ||||
| Nucleus accumbens core | 1.34 | 103 ± 14.5 | 106 ± 12.7 | 2.9 |
| Endopiriform nucleus | 1.10 | 93.4 ± 6.1 | 92.4 ± 8.9 | −1.1 |
| Medial habenula nucleus | −1.70 | 132 ± 16.2 | 126 ± 16.7 | −4.5 |
| Thalamus | −1.70 | 50.1 ± 7.4 | 49.6 ± 7.2 | −1.0 |
| Basolateral amygdala | −1.70 | 108 ± 18.3 | 109 ± 13.0 | 0.9 |
| Periaqueductal gray | −3.40 | 65.1 ± 14.1 | 69.7 ± 11.5 | 7.1 |
| Superficial gray layer of superior colliculus | −3.40 | 103 ± 9.0 | 93.3 ± 16.2 | −9.4 |
| Intermediate gray layer of superior colliculus | −3.40 | 80.0 ± 9.0 | 74.6 ± 14.8 | −6.8 |
| [3H]deltorphin-1 binding | ||||
| Olfactory bulb | 4.28 | |||
| Exterior plexiform layer | 64.2 ± 4.6 | 68.3 ± 6.3 | 6.4 | |
| Interior granular layer | 148 ± 20.3 | 160.0 ± 20.1 | 8.1 | |
| Nucleus accumbens core | 1.34 | 56.3 ± 4.8 | 60.4 ± 4.5 | 7.3 |
| Caudate-putamen | 1.10 | 82.2 ± 6.2 | 93.5 ± 2.9 | 13.7 |
| Olfactory tubercle | 1.10 | 80.0 ± 6.4 | 68.8 ± 7.5 | −14.0 |
| Thalamus | −1.70 | 25.5 ± 1.8 | 24 ± 1.1 | −5.9 |
| Basolateral amygdala | −1.70 | 74.1 ± 5.0 | 75.0 ± 8.6 | 1.2 |
| Presubiculum | −4.04 | 61.6 ± 6.3 | 50.8 ± 3.1 | −17.5 |
| [3H]CI-977 binding | ||||
| Claustrum | 1.94 | 114 ± 10.2 | 119 ± 14.5 | 4.4 |
| Nucleus accumbens core | 1.34 | 35.0 ± 4.7 | 28.9 ± 2.5 | −17.4 |
| Olfactory tubercle | 1.10 | 29.2 ± 3.5 | 35.2 ± 2.8 | 20.5 |
| Dorsal endopiriform nucleus | 1.10 | 125.5 ± 7.6 | 136.2 ± 15.4 | 8.5 |
| Ventral pallidum | 0.14 | 47.7 ± 2.4 | 46.0 ± 6.2 | −3.6 |
| Basolateral amygdala | −1.70 | 37.8 ± 5.6 | 38.6 ± 3.7 | 2.1 |
| Hypothalamus | −1.70 | 35.9 ± 4.4 | 32.6 ± 1.1 | −9.1 |
| Substantia nigra | −3.40 | 37.7 ± 2.3 | 31.8 ± 2.5 | −15.6 |
| Superficial gray of the superior colliculus | −3.40 | 7.3 ± 1.8 | 8.8 ± 3.2 | 20.5 |
| [3H]nociceptin binding | ||||
| Anterior olfactory bulb | 3.08 | 88.2 ± 6.4 | 83.9 ± 5.6 | −4.9 |
| Nucleus accumbens | 1.34 | 55.4 ± 2.2 | 55.1 ± 3.1 | −0.5 |
| Thalamus | −1.70 | 39.5 ± 1.4 | 37.1 ± 1.4 | −6.1 |
| Amygdala | −1.70 | 111 ± 8.0 | 105 ± 7.9 | −5.4 |
| Hippocampus | −2.46 | 60.0 ± 3.6 | 52.8 ± 2.0 | −12.0 |
| Periaqueductal gray | −3.40 | 48.6 ± 3.5 | 48.6 ± 5.8 | 0 |
| Superficial gray layer of superior colliculus | −3.40 | 78.2 ± 6.1 | 78.4 ± 1.9 | 0.3 |
| Intermediate gray layer of superior colliculus | −3.40 | 46.4 ± 2.0 | 47.2 ± 2.0 | 1.7 |
| Presubiculum | −3.40 | 75.8 ± 1.6 | 69.4 ± 3.1 | −8.4 |
The mean specific binding (n = 4) of [3H]DAMGO, [3H]deltorphin-1, [3H]CI-977, or [3H]nociceptin (femtomoles per milligram) ± SEM in brain regions of wild-type (+/+) and homozygous (−/−) A2A receptor knock-out mice. Measurements in the regions were performed at the bregma coordinates taken from the mouse atlas of Franklin and Paxinos (1997). Regional determinates were made from both left and right sides of the sections, which were cut 300 μm apart. The labeling was performed on sections from +/+ and −/− mice in a completely paired protocol. Specific binding of [3H]DAMGO, [3H]deltorphin-1, and [3H]nociceptin was >85% in all regions. Specific binding of [3H]CI-977 was >50% in all regions. The data show only regions expressing high receptor levels and regions associated with pain processing. Analysis of all 40–50 regions in which receptor measurements were made showed that comparison of genotypes was not statistically significant for any ligand (p > 0.05; ANOVA). The percentage (%) change in binding represents the change in binding levels in −/− brains compared with +/+. A minus sign indicates a percentage decrease in binding levels.
Table 2.
Quantitative autoradiography of δ receptor binding in wild-type (+/+) and homozygous (−/−) A2A mutant mice
| Region | Segments from rat atlas | [3H]deltorphin-1-specific binding (fmol/mg) | % Change in binding | |
|---|---|---|---|---|
| Wild-type (+/+) | Homozygous (−/−) | (−/−) | ||
| Cervical | C1 and C6 | |||
| Superficial layers (laminas I and II) | 30.0 ± 2.6 | 25.4 ± 2.6 | −15.3 | |
| Laminas III–VI | 15.5 ± 1.0 | 13.7 ± 0.8 | −11.6 | |
| Lamina X | 18.3 ± 2.0 | 18.0 ± 0.9 | −1.6 | |
| Ventral horn (laminas VII–IX) | 13.7 ± 0.9 | 12.4 ± 0.7 | −9.5 | |
| Dorsal horn (laminas I–VI) | 18.7 ± 1.3 | 15.4 ± 0.9 | −17.6 | |
| Thoracic | T3 and T6 | |||
| Superficial layers (laminas I and II) | 27.7 ± 1.8 | 24.9 ± 2.2 | −10.1 | |
| Laminas III–VI | 15.7 ± 1.0 | 15.4 ± 1.6 | −1.9 | |
| Lamina X | 16.7 ± 1.2 | 15.8 ± 1.5 | −5.4 | |
| Ventral horn (laminas VII–IX) | 14.9 ± 0.9 | 14.4 ± 1.1 | −3.4 | |
| Dorsal horn (laminas I–VI) | 19.3 ± 0.8 | 18.8 ± 2.0 | −2.6 | |
| Sacral | S4 | |||
| Superficial layers (laminas I and II) | 21.2 ± 1.3 | 17.2 ± 1.5 | −18.9 | |
| Laminas III–VI | 11.2 ± 0.9 | 12.0 ± 0.9 | 7.1 | |
| Lamina X | 11.2 ± 0.8 | 11.6 ± 0.5 | 3.6 | |
| Ventral horn (laminas VII–IX) | 10.9 ± 1.0 | 11.7 ± 0.4 | 7.3 | |
| Dorsal horn (laminas I–VI) | 14.2 ± 1.2 | 13.5 ± 0.7 | −4.9 | |
The mean specific binding (n = 4) of [3H]deltorphin-I (femtomoles per milligram) ± SEM in spinal cord regions of wild-type (+/+) and homozygous (−/−) A2A receptor knock-out mice. Measurements in the regions were performed at the bregma coordinates taken from the rat atlas ofPaxinos and Watson (1986). Regional determinates were made from both left and right sides of the sections, which were cut 300 μm apart. The labeling was performed on sections from +/+ and −/− mice in a completely paired protocol. Specific binding was >80% in all regions. Comparison of genotypes was statistically significant (p < 0.01; ANOVA). The percentage (%) change in binding represents the change in binding levels in −/− spinal cords compared with +/+. A minus sign indicates a percentage decrease in binding levels. The overall mean and median percentage changes across regions were −5.8 and −5.4%, respectively.
Table 3.
Quantitative autoradiography of κ receptor binding in wild-type (+/+) and homozygous (−/−) A2A mutant mice
| Region | Segments from rat atlas | [3H]CI-977-specific binding (fmol/mg) | % Change in binding | |
|---|---|---|---|---|
| Wild-type (+/+) | Homozygous (−/−) | (−/−) | ||
| Cervical | C1 and C6 | |||
| Superficial layers (laminas I and II) | 15.9 ± 1.7 | 15.7 ± 1.3 | −1.3 | |
| Laminas III–VI | 4.8 ± 0.9 | 4.5 ± 1.0 | −6.3 | |
| Lamina X | 9.8 ± 1.2 | 11.4 ± 1.1 | 16.3 | |
| Ventral horn (laminas VII–IX) | 2.5 ± 0.7 | 2.4 ± 0.6 | −4.0 | |
| Dorsal horn (laminas I–VI) | 7.4 ± 1.4 | 8.0 ± 1.1 | 8.1 | |
| Thoracic | T3 and T6 | |||
| Superficial layers (laminas I and II) | 16.9 ± 2.1 | 20.1 ± 1.4 | 18.9 | |
| Laminas III–VI | 7.0 ± 1.3 | 9.2 ± 1.0 | 31.4 | |
| Lamina X | 11.7 ± 0.9 | 11.8 ± 1.0 | 0.9 | |
| Ventral horn (laminas VII–IX) | 5.1 ± 1.1 | 6.1 ± 1.0 | 19.6 | |
| Dorsal horn (laminas I–VI) | 9.9 ± 1.4 | 12.7 ± 1.2 | 28.3 | |
| Sacral | S4 | |||
| Superficial layers (laminas I and II) | 17.2 ± 1.6 | 21.8 ± 2.8 | 26.7 | |
| Laminas III–VI | 8.4 ± 1.0 | 7.0 ± 2.4 | −16.7 | |
| Lamina X | 11.2 ± 1.1 | 13.4 ± 3.4 | 19.6 | |
| Ventral horn (laminas VII–IX) | 4.7 ± 0.8 | 2.4 ± 1.0 | −48.9 | |
| Dorsal horn (laminas I–VI) | 12.0 ± 1.1 | 13.7 ± 2.7 | 14.2 | |
The mean specific binding (n = 4) of [3H]CI-977 (femtomoles per milligram) ± SEM in spinal cord regions of wild-type (+/+) and homozygous (−/−) A2A receptor knock-out mice. Measurements in the regions were performed at the bregma coordinates taken from the rat atlas ofPaxinos and Watson (1986). Regional determinates were made from both left and right sides of the sections, which were cut 300 μm apart. The labeling was performed on sections from +/+ and −/− mice in a completely paired protocol. Specific binding was >45% in all regions. Comparison of genotypes was statistically significant (p < 0.05; ANOVA). The percentage (%) change in binding represents the change in binding levels in −/− spinal cords compared with +/+. A minus sign indicates a percentage decrease in binding levels. The overall mean and median percentage changes across regions were 7.1 and 14.2%, respectively.
Table 4.
Quantitative autoradiography of μ receptor binding in wild-type (+/+) and homozygous (−/−) A2A mutant mice
| Region | Segments from rat atlas | [3H]DAMGO-specific binding (fmol/mg) | % Change in binding | |
|---|---|---|---|---|
| Wild-type (+/+) | Homozygous (−/−) | (−/−) | ||
| Cervical | C1 and C6 | |||
| Superficial layers (laminas I and II) | 96.2 ± 4.1 | 95.9 ± 3.0 | −0.3 | |
| Laminas III–VI | 31.2 ± 2.2 | 30.2 ± 2.2 | −3.2 | |
| Lamina X | 27.2 ± 1.7 | 28.9 ± 2.4 | 6.3 | |
| Ventral horn (laminas VII–IX) | 18.9 ± 1.8 | 19.1 ± 2.1 | 1.1 | |
| Dorsal horn (laminas I–VI) | 43.0 ± 1.7 | 41.3 ± 3.1 | −4.0 | |
| Thoracic | T3 and T6 | |||
| Superficial layers (laminas I and II) | 99.5 ± 4.6 | 91.5 ± 3.6 | −8.0 | |
| Laminas III–VI | 30.5 ± 2.7 | 34.5 ± 3.0 | 13.1 | |
| Lamina X | 22.6 ± 2.4 | 18.8 ± 1.6 | −16.8 | |
| Ventral horn (laminas VII–IX) | 17.4 ± 3.0 | 20.5 ± 2.0 | 17.8 | |
| Dorsal horn (laminas I–VI) | 48.9 ± 2.6 | 48.8 ± 2.4 | −0.2 | |
| Sacral | S4 | |||
| Superficial layers (laminas I and II) | 91.5 ± 8.6 | 92.4 ± 4.9 | 1.0 | |
| Laminas III–VI | 31.6 ± 3.1 | 31.1 ± 2.6 | −1.6 | |
| Lamina X | 29.7 ± 4.5 | 29.4 ± 6.7 | −1.0 | |
| Ventral horn (laminas VII–IX) | 23.7 ± 1.7 | 19.7 ± 2.9 | −16.9 | |
| Dorsal horn (laminas I–VI) | 47.5 ± 4.2 | 44.6 ± 4.7 | −6.1 | |
The mean specific binding (n = 4) of [3H]DAMGO (femtomoles per milligram) ± SEM in spinal cord regions of wild-type (+/+) and homozygous (−/−) A2A receptor knock-out mice. Measurements in the regions were performed at the bregma coordinates taken from the rat atlas ofPaxinos and Watson (1986). Regional determinates were made from both left and right sides of the sections, which were cut 300 μm apart. The labeling was performed on sections from +/+ and −/− mice in a completely paired protocol. Specific binding was >90% in all regions. Comparison of genotypes was not statistically significant (p > 0.05; ANOVA). The percentage (%) change in binding represents the change in binding levels in −/− spinal cords compared with +/+. A minus sign indicates a percentage decrease in binding levels. The overall mean and median percentage changes across regions were −1 and −0.9%, respectively.
Table 5.
Quantitative autoradiography of ORL1 receptor binding in wild-type (+/+) and homozygous (−/−) A2A mutant mice
| Region | Segments from rat atlas | [3H]nociceptin-specific binding (fmol/mg) | % Change in binding | |
|---|---|---|---|---|
| Wild-type (+/+) | Homozygous (−/−) | (−/−) | ||
| Cervical | C1 and C6 | |||
| Superficial layers (laminas I and II) | 52.9 ± 3.6 | 55.8 ± 6.3 | 5.5 | |
| Laminas III–VII | 36.9 ± 1.7 | 36.5 ± 1.9 | −1.1 | |
| Lamina X | 46.8 ± 3.7 | 50.5 ± 2.8 | 7.9 | |
| Laminas VIII and IX | 20.4 ± 1.4 | 19.0 ± 2.1 | −6.9 | |
| Dorsal horn (laminas I–VI) | 40.8 ± 2.1 | 42.0 ± 2.7 | 2.9 | |
| Thoracic | T3 and T6 | |||
| Superficial layers (laminas I and II) | 54.6 ± 3.6 | 67.8 ± 8.5 | 24.2 | |
| Laminas III–VII | 45.9 ± 2.5 | 48.8 ± 2.6 | 6.3 | |
| Lamina X | 54.0 ± 2.8 | 57.7 ± 2.6 | 6.9 | |
| Laminas VIII and IX | 23.7 ± 1.0 | 23.1 ± 1 | −2.5 | |
| Dorsal horn (laminas I–VI) | 48.3 ± 3.3 | 54.4 ± 4.6 | 12.6 | |
| Sacral | S4 | |||
| Superficial layers (laminas I and II) | 66.3 ± 3.6 | 75.6 ± 11.2 | 14.0 | |
| Laminas III–VII | 50.5 ± 1.8 | 51.4 ± 2.6 | 1.8 | |
| Lamina X | 73.6 ± 6.6 | 82.1 ± 7.2 | 11.5 | |
| Laminas VIII and IX | 24.0 ± 1.7 | 22.4 ± 1.3 | −6.7 | |
| Dorsal horn (laminas I–VI) | 56.2 ± 2.3 | 59.2 ± 5.6 | 5.3 | |
The mean specific binding (n = 4) of [3H]nociceptin (femtomoles per milligram) ± SEM in spinal cord regions of wild-type (+/+) and homozygous (−/−) A2A receptor knock-out mice. Measurements in the regions were performed at the bregma coordinates taken from the rat atlas ofPaxinos and Watson (1986). Regional determinates were made from both left and right sides of the sections, which were cut 300 μm apart. The labeling was performed on sections from +/+ and −/− mice in a completely paired protocol. Specific binding was >90% in all regions. Comparison of genotypes was not statistically significant (p > 0.05; ANOVA). The percentage (%) change in binding represents the change in binding levels in −/− spinal cords compared with +/+. A minus sign indicates a percentage decrease in binding levels. The overall mean and median percentage changes across regions were 5.3 and 5.3%, respectively.
μ, δ, and κ opioid receptor-mediated antinociception in wild-type (+/+) and A2A receptor knock-out (−/−) mice
An antinociceptive time course and dose–response relationships were established for the μ agonist morphine (Fig.3), the δ agonist deltorphin-I (Fig.4), and the κ agonist CI-977 (Fig.5). The antinociceptive effect was at a maximum at 45, 15, and 10 min after injection for morphine, deltorphin-I, and CI-977, respectively, in both wild-type and knock-out mice. Comparison of antinociceptive dose–response relationships between +/+ and −/− mice showed that there was a significant decrease in δ-mediated antinociception (p < 0.05) and a significant increase in κ-mediated antinociception (p < 0.01) but no change in μ-mediated antinociception (p > 0.05).
Fig. 3.
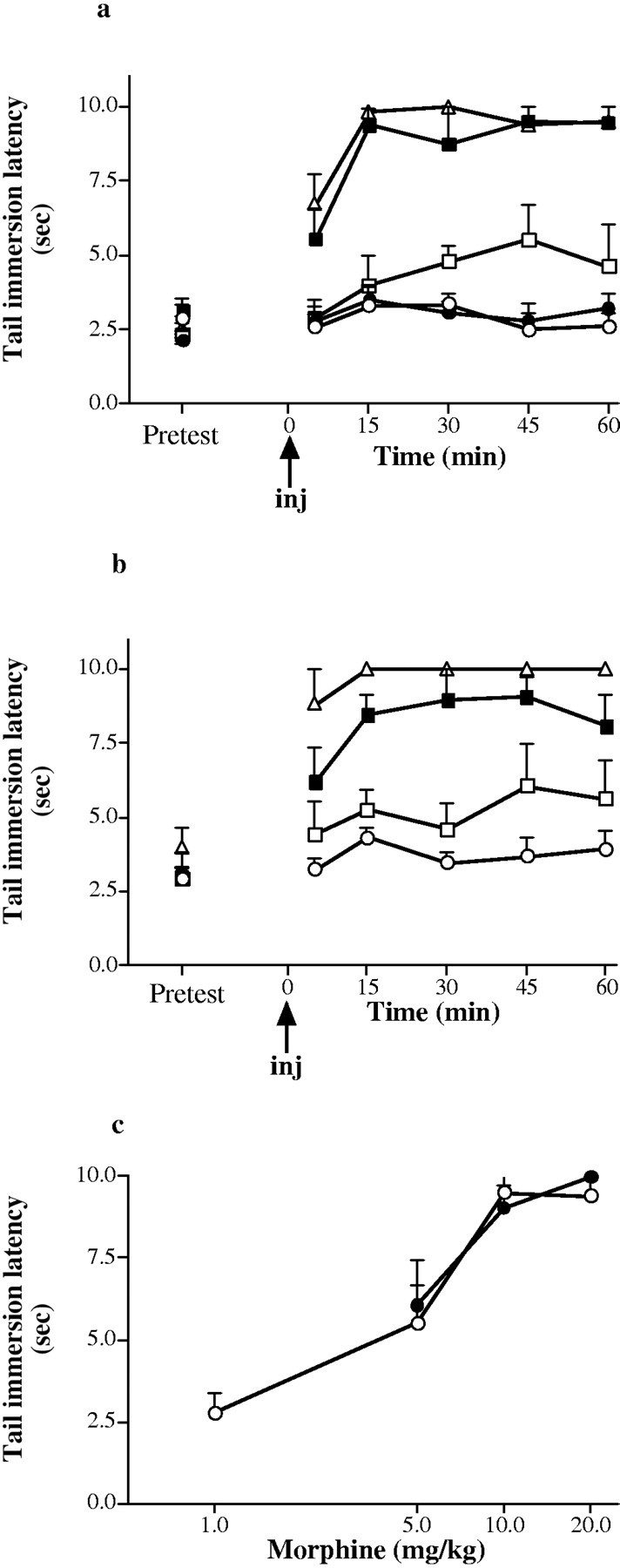
Dose-related antinociception induced by morphine sulfate in 12-week-old wild-type (+/+) and A2A receptor knock-out (−/−) mice. a, Time course of morphine-induced antinociception in the +/+ mice; b, time course of morphine-induced antinociception in the −/− mice measured as tail immersion latency at 55°C. ○, Saline control values; ●, 1 mg/kg morphine sulfate; ■, 5 mg/kg morphine sulfate; ▪, 10 mg/kg morphine sulfate; ▵, 20 mg/kg morphine sulfate. Doses (0.1 ml volume, i.p) were administered at 0 min.pretest, Pretest responses 20 min before injection.c, Dose–response relationship for morphine sulfate at peak antinociception (45 min after injection) in wild-type (○) and A2A receptor knock-out (●) mice. Values are means ± SEM of five determinations (n = 5). Differences in dose–response curves between genotypes were not statistically significant (p > 0.05; ANOVA). The difference in tail immersion latencies between +/+ and −/− saline-treated animals was statistically significant (p < 0.05; ANOVA).
Fig. 4.
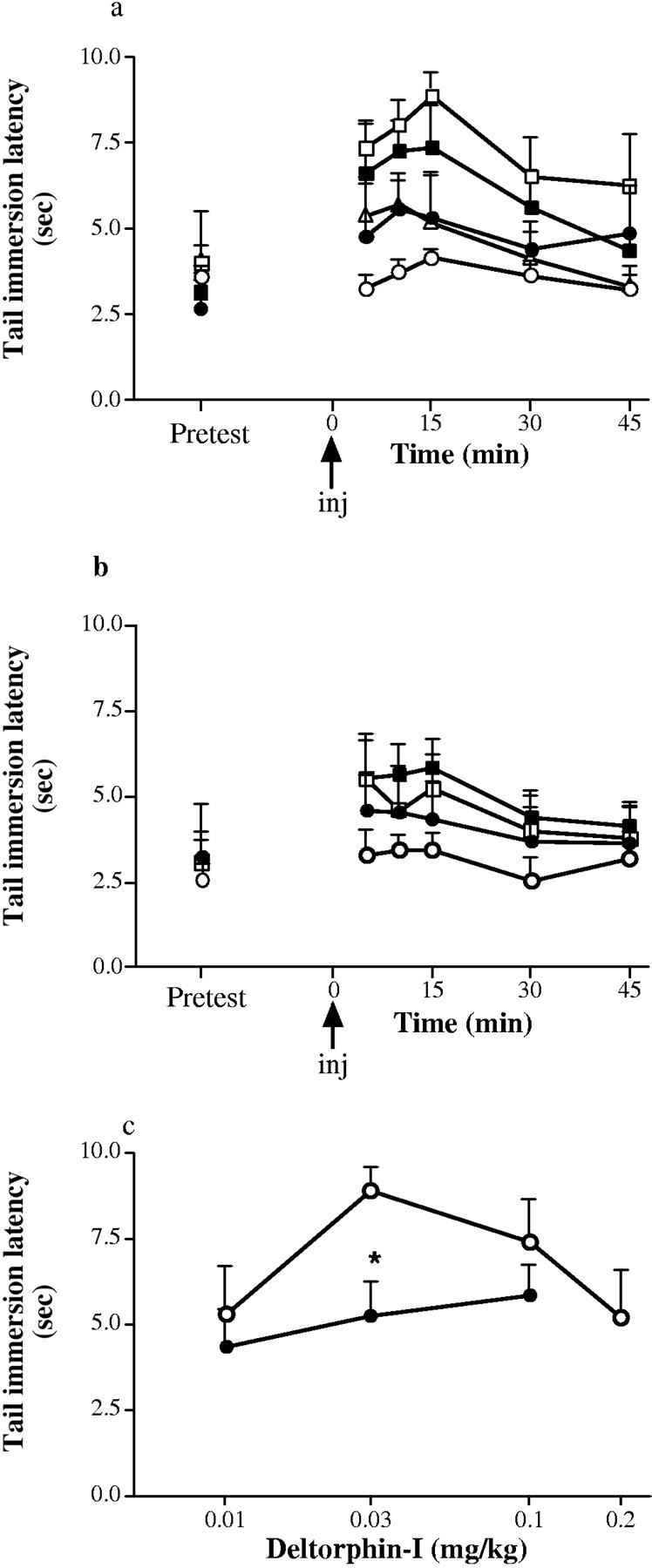
Dose-related antinociception induced by deltorphin-I in 12-week-old wild-type (+/+) and A2Areceptor knock-out (−/−) mice. a, Time course of deltorphin-I-induced antinociception in the +/+ mice; b, time course of deltorphin-I induced antinociception in the −/− mice measured as tail immersion latency at 52°C. ○ represents vehicle control values; ●, 0.01 mg/kg deltorphin-I; ■, 0.03 mg/kg deltorphin-I; ▪, 0.1 mg/kg deltorphin-I; ▵, 0.2 mg/kg deltorphin-I. Doses (0.1 ml volume, i.p) are administered at 0 min.pretest, Pretest responses 20 min before injection. c, Dose–response relationship for deltorphin-I at peak antinociception (15 min after injection) in wild-type (○) and A2A receptor knock-out (●) mice. Values are means ± SEM of five determinations (n = 5). Differences in dose–response curves between genotypes were statistically significant (p < 0.05; ANOVA); *p< 0.05 (Scheffe's post hoc analysis). The difference in tail immersion latency between +/+ and −/− vehicle-treated animals was not statistically significant (p > 0.05; ANOVA).
Fig. 5.
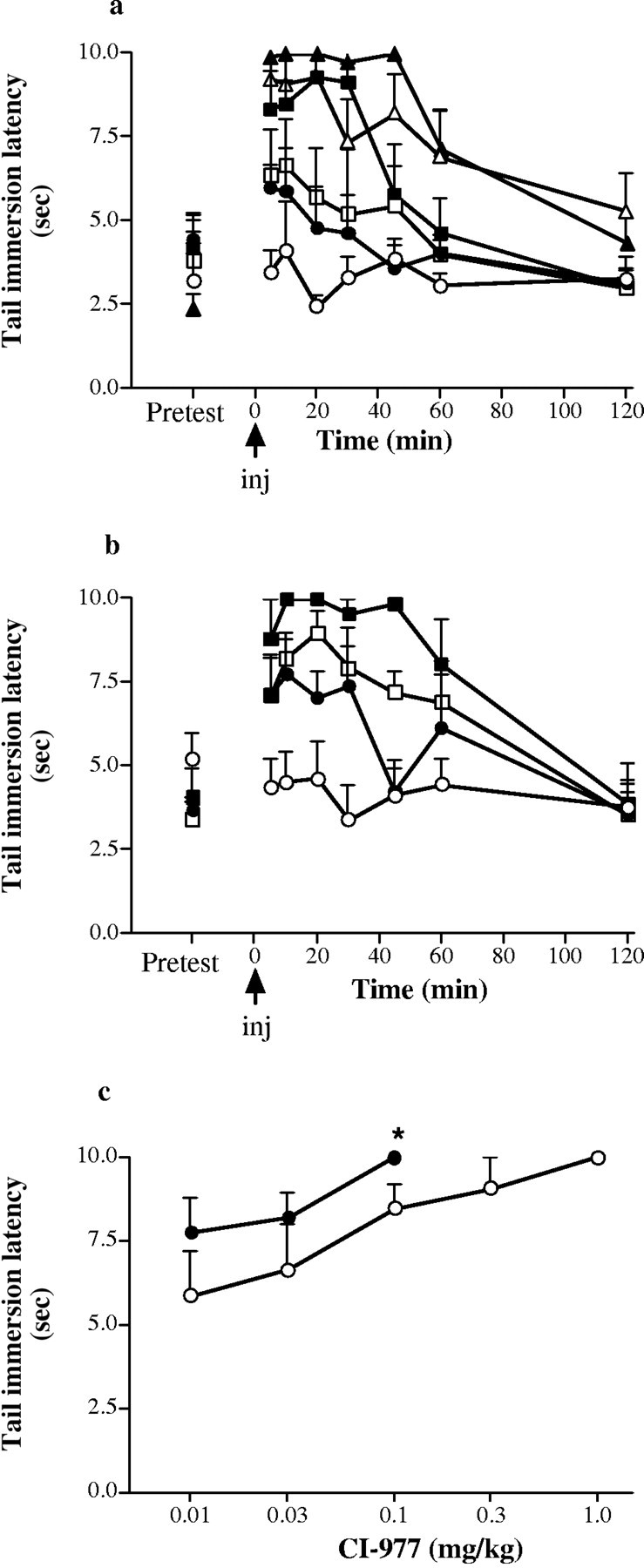
Dose-related antinociception induced by CI-977 in 12-week-old wild-type (+/+) and A2A receptor knock-out (−/−) mice. a, Time course of CI-977-induced antinociception in the +/+ mice; b, time course of CI-977-induced antinociception in the −/− mice measured as tail immersion latency at 52°C. ○ represents saline control values; ●, 0.01 mg/kg CI-977; ■, 0.03 mg/kg CI-977; ▪, 0.1 mg/kg CI-977; ▵, 0.3 mg/kg CI-977; ▴ 1 mg/kg CI-977. Doses (0.1 ml volume, i.p) are administered at 0 min. pretest, Pretest responses 20 min before injection. c, Dose–response relationship for CI-977 at peak antinociception (10 min after injection) in wild-type (○) and A2A receptor knock-out (●) mice. Values are means ± SEM of five determinations (n = 5). Differences in dose–response curves between genotypes were statistically significant (p < 0.05; ANOVA); *p< 0.05 (Scheffe's post hoc analysis). Differences in tail immersion latency between +/+ and −/− saline-treated animals were not statistically significant (p > 0.05; ANOVA).
Comparison of basal reactivity with thermal stimuli of 55°C between knock-out and wild-type animals showed that there was a significant increase in latency in knock-out animals (p < 0.05) (Fig. 6a). However, when the animals were exposed to a stimulus of 52°C, no significant difference in basal antinociception was found between genotypes (p > 0.05) (Fig. 6b). Similarly, a comparison of tail immersion latencies in vehicle-treated animals also demonstrated a significant increase in knock-out mice when they were exposed to a thermal stimuli of 55°C (p < 0.05) but not when they were exposed to 52°C (p > 0.05) (Figs.3-5). Finally, no significant change in basal nociceptive threshold (135.7 ± 12.6 gm for +/+ vs 157.9 ± 10.2 gm for −/−;n = 43) was found when +/+ and −/− mice were tested with the tail pressure test.
Fig. 6.
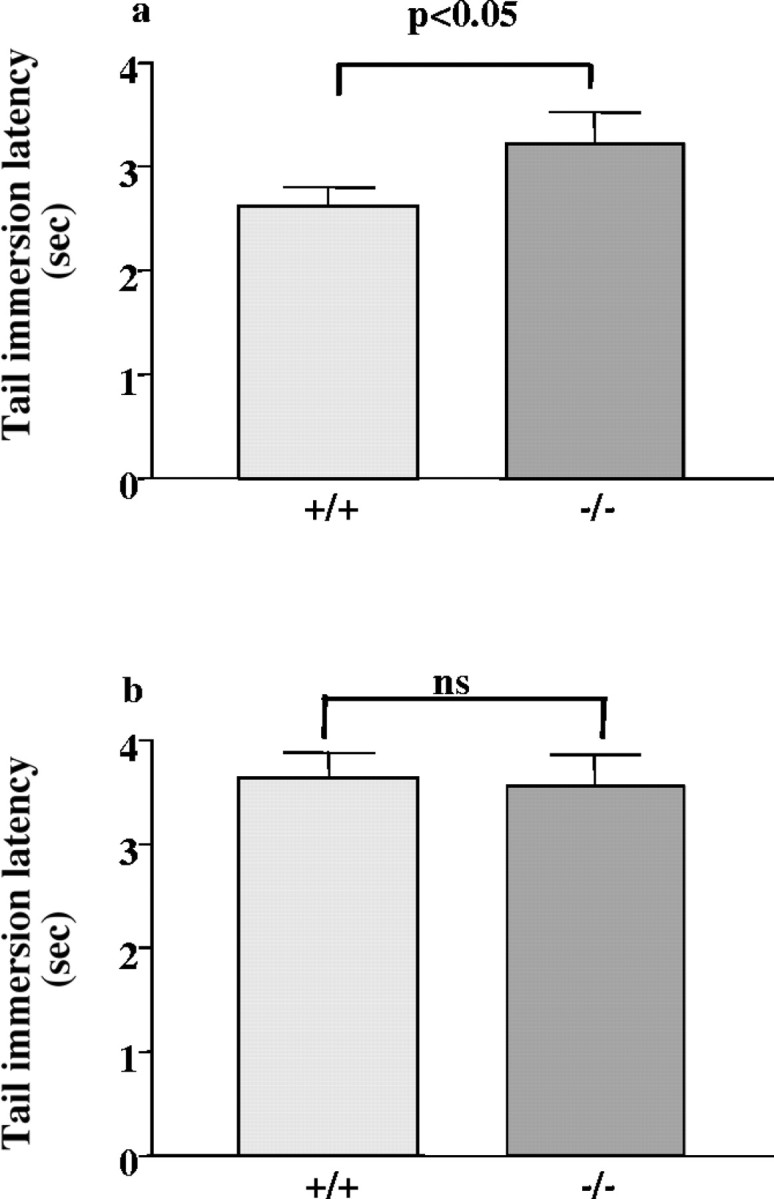
Tail immersion responses to acute thermal stimulus in wild-type (+/+) and A2A receptor knock-out (−/−) mice.a, Basal reactivity to a thermal stimulus of 55°C for wild-type (+/+) mice (n = 25) and A2Areceptor knock-out (−/−) mice (n = 20);b, basal reactivity to a thermal stimulus of 52°C for wild-type (+/+) mice (n = 55) and A2Areceptor knock-out (−/−) mice (n = 40). Error bars represent SEM. ns, Not significant.p < 0.05; significant difference between genotypes (Student's t test).
DISCUSSION
Although quantitative mapping of all four opioid receptors has been performed in the mouse brain (Clarke et al., 2001), the current study now reports the first complete quantitative autoradiography in spinal cord using highly selective ligands in this species. The qualitative pattern of μ, δ, κ, and ORL1 receptors in the spinal cord was similar to the studies that have been reported by others in the rat (Besse et al., 1990, 1991; Neal et al., 1999) and mouse (Narita et al., 1999; Mogil et al., 2000).
The lack of change of μ, δ, κ, or ORL1 binding in the brains of −/− mice clearly suggests that a direct A2A receptor interaction with any opioid receptor subtype is not relevant in the mouse brain. This is in agreement with a study that failed to find changes in A2A receptor binding in the striatum of chronically morphine-treated mice (Kaplan et al., 1994) in contrast with the rat, where A2Areceptors are downregulated (De Montis, 1992). Moreover, the decrease in proenkephalin mRNA, observed in A2A receptor knock-out mice striatum (Ledent et al., 1997), does not appear to trigger any changes in opioid receptor numbers. It is possible, however, that the decrease in proenkephalin mRNA in −/− mice does not result in a decrease in the levels of the transmitter itself. Indeed, enkephalin immunoreactivity is unaltered in another strain of A2A knock-out mice (Chen et al., 1999). The lack of change in nociceptin mRNA (Dassesse et al., 2000) in the striatum of −/− mice supports the lack of changes in ORL1 binding observed here in the brains of −/− mice of the same strain.
In contrast to the brain, significant changes in δ and κ receptors were observed in the spinal cord of −/− mice, with no change in μ and ORL1 binding. To investigate whether the small changes in binding had any behavioral relevance, μ-, δ-, and κ-mediated antinociception assays were performed. Deltorphin-I and CI-977 were used to enable direct correlation with the results from the binding studies. Morphine was used as a prototypic μ agonist and because the μ receptor is its primary target (Matthes et al., 1996). Tail immersion was used as an analgesic test to investigate changes in spinal rather than supraspinal antinociception, because this assay is classically considered to reflect the actions of drugs on pain transmission at spinal sites (DeLander et al., 1992). A temperature of 55°C was used for testing morphine antinociception because it has been shown that no supraspinal circuits are involved at that temperature (Ossipov et al., 1995); 52°C was also used to test deltorphin-I- and CI-977-mediated antinociception in common with previous studies (Millan, 1989; Crook et al., 1993; Kitchen et al., 1994, 1995; Matthes et al., 1998) and because these drugs have limited or no antinociceptive activity at higher intensity thermal stimuli.
In accordance with the autoradiography in the spinal cord, we showed a decrease and increase in δ- and κ-mediated antinociception, respectively, and no change in μ-mediated antinociception in A2A receptor knock-out mice. This strongly suggests that even relatively small changes in the receptor density in −/− mice result in significant changes in the antinociceptive responses. In wild-type mice, the dose–response curve to deltorphin-I was bell shaped. It is not clear why this occurs, but we have observed a similar profile with this peptide in neonatal rats (our unpublished observations).
The possibility that the changes in δ and κ receptors are caused by lack of spinal A2A receptors in −/− mice can be ruled out, because we have been unable to detect any A2A receptors in the spinal cord of wild-type mice. We have also shown the absence of A2Areceptors in spinal cord of another strain of mice (Bailey et al., 2002). This is at odds with the observation of DeLander and Keil (1994)that antinociception induced by intrathecal coadministration of the selective A2A agonist CGS 21680 and μ-, δ-, or κ-selective opioid agonists showed an additive, synergistic, or multiplicative interaction between them. However, the issue as to whether CGS21680 actually induces antinociception by acting on A2A receptors is debatable because significantly greater doses of CGS21680, as compared with a selective A1receptor agonist, were required to induce antinociception. Thus it is likely that the opioid–A2A receptor agonist interactions observed in that study were caused by interactions at an A1 receptor at which CGS21680 has lesser potency (DeLander and Keil, 1994). However, expression of the A2A receptor gene has been reported in the dorsal root ganglia in mouse (Kaelin-Lang et al., 1998), and A2A receptors are present on sensory nerve terminals in the periphery. It has been shown that A2A receptors play a role in peripheral regulation of pain. Local administration of a selective A2A receptor agonist, CGS21680, has been reported to produce mechanical hyperalgesia (Khasar et al., 1995) and enhance pain responses in the low-concentration formalin model in rat (Doak and Sawynok, 1995). That action has been proposed to result from the stimulation of adenylate cyclase resulting in an increase in cAMP levels in the sensory nerve terminal (Taiwo and Levine, 1991; Khasar et al., 1995). As a result, the higher nociceptive threshold observed in A2A knock-out mice has been suggested to be attributable to the peripheral lack of A2Areceptors (Ledent et al., 1997). Accordingly, the changes in δ and κ opioid receptor-mediated antinociception and binding in the superficial layers of the spinal cord of −/− mice might be the result of the lack of A2A receptors in sensory nerve terminals. Activation of μ, δ, and κ opioid receptors located on the synaptic terminals of primary afferent fibers has been proposed as the basis for the depressive effects of opioid agonists on pronociceptive information coming from the peripheral nerve terminals (Besson and Chaouch, 1987). Thus it seems possible that the changes in δ and κ opioid-mediated antinociception and binding in A2A knock-out mice are caused by changes occurring in peripheral pain transmission as a consequence of lack of A2A receptors at sensory terminals. Because A2A receptors are strictly localized solely in the striatum of mouse brain and are not expressed in any major pain circuitry, it is highly unlikely that the lack of supraspinal A2A receptors could be the cause of the changes in the spinal opioid receptors and opioid-mediated antinociception in −/− animals. Why the changes in δ and κ receptor-mediated antinociception and binding are in opposition and why no change in μ receptor-mediated antinociception and binding was observed in −/− mice are not clear. However, it implies the presence of different spinal mechanisms of opioid receptor subtypes in modulating peripheral pain. Nonetheless, this study strongly suggests an interaction between peripheral A2A receptors and spinal δ and κ receptors but not μ receptors in regulating pain.
To check whether the changes observed in the opioid-mediated antinociception experiments were actually caused by opioid-mediated antinociception and not changes in basal pain sensitivity in −/− mice, a comparison of basal nociceptive latencies was performed between genotypes. The significant increase in tail withdrawal latency in −/− mice when they are exposed to 55°C water, the temperature used for morphine-mediated antinociception, is in agreement with the findings ofLedent et al. (1997) and the suggestion that peripheral A2A receptors enhance pain. However, it is known that blood flow influences nociceptive thresholds. A2A receptor knock-out mice have been shown to have high blood pressure (Ledent et al., 1997), which could reduce the skin temperature of the tail (Coupar and Tran, 2002) and lead to apparent antinociception (Hole and Tjolsen, 1993). Indeed, no significant change in basal nociceptive threshold was observed when +/+ and −/− mice were tested with the tail pressure test, a nociceptive response that would not be influenced by blood flow. In any case, the differences in δ-and κ-mediated antinociception shown in this paper could not be attributed to differences in blood flow or tail temperature because there was no significant difference in basal tail immersion latencies between genotypes when tested at 52°C. The lack of significant difference in nociceptive threshold between genotypes in the response to a 52°C stimulus could be caused by the reduced importance of peripheral A2A receptors at the lower temperature or to the involvement of supraspinal circuits, which are absent when the stimulus is 55°C (Ossipov et al., 1995).
In conclusion, deletion of the A2A receptor gene causes changes in δ and κ receptor-mediated antinociception and binding in the spinal cord but not in the brain of mutant mice. This supports a functional interaction between the spinal opioid and the peripheral adenosine system in control of pain pathways.
Footnotes
This study was supported by a University of Surrey Research Scholarship and the Wellcome Trust (057088).
Correspondence should be addressed to Prof. Ian Kitchen, Pharmacology Group School of Biomedical and Life Sciences, University of Surrey, Guildford, Surrey, GU2 7XH UK. E-mail:I.Kitchen@surrey.ac.uk.
This study was supported by a University of Surrey Research Scholarship and the Wellcome Trust (057088).
Correspondence should be addressed to Prof. Ian Kitchen, Pharmacology Group School of Biomedical and Life Sciences, University of Surrey, Guildford, Surrey, GU2 7XH UK. E-mail:I.Kitchen@surrey.ac.uk.
REFERENCES
- 1.Bailey A, Matthes H, Kieffer B, Slowe S, Hourani SMO, Kitchen I. Quantitative autoradiography of adenosine receptors and NBTI-sensitive adenosine transporters in the brains and spinal cords of mice deficient in the μ-opioid receptor gene. Brain Res. 2002;943:68–79. doi: 10.1016/s0006-8993(02)02536-2. [DOI] [PubMed] [Google Scholar]
- 2.Besse D, Lombard MC, Zajac JM, Roques BP, Besson A. Pre- and postsynaptic distribution of μ, δ and κ opioid receptors in the superficial layers of the cervical dorsal horn of the rat spinal cord. Brain Res. 1990;521:15–22. doi: 10.1016/0006-8993(90)91519-m. [DOI] [PubMed] [Google Scholar]
- 3.Besse D, Lombard MC, Besson JM. Autoradiographic distribution of mu, delta and kappa opioid binding sites in the superficial dorsal horn, over the rostrocaudal axis of the rat spinal cord. Brain Res. 1991;548:287–291. doi: 10.1016/0006-8993(91)91134-m. [DOI] [PubMed] [Google Scholar]
- 4.Besson A, Chaouch A. Peripheral and spinal mechanisms of nociception. Physiol Rev. 1987;67:67–186. doi: 10.1152/physrev.1987.67.1.67. [DOI] [PubMed] [Google Scholar]
- 5.Cahill CM, White TD, Sawynok J. Spinal opioid receptors and adenosine release: neurochemical and behavioural characterization of opioid subtypes. J Pharmacol Exp Ther. 1995;275:84–93. [PubMed] [Google Scholar]
- 6.Cahill CM, White TD, Sawynok J. Synergy between mu/delta-opioid receptors mediates adenosine release from spinal cord synaptosomes. Eur J Pharmacol. 1996;298:45–49. doi: 10.1016/0014-2999(95)00775-x. [DOI] [PubMed] [Google Scholar]
- 7.Chen J-F, Huang Z, Ma J, Zhu J, Moratella R, Standaert D, Moskowitz MA, Fink JS, Schwarzschild MA. A2A adenosine receptor deficiency attenuates brain injury induced by transient focal ischemia in mice. J Neurosci. 1999;19:9192–9200. doi: 10.1523/JNEUROSCI.19-21-09192.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clarke S, Chen Z, Hsu M-S, Pintar JE, Hill R, Kitchen I. Quantitative autoradiographic mapping of the ORL1, μ-, δ- and κ1-receptors in the brains of knockout mice lacking the ORL1 receptor gene. Brain Res. 2001;906:13–24. doi: 10.1016/s0006-8993(01)02531-8. [DOI] [PubMed] [Google Scholar]
- 9.Coupar IM, Tran BLT. Effects of adenosine agonists on consumptive behaviour and body temperature. J Pharm Pharmacol. 2002;54:289–294. doi: 10.1211/0022357021778330. [DOI] [PubMed] [Google Scholar]
- 10.Crook TJ, Kitchen I, Hill R. Differential antagonism of the antinociceptive effects of selective δ-opioid receptor agonists by the δ-antagonist naltriben in post-weanling rats. Pharmacol Commun. 1993;3:351–356. doi: 10.1111/j.1476-5381.1992.tb12785.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dassesse D, Ledent C, Meunier JC, Parmentier M, Schiffmann SN. Regulation of nociceptin mRNA expression in the septum by dopamine and adenosine systems. NeuroReport. 2000;11:3243–3246. doi: 10.1097/00001756-200009280-00038. [DOI] [PubMed] [Google Scholar]
- 12.DeLander GE, Keil GJ. Antinociception induced by intrathecal coadministration of selective adenosine receptor and selective opioid agonists in mice. J Pharmacol Exp Ther. 1994;268:943–951. [PubMed] [Google Scholar]
- 13.DeLander GE, Mosberg HI, Porreca F. Involvement of adenosine in antinociception produced by spinal or supraspinal receptor-selective opioid agonists: dissociation from gastrointestinal effects in mice. J Pharmacol Exp Ther. 1992;263:1097–1104. [PubMed] [Google Scholar]
- 14.De Montis MG. Decreased adenosine A2 receptor function in morphine dependent rats. Pharmacol Res. 1992;25:232–233. [Google Scholar]
- 15.Doak GJ, Sawynok J. Complex role of peripheral adenosine in the genesis of the response to subcutaneous formalin in the rat. Eur J Pharmacol. 1995;281:311–318. doi: 10.1016/0014-2999(95)00257-l. [DOI] [PubMed] [Google Scholar]
- 16.Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Academic; San Diego: 1997. [Google Scholar]
- 17.Fredholm BB, Vernet L. Morphine increases depolarization induced purine release from rat cortical slices. Acta Physiol Scand. 1978;104:502–504. doi: 10.1111/j.1748-1716.1978.tb06308.x. [DOI] [PubMed] [Google Scholar]
- 18.Hole K, Tjolsen A. The tail-flick and formalin tests in rodents: changes in skin temperature as a confounding factor. Pain. 1993;53:247–254. doi: 10.1016/0304-3959(93)90220-J. [DOI] [PubMed] [Google Scholar]
- 19.Janssen PAJ, Niemeggers CJE, Dony JGH. The inhibitory effect of fentanyl and other morphine-like analgesics on the warm-water induced tail withdrawal reflex. Arzneimittelforschung. 1963;13:502–504. [PubMed] [Google Scholar]
- 20.Jenck F, Moreau JL, Martin JR, Kilpatrick GJ, Reinscheid RK, Monsma FJJ, Nothacker HP, Civelli O. Orphanin FQ acts as an anxiolytic to attenuate behavioral responses to stress. Proc Natl Acad Sci USA. 1997;94:14854–14858. doi: 10.1073/pnas.94.26.14854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaelin-Lang A, Lauterburg T, Burgunder J-M. Expression of adenosine A2a receptor gene in rat dorsal root and autonomic ganglia. Neurosci Lett. 1998;246:21–24. doi: 10.1016/s0304-3940(98)00216-x. [DOI] [PubMed] [Google Scholar]
- 22.Kaplan GB, Leite-Morris KA, Sears MT. Alterations of adenosine A1 receptors in morphine dependence. Brain Res. 1994;657:347–350. doi: 10.1016/0006-8993(94)90990-3. [DOI] [PubMed] [Google Scholar]
- 23.Khasar SG, Wang J-F, Taiwo YO, Heller PH, Green PG, Levine JD. Mu-opioid agonist enhancement of prostaglandin-induced hyperalgesia in the rat: a G-protein βγ subunit-mediated effect? Neuroscience. 1995;67:189–195. doi: 10.1016/0306-4522(94)00632-f. [DOI] [PubMed] [Google Scholar]
- 24.Kitchen I. Modification of an analgesy meter for paw-pressure antinociceptive testing in neonatal rats. J Pharmacol Methods. 1984;12:255–258. doi: 10.1016/0160-5402(84)90011-1. [DOI] [PubMed] [Google Scholar]
- 25.Kitchen I, Crook TJ, Muhammad BY, Hill R. Evidence that weaning stimulates the developmental expression of a δ-opioid receptor subtype in the rat. Dev Brain Res. 1994;78:147–150. doi: 10.1016/0165-3806(94)90020-5. [DOI] [PubMed] [Google Scholar]
- 26.Kitchen I, Kelly MDW, DeCabo C. κ- and δ-opioid receptor mediated antinociception in preweanling rats. Analgesia. 1995;1:512–515. [Google Scholar]
- 27.Kitchen I, Slowe SJ, Matthes HW, Kieffer B. Quantitative autoradiographic mapping of μ-, δ- and κ-opioid receptors in knockout mice lacking the μ-opioid receptor gene. Brain Res. 1997;778:73–88. doi: 10.1016/s0006-8993(97)00988-8. [DOI] [PubMed] [Google Scholar]
- 28.Ledent C, Vaugeois JM, Schiffmann SN, Pedrazzini T, El Yacoubi M, Vanderhaeghen JJ, Costentin J, Heath JK, Vassart G, Parmentier M. Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2a receptor. Nature. 1997;388:674–678. doi: 10.1038/41771. [DOI] [PubMed] [Google Scholar]
- 29.Lindskog M, Svenningsson P, Fredholm BB, Greegard P, Fisone G. μ- and δ-opioid receptor agonists inhibit DARPP-32 phosphorylation in distinct populations of striatal projection neurons. Eur J Pharmacol. 1999;11:2182–2186. doi: 10.1046/j.1460-9568.1999.00597.x. [DOI] [PubMed] [Google Scholar]
- 30.Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, Befort K, Dierich A, Le Meur M, Dolle P, Tzavara E, Hanoune J, Roques BP, Kieffer BL. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the μ-opioid-receptor gene. Nature. 1996;383:819–823. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- 31.Matthes HW, Smadja C, Valverde O, Foutz AS, Boudinot E, Denavit-Saubié M, Vonesch J-L, Severini C, Negri L, Roques BP, Maldonado R, Kieffer B. Activity of the δ-opioid receptor is partially reduced while activity of the κ-receptor is maintained in mutant mice lacking the μ-receptor. J Neurosci. 1998;18:7285–7295. doi: 10.1523/JNEUROSCI.18-18-07285.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Millan MJ. Kappa-opioid receptor-mediated antinociception in the rat. I. Comparative actions of mu- and kappa-opioids against noxious thermal, pressure and electrical stimuli. J Pharmacol Exp Ther. 1989;251:334–341. [PubMed] [Google Scholar]
- 33.Mogil JS, Grisel JE, Reinscheid RK, Civelli O, Belknap JP, Grandy DK. Orphanin FQ is a functional anti-opioid peptide. Neuroscience. 1996;75:333–337. doi: 10.1016/0306-4522(96)00338-7. [DOI] [PubMed] [Google Scholar]
- 34.Mogil JS, Grisel JE, Hayward MD, Bales JR, Rubinstein M, Belknap JK, Low MJ. Disparate spinal and supraspinal opioid antinociceptive responses in beta-endorphin-deficient mice. Neuroscience. 2000;101:709–717. doi: 10.1016/s0306-4522(00)00422-x. [DOI] [PubMed] [Google Scholar]
- 35.Narita M, Mizoguchi H, Oji DE, Dun NJ, Hwang BH, Nagase H, Tseng LF. Identification of the G-protein-coupled ORL1 receptor in the mouse spinal cord by [35S]-GTPγS binding and immunohistochemistry. Br J Pharmacol. 1999;128:1300–1306. doi: 10.1038/sj.bjp.0702907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Neal CR, Jr, Mansour A, Reinscheid R, Nothacker HP, Civelli O, Akil H, Watson SJ., Jr Opioid receptor-like (ORL1) receptor distribution in the rat central nervous system: comparison of ORL1 receptor mRNA expression with (125)I-[(14)Tyr]-orphanin FQ binding. J Comp Neurol. 1999;412:563–605. [PubMed] [Google Scholar]
- 37.Ossipov MH, Kovelowski CJ, Nichols ML, Hruby VJ, Porreca F. Characterization of supraspinal antinociceptive actions of opioid delta agonists in the rat. Pain. 1995;62:287–293. doi: 10.1016/0304-3959(94)00231-3. [DOI] [PubMed] [Google Scholar]
- 38.Paxinos G, Watson C. The rat brain in stereotaxic coordinates, Ed 2. Academic; San Diego: 1986. [DOI] [PubMed] [Google Scholar]
- 39.Phillis JW, Jiang ZG, Chelack BJ, Wu PH. Morphine enhances adenosine release from the in vivo rat cerebral cortex. Eur J Pharmacol. 1980;65:97–100. doi: 10.1016/0014-2999(80)90215-0. [DOI] [PubMed] [Google Scholar]
- 40.Sawynok J. Adenosine receptor activation and nociception. Eur J Pharmacol. 1998;347:1–11. doi: 10.1016/s0014-2999(97)01605-1. [DOI] [PubMed] [Google Scholar]
- 41.Sawynok J, Sweeney MI, White TD. Adenosine release may mediate spinal analgesia by morphine. Trends Pharmacol Sci. 1989;10:186–189. doi: 10.1016/0165-6147(89)90235-6. [DOI] [PubMed] [Google Scholar]
- 42.Slowe SJ, Simonin F, Kieffer B, Kitchen I. Quantitative autoradiography of μ-, δ- and κ1 opioid receptors in κ-opioid receptor knockout mice. Brain Res. 1999;818:335–345. doi: 10.1016/s0006-8993(98)01201-3. [DOI] [PubMed] [Google Scholar]
- 43.Stone TW. The effects of morphine and methionine-enkephalin on the release of purines from cerebral cortex slices of rats and mice. Br J Pharmacol. 1981;74:171–176. doi: 10.1111/j.1476-5381.1981.tb09970.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Svenningsson P, Le Moine C, Fisone G, Fredholm BB. Distribution, biochemistry and function of striatal adenosine A2A receptors. Prog Neurobiol. 1999;59:355–396. doi: 10.1016/s0301-0082(99)00011-8. [DOI] [PubMed] [Google Scholar]
- 45.Sweeney MI, White TD, Jhamandas KH, Sawynok J. Morphine releases endogenous adenosine from the spinal cord in vivo. Eur J Pharmacol. 1987;141:169–170. doi: 10.1016/0014-2999(87)90428-6. [DOI] [PubMed] [Google Scholar]
- 46.Sweeney MI, White TD, Sawynok J. Morphine, capsaicin and K+ release purines from capsaicin-sensitive primary afferent nerve terminals in the spinal cord. J Pharmacol Exp Ther. 1989;248:447–454. [PubMed] [Google Scholar]
- 47.Taiwo YO, Levine JD. Further confirmation of the role of adenyl cyclase and of cAMP-dependent protein kinase in primary afferent hyperalgesia. Neuroscience. 1991;44:131–135. doi: 10.1016/0306-4522(91)90255-m. [DOI] [PubMed] [Google Scholar]