

Abstract
Calmodulin (CaM) was identified as a KCNQ2 and KCNQ3 potassium channel-binding protein, using a yeast two-hybrid screen. CaM is tethered constitutively to the channel, in the absence or presence of Ca2+, in transfected cells and also coimmunoprecipitates with KCNQ2/3 from mouse brain. The structural elements critical for CaM binding to KCNQ2 lie in two conserved motifs in the proximal half of the channel C-terminal domain. Truncations and point mutations in these two motifs disrupt the interaction. The first CaM-binding motif has a sequence that conforms partially to the consensus IQ motif, but both wild-type CaM and a Ca2+-insensitive CaM mutant bind to KCNQ2. The voltage-dependent activation of the KCNQ2/3 channel also shows no Ca2+ sensitivity, nor is it affected by overexpression of the Ca2+-insensitive CaM mutant. On the other hand, KCNQ2 mutants deficient in CaM binding are unable to generate detectable currents when coexpressed with KCNQ3 in CHO cells, although they are expressed and targeted to the cell membrane and retain the ability to assemble with KCNQ3. A fusion protein containing both of the KCNQ2 CaM-binding motifs competes with the full-length KCNQ2 channel for CaM binding and decreases KCNQ2/3 current density in CHO cells. The correlation of CaM binding with channel function suggests that CaM is an auxiliary subunit of the KCNQ2/3 channel.
Keywords: calmodulin, KCNQ channels, M current, IQ motif, channel modulation, auxiliary subunit, Ca2+-independent interaction
KCNQ2 and KCNQ3 are two members of the KCNQ potassium channel family that are expressed exclusively in the nervous system. In addition to forming functional homomeric channels, KCNQ2 and KCNQ3 subunits coassemble to form a heteromeric channel that underlies the neuronal M current (Wang et al., 1998). Consistent with the proposal that the M current plays a critical role in controlling neuronal excitability, mutations in KCNQ2 and KCNQ3 genes have been linked to an autosomal dominant human epilepsy (Biervert et al., 1998;Charlier et al., 1998; Singh et al., 1998). The M current is modulated richly by a variety of receptor systems via poorly understood intracellular signaling mechanisms (Brown and Adams, 1980; Marrion, 1997; Selyanko et al., 2000), and KCNQ2/3 channel activity also is strongly suppressed by muscarinic agonists in both neurons and heterologous cells (Selyanko et al., 2000; Shapiro et al., 2000).
The recent cloning of five mammalian KCNQ genes establishes a voltage-dependent potassium (Kv) channel family evolutionarily distinct from other Kv channels. Most notably, KCNQ channels possess a C-terminal domain after the end of the sixth transmembrane domain, which is much longer than those of most other Kv channels (Kubisch et al., 1999; Lerche et al., 2000; Schroeder et al., 2000). Although the function of the C-terminal domain currently is unknown, it appears to be indispensable for channel function because several loss-of-function KCNQ2 mutations identified in epileptic patients involve truncations or alterations in this region of the channel (Biervert et al., 1998; Singh et al., 1998; Lerche et al., 1999). KCNQ2 splice variants missing an intact C-terminal tail also fail to give rise to measurable current when expressed in heterologous systems (Nakamura et al., 1998; Smith et al., 2001). One possibility is that the long C-terminal tail domain provides the sites where other signaling proteins interact with the channel and modulate channel activity, as has been shown for KCNQ1. This cardiac channel interacts with an adaptor protein via a C-terminal domain leucine zipper motif, thus recruiting protein kinase A (PKA) and protein phosphatase 1 into a signaling complex that is required for β-adrenergic receptor modulation of the channel (Marx et al., 2002). KCNQ2 and KCNQ3 may interact with cytoskeleton, because they are found in brain in a Triton X-100 insoluble protein complex along with tubulin and a PKA subunit (Cooper et al., 2000). There is also evidence that a single membrane-spanning protein, KCNE2, associates with these two channels (Tinel et al., 2000).
The universal cellular calcium sensor calmodulin (CaM) is a small protein with four EF-hand-type Ca2+-binding sites. Evidence that ion channels are among the numerous target proteins regulated by CaM binding has been accumulating in recent years. In a number of Ca2+-permeant channels CaM was suggested to mediate negative feedback processes that regulate Ca2+ entry into the cell (Levitan, 1999;Saimi and Kung, 2002). CaM was shown to be tethered constitutively to two types of calcium-activated potassium channels [small (SK) and intermediate (IK) conductance] and to confer calcium dependence to these channels (Xia et al., 1998; Fanger et al., 1999). For a voltage-dependent potassium channel, human ether à go-go (EAG), CaM was found to bind the channel in a Ca2+-dependent manner and inhibit its activity (Schonherr et al., 2000).
In this study we describe and characterize the interaction of CaM with KCNQ2/3 and identify two conserved CaM-binding sites in the C-terminal domain of the channel protein. Via mutagenesis and competition experiments we show that the Ca2+-sensing function of CaM is not necessary for it to bind to the channel. Instead, the Ca2+-independent interaction itself seems to be essential for channel function.
MATERIALS AND METHODS
Yeast two-hybrid screen and interaction assay. A yeast two-hybrid screen was performed according to a standard protocol (Schopperle et al., 1998). The coding sequence of the last 550 amino acids of the mouse KCNQ2 channel and that of the last 512 amino acids of the mouse KCNQ3 channel were cloned into the bait vector, pEG202. The baits were screened against a mouse brain cDNA library subcloned into the prey vector, pJG45 (Matchmaker cDNA library, Clontech, Palo Alto, CA). Approximately 3,400,000 yeast cotransformants were screened for KCNQ2-interacting clones and ∼1,000,000 for KCNQ3 interactors. True positive clones were selected on plates lacking leucine, tryptophan, histidine, and uracil and were assayed for β-galactosidase activity on plates containing X-gal. Library plasmids of the interactor clones were isolated from yeast cells, and the sequences of their library inserts were determined by DNA sequencing.
For the interaction assay the indicated KCNQ2 C-terminal-fragment coding sequences were subcloned into the bait vector, pEG202, as fusions with the LexA DNA-binding domain. CaM was fused to the B42 transcription activation domain in the prey vector pJG45. Positive interactions were identified as blue colonies grown on the selection plates (medium lacking leucine, tryptophan, histidine, and uracil) containing X-gal on the third day of plating.
DNA constructs and mutagenesis. Mouse KCNQ2 was amplified by PCR from a mouse brain cDNA library (Marathon-ready cDNA library, Clontech) by using the following primers designed according to the sequence of rat KCNQ2 (GenBank accession number AF087453): 5′-CGCCGCCGGCACCATGGTGCAAAAGTCGCG-3′ and 5′-CTTCCTAGGCCCTGCCCAAGCCACATCTCCAAAGGG-3′. The PCR product was TA-cloned into the mammalian expression vector pCDNA3.1/V5/His-TOPO (Invitrogen, San Diego, CA) to tag the channel with a V5 epitope at the C terminus. The complete coding sequence of mouse KCNQ2 has been deposited to GenBank (accession number AF490773). A mouse KCNQ3 fragment including the C-terminal sequence was PCR-amplified from the same library with the following primers: 5′-GGCGCGCGGATCATGGCATTGGAGTTCCCG-3′ and 5′-AGTGGGCTTGTTGGAAGGGGTCCATATGGAATC-3′. The C-terminal sequence, which is 99.4% identical to that of the rat KCNQ3 (GenBank accession number AF091247) on the amino acid level, then was subcloned into pEG202 for the two-hybrid screen. Full-length rat KCNQ3 cDNA was the kind gift of Dr. David McKinnon (State University of New York at Stony Brook, Stony Brook, NY) (Wang et al., 1998). It also was tagged with a V5 epitope in pCDNA3.1/V5/His-TOPO. For KCNQ2/3 channel recordings, the mouse KCNQ2 and rat KCNQ3 cDNAs were cloned into pIRES2-EGFP, a bicistronic vector that allows for coexpression of both subunits in the same cell (Clontech). This was done by inserting KCNQ2 cDNA into the multicloning site of the vector and replacing enhanced green fluorescent protein (EGFP) cDNA in the original vector with KCNQ3 cDNA. CaM cDNA (the kind gift of Dr. John Lowenstein, Brandeis University, Waltham, MA) was subcloned into the two-hybrid prey vector pJG45 and a modified version of the mammalian expression vector pCDNA3 to give it a hemagglutinin (HA) epitope tag at the N terminus. For the expression of GST fusion proteins in mammalian cells, the indicated KCNQ2 sequences were cloned into the pEBG-1 vector (kindly provided by Dr. Joseph Avruch, Harvard Medical School, Boston, MA).
Site-directed mutagenesis was done via the Quik-Change system (Stratagene, La Jolla, CA) according to the manufacturer's instructions. The final cDNAs were sequenced through the entire coding region to ensure that there were no additional mutations introduced by PCR except at the desired sites (DNA sequencing facility, University of Pennsylvania, Philadelphia, PA).
Antibody preparation, coimmunoprecipitation, and Western blotting. The anti-mouse KCNQ2 antibody was generated by immunizing rabbits with a GST fusion protein containing the C-terminal 42 amino acids of the cloned channel. Specific anti-mKCNQ2 antibody was affinity-purified by Sepharose beads coupled with a maltose-binding protein (New England Biolabs, Beverly, MA) fusion containing the same mKCNQ2 C-terminal sequence. This antibody recognizes KCNQ3 protein as well. Other antibodies were purchased from Upstate Biotechnology (Lake Placid, NY; anti-CaM), Santa Cruz Biotechnology (Santa Cruz, CA; anti-GST, anti-β-tubulin), and Invitrogen (anti-V5, anti-Myc).
Coimmunoprecipitation and Western blotting were done as described previously (Wang et al., 1999). tsA201 cells were maintained in DMEM supplemented with 10% fetal bovine serum (FBS). At 48 hr after transfection with a calcium phosphate protocol, the cells were lysed in a buffer containing 1% CHAPS and (in mm) 20 Tris-HCl, pH 7.5, 10 EDTA, 120 NaCl, 50 KCl, 50 NaF, 2 DTT, and protease inhibitors (1 mm PMSF plus 1 μg/ml each aprotinin, leupeptin, and pepstatin A). Cleared lysates were incubated for 2 hr at 4°C with the appropriate antibodies, and the immunocomplexes were precipitated with protein A/G-agarose beads (Santa Cruz Biotechnology).
Proteins in the cell lysates or immunoprecipitates were separated on polyacrylamide gels and transferred to nitrocellulose membranes. The blots were blocked with 5% nonfat milk in TBST (10 mmTris-HCl, pH 7.5, 150 mm NaCl, 0.1% Tween 20) and then incubated with the appropriate primary antibodies in blocking buffer at 4°C overnight. After three washes with TBST the blots were incubated with horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG (Amersham Biosciences, Arlington Heights, IL) for 1 hr at room temperature. Proteins were visualized with an enhanced chemiluminescence detection system (Amersham Biosciences).
Membrane preparation from tissues and affinity purification of KCNQ channels. For crude membrane preparation the stripped mouse brains and mouse pancreas (Pel-Freez Biologicals, Rogers, AK) were ground to fine powder while being kept frozen in liquid nitrogen. The powders were homogenized further by using a glass homogenizer with five strokes in a buffer containing (in mm) 2.5 KCl, 250 sucrose, and 25 HEPES, pH 7.4, with complete protease inhibitors and 2 mm DTT. The homogenates were centrifuged at 1000 ×g for 10 min, and the supernatants were centrifuged again for 1 hr at 150,000 × g. The pellets were the crude membrane extracts.
Immunoaffinity purification of KCNQ2/3 channels from tissue extracts was done after a protocol by Cooper and colleagues (Cooper et al., 2000). Briefly, the membrane fractions were resuspended in a lysis buffer containing 1% Triton X-100 plus (in mm) 20 Tris-HCl, pH 7.5, 10 EDTA, 120 NaCl, 50 KCl, and 50 NaF with 2 DTT and complete protease inhibitors. The protein concentrations of the lysates were adjusted to 2–3 mg/ml (DC protein assay, Bio-Rad, Hercules, CA). Polyclonal KCNQ2/3 antibody, which was coupled covalently to Sepharose beads, was added to the lysates and incubated at 4°C overnight. Then the beads were recovered and washed five times by using 200 bead volumes of lysis buffer. Proteins bound to the beads were eluted with sample loading buffer.
Cell surface biotinylation. tsA 201 cells were washed three times with PBS at 2 d after transfection and then were incubated with 2 ml of 0.5 mg/ml Sulfo-NHS-LC-Biotin (Pierce, Rockford, IL) in PBS, pH 8.0, at room temperature for 30 min with gentle agitation. This reagent is cell membrane-impermeable because of its charged sulfate group, and it does not label an overexpressed intracellular protein, 14-3-3, used as a negative control for this assay in our laboratory. After the labeling reaction the reagent was removed by washing the cells three times with PBS. Cells were harvested and lysed in the lysis buffer as described previously. Then 20 μl of ImmunoPure immobilized streptavidin beads (Pierce) was added to each cell lysate to isolate the biotinylated proteins. Channels in total cell lysates and streptavidin precipitates were analyzed by Western blotting with the anti-V5 antibody. To confirm that the biotinylation reagent did not leak into the cell and label intracellular proteins, we stripped and reprobed the same blots with an anti-β-tubulin antibody (Joiner et al., 2001).
Emission fluorescence spectroscopy. Tryptophan fluorescence of the IQ-2 peptide (see Fig. 7A), alone or with increasing amounts of CaM, was measured with the PerkinElmer LS50B luminescence spectrometer (PerkinElmer Life Sciences, Emeryville, CA). The excitation wavelength was 295 nm, and emission spectra were recorded from 300 to 400 nm. Both excitation and emission bandwidths were 5 nm. The concentration of IQ-2 peptide was 1.6 μm in 30 mm Tris-HCl, pH 7.5, and 50 mm NaCl, with a 1 mmconcentration of either CaCl2 or EGTA. For the titration experiments a small volume of concentrated CaM was added to the buffer containing the IQ peptide. The final fluorescence data were obtained by subtracting the buffer fluorescence from those of the peptide and peptide plus CaM and correcting for changes in the sample volume caused by the CaM additions. The binding was described as: IQ + CaM ↔ IQ·CaM.
Fig. 7.
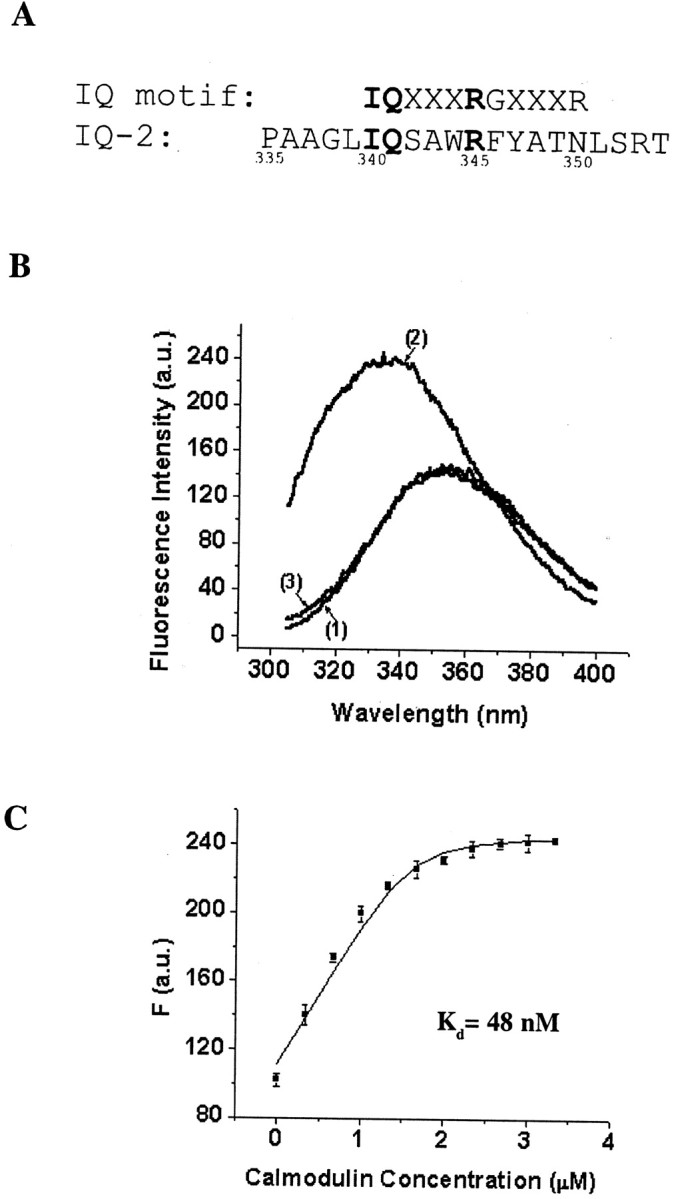
Fluorescence emission assay of the Ca2+-dependent interaction between an IQ-2 peptide and CaM. A, Alignment of the KCNQ2 IQ-2 sequence (bottom) with the canonical IQ motif (top), where X is any amino acid. Residues that are identical in the two sequences are highlighted inbold. B, Representative fluorescence emission spectra of 1.6 μm IQ-2 peptide alone in the presence of 1 mm Ca2+ or 1 mm EGTA (curve 1), 1.6 μm IQ-2 peptide with 3.33 μm CaM in the presence of 1 mm Ca2+ (curve 2), or 1 mm EGTA (curve 3). In the presence of 1 mm Ca2+ the addition of CaM blue-shifts the maximum emission wavelength and increases the fluorescence intensity (presented in arbitrary units, a.u.).C, Fluorescence titration of the IQ-2 peptide with CaM in the presence of 1 mm Ca2+. Fluorescence data from three independent experiments (mean ± SEM) are plotted against the CaM concentration. The line is the fit to a simple binding model: IQ-2 + CaM ↔ IQ-2·CaM. The dissociation constant, KD, is estimated at 48 nm. The quantum efficiencies of IQ-2 and IQ·CaM,FIQ, andFIQ·CaM are estimated to be 69.72 μm−1 and 154.65 μm−1, respectively.
The dissociation constant KD for this reaction is:
| Equation 1 |
where [IQ]0 is the initial concentration of the peptide, [CaM]0 is the initial concentration of CaM, and [IQ·CaM] is the concentration of the IQ·CaM complex, respectively. Because the observed fluorescence is attributed to a single tryptophan residue in the IQ-2 peptide, the fluorescence intensity F is:
| Equation 2 |
where FIQ andFIQ·CaM are the quantum efficiencies of IQ-2 and IQ·CaM. Experimental fluorescence intensity data given as a function of [CaM] were fit by combining equations 1 and 2, using a nonlinear least squares program (Origin, Microcal, Northampton, MA) to give the estimated value for KD,FIQ, andFIQ·CaM.
Electrophysiological recordings. Chinese hamster ovary (CHO) cells were maintained in Ham's F-12 medium with 10% FBS and transfected with Lipofectamine Plus (Invitrogen, San Diego, CA) according to the manufacturer's instructions. To identify transfected cells, we cotransfected the cells with channel constructs and EGFP; only cells with bright green fluorescence were used for recordings.
At 1–3 d after transfection the KCNQ2/3 currents were recorded in the whole-cell configuration with an Axopatch 200A amplifier (Axon Instruments, Union City, CA). Pipette electrodes were pulled from borosilicate glass and had resistances of 2–4 MΩ. The bath solution consisted of (in mm): 105 NaCl, 30 KCl, 2 MgCl2, 1.8 CaCl2, 10 HEPES, pH 7.2. The electrode solution consisted of (in mm): 10 NaCl, 130 KCl, 0.5 MgCl2, 5 EGTA, 10 HEPES, pH 7.2. For internal solutions containing 100 nm and 10 μm free Ca2+, appropriate amounts of CaCl2 were added to the internal solution according to calculations from Equal software (Biosoft, Cambridge, UK). Cells were held at −80 mV and depolarized to +80 mV in 10 mV steps for 1 sec. Data were acquired and analyzed with pClamp 7 software (Axon Instruments). To construct the activation curves, we fit relative tail currents (Irel) to the Boltzmann equation:
| Equation 3 |
where V0.5 is the half-maximum activation voltage and k is the slope factor. All results are shown as mean ± SEM; statistical significance was assessed by one-way ANOVA (Origin).
RESULTS
Two-hybrid screen that uses KCNQ2 and KCNQ3 C termini as bait
The cloned mouse KCNQ2 channel has a long C-terminal domain after the putative S6 transmembrane segment. This domain shares little homology with its counterparts in other Kv channels and contains no obvious structural motifs. To search for proteins interacting with KCNQ channels, we conducted a yeast two-hybrid screen by using the last 550 amino acids of the KCNQ2 channel as bait. We screened ∼3.4 × 106 yeast cotransformants and identified multiple positive interactor clones. All of these clones contain overlapping cDNA inserts of two mouse genes, M27844 and M19381, encoding the same protein CaM. All of the cDNA inserts contain the complete coding sequence for CaM but differ in the length of their 5′- and 3′-untranslated regions. In an independent screen with the entire C-terminal domain of the mouse KCNQ3 channel as bait, CaM also was identified as a positive interactor.
CaM associates with KCNQ channels both in transfected cells and in mouse brain
To confirm the results from the two-hybrid screen, we expressed full-length KCNQ channels and CaM in the tsA 201 mammalian cell line and tested the interaction by coimmunoprecipitation experiments. Overexpressed CaM, tagged with a HA epitope, is detected in KCNQ2 channel immunoprecipitates when both proteins are transfected together (Fig. 1A, bottom panel, lane 2), but not when CaM is transfected with the vector (Fig. 1A, bottom panel,lane 1). Endogenous CaM is also present in the channel immunoprecipitates, as shown by an 18 kDa band on the blot that is recognized by a specific monoclonal antibody against CaM, even when no exogenous CaM is introduced into the cells (data not shown). In the reciprocal experiment KCNQ2 is found in the CaM immunoprecipitates (data not shown). Using the same strategy, we also are able to confirm the interaction of CaM and the KCNQ3 channel in transfected cells (Fig.1A, bottom panel, lane 3). We fail to detect any interaction by coimmunoprecipitation when we mix together two lysates from cells transfected with KCNQ2 or CaM individually, suggesting that the interaction between KCNQ2 and CaM might occur early in the biogenesis pathway.
Fig. 1.
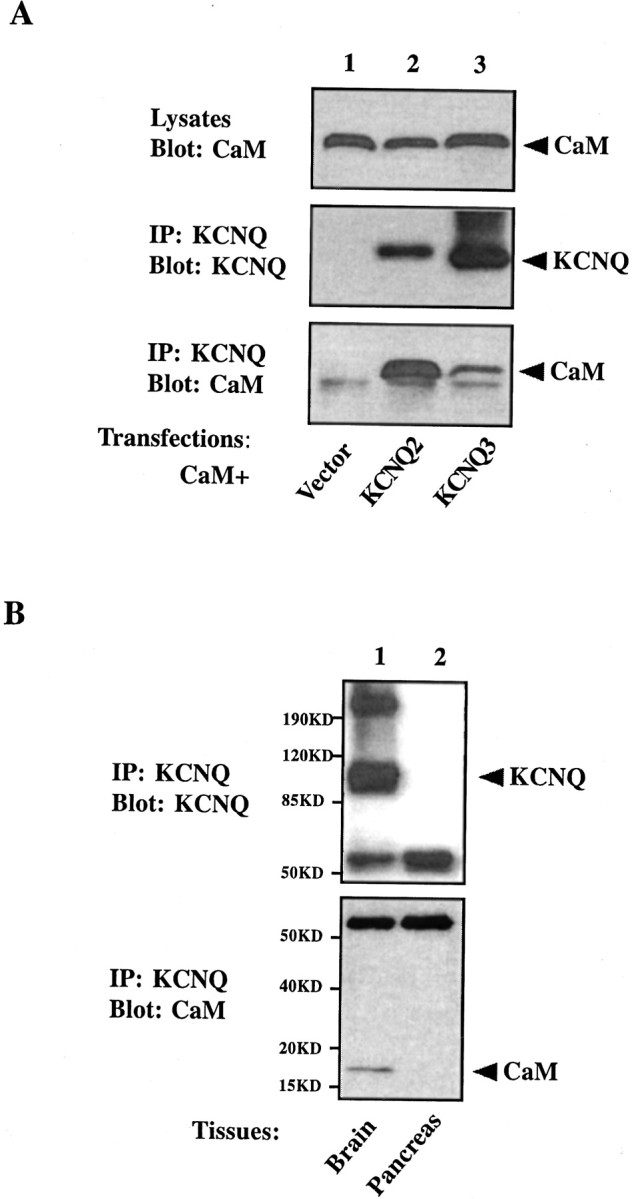
CaM coimmunoprecipitates with KCNQ2 and KCNQ3 channels. A, tsA 201 cells were transfected with HA-tagged CaM together with one of following constructs: vector (lane 1), V5-tagged KCNQ2 (lane 2), or V5-tagged KCNQ3 (lane 3). Cell lysates were probed for the expression of CaM with the anti-HA antibody (top panel). Channel immunoprecipitates were probed with the anti-V5 antibody to confirm the precipitation of channels (middle panel) and were probed with the anti-HA antibody to detect CaM pulled down with the channels (bottom panel). CaM is present in a KCNQ channel immunoprecipitate, but not in that from vector-transfected cells.B, CaM copurifies with KCNQ2 and KCNQ3 channels from mouse brain. KCNQ2 and KCNQ3 channels were immunopurified from the crude membrane fraction of mouse brain (lane 1), but not pancreas (lane 2), with anti-KCNQ2/3 antibody-coupled Sepharose beads (top panel). CaM is detected in the channel immunoprecipitates from brain, but not pancreas, when probed with a specific monoclonal antibody against CaM (bottom panel).
To look for the existence of such a KCNQ2/3–CaM complex in the native neuronal environment, we asked whether CaM copurifies with the channel when KCNQ2/3 is immunoprecipitated from mouse brain lysates. Consistent with a previous report, most KCNQ channels are found in a 1% Triton X-100 insoluble subcellular fraction (Cooper et al., 2000). When the channels are pulled down from this fraction with a polyclonal antibody that recognizes both KCNQ2 and KCNQ3 subunits, a 95 kDa protein band that is present in the brain membrane prep (data not shown) is now enriched in the immunoprecipitates (Fig. 1B,top panel, lane 1). This band is indeed KCNQ protein because it is absent in the membrane prep and immunoprecipitates prepared from a control tissue, mouse pancreas (Fig.1B, top panel, lane 2), which has been shown to lack KCNQ2/3 mRNA (Biervert et al., 1998; Schroeder et al., 1998). CaM immunoreactivity is detected in the channel immunoprecipitates from brain, but not pancreas (Fig.1B, bottom panel). Therefore, the interaction of KCNQ channels and CaM occurs not only in heterologous cells but also in mouse brain.
CaM is tethered constitutively to KCNQ channels
The yeast two-hybrid system was shown to detect only Ca2+-independent interactions between CaM and the SK2 channel (Keen et al., 1999), suggesting that the CaM-KCNQ interaction in yeast is also Ca2+-independent. We have two additional lines of evidence that suggest CaM is tethered to KCNQ channel tails constitutively. First, coimmunoprecipitation of CaM and KCNQ2 persists even when the cells are lysed with a buffer containing a 1 mm (data not shown) or 10 mm (Fig.2, bottom panel, lane 1) concentration of the Ca2+ chelator EGTA. Second, we made a Ca2+-insensitive CaM (Putkey et al., 1989; Geiser et al., 1991) by replacing the first aspartate residue in each of its four EF-hand Ca2+-binding motifs with an alanine and tested its ability to coimmunoprecipitate with KCNQ2. The mutant, CaM1234, is indeed unable to bind Ca2+, because it loses the Ca2+-dependent mobility shift characteristic of wild-type CaM on a protein gel (Fig. 2, top panel) (Geiser et al., 1991; Xia et al., 1998). This mutant still coimmunoprecipitates robustly with the KCNQ2 channel, although to a somewhat reduced extent when compared with wild-type CaM (Fig. 2,bottom panel). Clearly, CaM binds to KCNQ2 independent of its Ca2+-binding ability.
Fig. 2.

Both Ca2+-CaM and apo-CaM coimmunoprecipitate with KCNQ2. tsA 201 cells were transfected with V5-tagged KCNQ2 together with one of two HA-tagged CaM constructs: wild type (lanes 1, 2) or Ca2+-insensitive mutant (CaM1234; lanes 3, 4). Cell lysates were probed for the expression of CaMs with the anti-HA antibody (top panel). KCNQ2 was precipitated with a polyclonal antibody against KCNQ2/3 in the presence of either 10 mm EGTA (lanes 1, 3) or 1 mm Ca2+ (lanes 2, 4), as indicated on the top of the blots. Channel immunoprecipitates were eluted with sample loading buffer containing 2 mm EGTA (for precipitates prepared in the presence of 10 mm EGTA) or 5 mmCa2+ (for precipitates prepared in the presence of 1 mm Ca2+) and were used for Western blot. Blots were probed for the channel by using anti-V5 antibody (middle panel) and for CaMs by using anti-HA antibody (bottom panel). A doublet usually shows up in the channel blot (middle panel) with longer exposure. Both bands represent overexpressed KCNQ2 protein because they are absent in vector-transfected cells. CaM is detected in all immunoprecipitates. Ca2+-loaded CaM (lane 2) shows greater mobility than the apo-CaM (CaM1234and wild-type CaM in the presence of EGTA) on the blots.
Voltage-dependent activation of the KCNQ2/3 channel shows no Ca2+ sensitivity
Recent years have seen the rediscovery of CaM as a key regulator of ion channel function by virtue of its constitutive tethering to the channels and its well established function of Ca2+ sensing (Saimi and Kung, 2002). In those examples the tethered CaM mediates some Ca2+-sensitive property of the channel, because the Ca2+-insensitive CaM mutants can act as dominant negatives for these effects (Xia et al., 1998;Peterson et al., 1999; Zuhlke et al., 1999). There has long been controversy about whether the native M current is modulated by intracellular Ca2+, and the heteromeric KCNQ2/3 channel has not been reported to show Ca2+ dependence. We tested the voltage-dependent activation of KCNQ2/3 channels expressed in CHO cells, using whole-cell patch recording with a range of Ca2+ concentrations in the pipette. No significant difference was detected in the half-maximal activation voltage and slope factor for activation with 0 (5 mm EGTA), 100 nm, or 10 μm free intracellular Ca2+ (Table1). One possible explanation for the lack of effect is that there is insufficient endogenous CaM to interact with the overexpressed channel. This seems not to be the case, because coexpressing either wild-type CaM or CaM1234together with the channel has no effects on the voltage sensitivity of KCNQ2/3 (Table 1), although our biochemical data demonstrate that both of them bind to the channel tail (Fig. 2). The kinetics for activation and deactivation also remains the same under all conditions (data not shown).
Table 1.
Steady-state activation of the KCNQ2/3 channel under different recording conditions
| Transfections | Recording conditions | V0.5 (mV) | k(mV) | n |
|---|---|---|---|---|
| 5 mmEGTA | −16.70 ± 1.73 | 12.15 ± 0.62 | 12 | |
| IR-Q2Q3 | 100 nmCa2+ | −19.18 ± 3.32 | 13.36 ± 1.04 | 5 |
| 10 μmCa2+ | −17.80 ± 0.86 | 13.38 ± 0.42 | 12 | |
| 5 mmEGTA | −17.52 ± 1.94 | 13.56 ± 0.71 | 9 | |
| IR-Q2Q3 + CaM | 100 nmCa2+ | −19.56 ± 1.02 | 13.35 ± 0.61 | 7 |
| 10 μmCa2+ | −21.46 ± 0.75 | 12.35 ± 0.80 | 6 | |
| 5 mm EGTA | −18.62 ± 1.56 | 13.89 ± 0.66 | 7 | |
| IR-Q2Q3 + CaM1234 | 100 nmCa2+ | −24.48 ± 0.88* | 12.66 ± 0.46 | 7 |
| 10 μ mCa2+ | −21.84 ± 0.93 | 13.79 ± 0.79 | 10 |
IR-Q2Q3, a bicistronic vector expressing both KCNQ2 and KCNQ3 subunits, was transfected into CHO cells alone (top) or together with wild-type CaM (middle) or CaM1234 (bottom). Tail currents were recorded from transfected cells at three different intracellular Ca2+ concentrations. Data were fit to the Boltzmann equation. The half-maximal voltage for activation (V0.5), the slope factor (k), and the number of cells recorded for each condition (n) are listed. Overexpression of wild-type or mutant CaMs or varying intracellular Ca2+ concentration does not cause a marked change inV0.5 and k of the KCNQ2/3 channel. Cotransfection of CaM1234 slightly increasesV0.5 when recorded with 100 nM Ca2+on the inside (*p < 0.05). The values forV0.5 and k listed in the table agree with previous reports on KCNQ2/3 channels expressed in CHO cells (Selyanko et al., 2000; Tatulian et al., 2001).
Two binding sites are required for the constitutive tethering of CaM to KCNQ2
We adopted a truncation analysis strategy to identify binding domains for CaM in the KCNQ2 C-terminal tail. The original two-hybrid bait, consisting of amino acids 321–870, was shortened progressively from either its N or C terminus, and the resultant truncation mutants were tested against CaM in a two-hybrid interaction assay. Deletion of over one-half of the tail from the C-terminal end (amino acids 568–870) leaves the interaction intact (Fig.3). However, further truncation at position 535 completely abolishes the interaction, indicating that amino acids 536–567, which we call site 2, are important for the channel to bind to CaM (Fig. 3). In contrast, the N terminus of the tail tolerates little truncation. Removal of just 38 residues (amino acids 321–358) from the positive interactor, Q2CΔ31, takes away its ability to bind CaM (Fig. 3). This region, named site 1, begins immediately after the S6 transmembrane domain and includes an IQ-2 peptide sequence (see Fig. 7A). The truncation analysis shows that the two CaM-binding motifs, site 1 and site 2, are both necessary for KCNQ–CaM interaction, and removal of either one will disrupt the association. Furthermore, the fragment containing both sites, Q2CΔ31, also interacts with the Ca2+-insensitive CaM mutant CaM1234 in the two-hybrid system (data not shown), suggesting that it is the minimum sequence mediating the Ca2+-independent constitutive interaction of CaM and KCNQ2.
Fig. 3.

Truncation analysis reveals two sites in the KCNQ2 C-terminal domain that are necessary for CaM–KCNQ2 interaction. KCNQ2 C-terminal fragments (shown as boxes with amino acidnumbers) were tested against CaM in the two-hybrid system for LacZ reporter gene expression (positive interactions are indicated by plus signs). Further truncation from either end of Q2CΔ31 (amino acids 321–567) abolished the interaction. The locations of the two putative CaM-binding sites, site 1 (amino acids 321–358; hatched boxes) and site 2 (amino acids 536–567; filled boxes), are indicated in each fragment.
To confirm further the necessity and sufficiency of these two sites in mediating the interaction, we constructed a GST fusion protein containing these sites, GST-Q2CΔ31 (Fig.4A), and tested its ability to bind CaM by coimmunoprecipitation. Two other fusion proteins were constructed also (Fig. 4A): one is missing site 2 and the other is identical to GST-Q2CΔ31 except for a point mutation (R345E) in site 1 that disrupts the interaction (see below). Not surprisingly, GST-Q2CΔ31 is the only protein among the three that coimmunoprecipitates with CaM (Fig. 4B, bottom panel).
Fig. 4.

A GST fusion protein containing both binding sites coimmunoprecipitates with CaM. A, Schematic view of the fusion protein constructs. GST is shown as an oval. KCNQ2 C-terminal fragments are presented as lines with amino acid numbers. The locations of site 1 (hatched boxes) and site 2 (filled boxes) are indicated. The position of a mutation introduced in site 1, R345E, is indicated by an arrow.B, tsA 201 cells were transfected with HA-tagged CaM together with GST (lane 1), GST-Q2CΔ31 (lane 2), GST-Q2CΔ31(R345E) (lane 3), and GST-Q2CΔ4 (lane 4). Cell lysates were probed for the expression of CaM by using an anti-HA antibody (top panel). GST immunoprecipitates were probed for GST with anti-GST (middle panel) and for CaM with anti-HA (bottom panel). The fusion protein containing both sites, GST-Q2CΔ31, coimmunoprecipitates with CaM, whereas the one lacking site 2, GST-Q2CΔ4, does not. A single amino acid mutation (R345E) in site 1 also disrupts the interaction.
Mutations in site 1 and site 2 disrupt the association of CaM with KCNQ2
The interaction between KCNQ2 and CaM can be disrupted by point mutations in both site 1 and site 2. When a single positively charged residue in site 1 is changed to a negatively charged one (R345E), CaM no longer can be detected in the immunoprecipitates of the mutant channel (Fig. 5A, bottom panel, lane 2). Point mutation of another conserved residue in site 1, I340E, also eliminate the CaM-binding ability of the channel (data not shown). Additionally, binding is abolished when the charges of two conserved basic residues in site 2 are reversed (K553E/R554E) (Fig. 5A, bottom panel, lane 3).
Fig. 5.
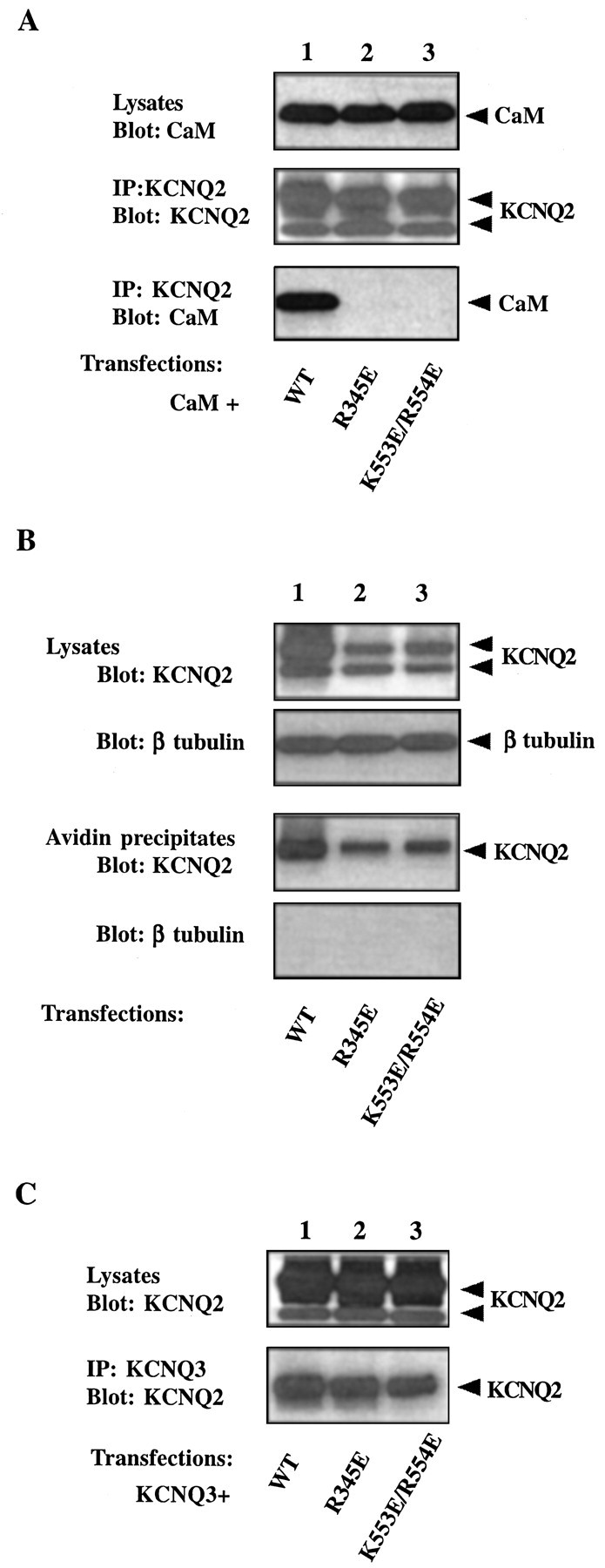
Properties of two KCNQ2 channels with mutations in CaM-binding sites. A, R345E and K553E/R554E KCNQ2 do not coimmunoprecipitate with CaM. HA-tagged CaM was coexpressed with one of the following V5-tagged KCNQ2 constructs in tsA 201 cells: wild type (lane 1), R345E (lane 2), or K553E/R554E (lane 3). Cell lysates were probed for the expression of CaM with anti-HA (top panel). KCNQ2 channel immunoprecipitates were probed for the channel with anti-V5 (middle panel) and for CaM with anti-HA (bottom panel). CaM is detected in immunoprecipitates of wild-type, but not mutant, channels. B, R345E and K553E/R554E KCNQ2 are expressed on the cell surface. tsA 201 cells were transfected with V5-tagged KCNQ2 channels. Cell surface proteins were biotinylated by a membrane-impermeable reagent and isolated by streptavidin beads. Channel immunoreactivity in the lysates (first panel) and in the streptavidin precipitates (third panel) was detected with the anti-V5 antibody. The proportions of mutant channels (lanes 2, 3) targeted to the surface are comparable with those of the wild-type KCNQ2 (lane 1). The biotinylation reagent did not label an intracellular protein, β-tubulin (secondand fourth panels), confirming its specificity for cell surface proteins. C, R345E and K553E/R554E KCNQ2 still coimmunoprecipitate with KCNQ3. C-Myc-tagged KCNQ3 channel was transfected together with one of the V5-tagged KCNQ2 channels into tsA 201 cells. The KCNQ3 channel was pulled down with anti-Myc antibody, and the lysates (top panel) and immunoprecipitates (bottom panel) were probed for KCNQ2 with anti-V5. Both wild-type (lane 1) and mutant KCNQ2 channels (lanes 2, 3) immunoprecipitate with KCNQ3. Note that in B and C thetop band of the KCNQ2 doublet in the cell lysates is enriched in the streptavidin or anti-KCNQ3 precipitates.
To summarize, our biochemical data demonstrate that CaM is tethered to the KCNQ2/3 channel in a Ca2+-independent manner via two binding motifs, site 1 and site 2, which are located in the proximal portion of the C-terminal domain of the channel. We then asked what the functional consequence of this constitutive interaction is.
Correlation between CaM binding and channel function
During our patch-clamp recordings we noticed that two KCNQ2 mutants deficient in CaM binding, R345E and K553E/R554E, do not give rise to detectable currents when coexpressed with KCNQ3 in CHO cells. These mutants are expressed at a somewhat reduced level compared with the wild-type channel (Fig. 5A, middle panel,B,C, top panels), but this difference in expression cannot account for the complete loss of channel activity. We tested two other possibilities that could explain a nonfunctional channel. We first asked whether mutant channels could make it to the cell membrane surface. We labeled the total membrane proteins with a membrane-impermeable biotinylation reagent and isolated membrane proteins with streptavidin beads. The ratio of channels on the membrane to those in the total lysates is not significantly different in wild-type and the two mutant channels (Fig. 5B), indicating that the membrane targeting of the mutants is not altered. We then tested whether the mutants can still coassemble with KCNQ3. When KCNQ3 is immunoprecipitated from the cotransfected cells, the mutant KCNQ2 proteins are also present in the precipitates, and quantification shows no marked difference from wild type (Fig. 5C). Thus the lack of channel activity cannot be explained by their inability to form heteromers. Despite the fact that some small quantitative differences do exist in the expression of wild-type and mutant channels, the correlation between the ability of a channel to bind CaM and its function can be extended to several other KCNQ2 mutants bearing mutations in one of the binding domains (Table2). In all cases the mutants are more or less normal in terms of their surface expression and their heteromerization with KCNQ3. All those that can bind CaM give easily detectable currents when coexpressed with KCNQ3, whereas those that cannot bind produce no current. Hence we hypothesize that CaM binding is necessary for a functional KCNQ2/3 channel.
Table 2.
Properties of KCNQ2 mutant channels
| KCNQ2 mutants | Interaction with CaM | KCNQ2/3 activity | Coassembly with KCNQ3 | Membrane localization |
|---|---|---|---|---|
| R345E | − | −(2/30) | + | + |
| I340E | − | −(0/21) | + | + |
| M566T/D567Y | + | +(15/17) | + | + |
| ΔDVMD | + | +(12/17) | + | + |
| A4 | + | +(4/4) | + | + |
| K555E/K557E | + | +(17/21) | + | + |
| K553E/R554E | − | −(2/27) | + | + |
| K553E | + | +(5/5) | + | + |
| R554E | + | +(6/6) | + | + |
Interaction with CaM and coassembly with KCNQ3 were assayed by coimmunoprecipitation. Membrane localization was assayed by surface biotinylation. KCNQ2/3 activity was measured from CHO cells cotransfected with the KCNQ2 mutants and KCNQ3 subunit cloned into the bicistronic pIRES vector. Numbers of cells that gave measurable currents, n, of the total numbers of cells recorded,N, are shown in parentheses (n/N). With wild-type KCNQ2, 130 of 130 cells exhibited currents. For all assays the positive results are indicated by a plus sign without specifying any quantitative differences between mutants. R345E and I340E are mutated in the CaM binding site 1, whereas the rest have mutations in site 2. A4 represents K553A/R554A/K555A/K557A. ΔDVMD has a 4-amino-acid deletion from D564 to D567.
A fusion protein that decreases CaM binding to KCNQ2 reduces channel activity
It is nearly impossible to find a CaM-free system to test this hypothesis, because removal of such an important molecule from any cell is generally lethal. We therefore sought a molecule that would reduce the degree of CaM binding to the KCNQ2/3 channel. Because less functional CaM–KCNQ would be available in the cell in the presence of this competitor, our hypothesis predicts it would decrease channel activity. GST-Q2CΔ31 could be such a candidate molecule, because it contains both CaM-binding motifs and associates with CaM in transfected cells (Fig. 4). GST-Q2CΔ31 indeed is capable of competing with the full-length KCNQ2 channel for binding to endogenous CaM, as shown by less CaM in KCNQ2 immunoprecipitates when GST-Q2CΔ31 is cotransfected into the cells (Fig.6A, top panel, lane 1). Consistent with our hypothesis, when GST-Q2CΔ31 is coexpressed with the KCNQ2/3 channel in CHO cells, it significantly reduces the current density (Fig. 6B,C) compared with two other fusion proteins that do not compete for CaM binding (Fig. 6A). These data support the hypothesis that the constitutive tethering of CaM to the KCNQ2/3 channel is essential for the channel function.
Fig. 6.
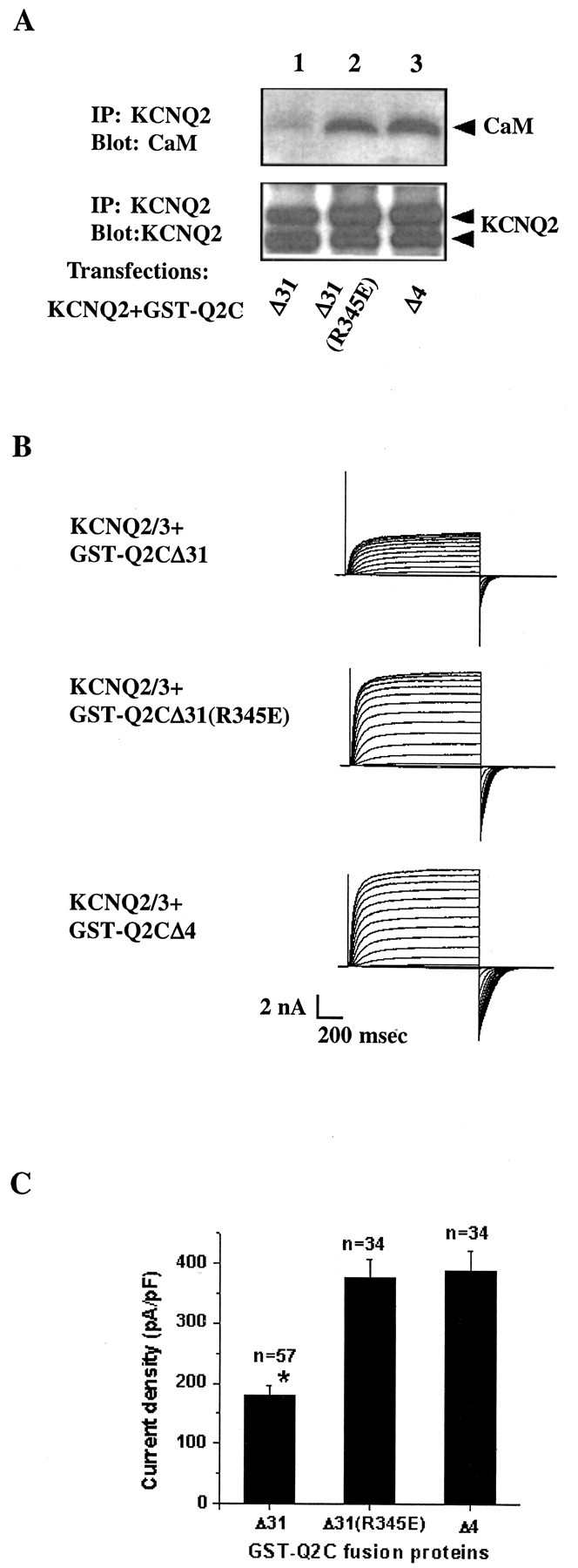
A two-site fusion protein decreases CaM binding to KCNQ2 and reduces channel activity. A, GST-Q2CΔ31 competes with the full-length KCNQ2 channel for binding to endogenous CaM. tsA 201 cells were transfected with V5-tagged KCNQ2 together with one of the GST fusion constructs in a 1:1.5 molar ratio: GST-Q2CΔ31 (lane 1), GST-Q2CΔ31(R345E) (lane 2), or GST-Q2CΔ4 (lane 3). KCNQ2 immunoprecipitates were probed for the channel with an anti-V5 antibody (bottom panel) and endogenous CaM with a monoclonal anti-CaM antibody (toppanel). In cells that were transfected with GST-Q2CΔ31 (lane 1), there is considerably less CaM in the KCNQ2 channel immunoprecipitates compared with cells in which the two other fusion proteins were transfected (lanes 2, 3). Similar results were obtained when the fusion proteins were cotransfected with both KCNQ2 and KCNQ3 subunits (data not shown). Note that these GST fusion proteins are expressed at comparable levels in tsA 201 cells (Fig. 4B, middle panel). B, Representative recording traces from three CHO cells cotransfected with IR-Q2Q3 and one of the GST fusion protein constructs in a 1:1.5 molar ratio. The membrane capacitances were comparable in these three cells. C, GST-Q2CΔ31 reduces the current density of the KCNQ2/3 channel in CHO cells. Whole-cell tail currents after a hyperpolarizing step from + 20 to −80 mV were recorded 2–3 d after transfection and normalized to membrane capacitance to give the current density in that cell. GST-Q2CΔ31 significantly reduces KCNQ2/3 current density; *p < 0.05.
A role for Ca2+?
Many studies have shown that CaM binds to a relatively small stretch of sequences on its target proteins, such as Baa (basic amphiphilic α) helices and IQ motifs (Rhoads and Frieldberg, 1997). A careful inspection of the C-terminal amino acid sequence of KCNQ2 reveals that amino acids 335–354 within site 1 resemble an IQ motif (Fig. 7A), which is loosely defined as IQxxxRGxxxR. Although it was proposed originally as a consensus sequence mediating Ca2+-independent binding, more and more IQ motifs have been found to bind CaM in a Ca2+-dependent manner, with varying Ca2+ sensitivities (Jurado et al., 1999). We thus used a 20-amino-acid synthetic peptide, IQ-2, corresponding in sequence to a portion of site 1 in KCNQ2 (Fig. 7A), in a gel mobility shift assay. In the presence of Ca2+ the preincubation of CaM and IQ-2 shifts the CaM band to a higher position on a SDS-PAGE gel, and this mobility shift does not happen in the absence of Ca2+ (data not shown), suggesting the formation of an IQ–Ca2+·CaM complex.
Because the IQ-2 peptide contains a single tryptophan residue (W344) whereas CaM has none, the intrinsic fluorescence of the peptide can be followed as a sensitive measure for any perturbation of the microenvironment around W344 because of the conformational changes caused by the CaM–peptide interaction (Keen et al., 1999). Adding CaM to peptide in the absence of Ca2+ (Fig.7B, curve 3) does not change the emission spectrum of the peptide, which peaks at 355 nm (Fig. 7B,curve 1). In the presence of Ca2+ the CaM addition blue-shifts the emission maximum to 338 nm and increases fluorescence intensity by a factor of 1.6 after the reaction reaches saturation (Fig.7B, curve 2). These results suggest that Ca2+-loaded CaM binds to the IQ-2 peptide and, in doing so, introduces W344 into a more hydrophobic environment. To quantify the binding reaction, we applied CaM titration to the peptide. Fitting the experimental results to a simple binding model in which IQ-2 binds to CaM with a 1:1 stoichiometry, we estimated the apparent dissociation constant at 48 nm (Fig.7C).
DISCUSSION
The cytoplasmic domains of potassium channels greatly influence the expression, membrane targeting, subcellular localization, and gating properties of the pore-forming subunit. One common mechanism by which they accomplish these diverse functions is to interact with other modulatory proteins (Holmes et al., 1996; Schopperle et al., 1998;Trimmer, 1998; Sheng and Kim, 1999). The recently cloned neuronal M current KCNQ2/3 channel has a C-terminal tail >500 amino acids in length, with functions that are yet to be identified. These may be especially important, as suggested by the observations that the tail domain is subject to extensive alternative splicing and is the site of several pathogenic mutations (Biervert et al., 1998; Nakamura et al., 1998; Singh et al., 1998; Pan et al., 2001; Smith et al., 2001). In this study we demonstrate that CaM is tethered to the KCNQ2 and KCNQ3 channels via two motifs in the proximal portion of the C-terminal domain, and the tethering is necessary for channel function. Intriguingly, these two motifs are conserved both within the mammalian KCNQ family and across species. While this paper was under review,Yus-Nájera et al. (2002) reported the binding of CaM to mammalian KCNQ1–5. Although these authors did not examine native tissue or the functional correlates of the binding interaction, their conclusions about CaM association with the C-terminal tail domain of KCNQ agree with ours.
The biochemical interaction described here for CaM and KCNQ channels displays similarities to what is known for CaM–SK interactions. Our results show that several conserved positively charged residues are important for the Ca2+-independent binding of CaM to KCNQ2. Similar results were obtained for the CaM binding to SK (Keen et al., 1999). More importantly, both cases feature a constitutive association and a Ca2+-dependent component of interaction. In SK2, amino acids 390–487 in the proximal portion of the C-terminal tail were identified as the CaM-binding domain (CaMBD) (Xia et al., 1998). Several different assays show that CaMBD binds to CaM in the absence or presence of Ca2+, whereas a subregion (amino acids 423–487) shows only Ca2+-dependent binding (Xia et al., 1998). We demonstrated by coimmunoprecipitation and two-hybrid assay that CaM is bound constitutively to KCNQ2 and KCNQ3 channels. The minimum KCNQ2 sequence necessary for CaM binding is a stretch of 247 amino acids (from 321 to 567) beginning immediately after the S6 transmembrane domain. In the yeast two-hybrid system CaM can still bind to this region even if all four of its Ca2+-binding sites are mutated, suggesting that it is the site where CaM tethers to the channel independently of Ca2+. However, there is an IQ motif (amino acids 335–354) within this minimum sequence that can bind CaM onlyin vitro in the presence Ca2+. CaM also has been shown to tether constitutively to some voltage-dependent calcium channels and binds to IQ motifs in those channels in the presence of Ca2+ (Peterson et al., 1999).
IQ motifs generally are predicted to be amphipathic α-helices that bind to their targets with different levels of Ca2+ dependence. Our fluorescence measurement shows that an IQ-2 peptide from KCNQ2 binds to CaM only in the presence of Ca2+, but the precise Ca2+ dependence of this binding was not investigated further. Of course, the Ca2+affinity of CaM in the CaM–peptide complex may not be the same as that in the context of the full-length channel protein, because the channel has additional sequences that also contribute to CaM binding. Therefore, it is hard to predict on the basis of our biochemical data whether the IQ-2 motif binds to CaM in response to a rise in intracellular Ca2+ and whether the Ca2+-dependent interaction we see in vitro is of any significance in terms of channel function. The recent x-ray crystal structure of an SK2 CaMBD–CaM complex provides a detailed view of both Ca2+-dependent and -independent interactions and suggests a model for the mechanistic coupling between Ca2+ binding to CaM and channel gating (Schumacher et al., 2001). The complete understanding of how CaM is bound to KCNQ channels may have to await the elucidation of a similar structure.
We tested whether KCNQ2 mutants that are deficient in CaM binding fail to produce measurable currents because they are unable to reach the cell surface. Because CaM seems to be associated with the KCNQ2 channel early in the biosynthetic pathway, it might be involved in the membrane trafficking process of the channel, as has been proposed for SK channels (Joiner et al., 2001). Our data argue against this possibility. The R345E and K553E/R554E mutant channels completely lose their abilities to interact with CaM, but we did not detect any marked difference in their surface expression compared with the wild-type channel. It is interesting in this context that a human KCNQ2 mutant that causes BFNC, KCNQ2 (1600ins5bp), truncates the protein at the far end of the CaM-binding site 2. This mutant was shown to have no surface expression in Xenopus oocytes, indicating that at least some domains responsible for membrane targeting are located downstream of site 2 (Schwake et al., 2000).
KCNQ channels often function as heteromultimers in vivo. For example, the KCNQ2/3 heteromeric channel gives rise to a current that is 10-fold bigger than that of the homomeric channels (Wang et al., 1998; Yang et al., 1998). It seemed possible that the KCNQ2 mutant channels deficient in CaM binding lose their ability to assemble with KCNQ3, and the small currents given by homomeric channels elude our detection. Such an argument is in conflict with two lines of evidence. First, all of our KCNQ2 mutants still can coimmunoprecipitate with KCNQ3. Thus the CaM-binding ability of the channel can be dissociated from its ability to coassemble with KCNQ3. Second, functional interactions have been shown for KCNQ3 with all KCNQ family members except KCNQ1, but other members seem not to mix with each other (Kubisch et al., 1999; Lerche et al., 2000; Schroeder et al., 2000). Therefore, the structural element that mediates the heteromeric assembly is more likely to be unique to the KCNQ3 sequence rather than common to all KCNQs, as the conserved CaM-binding motifs are. Although we did not test whether the CaM-binding deficient KCNQ mutants are normal in their homomeric assembly, mutational studies in the KCNQ1 channel have identified a C-terminal domain downstream of site 2 as the cytoplasmic assembly domain. Deletion of a region homologous to site 2 in KCNQ1 produces no functional channel in a heterologous expression system, but this mutant appears to retain the ability to coassemble with other subunits, because it can suppress functional expression of wild-type channels in a dominant negative manner (Schmitt et al., 2000).
The correlation between CaM binding and channel function in our mutagenesis studies is obvious. The major defect these KCNQ2 mutants exhibit is in their ability to associate with CaM, but not in the other properties we assayed. On the other hand, we did not detect any Ca2+-dependent influence on channel activity. We thus hypothesize that CaM binding, but not its Ca2+-sensing ability, is necessary for a functional KCNQ2/3 channel. This hypothesis predicts that, if we reduce the level of CaM bound to KCNQ channels, we should decrease channel activity. Indeed, a fusion protein consisting of the CaM-binding domain of KCNQ2, GST-Q2CΔ31, decreases CaM binding to the channel, presumably by competing for endogenous CaM. When this fusion protein is coexpressed with the KCNQ2/3 channel, the current density is reduced significantly, although KCNQ2 is still targeted to the plasma membrane (data not shown). Furthermore, CaM-null flies exhibit a phenotype consistent with our hypothesis. Maternal deposits of CaM allow these flies to survive to the larval stage, but the larvae are overexcitable, as would be expected from the loss of a potassium current that normally opposes neuronal excitability (Heiman et al., 1996). It also is interesting that four single amino acid substitutions in the equivalent of site 1 in KCNQ1 and two in site 2 are found in patients with long-QT syndrome (Splawski et al., 2000). It seems possible that channel dysfunction in these mutants also is caused by their inabilities to bind CaM.
Our results illustrate that Ca2+-free apo-CaM itself is likely to have important functions. Indeed, this is not the only case in which a calcium-binding protein like CaM modulates its targets in the absence of Ca2+binding. Apo-CaM has been shown to bind and modulate several target proteins, including the inositol trisphosphate receptor and ryanodine receptor 1 (Patel et al., 1997; Rodney et al., 2000). Recently, a neuronal Ca2+-binding protein that shares nearly 56% identity with CaM was shown to modulate the calcium channel Cav2.1 in the absence of Ca2+ (Lee et al., 2002).
Taken together, our data are consistent with the hypothesis that the binding of CaM to KCNQ channels is necessary for channel function. It is conceivable that the interaction between the KCNQ C-terminal domain and CaM is dynamic and regulated by intracellular signaling pathways. For example, there are multiple PKC consensus substrate sites within site 2, and their phosphorylation state may affect CaM binding. It also remains a distinct possibility that other signaling molecules might interact with the channel tail by using sequences adjacent to or overlapping the CaM-binding domains, causing changes in the CaM channel interaction and hence in channel function. Competitive binding of CaM and other proteins to a common binding domain has been shown for NMDA receptors, metabotropic glutamate receptors, and the Trp3 channel (Wyszynski et al., 1997; El Far et al., 2001; Zhang et al., 2001). Therefore, regulating the binding of CaM to KCNQ channels may provide a novel mechanism for modulating channel activity. It will be intriguing to see whether this mechanism is used by the multiple receptor types that are known to modulate neuronal M current.
Footnotes
This work was supported by a grant from the National Institutes of Health. We are grateful to David McKinnon for rKCNQ3 cDNA, John Lowenstein for CaM cDNA, and Ping Jin for subcloning of CaM. We thank Carol Deutsch, Yi Zhou, Angela Jaramillo, and Lindsey Ciali for critical comments on this manuscript and Zhe Lu, Brian Salzberg, and Levitan laboratory members for helpful suggestions and discussion.
Correspondence should be addressed to Dr. Irwin Levitan, Department of Neuroscience, University of Pennsylvania School of Medicine, 3450 Hamilton Walk, Philadelphia, PA 19104. E-mail:levitani@mail.med.upenn.edu.
REFERENCES
- 1.Biervert C, Schroeder BC, Kubisch C, Berkovic SF, Propping P, Jentsch TJ, Steinlein OK. A potassium channel mutation in neonatal human epilepsy. Science. 1998;279:403–406. doi: 10.1126/science.279.5349.403. [DOI] [PubMed] [Google Scholar]
- 2.Brown DA, Adams PR. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature. 1980;283:673–676. doi: 10.1038/283673a0. [DOI] [PubMed] [Google Scholar]
- 3.Charlier C, Singh NA, Ryan SG, Lewis TB, Reus BE, Leach RJ, Leppert M. A pore mutation in a novel KQT-like potassium channel gene in an idiopathic epilepsy family. Nat Genet. 1998;18:53–55. doi: 10.1038/ng0198-53. [DOI] [PubMed] [Google Scholar]
- 4.Cooper EC, Aldape KD, Abosch A, Barbaro NM, Berger MS, Peacock WS, Jan YN, Jan LY. Colocalization and coassembly of two human brain M-type potassium channel subunits that are mutated in epilepsy. Proc Natl Acad Sci USA. 2000;97:4914–4919. doi: 10.1073/pnas.090092797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.El Far O, Bofill-Cardona E, Airas JM, O'Connor V, Boehm S, Freissmuth M, Nanoff C, Betz H. Mapping of calmodulin and G-βγ binding domains within the C-terminal region of the metabotropic glutamate receptor 7A. J Biol Chem. 2001;276:30662–30669. doi: 10.1074/jbc.M102573200. [DOI] [PubMed] [Google Scholar]
- 6.Fanger CM, Ghanshani S, Logsdon NJ, Rauer H, Kalman K, Zhou J, Beckingham K, Chandy KG, Cahalan MD, Aiyar J. Calmodulin mediates calcium-dependent activation of the intermediate conductance KCa channel, IKCa1. J Biol Chem. 1999;274:5746–5754. doi: 10.1074/jbc.274.9.5746. [DOI] [PubMed] [Google Scholar]
- 7.Geiser JR, Tuinen DV, Brockerhoff SE, Neff MM, Davis TN. Can calmodulin function without binding calcium? Cell. 1991;65:949–959. doi: 10.1016/0092-8674(91)90547-c. [DOI] [PubMed] [Google Scholar]
- 8.Heiman RG, Atkinson RC, Andruss BF, Bolduc C, Kovalick GE, Beckingham K. Spontaneous avoidance behavior in Drosophila null for calmodulin expression. Proc Natl Acad Sci USA. 1996;93:2420–2425. doi: 10.1073/pnas.93.6.2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holmes TC, Fadool DA, Ren R, Levitan IB. Association of Src tyrosine kinase with a human potassium channel mediated by SH3 domain. Science. 1996;274:2089–2091. doi: 10.1126/science.274.5295.2089. [DOI] [PubMed] [Google Scholar]
- 10.Joiner WJ, Khanna R, Schlichter LC, Kaczmarek LK. Calmodulin regulates assembly and trafficking of SK4/IK1 Ca2+-activated K+ channels. J Biol Chem. 2001;276:37980–37985. doi: 10.1074/jbc.M104965200. [DOI] [PubMed] [Google Scholar]
- 11.Jurado LA, Chockalingam PS, Jarrett HW. Apocalmodulin. Physiol Rev. 1999;79:661–682. doi: 10.1152/physrev.1999.79.3.661. [DOI] [PubMed] [Google Scholar]
- 12.Keen JE, Khawaled R, Farrens DL, Neelands T, Rivard A, Bond CT, Janowsky A, Fakler B, Adelman JP, Maylie J. Domains responsible for constitutive and Ca2+-dependent interactions between calmodulin and small conductance Ca2+-activated potassium channels. J Neurosci. 1999;19:8830–8838. doi: 10.1523/JNEUROSCI.19-20-08830.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kubisch C, Schroeder BC, Friedrich T, Lutjohann B, El Amraoui A, Marlin S, Petit C, Jentsch TJ. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell. 1999;96:437–446. doi: 10.1016/s0092-8674(00)80556-5. [DOI] [PubMed] [Google Scholar]
- 14.Lee A, Westenbroek RE, Haeseleer F, Palczewski K, Scheuer T, Catterall WA. Differential modulation of Cav2.1 channels by calmodulin and Ca2+-binding protein 1. Nat Neurosci. 2002;5:210–217. doi: 10.1038/nn805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lerche C, Scherer CR, Seebohm G, Derst C, Wei AD, Busch AE, Steinmeyer K. Molecular cloning and functional expression of KCNQ5, a potassium channel subunit that may contribute to neuronal M current diversity. J Biol Chem. 2000;275:22395–22400. doi: 10.1074/jbc.M002378200. [DOI] [PubMed] [Google Scholar]
- 16.Lerche H, Biervert C, Alekov AK, Schleithoff L, Lindner M, Klinger W, Bretschneider F, Mitrovic N, Jurkat-Rott K, Bode H, Lehmann-Horn F, Steinlein OK. A reduced K+ current due to a novel mutation in KCNQ2 causes neonatal convulsions. Ann Neurol. 1999;46:305–312. doi: 10.1002/1531-8249(199909)46:3<305::aid-ana5>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 17.Levitan IB. It is calmodulin after all! Mediator of the calcium modulation of multiple ion channels. Neuron. 1999;22:645–648. doi: 10.1016/s0896-6273(00)80722-9. [DOI] [PubMed] [Google Scholar]
- 18.Marrion NV. Control of M current. Annu Rev Physiol. 1997;59:483–504. doi: 10.1146/annurev.physiol.59.1.483. [DOI] [PubMed] [Google Scholar]
- 19.Marx SO, Kurokawa J, Reiken S, Motoike H, D'Armiento J, Marks AR, Kass RS. Requirement of a macromolecular signaling complex for β-adrenergic receptor modulation of the KCNQ1–KCNE1 potassium channel. Science. 2002;295:496–499. doi: 10.1126/science.1066843. [DOI] [PubMed] [Google Scholar]
- 20.Nakamura M, Watanabe H, Kubo Y, Yokoyama M, Matsumoto T, Sasai H, Nishi Y. KQT2, a new putative potassium channel family produced by alternative splicing. Isolation, genomic structure, and alternative splicing of the putative potassium channels. Receptors Channels. 1998;5:255–271. [PubMed] [Google Scholar]
- 21.Pan Z, Selyanko AA, Hadley JK, Brown DA, Dixon JE, McKinnon D. Alternative splicing of KCNQ2 potassium channel transcripts contributes to the functional diversity of M currents. J Physiol (Lond) 2001;531:347–358. doi: 10.1111/j.1469-7793.2001.0347i.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patel S, Morris SA, Adkins CE, O'Beirne G, Taylor CW. Ca2+-independent inhibition of inositol trisphosphate receptors by calmodulin: redistribution of calmodulin as a possible means of regulating Ca2+ mobilization. Proc Natl Acad Sci USA. 1997;94:11627–11632. doi: 10.1073/pnas.94.21.11627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peterson BZ, DeMaria CD, Adelman JP, Yue DT. Calmodulin is the Ca2+ sensor for Ca2+-dependent inactivation of L-type calcium channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 24.Putkey JA, Sweeney HL, Campbell ST. Site-directed mutation of the trigger calcium-binding sites in cardiac troponin C. J Biol Chem. 1989;264:12370–12378. [PubMed] [Google Scholar]
- 25.Rhoads AR, Frieldberg F. Sequence motifs for calmodulin recognition. FASEB J. 1997;11:331–340. doi: 10.1096/fasebj.11.5.9141499. [DOI] [PubMed] [Google Scholar]
- 26.Rodney GG, Williams BY, Strasburg GM, Beckingham K, Hamilton SL. Regulation of RYR1 activity by Ca2+ and calmodulin. Biochemistry. 2000;39:7807–7812. doi: 10.1021/bi0005660. [DOI] [PubMed] [Google Scholar]
- 27.Saimi Y, Kung C. Calmodulin as an ion channel subunit. Annu Rev Physiol. 2002;64:289–311. doi: 10.1146/annurev.physiol.64.100301.111649. [DOI] [PubMed] [Google Scholar]
- 28.Schmitt N, Schwarz M, Peretz A, Abitbol I, Attali B, Pongs O. A recessive C-terminal Jervell and Lange-Nielsen mutation of the KCNQ1 channel impairs subunit assembly. EMBO J. 2000;19:332–340. doi: 10.1093/emboj/19.3.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schonherr R, Lober K, Heinemann SH. Inhibition of human ether à go-go potassium channels by Ca2+/calmodulin. EMBO J. 2000;19:3263–3271. doi: 10.1093/emboj/19.13.3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schopperle WM, Holmqvist MH, Zhou Y, Wang J, Wang Z, Griffith LC, Keselman I, Kusinitz F, Dagan D, Levitan IB. Slob, a novel protein that interacts with the Slowpoke calcium-dependent potassium channel. Neuron. 1998;20:565–573. doi: 10.1016/s0896-6273(00)80995-2. [DOI] [PubMed] [Google Scholar]
- 31.Schroeder BC, Kubisch C, Stein V, Jentsch TJ. Moderate loss of function of cyclic-AMP-modulated KCNQ2/KCNQ3 K+ channels causes epilepsy. Nature. 1998;396:687–690. doi: 10.1038/25367. [DOI] [PubMed] [Google Scholar]
- 32.Schroeder BC, Hechenberger M, Weinreich F, Kubisch C, Jentsch TJ. KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J Biol Chem. 2000;275:24089–24095. doi: 10.1074/jbc.M003245200. [DOI] [PubMed] [Google Scholar]
- 33.Schumacher MA, Rivard AF, Bachinger HP, Adelman JP. Structure of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nature. 2001;410:1120–1124. doi: 10.1038/35074145. [DOI] [PubMed] [Google Scholar]
- 34.Schwake M, Pusch M, Kharkovets T, Jentsch TJ. Surface expression and single channel properties of KCNQ2/KCNQ3, M-type K+ channels involved in epilepsy. J Biol Chem. 2000;275:13343–13348. doi: 10.1074/jbc.275.18.13343. [DOI] [PubMed] [Google Scholar]
- 35.Selyanko AA, Hadley JK, Wood IC, Abogadie FC, Jentsch TJ, Brown DA. Inhibition of KCNQ1–4 potassium channels expressed in mammalian cells via m1 muscarinic acetylcholine receptors. J Physiol (Lond) 2000;522[Pt 3]:349–355. doi: 10.1111/j.1469-7793.2000.t01-2-00349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shapiro MS, Roche JP, Kaftan EJ, Cruzblanca H, Mackie K, Hille B. Reconstitution of muscarinic modulation of the KCNQ2/KCNQ3 K+ channels that underlie the neuronal M current. J Neurosci. 2000;20:1710–1721. doi: 10.1523/JNEUROSCI.20-05-01710.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sheng M, Kim E. Ion channel associated proteins. Curr Opin Neurobiol. 1999;6:602–608. doi: 10.1016/s0959-4388(96)80091-2. [DOI] [PubMed] [Google Scholar]
- 38.Singh NA, Charlier C, Stauffer D, DuPont BR, Leach RJ, Melis R, Ronen GM, Bjerre I, Quattlebaum T, Murphy JV, McHarg ML, Gagnon D, Rosales TO, Peiffer A, Anderson VE, Leppert M. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nat Genet. 1998;18:25–29. doi: 10.1038/ng0198-25. [DOI] [PubMed] [Google Scholar]
- 39.Smith JS, Iannotti CA, Dargis P, Christian EP, Aiyar J. Differential expression of KCNQ2 splice variants: implications to M current function during neuronal development. J Neurosci. 2001;21:1096–1103. doi: 10.1523/JNEUROSCI.21-04-01096.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Splawski I, Shen J, Timothy KW, Lehmann MH, Priori S, Robinson JL, Moss AJ, Schwartz PJ, Towbin JA, Vincent GM, Keating MT. Spectrum of mutations in long-QT syndrome genes: KVLQT1, HERG, SCN5A, KCNE1, and KCNE2. Circulation. 2000;102:1178–1185. doi: 10.1161/01.cir.102.10.1178. [DOI] [PubMed] [Google Scholar]
- 41.Tatulian L, Delmas P, Abogadie FC, Brown DA. Activation of expressed KCNQ potassium currents and native neuronal M-type potassium currents by the anti-convulsant drug retigabine. J Neurosci. 2001;21:5535–5545. doi: 10.1523/JNEUROSCI.21-15-05535.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tinel N, Diochot S, Lauritzen I, Barhanin J, Lazdunski M, Borsotto M. M-type KCNQ2–KCNQ3 potassium channels are modulated by the KCNE2 subunit. FEBS Lett. 2000;480:137–141. doi: 10.1016/s0014-5793(00)01918-9. [DOI] [PubMed] [Google Scholar]
- 43.Trimmer JS. Regulation of ion channel expression by cytoplasmic subunits. Curr Opin Neurobiol. 1998;8:370–374. doi: 10.1016/s0959-4388(98)80063-9. [DOI] [PubMed] [Google Scholar]
- 44.Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- 45.Wang J, Zhou Y, Wen H, Levitan IB. Simultaneous binding of two protein kinases to a calcium-dependent potassium channel. J Neurosci. 1999;19:RC4. doi: 10.1523/JNEUROSCI.19-10-j0005.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wyszynski M, Lin J, Rao A, Nigh E, Beggs AH, Craig AM, Sheng M. Competitive binding of α-actinin and calmodulin to the NMDA receptor. Nature. 1997;385:439–442. doi: 10.1038/385439a0. [DOI] [PubMed] [Google Scholar]
- 47.Xia XM, Fakler B, Rivard A, Wayman G, Johnson-Pais T, Keen JE, Ishii T, Hirschberg B, Bond CT, Lutsenko S, Maylie J, Adelman JP. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature. 1998;395:503–507. doi: 10.1038/26758. [DOI] [PubMed] [Google Scholar]
- 48.Yang WP, Levesque PC, Little WA, Conder ML, Ramakrishnan P, Neubauer MG, Blanar MA. Functional expression of two KvLQT1-related potassium channels responsible for an inherited idiopathic epilepsy. J Biol Chem. 1998;273:19419–19423. doi: 10.1074/jbc.273.31.19419. [DOI] [PubMed] [Google Scholar]
- 49.Yus-Nájera E, Santana-Castro I, Villarroel A. The identification and characterization of a non-continuous calmodulin binding site in non-inactivating voltage-dependent KCNQ potassium channels. J Biol Chem. 2002;277:28545–28553. doi: 10.1074/jbc.M204130200. [DOI] [PubMed] [Google Scholar]
- 50.Zhang Z, Tang J, Tikunova S, Johnson JD, Chen Z, Qin N, Dietrich A, Stefani E, Birnbaumer L, Zhu MX. Activation of Trp3 by inositol 1,4,5-trisphosphate receptors through displacement of inhibitory calmodulin from a common binding domain. Proc Natl Acad Sci USA. 2001;98:3168–3173. doi: 10.1073/pnas.051632698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zuhlke RD, Pitt GS, Deisseroth K, Tsien RW, Reuter H. Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature. 1999;399:159–162. doi: 10.1038/20200. [DOI] [PubMed] [Google Scholar]