

Abstract
Sympathetic neuronal apoptosis after nerve growth factor (NGF) deprivation requires the activation of two events: a protein synthesis-dependent, Bax-dependent release of mitochondrial cytochromec and a protein synthesis-independent, Bax-independent development of competence. Unlike in most cells, cytosolic cytochromec is not sufficient to induce cell death in NGF-maintained sympathetic neurons but can do so in neurons that have developed competence. We report that cytosolic cytochromec-induced apoptosis in competent sympathetic neurons is completely dependent on caspase-9. In addition, the neuroprotective agents KCl and chlorophenylthio-cAMP are potent inhibitors of the development-of-competence pathway in NGF-deprived sympathetic neurons. We also find that the development of competence is reversible. Readdition of NGF reverses competence, and neurons can regain their resistance to cytosolic cytochrome c. Importantly, we examined the mechanism of development of competence and report that the inability of cytochrome c to activate caspases in NGF-maintained sympathetic neurons can be overcome with exogenous Smac that inhibits the inhibitor of apoptosis (IAP) family of proteins. Microinjection of cytochrome c and Smac, but neither alone, induces rapid cell death in NGF-maintained neurons. These data suggest that development of competence may be the result of the loss of the function of one or more members of the IAP family of caspase inhibitors that is needed before cytochrome c can activate caspases and induce cell death in neurons.
Keywords: apoptosis, IAPs, cytochrome c, NGF, Smac, caspases
Neuronal death by apoptosis occurs extensively during development and is also observed in numerous pathological situations (Oppenheim, 1991; Deshmukh, 1998; Mattson, 2000; Yuan and Yankner, 2000). The mechanism of neuronal apoptosis has been widely studied in sympathetic neurons that are dependent on nerve growth factor (NGF) for survival and undergo apoptosis within 24–48 hr after NGF removal in culture (Martin et al., 1988; Edwards et al., 1991; Deckwerth and Johnson, 1993; Deshmukh and Johnson, 1997). This death is prevented by inhibitors of macromolecular synthesis, such as cycloheximide (Martin et al., 1988), by depolarizing concentrations of potassium (Scott and Fisher, 1970; Franklin and Johnson, 1992) and by cAMP analogs that raise intracellular cAMP levels (Rydel and Greene, 1988). Sympathetic neuronal death is also dependent on Bax and caspases. Bax-deficient (Deckwerth et al., 1996) or caspase-inhibited (Deshmukh et al., 1996; Troy et al., 1996; McCarthy et al., 1997;Deshmukh et al., 2000) sympathetic neurons do not undergo apoptosis after NGF deprivation.
Recent studies have examined the mechanism by which caspases are activated during apoptosis. The critical event for caspase activation in most cells appears to be the Bcl-2-family-protein-regulated translocation of cytochrome c from the mitochondria to cytosol (Hengartner, 2000). Once in the cytosol, cytochromec presumably binds to Apaf-1 and promotes its multimerization. The subsequent recruitment of procaspase-9 to this complex induces the activation of caspase-9 and rapid apoptosis (Budihardjo et al., 1999). Consistent with this model, cytosolic microinjection of cytochrome c is sufficient to induce a rapid, caspase-dependent death in many cell lines (Li et al., 1997;Brustugun et al., 1998; Juin et al., 1999; Chang et al., 2000).
In contrast, in sympathetic neurons, the mitochondrial release of cytochrome c is necessary but not sufficient to induce cell death. NGF-deprived sympathetic neurons exhibit a Bax-dependent loss of cytochrome c from the mitochondria that is necessary for cell death (Deshmukh and Johnson, 1998; Neame et al., 1998; Martinou et al., 1999). However, NGF-maintained sympathetic neurons are remarkably resistant to cytosolic microinjection of cytochrome c, indicating that the cytosolic translocation of cytochrome cis not sufficient to induce cell death in these neurons (Deshmukh and Johnson, 1998; Neame et al., 1998). Importantly, NGF deprivation induces another event, called the development of competence, which is needed along with cytochrome c to induce cell death in sympathetic neurons. Sympathetic neurons that are deprived of NGF for a period, but kept alive by a block in the cytochromec-release pathway either by addition of cycloheximide or Bax deficiency, die rapidly when exposed to cytosolic cytochromec (Deshmukh and Johnson, 1998). Thus, NGF deprivation activates two pathways in sympathetic neurons: a macromolecular synthesis-dependent, Bax-dependent pathway that leads to cytochromec release from the mitochondria and a macromolecular synthesis-independent, Bax-independent pathway that leads to the development of competence (Deshmukh and Johnson, 1998). Activation of both pathways is needed for apoptosis in sympathetic neurons. In this study, we have examined the molecular mechanism of the development-of-competence pathway.
MATERIALS AND METHODS
Reagents. All reagents were purchased from Sigma (St. Louis, MO) unless otherwise stated. Collagenase and trypsin were purchased from Worthington Biochemical Corp. (Freehold, NJ). The phosphatidylinositol 3-kinase (PI-3-kinase) inhibitor 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) was purchased from Biomol (Plymouth Meeting, PA). The caspase inhibitor boc-aspartyl(OMe)-fluoromethylketone (BAF) was purchased from Enzyme Systems Products (Livermore, CA). Untimed-pregnant (ICR) mice were purchased from Harlan Sprague Dawley (Indianapolis, IN).
Sympathetic neuronal cultures. Primary cultures of sympathetic neurons from the superior cervical ganglion were prepared from postnatal day 1 (P1) mice essentially as described previously for rats (Johnson and Argiro, 1983; Deshmukh et al., 1996). Briefly, the dissected ganglia were treated with collagenase (1 mg/ml) and then trypsin (2.5 mg/ml) for 30 min each at 37°C. The ganglia were triturated, and the dissociated cells were plated on collagen-coated dishes in NGF-containing medium (AM50). This medium contained Eagle's minimum essential medium with Earle's salts (Invitrogen, Gaithersburg, MD) with the addition of 50 ng/ml 2.5S NGF, 10% fetal calf serum, 2 mm glutamine, 100 μg/ml penicillin, and 100 μg/ml streptomycin; 20 μmfluorodeoxyuridine, 20 μm uridine, and 3.3 μg/ml aphidicolin were also included to reduce the number of non-neuronal cells. ICR outbred mice (Harlan Sprague Dawley) were used for all experiments except those involving Bax-deficient and caspase-9-deficient sympathetic neurons. The genetic background of Bax-deficient and caspase-9-deficient mice was C57BL/6; wild-type littermates were used as controls in these experiments.
Bax−/− and caspase-9−/− mice. Breeding and genotyping of Bax-deficient mice have been described previously (Knudson et al., 1995; Deckwerth et al., 1996). Bax-deficient sympathetic neurons were isolated from P1 mice. Breeding and genotyping of caspase-9-deficient mice were performed as described previously (Kuida et al., 1998). Caspase-9-deficient sympathetic neurons were isolated from embryonic day 17 mice because of lethality associated with caspase-9 deficiency at later ages; these cultures were maintained in NGF-containing medium for 6–7 d (instead of the normal 4–5 d) before subjecting them to the experimental conditions.
Culture conditions. Sympathetic neuronal cultures were grown in NGF-containing medium (AM50) for 4–5 d and then either maintained in AM50 or treated as follows. For NGF deprivation, cultures were rinsed twice with medium lacking NGF (AM0: AM50 medium without NGF), followed by the addition of AM0 containing goat anti-NGF-neutralizing antibody (Ruit et al., 1992). Other conditions required the addition of 1 μg/ml cycloheximide, 0.1 μg/ml actinomycin D, 400 μmchlorophenylthio-cAMP (CPTcAMP), or 35 mm KCl to the anti-NGF-containing medium. For experiments in which NGF or other agents were readded to cultures after NGF-deprived neurons had developed competence, cultures were rinsed three times with the AM0 medium and then incubated in the appropriate medium.
Microinjection of cytochrome c and quantitation of cell death. Our method for microinjecting sympathetic neurons with cytochrome c and assessing cell death after the microinjections has been described previously (Deshmukh and Johnson, 1998). Briefly, sympathetic neuronal cultures (1500–3000 cells), grown in the appropriate medium on collagen-coated, 35 mm dishes (Corning, Corning, NY), were switched to Leibovitz's L-15 medium (Invitrogen) containing 100 μg/ml penicillin and 100 μg/ml streptomycin just before injections. To identify the injected cells, the microinjection solution (in mm: 100 KCl and 10 KPi, pH 7.4) contained rhodamine dextran (4 mg/ml). Microinjection solution containing rhodamine dextran (dye) alone or dye plus cytochrome c (5–25 mg/ml) was injected into the cytoplasm of neurons by using Femtotips needles (Eppendorf, Inc., Madison, WI). Immediately after the injections, the number of viable cells injected was determined by counting the number of rhodamine-positive cells that had intact, phase-bright cell bodies. Cultures were then switched to the appropriate medium, and, at various times after injections, the number of viable, injected neurons remaining was determined by using the same counting criterion.
Immunohistochemical assessment of caspase-3 activation.Neuronal cultures were immunostained as described previously (Deshmukh and Johnson, 1998). Briefly, sympathetic neurons (1500–3000 cells) were grown on collagen-coated, 35 mm dishes (Corning) in the appropriate medium. Neurons were microinjected with cytochromec along with a nonspecific mouse monoclonal antibody (IgG) to allow identification of the injected cells. (The rhodamine dextran dye could not be used to mark the injected cells in these experiments because this dye becomes cell permeable during the immunohistochemical procedure.) After the microinjections, cultures were washed once with PBS and then fixed with freshly made 4% paraformaldehyde in PBS for 30 min at 4°C. Cultures were then washed three times with Tris-buffered saline (TBS; 100 mm Tris-HCl, pH 7.6, and 0.9% NaCl), exposed to blocking solution (TBS containing 5% goat serum and 0.3% Triton X-100) for 30 min at room temperature, and incubated in anti-active caspase-3 antibody (CM1 antibody kindly provided by Dr. Anu Srinivasan, IDUN Pharmaceuticals, San Diego, CA) solution overnight at 4°C. The primary antibody was diluted 1:5000 in TBS containing 1% goat serum and 0.3% Triton X-100. Cultures were then washed three times with TBS and incubated in an anti-rabbit Alexa Fluor 488-conjugated (Molecular Probes, Inc., Eugene, OR) secondary antibody solution in TBS containing 1% goat serum and 0.3% Triton X-100 for 2–4 hr at 4°C. Cultures were also incubated with an anti-mouse Cy3-conjugated (Jackson ImmunoResearch, West Grove, PA) secondary antibody solution to label the microinjected cells. After the secondary antibody incubation, cultures were washed twice in TBS and stained with the nuclear dye bisbenzimide (Hoechst 33258 used at 1 μg/ml; Molecular Probes, Inc.) for 15 min at room temperature. After washing twice with TBS and adding one drop of mounting medium (50% glycerol, 0.1% paraphenylenediamine in PBS) on the cells, a glass coverslip was placed on the cells, and the samples were examined by fluorescence microscopy.
Western blot analysis. Sympathetic neurons (5000–10,000 cells) were washed twice with ice-cold TBS and then lysed with the addition of SDS-PAGE sample buffer. Samples were then boiled for 5 min and resolved by SDS-PAGE (12% Tris-glycine gels; Novex, San Diego, CA). The proteins were then transferred to polyvinyl difluoride membranes (Millipore, Burlington, MA) and washed with TBS containing 0.1% Tween 20 (TBST). After incubation of the membrane with a blocking solution (5% nonfat milk in TBST) for 1 hr at room temperature, the membrane was then incubated in primary antibody (in blocking solution) overnight at 4°C. Immunoblots were then washed three times with TBST and incubated in the appropriate secondary antibody (in blocking solution) for 2 hr at room temperature. Proteins were visualized by using the SuperSignal Chemiluminescent Detection system (Pierce, Rockford, IL) according to the manufacturer's instructions. The anti-phospho-Akt antibody (S473; New England Biolabs, Inc., Beverly, MA) was used at 1:1000 dilution, and the anti-tubulin antibody (Sigma) was used at 1:50,000 dilution.
RESULTS
Staurosporine induces the development of competence in sympathetic neurons
Apoptosis in NGF-deprived sympathetic neurons requires both the release of cytochrome c and the development of competence (Deshmukh and Johnson, 1998). To determine whether the development-of-competence pathway is also activated in other models of sympathetic neuronal apoptosis, we examined whether sympathetic neurons treated with 100 nm staurosporine (STS) developed competence. Like NGF deprivation, 100 nm STS treatment induces apoptosis with cytochrome c release and caspase activation in sympathetic neurons (Deshmukh and Johnson, 2000). Therefore, we asked whether cytosolic microinjection of cytochromec was able to induce cell death in 100 nm STS-treated neurons. These experiments were done in Bax-deficient neurons to prevent any cell death that would otherwise occur with 100 nm STS treatment alone. Microinjection of cytochrome c induced rapid cell death in 100 nm STS-treated but not NGF-maintained sympathetic neurons (Fig. 1). Microinjection with the rhodamine dextran dye alone did not induce any cell death in 100 nm STS-treated neurons, indicating that these neurons did not die simply because of the microinjection or because of treatment with 100 nm STS alone (Fig. 1). The observation that 100 nm STS treatment induced competence and permitted cytochrome c to induce cell death in sympathetic neurons indicates that the development of competence is not unique to the apoptotic pathway induced with NGF deprivation.
Fig. 1.

Activation of the apoptotic pathway with 100 nm STS induces the development of competence in NGF-maintained sympathetic neurons. Bax-deficient sympathetic neurons were maintained in NGF (+NGF), deprived of NGF (−NGF), or maintained in NGF in the presence of 100 nm STS (+NGF+STS) for 36 hr. Bax deficiency permits these neurons to be deprived of NGF or treated with 100 nm STS yet remain alive, because the Bax is required for the release of cytochrome c under these conditions. Neurons were then microinjected with cytochrome c (10 mg/ml) and rhodamine dextran (C) or rhodamine dextran alone (R), and the number of cells injected was determined after the microinjections (time 0 hr) by counting using a fluorescent microscope. At 3, 6, and 12 hr after the injections, the number of microinjected cells that remained viable was determined and expressed as a percentage of the original number of microinjected cells. Microinjection of cytochrome c, but not rhodamine dextran alone, induces rapid cell death in 100 nm STS-treated neurons. Results are means ± SEM for three experiments with ∼100 cells counted for each time point per experiment.
Depolarizing concentration of KCl or CPTcAMP inhibits the development-of-competence pathway
Although NGF deprivation activates both the cytochromec release and the development-of-competence pathways, whereas the pathway leading to cytochrome c release is dependent on macromolecular synthesis and Bax function, the development-of-competence pathway requires neither macromolecular synthesis nor Bax function (Deshmukh and Johnson, 1998). Therefore, these two pathways must diverge after NGF deprivation at a point that is upstream of the macromolecular synthesis- and Bax-dependent steps during sympathetic neuronal apoptosis.
To determine the point at which these two pathways diverge after NGF removal and to characterize the components of the signaling pathway that leads to the development of competence, we examined whether neuroprotective agents such as a depolarizing concentration of K+ (35 mm KCl) or a cAMP analog (400 μm CPTcAMP) that prevents sympathetic neuronal apoptosis (Rydel and Greene, 1988; Edwards et al., 1991;Deckwerth and Johnson, 1993) block the development-of-competence pathway. Microinjection of cytochrome c did not induce cell death in NGF-deprived, 35 mm KCl-treated neurons or NGF-deprived, 400 μm CPTcAMP-treated neurons (Fig. 2). Control cells that were deprived of NGF in the presence of cycloheximide developed competence and died rapidly after microinjection of cytochrome c. Thus, both KCl and CPTcAMP inhibited the development-of-competence pathway in NGF-deprived sympathetic neurons.
Fig. 2.

KCl and cAMP block the development of competence in NGF-deprived sympathetic neurons. Mouse sympathetic neurons were deprived of NGF in the presence of 1 μg/ml cycloheximide (−NGF+CHX), 35 mm KCl (−NGF+KCl), or 400 μm CPTcAMP (−NGF+CPTcAMP) for 36 hr. Neurons were then microinjected with cytochrome c (10 mg/ml), and the viability of the cells at 3, 6, and 12 hr after the injections was determined as described in the legend to Figure 1. Although the NGF-deprived, cycloheximide-saved cells developed competence (microinjection of cytochrome c induced rapid cell death), both KCl and CPTcAMP prevented the development of competence in these neurons (microinjection of cytochrome c did not induce cell death). Data shown are mean ± range of two experiments with ∼100 cells counted for each time point per experiment.
Inhibition of the PI-3-kinase signaling pathway leads to the partial development of competence
We subsequently examined whether NGF signaling through the PI-3-kinase pathway was essential for keeping the development-of-competence pathway inhibited. Signaling through the PI-3-kinase pathway has been reported to be important for maintaining neuronal survival (see Discussion). Therefore, we examined whether inhibiting the PI-3-kinase signaling pathway with LY294002 (Vlahos et al., 1994) was sufficient to induce the development of competence in NGF-maintained sympathetic neurons.
Bax-deficient sympathetic neurons were treated with 50 μmLY294002 for ≥48 hr and then microinjected with cytochromec to assess whether these neurons had developed competence. Bax-deficient neurons were used in these experiments to prevent any cell death that would otherwise occur with LY294002 treatment alone. Microinjection of cytochrome c but not dye alone induced cell death in the PI-3-kinase-inhibited sympathetic neurons (Fig.3A). However, the kinetics and extent of cell death induced by cytochrome c microinjection in LY294002-treated neurons were reduced compared with the cell death induced by cytochrome c microinjection in the control, NGF-deprived neurons. Although microinjection of cytochromec induced 60–70% of neurons to die by 3 hr after the microinjections in NGF-deprived neurons that had developed competence, only 20% of the LY294002-treated neurons died by 3 hr after microinjection of cytochrome c (Fig. 3A). Even 12 hr after the cytochrome c microinjections, although all of the NGF-deprived neurons had died, 25% of the LY294002-treated, cytochrome c-microinjected neurons still remained viable. The reduced cell death observed in LY294002-treated, cytochromec-microinjected neurons was not caused by partial inhibition of the PI-3-kinase-signaling pathway with LY294002, because 50 μm LY294002 was effective in completely inhibiting the phosphorylation of Akt, one of the downstream targets of the PI-3-kinase-signaling pathway, in these cells (Fig. 3B) (Tsui-Pierchala et al., 2000). The observation that inhibiting PI-3-kinase signaling with LY294002 induced only partial development of competence indicates that NGF signals to inhibit the development-of-competence pathway by using both PI-3-kinase-dependent and PI-3-kinase-independent mechanisms.
Fig. 3.

Inhibition of the PI-3-kinase signaling pathway induces partial development of competence in NGF-maintained sympathetic neurons. A, Bax-deficient sympathetic neurons were maintained in NGF (+NGF), deprived of NGF (−NGF), or maintained in NGF in the presence of a 50 μm concentration of the PI-3-kinase inhibitor LY294002 (+NGF+LY) for 36 hr. Bax deficiency permits these neurons to be deprived of NGF or to be treated with LY294002 yet remain alive to allow for the microinjection experiments. Neurons were then microinjected with cytochrome c (10 mg/ml) and rhodamine dextran (C) or rhodamine dextran alone (R), and the number of cells injected was determined as described in the legend to Figure 1. Results are the means ± SEM for three experiments with ∼100 cells counted for each time point per experiment. B, A 50 μm concentration of LY294002 inhibits the PI-3-kinase signaling-mediated phosphorylation of its substrate Akt in NGF-maintained sympathetic neurons. Western immunoblots show lysates of NGF-maintained sympathetic neurons either alone or in the presence of 50 μm LY294002 for 36 hr that were probed with an anti-phospho-specific Akt (α p-Akt) antibody to assess the inhibition of the PI-3-kinase signaling in neurons. Lysates were also probed with an anti-tubulin (α Tubulin) antibody as a loading control.
Cytochrome c-induced death in competent sympathetic neurons uses the caspase-9-dependent apoptotic pathway
Cell death induced by microinjection of cytochrome c in competent sympathetic neurons is caspase dependent, because the pan-caspase inhibitor BAF blocks this death (Deshmukh and Johnson, 1998). Although several in vitro studies indicate that cytochrome c induces caspase activation via the Apaf-1 and caspase-9-mediated pathway (Budihardjo et al., 1999), whether microinjected cytochrome c activates this pathway in sympathetic neurons is unclear. Sympathetic neuronal apoptosis after NGF deprivation requires caspase-9 (Deshmukh et al., 2000). Therefore, we examined whether sympathetic neuronal death induced by microinjection of cytochrome c was also caspase-9 dependent. Sympathetic neurons from caspase-9-deficient mice (−/−) or the wild-type littermates (+/+) were either maintained in NGF or deprived of NGF in the presence of cycloheximide for 48 hr and then microinjected with cytochrome c. Microinjection of cytochrome c induced rapid cell death in the NGF-deprived, cycloheximide-treated neurons obtained from wild-type mice (Fig.4). However, microinjection of cytochromec was incapable of inducing cell death in NGF-deprived, cycloheximide-treated neurons that were deficient in caspase-9 (Fig.4). As expected, microinjection of cytochrome c did not induce cell death in NGF-maintained neurons from either wild-type or caspase-9-deficient mice.
Fig. 4.

Caspase-9 is required for cytochromec-induced cell death in sympathetic neurons. Sympathetic neurons from caspase-9-deficient mice (−/−) or their wild-type littermates (+/+) were either maintained in NGF (+NGF) or deprived of NGF in the presence of 1 μg/ml cycloheximide (−NGF+CHX) for 36 hr. Cells were then microinjected with cytochrome c (10 mg/ml), and the number of cells remaining viable 3, 6, and 10 hr after the injections was determined as described in the legend to Figure 1. Microinjection of cytochrome c induces rapid cell death in competent, wild-type (+/+) but not caspase-9-deficient (−/−) neurons. Data shown are means ± range of two experiments with ∼100 cells counted for each time point per experiment.
We also examined whether microinjected cytochrome c induced cell death in competent sympathetic neurons by activating caspase-3. Activation of caspase-3 was determined by immunostaining with the CM1 antibody that recognizes the activated but not the zymogen form of caspase-3 (Srinivasan et al., 1998). Microinjection of cytochromec produced CM1 staining in competent, NGF-deprived, cycloheximide-treated neurons but not in NGF-maintained sympathetic neurons (Fig. 5). Together, these experiments indicate that cytosolic cytochrome c induced cell death in competent sympathetic neurons by activating a caspase-9-dependent, caspase-3-mediated apoptotic pathway.
Fig. 5.

Caspase-3 is activated in the NGF-deprived, cycloheximide-treated, competent neurons but not in the NGF-maintained neurons after microinjection with cytochrome c. Mouse sympathetic neurons were either maintained in NGF (+NGF) or deprived of NGF in the presence of 1 μg/ml cycloheximide (−NGF+CHX). Cells were microinjected with cytochrome c (10 mg/ml) along with a nonspecific (IgG) mouse monoclonal antibody to mark the injected cells. Four hours later, the injected cells were fixed and incubated with anti-activated-caspase-3 (CM1) antibodies (rabbit). Anti-mouse (for IgG) and anti-rabbit (for CM1) secondary antibodies were then used to immunostain the cells. Photomicrographs of representative cells (arrows mark the injected cells) show caspase-3 activation in competent (−NGF+CHX) but not in NGF-maintained (+NGF) neurons after microinjection of cytochrome c. Scale bar, 20 μm.
Readdition of NGF to competent sympathetic neurons restores resistance to cytosolic cytochrome c: reversal of competence is slow and requires new protein synthesis
NGF-maintained sympathetic neurons develop competence only after 24–36 hr of NGF deprivation and by a mechanism that does not require protein synthesis (Deshmukh and Johnson, 1998). To determine whether this process is reversible, we examined whether readdition of NGF to competent neurons results in the loss of competence and promotes resistance to cytosolic cytochrome c. NGF-deprived, cycloheximide-saved sympathetic neurons that had developed competence were washed and re-exposed to NGF-containing medium. At various times after NGF readdition, neurons were microinjected with cytochromec to determine whether these neurons had developed resistance to cytosolic cytochrome c. Readdition of NGF to competent neurons resulted in complete loss of competence (Fig.6). However, the reversal of competence was slow, and neurons regained complete resistance to cytosolic cytochrome c only after 12–24 hr of NGF readdition (Fig.6). We also examined whether competence could be reversed with the addition of KCl or CPTcAMP. Like NGF readdition, exposure to 35 mm KCl or 400 μm CPTcAMP also induced the reversal of competence and promoted the resistance to cytosolic cytochrome c by a similar time course (Fig.6).
Fig. 6.

Time course of loss of competence with readdition of NGF, KCl, or cAMP. Mouse sympathetic neurons that had developed competence (NGF deprived and cycloheximide treated for 36 hr) were treated with the addition of NGF (+NGF), 400 μm CPTcAMP (+cAMP), or 35 mmKCl (+KCl). At the times indicated after the addition of these agents, cells were microinjected with cytochromec (10 mg/ml) to examine the competence state of these cells. Competence was assessed by examining survival 6 hr after microinjection of cytochrome c as described in the legend to Figure 1. Note that at time 0 (fully competent cells), only 10% of the injected cells survive injection of cytochromec. However, 12 hr after the addition of NGF (or KCl or CPTcAMP), ∼80% of the cells have lost competence and survive microinjection of cytochrome c. Data shown are means ± range of two experiments with ∼100 cells counted for each time point per experiment.
To determine whether the reversal of competence required new protein synthesis, we examined whether macromolecular synthesis inhibitors prevented the ability of NGF to reverse competence. NGF was readded to NGF-deprived, competent neurons alone or in the presence of RNA or protein synthesis inhibitor, actinomycin D, or cycloheximide, respectively. Competent sympathetic neurons that were treated with NGF readdition alone reversed competence and became resistant to cytosolic cytochrome c by 12 hr after NGF readdition (Figs. 6,7A). However, RNA or protein synthesis inhibitor blocked the ability of NGF to reverse competence. Neurons that were treated with NGF readdition in the presence of either actinomycin D or cycloheximide remained sensitive to cytosolic cytochrome c and died rapidly after microinjection of cytochrome c even after 12 hr of NGF readdition (Fig.7A). Actinomycin D or cycloheximide also prevented the ability of KCl and CPTcAMP to reverse competence. Competent neurons that were treated with KCl or CPTcAMP for 12 hr in the presence of actinomycin D or cycloheximide remained sensitive to cytochromec and died rapidly with cytosolic microinjection of cytochrome c (Fig. 7B,C).
Fig. 7.
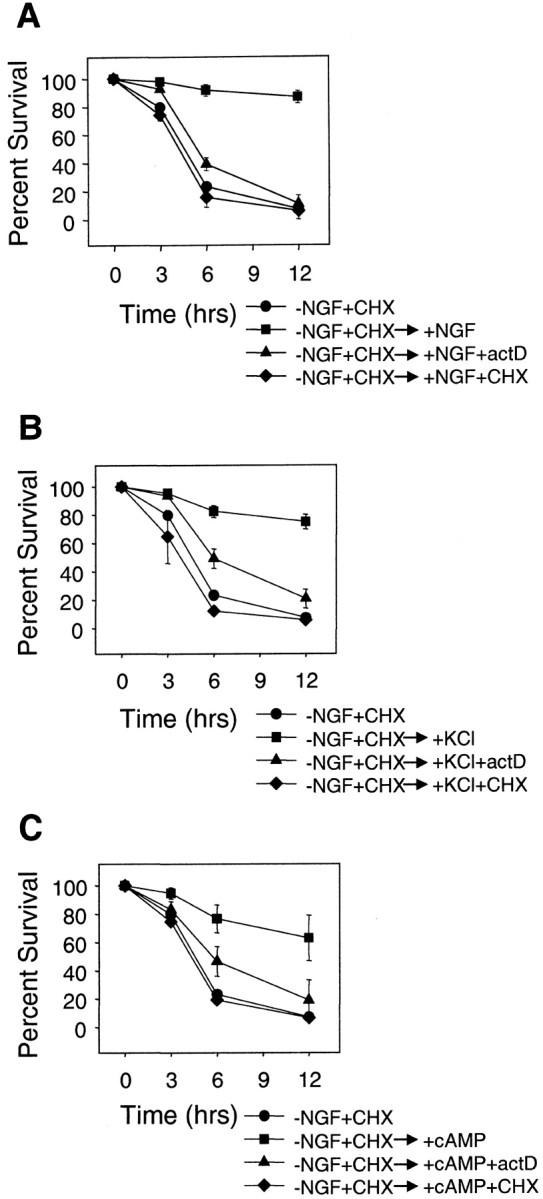
Reversal of competence requires macromolecular synthesis. A, Mouse sympathetic neurons that had developed competence (NGF deprived and cycloheximide treated for 36 hr) were treated for 12 hr with NGF alone (+NGF), NGF with 0.1 μg/ml actinomycin-D (+NGF+actD), or NGF with 1 μg/ml cycloheximide (+NGF+CHX). Cells were then microinjected with cytochrome c, and the competence state was determined by assessing the survival as described in the legend to Figure 1.B, Mouse sympathetic neurons that had developed competence (NGF deprived and cycloheximide treated for 36 hr) were treated for 12 hr with 35 mm KCl alone (+KCl), KCl in the presence of 0.1 μg/ml actinomycin D (+KCl+actD), or 1 μg/ml cycloheximide (+KCl+CHX), and the competence status of these cells was assessed exactly as described in A.C, Mouse sympathetic neurons that had developed competence (NGF deprived and cycloheximide treated for 36 hr) were treated for 12 hr with 400 μm CPTcAMP alone (+cAMP), CPTcAMP in the presence of 0.1 μg/ml actinomycin D (+cAMP+actD), or 1 μg/ml cycloheximide (+cAMP+CHX), and the competence status of these cells was assessed exactly as described in A and Figure1. As seen in Figure 6, the addition of NGF, KCl, or CPTcAMP reversed competence by 12 hr after factor addition. However, RNA and protein synthesis inhibitors (actinomycin-D and cycloheximide, respectively) blocked the ability of NGF, KCl, or CPTcAMP to reverse competence, because these cells remained susceptible to microinjection of cytochrome c. Data shown are means ± range of two to three experiments with ∼100 cells counted for each time point per experiment.
Thus, the competence state of sympathetic neurons was reversible. However, although the development of competence was not protein synthesis dependent, the reversal of competence surprisingly required new protein synthesis. These results are consistent with the model in which the synthesis of a protein that inhibits cytochromec-mediated caspase activation (either directly or indirectly) is needed for the reversal of competence. Consequently, the development of competence may involve the inactivation or degradation of such a caspase inhibitory protein.
Cytosolic microinjection of Smac induces competence and permits cytochrome c-mediated cell death in NGF-maintained sympathetic neurons
We considered the possibility that the caspase-inhibitory protein regulating competence in sympathetic neurons might belong to the inhibitor of apoptosis (IAP) family of proteins. IAP proteins such as X-linked IAP (XIAP), inhibitor of apoptosis-1 (cIAP-1), and cIAP-2 bind to caspases and are potent inhibitors of their activation in vitro (Deveraux and Reed, 1999). Overexpression of these proteins prevents apoptosis in many cells, including chicken sympathetic neurons (Wiese et al., 1999) and cerebellar granule neurons (Simons et al., 1999). Thus, the inability of cytosolic cytochrome c to induce cell death in NGF-maintained sympathetic neurons could be attributable to caspase inhibition by the IAP family of proteins.
To examine this, we used the recently identified Smac protein that can bind to and inhibit IAP function (Du et al., 2000; Verhagen et al., 2000). We asked whether cytosolic microinjection of mature Smac would permit cytochrome c to induce cell death in NGF-maintained sympathetic neurons. Because mature Smac can bind to and inhibit several IAPs in vitro, microinjection of Smac in the cytosol allowed us potentially to inhibit multiple IAPs in these neurons, thereby overcoming any complexity caused by functional redundancy of IAPs in these cells. Cytosolic microinjection of mature Smac alone was insufficient to induce cell death in NGF-maintained sympathetic neurons (Fig.8A). As expected, cytosolic microinjection of cytochrome c alone also did not induce cell death in NGF-maintained sympathetic neurons. However, coinjection of cytochrome c and Smac remarkably induced a rapid, caspase-dependent death in these NGF-maintained neurons (Fig.8A). Thus, exogenous cytosolic mature Smac was sufficient to induce competence rapidly and permit cytochromec-mediated cell death in NGF-maintained sympathetic neurons. Cytosolic microinjection of Smac and cytochrome c also induced rapid cell death in KCl- or CPTcAMP-maintained neurons (data not shown). The ability of Smac to convert rapidly NGF-maintained neurons from being resistant to being sensitive to cytosolic cytochromec indicates that exogenous cytosolic mature Smac induced events in these neurons that were functionally indistinguishable from those induced by the development-of-competence pathway after 24–36 hr of NGF deprivation.
Fig. 8.

Cytosolic microinjection of Smac induces competence in NGF-maintained sympathetic neurons. A, NGF-maintained mouse sympathetic neurons were microinjected with cytochrome c (15 mg/ml) alone (Cytc), Smac (0.6 mg/ml) alone (Smac), or cytochromec and Smac (Cytc+Smac). Parallel cultures of sympathetic neurons that were deprived of NGF in the presence of cycloheximide for 36 hr were injected with cytochrome c[−NGF+CHX(Cytc)] as a positive control for competence. Viability of microinjected cells 3, 6, and 12 hr after these injections was determined as described in the legend to Figure 1. Some NGF-maintained cultures that were injected with cytochromec and Smac together were also treated with the pan-caspase inhibitor BAF (50 μm) to examine whether the cell death induced by cytochrome c and Smac was caspase dependent. Microinjection of Smac and cytochrome c, but neither alone, induced rapid cell death in NGF-maintained sympathetic neurons. B, Microinjection of Smac β does not induce competence in sympathetic neurons. NGF-maintained mouse sympathetic neurons were microinjected with cytochrome c (15 mg/ml) and either Smac (0.6 mg/ml) or Smac β (0.6 mg/ml). Control cells were also microinjected with cytochrome c, Smac, or Smac β alone. Viability of microinjected cells 24 hr after the injections is shown. Unlike Smac, Smac β is not able to promote caspase activation with cytochrome c in these neurons. Data shown are means ± range of two experiments with ∼100 cells counted for each time point per experiment.
To determine whether the N-terminal IAP-inhibiting domain of mature Smac was important for its ability to permit cytochrome c to activate caspases in sympathetic neurons, we examined whether microinjection of Smac β, an alternatively spliced form of Smac that lacks this N-terminal IAP-binding domain, could also promote cytochromec-mediated caspase activation in these neurons. Smac β is identical to mature Smac except in the first seven amino acids; this sequence corresponds to MKSDFYF in Smac β and AVPIA in mature Smac (Srinivasula et al., 2000; Roberts et al., 2001). Because the N-terminal AVPI motif in mature Smac is critical for interacting with the baculovirus IAP repeat-3 (BIR-3) domain of IAPs and for relieving the XIAP–caspase-9 inhibition (Liu et al., 2000; Wu et al., 2000), Smac β is unable to interact with the BIR-3 domain of IAPs and, as a consequence, is ineffective in blocking the XIAP inhibition of caspase-9 (Srinivasula et al., 2000). Unlike mature Smac, microinjection of Smac β and cytochrome c was unable to promote any significant cell death in NGF-maintained sympathetic neurons even 24 hr after the injections (Fig. 8B). As expected, microinjection of Smac, Smac β, or cytochrome calone also did not promote any cell death in these neurons (Fig.8B). Thus, the N-terminal IAP (BIR-3)-interacting domain of mature Smac was essential for promoting cytochromec-mediated caspase activation in NGF-maintained sympathetic neurons. These results indicate that the inability of cytochromec alone to activate caspases in these neurons was attributable to the presence of an IAP family protein(s) that could be overcome with exogenous cytosolic microinjection of mature Smac.
DISCUSSION
Signaling mechanism of the development-of-competence pathway
NGF deprivation activates the cytochrome c release and development-of-competence pathways in sympathetic neurons, both of which are required for apoptosis in these cells. These two pathways must diverge at some point after NGF deprivation, because whereas the cytochrome c release pathway is dependent on macromolecular synthesis and Bax function, the development-of-competence pathway requires neither macromolecular synthesis nor Bax function (Deshmukh and Johnson, 1998). We have determined that the neuroprotective agents KCl and CPTcAMP block the development of the competence pathway in NGF-deprived neurons (Fig. 2). Because KCl and CPTcAMP also inhibit the cytochrome c-release pathway after NGF deprivation (Putcha et al., 1999), both KCl and CPTcAMP most likely act at a point before the divergence of these two pathways to prevent sympathetic neuronal apoptosis.
We also examined the specific importance of the PI-3-kinase/Akt-signaling pathway in regulating competence in sympathetic neurons because of the reported survival-promoting importance of this pathway in neurons (Datta et al., 1999). Our results show that inhibition of the endogenous PI-3-kinase-signaling pathway with LY294002 induced partial activation of the development-of-competence pathway in NGF-maintained sympathetic neurons (Fig. 3). Thus, the PI-3-kinase-signaling pathway appeared critical, but only in part, for keeping the development-of-competence pathway inhibited in NGF-maintained sympathetic neurons. Overexpression of activated Akt (one of the downstream targets of the PI-3-kinase-signaling pathway) prevents apoptosis after cytochromec release in hybrid motor neuron 1 cells (Zhou et al., 2000). Thus, signaling through the PI-3-kinase/Akt-pathway may post-translationally modify components of the apoptosome complex and prevent caspase activation. Alternatively, because the PI-3-kinase-signaling pathway is important for maintaining the trophic and metabolic status of NGF-maintained sympathetic neurons (Tsui-Pierchala et al., 2000), PI-3-kinase signaling may prevent caspase activation indirectly by sustaining the macromolecular synthesis of proteins such as the IAPs (see below). Consistent with this hypothesis, PI-3-kinase-signaling pathway was shown to be important for the continued expression of ITA, an IAP family protein, in NGF-maintained chicken sensory and sympathetic neurons (Wiese et al., 1999). In the absence of NGF or if the PI-3-kinase pathway is inhibited, the expression of such proteins may decrease, thereby permitting cytochrome c-mediated caspase activation in these neurons.
Importance of the caspase-9-dependent pathway in sympathetic neuronal apoptosis
Our results show that in the absence of caspase-9, microinjected cytochrome c was unable to induce any cell death in competent sympathetic neurons (Fig. 4). These results indicate that caspase-9 was the only caspase capable of being activated by cytosolic cytochrome c to induce cell death in sympathetic neurons. These data are consistent with our previous observation that caspase-9-deficient sympathetic neurons are unable to undergo apoptosis after NGF deprivation (Deshmukh et al., 2000).
Our results also show that cytosolic cytochrome c induced caspase-3 activation in competent sympathetic neurons (Fig. 5). Activation of caspase-3 presumably occurred from a direct activation by caspase-9 in these neurons. Importantly, we did not detect any activated caspase-3 in NGF-maintained neurons microinjected with cytosolic cytochrome c (Fig. 5). These results indicate that the inability of cytosolic cytochrome c to induce cell death in NGF-maintained sympathetic neurons was because of a specific block either at, or upstream of, caspase-3 activation in these neurons.
Reversal of competence requires new protein synthesis
NGF deprivation induces the development of competence by a process that takes ∼24 hr and does not require protein synthesis (Deshmukh and Johnson, 1998). We report that this state of competence was reversible if NGF (or KCl or CPTcAMP) was added to the NGF-deprived, competent neurons (Fig. 6). Notably, although the development of competence does not require protein synthesis, the reversal of competence was completely dependent on new protein synthesis (Fig. 7). Addition of cycloheximide or actinomycin D completely prevented the ability of NGF, KCl, or CPTcAMP to reverse competence, and neurons remained sensitive to cytosolic microinjection of cytochromec. Inhibiting protein synthesis with cycloheximide was consistently more effective in preventing the reversal of competence than inhibiting RNA synthesis with actinomycin D (Fig. 7). This is not surprising, because cycloheximide would inhibit all de novoprotein synthesis, whereas actinomycin D would not be able to inhibit the synthesis of proteins that use mRNAs already present in cells.
The requirement of macromolecular synthesis for reversal of competence indicates that competent sympathetic neurons need to synthesize some protein to regain resistance to cytosolic cytochrome c. Such a protein must prevent cytochrome c-mediated caspase activation either directly or indirectly in these NGF-treated neurons.
Cytosolic Smac is sufficient to induce competence in sympathetic neurons
The remarkable ability of microinjected, cytosolic mature Smac to induce competence and permit cytochrome c to induce cell death in NGF-maintained neurons (Fig. 8) has two important implications. First, these results indicate that the absolute resistance of NGF-maintained sympathetic neurons to cytosolic cytochrome c is caused by a block in caspase activation by a Smac-inhibitable, IAP family protein(s). Second, that Smac and cytochrome c induced caspase-dependent cell death in NGF-maintained neurons only when microinjected together, but neither alone, indicates that simply overcoming the IAP inhibition with exogenous Smac is not sufficient to activate caspases in these neurons. Our results also show that unlike mature Smac, Smac β was ineffective in inducing competence (Fig. 8B). Because Smac β specifically lacks the IAP BIR-3-interacting domain, and because the BIR-3 domain has been implicated in inhibiting caspase-9 (Shi, 2002), these results point to IAP inhibition of caspase-9 as an important point of regulation of caspase activation in sympathetic neurons. We note that the ability of endogenous IAPs to inhibit caspase-9 activation in sympathetic neurons has also been implicated in another recent report (Troy et al., 2001). Together, these results indicate that caspase activation in sympathetic neurons requires both the accumulation of cytochrome c in the cytosol and the elimination of some IAP-like protein that otherwise keeps caspases inhibited in these neurons.
How might NGF deprivation induce competence and permit caspase activation in sympathetic neurons? One consequence of NGF deprivation is a dramatic reduction in the global rate of protein synthesis in these neurons (Deckwerth and Johnson, 1993). Therefore, NGF deprivation may induce competence by reducing the levels of IAPs simply as a result of a fall in global protein synthesis rates. This seems unlikely to be the major mechanism, because inhibiting protein synthesis alone for 24 hr is not sufficient to induce competence in NGF-maintained sympathetic neurons (Deshmukh and Johnson, 1998). More likely, the degradation or inhibition of IAPs in competent neurons is an active process that is triggered with NGF deprivation. For example, the NGF deprivation-induced development-of-competence pathway may activate post-translational mechanisms that target the IAPs for degradation or somehow prevent the interactions of IAPs with caspases. The proteasome-mediated degradation of IAPs has been observed in thymocytes undergoing apoptosis (Yang et al., 2000).
The development-of-competence pathway may also achieve inhibition of IAPs by promoting the cytosolic translocation of endogenous Smac or other proteins such as HtrA2 that inhibit IAPs (Suzuki et al., 2001;Hegde et al., 2002; Martins et al., 2002; Verhagen et al., 2002). If this were so, such proteins must translocate to the cytosol under conditions in which neurons develop competence and cytochromec remains sequestered in the mitochondria (i.e., in the NGF-deprived, cycloheximide-treated or the NGF-deprived, Bax-deficient neurons). However, this seems unlikely, because Smac (or HtrA2) appears to be released only with or after cytochrome c in the experimental models in which this has been examined (Du et al., 2000;Verhagen et al., 2000; Adrain et al., 2001; Suzuki et al., 2001).
The observation that microinjection of cytosolic Smac was sufficient to permit cytochrome c to activate caspases raises an important question of whether the competence pathway and the release of Smac (or HtrA2) from the mitochondria are redundant mechanisms for inhibiting the IAPs in these neurons. We speculate that the critical function of IAPs in regulating caspase activation in sympathetic neurons is a failsafe mechanism that would prevent unwanted caspase activation if any accidentally damaged mitochondria (or during normal mitochondrial turnover) release cytochrome c in neurons. For such a mechanism to be effective, the IAPs must also withstand any inhibition with Smac that may be released along with cytochrome c in such accidental situations. In contrast, situations such as NGF withdrawal that activate the physiological programmed cell death pathway must completely eliminate the IAP block and permit cytochromec to activate caspases and induce apoptosis. Therefore, we propose that the competence pathway that is activated after NGF deprivation must be the predominant mechanism that removes the IAPs (probably by promoting its degradation), and that Smac, if released along with cytochrome c, would be a secondary mechanism that would ensure the complete and rapid elimination of IAPs so as to allow cytochrome c to activate caspases and induce apoptosis. The recent report describing the lack of any significant defect in the Smac knock-out mice is consistent with our hypothesis and argues against the critical importance of Smac per se in promoting caspase activation (Okada et al., 2002). In our microinjection studies, however, Smac may have been sufficient to permit caspase activation, most likely because the concentration of Smac used in these experiments was significantly greater than the intracellular concentration of Smac in these neurons.
This stringent regulation of caspase activation beyond cytochromec release in sympathetic neurons provides a strong rationale for the functional existence of IAPs and their inhibitors in these neurons. The sufficiency of microinjected cytochrome c alone to induce cell death in most other cells in which this has been examined argues against this being a generalized mechanism by which caspase activation is regulated in most cells (Li et al., 1997;Brustugun et al., 1998; Juin et al., 1999; Chang et al., 2000). As discussed above, in NGF-maintained sympathetic neurons, such a regulation provides a failsafe mechanism that prevents any accidental activation of caspases and thereby ensures the long-term survival of these neurons. Other postmitotic cells that also need to last for the lifetime of the organism may also regulate caspase activation beyond cytochrome c release by a similar mechanism.
Footnotes
We thank Keisuke Kuida for supplying us with caspase-9 knock-out mice and Stanley Korsmeyer for the Bax knock-out mice. We thank Anu Srinivasan for the anti-active caspase-3 (CM1) antibodies. We also thank Girish Putcha, Louis Chang, Patricia Osborne, and members of the Deshmukh lab for useful discussions and critical review of this manuscript.
Correspondence should be addressed to Mohanish Deshmukh, 7109E Neuroscience Research Building, University of North Carolina, Chapel Hill, NC 27599-7090. E-mail: mohanish@med.unc.edu.
REFERENCES
- 1.Adrain C, Creagh EM, Martin SJ. Apoptosis-associated release of Smac/DIABLO from mitochondria requires active caspases and is blocked by Bcl-2. EMBO J. 2001;20:6627–6636. doi: 10.1093/emboj/20.23.6627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brustugun OT, Fladmark KE, Doskeland SO, Orrenius S, Zhivotovsky B. Apoptosis induced by microinjection of cytochrome c is caspase-dependent and is inhibited by Bcl-2. Cell Death Differ. 1998;5:660–668. doi: 10.1038/sj.cdd.4400399. [DOI] [PubMed] [Google Scholar]
- 3.Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol. 1999;15:269–290. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- 4.Chang SH, Phelps PC, Berezesky IK, Ebersberger MJ, Trump BF. Studies on the mechanisms and kinetics of apoptosis induced by microinjection of cytochrome c in rat kidney tubule epithelial cells (NRK-52E). Am J Pathol. 2000;156:637–649. doi: 10.1016/S0002-9440(10)64768-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Datta SR, Brunet A, Greenberg ME. Cellular survival: a play in three Akts. Genes Dev. 1999;13:2905–2927. doi: 10.1101/gad.13.22.2905. [DOI] [PubMed] [Google Scholar]
- 6.Deckwerth TL, Johnson EM., Jr Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J Cell Biol. 1993;123:1207–1222. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Deckwerth TL, Elliott JL, Knudson CM, Johnson EM, Jr, Snider WD, Korsmeyer SJ. Bax is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- 8.Deshmukh M. Caspases in ischaemic brain injury and neurodegenerative disease. Apoptosis. 1998;3:387–394. doi: 10.1023/a:1009602401251. [DOI] [PubMed] [Google Scholar]
- 9.Deshmukh M, Johnson EM., Jr Programmed cell death in neurons: focus on the pathway of nerve growth factor deprivation-induced death of sympathetic neurons. Mol Pharmacol. 1997;51:897–906. doi: 10.1124/mol.51.6.897. [DOI] [PubMed] [Google Scholar]
- 10.Deshmukh M, Johnson EM., Jr Evidence of a novel event during neuronal death: development of competence-to-die in response to cytoplasmic cytochrome c. Neuron. 1998;21:695–705. doi: 10.1016/s0896-6273(00)80587-5. [DOI] [PubMed] [Google Scholar]
- 11.Deshmukh M, Johnson EM., Jr Staurosporine-induced neuronal death: multiple mechanisms and methodological implications. Cell Death Differ. 2000;7:250–261. doi: 10.1038/sj.cdd.4400641. [DOI] [PubMed] [Google Scholar]
- 12.Deshmukh M, Vasilakos J, Deckwerth TL, Lampe PA, Shivers BD, Johnson EM., Jr Genetic and metabolic status of NGF-deprived sympathetic neurons saved by an inhibitor of ICE-family proteases. J Cell Biol. 1996;135:1341–1354. doi: 10.1083/jcb.135.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deshmukh M, Kuida K, Johnson EM., Jr Caspase inhibition extends the commitment to neuronal death beyond cytochrome c release to the point of mitochondrial depolarization. J Cell Biol. 2000;150:131–143. doi: 10.1083/jcb.150.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deveraux QL, Reed JC. IAP family proteins–suppressors of apoptosis. Genes Dev. 1999;13:239–252. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- 15.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 16.Edwards SN, Buckmaster AE, Tolkovsky AM. The death programme in cultured sympathetic neurones can be suppressed at the posttranslational level by nerve growth factor, cyclic AMP, and depolarization. J Neurochem. 1991;57:2140–2143. doi: 10.1111/j.1471-4159.1991.tb06434.x. [DOI] [PubMed] [Google Scholar]
- 17.Franklin JL, Johnson EM., Jr Suppression of programmed neuronal death by sustained elevation of cytoplasmic calcium. Trends Neurosci. 1992;15:501–508. doi: 10.1016/0166-2236(92)90103-f. [DOI] [PubMed] [Google Scholar]
- 18.Hegde R, Srinivasula SM, Zhang Z, Wassell R, Mukattash R, Cilenti L, DuBois G, Lazebnik Y, Zervos AS, Fernandes-Alnemri T, Alnemri ES. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J Biol Chem. 2002;277:432–438. doi: 10.1074/jbc.M109721200. [DOI] [PubMed] [Google Scholar]
- 19.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 20.Johnson MI, Argiro V. Techniques in the tissue culture of rat sympathetic neurons. Methods Enzymol. 1983;103:334–347. doi: 10.1016/s0076-6879(83)03022-0. [DOI] [PubMed] [Google Scholar]
- 21.Juin P, Hueber AO, Littlewood T, Evan G. c-Myc-induced sensitization to apoptosis is mediated through cytochrome c release. Genes Dev. 1999;13:1367–1381. doi: 10.1101/gad.13.11.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Knudson CM, Tung KS, Tourtellotte WG, Brown GA, Korsmeyer SJ. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science. 1995;270:96–99. doi: 10.1126/science.270.5233.96. [DOI] [PubMed] [Google Scholar]
- 23.Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase 9. Cell. 1998;94:325–337. doi: 10.1016/s0092-8674(00)81476-2. [DOI] [PubMed] [Google Scholar]
- 24.Li F, Srinivasan A, Wang Y, Armstrong RC, Tomaselli KJ, Fritz LC. Cell-specific induction of apoptosis by microinjection of cytochrome c. Bcl-XL has activity independent of cytochrome c release. J Biol Chem. 1997;272:30299–30305. doi: 10.1074/jbc.272.48.30299. [DOI] [PubMed] [Google Scholar]
- 25.Liu Z, Sun C, Olejniczak ET, Meadows RP, Betz SF, Oost T, Herrmann J, Wu JC, Fesik SW. Structural basis for binding of Smac/DIABLO to the XIAP BIR3 domain. Nature. 2000;408:1004–1008. doi: 10.1038/35050006. [DOI] [PubMed] [Google Scholar]
- 26.Martin DP, Schmidt RE, DiStefano PS, Lowry OH, Carter JG, Johnson EM., Jr Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J Cell Biol. 1988;106:829–844. doi: 10.1083/jcb.106.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinou I, Desagher S, Eskes R, Antonsson B, Andre E, Fakan S, Martinou JC. The release of cytochrome c from mitochondria during apoptosis of NGF-deprived sympathetic neurons is a reversible event. J Cell Biol. 1999;144:883–889. doi: 10.1083/jcb.144.5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martins LM, Iaccarino I, Tenev T, Gschmeissner S, Totty NF, Lemoine NR, Savopoulos J, Gray CW, Creasy CL, Dingwall C, Downward J. The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a reaper-like motif. J Biol Chem. 2002;277:439–444. doi: 10.1074/jbc.M109784200. [DOI] [PubMed] [Google Scholar]
- 29.Mattson MP. Apoptosis in neurodegenerative disorders. Nat Rev Mol Cell Biol. 2000;1:120–129. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- 30.McCarthy MJ, Rubin LL, Philpott KL. Involvement of caspases in sympathetic neuron apoptosis. J Cell Sci. 1997;110:2165–2173. doi: 10.1242/jcs.110.18.2165. [DOI] [PubMed] [Google Scholar]
- 31.Neame SJ, Rubin LL, Philpott KL. Blocking cytochrome c activity within intact neurons inhibits apoptosis. J Cell Biol. 1998;142:1583–1593. doi: 10.1083/jcb.142.6.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okada H, Suh WK, Jin J, Woo M, Du C, Elia A, Duncan GS, Wakeham A, Itie A, Lowe SW, Wang X, Mak TW. Generation and characterization of Smac/DIABLO-deficient mice. Mol Cell Biol. 2002;22:3509–3517. doi: 10.1128/MCB.22.10.3509-3517.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 34.Putcha GV, Deshmukh M, Johnson EM., Jr BAX translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, BCL-2, and caspases. J Neurosci. 1999;19:7476–7485. doi: 10.1523/JNEUROSCI.19-17-07476.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roberts DL, Merrison W, MacFarlane M, Cohen GM. The inhibitor of apoptosis protein-binding domain of Smac is not essential for its proapoptotic activity. J Cell Biol. 2001;153:221–228. doi: 10.1083/jcb.153.1.221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ruit KG, Elliott JL, Osborne PA, Yan Q, Snider WD. Selective dependence of mammalian dorsal root ganglion neurons on nerve growth factor during embryonic development. Neuron. 1992;8:573–587. doi: 10.1016/0896-6273(92)90284-k. [DOI] [PubMed] [Google Scholar]
- 37.Rydel RE, Greene LA. cAMP analogs promote survival and neurite outgrowth in cultures of rat sympathetic and sensory neurons independently of nerve growth factor. Proc Natl Acad Sci USA. 1988;85:1257–1261. doi: 10.1073/pnas.85.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scott BS, Fisher KC. Potassium concentration and number of neurons in cultures of dissociated ganglia. Exp Neurol. 1970;27:16–22. doi: 10.1016/0014-4886(70)90197-4. [DOI] [PubMed] [Google Scholar]
- 39.Shi Y. Mechanisms of caspase activation and inhibition during apoptosis. Mol Cell. 2002;9:459–470. doi: 10.1016/s1097-2765(02)00482-3. [DOI] [PubMed] [Google Scholar]
- 40.Simons M, Beinroth S, Gleichmann M, Liston P, Korneluk RG, MacKenzie AE, Bahr M, Klockgether T, Robertson GS, Weller M, Schulz JB. Adenovirus-mediated gene transfer of inhibitors of apoptosis protein delays apoptosis in cerebellar granule neurons. J Neurochem. 1999;72:292–301. doi: 10.1046/j.1471-4159.1999.0720292.x. [DOI] [PubMed] [Google Scholar]
- 41.Srinivasan A, Roth KA, Sayers RO, Shindler KS, Wong AM, Fritz LC, Tomaselli KJ. In situ immunodetection of activated caspase-3 in apoptotic neurons in the developing nervous system. Cell Death Differ. 1998;5:1004–1016. doi: 10.1038/sj.cdd.4400449. [DOI] [PubMed] [Google Scholar]
- 42.Srinivasula SM, Datta P, Fan XJ, Fernandes-Alnemri T, Huang Z, Alnemri ES. Molecular determinants of the caspase-promoting activity of Smac/DIABLO and its role in the death receptor pathway. J Biol Chem. 2000;275:36152–36157. doi: 10.1074/jbc.C000533200. [DOI] [PubMed] [Google Scholar]
- 43.Suzuki Y, Imai Y, Nakayama H, Takahashi K, Takio K, Takahashi R. A serine protease, HtrA2, is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell. 2001;8:613–621. doi: 10.1016/s1097-2765(01)00341-0. [DOI] [PubMed] [Google Scholar]
- 44.Troy CM, Stefanis L, Prochiantz A, Greene LA, Shelanski ML. The contrasting roles of ICE family proteases and interleukin-1β in apoptosis induced by trophic factor withdrawal and by copper/zinc superoxide dismutase down-regulation. Proc Natl Acad Sci USA. 1996;93:5635–5640. doi: 10.1073/pnas.93.11.5635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Troy CM, Rabacchi SA, Hohl JB, Angelastro JM, Greene LA, Shelanski ML. Death in the balance: alternative participation of the caspase-2 and -9 pathways in neuronal death induced by nerve growth factor deprivation. J Neurosci. 2001;21:5007–5016. doi: 10.1523/JNEUROSCI.21-14-05007.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsui-Pierchala BA, Putcha GV, Johnson EM., Jr Phosphatidylinositol 3-kinase is required for the trophic, but not the survival-promoting, actions of NGF on sympathetic neurons. J Neurosci. 2000;20:7228–7237. doi: 10.1523/JNEUROSCI.20-19-07228.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 48.Verhagen AM, Silke J, Ekert PG, Pakusch M, Kaufmann H, Connolly LM, Day CL, Tikoo A, Burke R, Wrobel C, Moritz RL, Simpson RJ, Vaux DL. HtrA2 promotes cell death through its serine protease activity and its ability to antagonize inhibitor of apoptosis proteins. J Biol Chem. 2002;277:445–454. doi: 10.1074/jbc.M109891200. [DOI] [PubMed] [Google Scholar]
- 49.Vlahos CJ, Matter WF, Hui KY, Brown RF. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002). J Biol Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- 50.Wiese S, Digby MR, Gunnersen JM, Gotz R, Pei G, Holtmann B, Lowenthal J, Sendtner M. The anti-apoptotic protein ITA is essential for NGF-mediated survival of embryonic chick neurons. Nat Neurosci. 1999;2:978–983. doi: 10.1038/14777. [DOI] [PubMed] [Google Scholar]
- 51.Wu G, Chai J, Suber TL, Wu JW, Du C, Wang X, Shi Y. Structural basis of IAP recognition by Smac/DIABLO. Nature. 2000;408:1008–1012. doi: 10.1038/35050012. [DOI] [PubMed] [Google Scholar]
- 52.Yang Y, Fang SY, Jensen JP, Weissman AM, Ashwell JD. Ubiquitin protein ligase activity of IAPs and their degradation in proteasomes in response to apoptotic stimuli. Science. 2000;288:874–877. doi: 10.1126/science.288.5467.874. [DOI] [PubMed] [Google Scholar]
- 53.Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- 54.Zhou H, Li XM, Meinkoth J, Pittman RN. Akt regulates cell survival and apoptosis at a postmitochondrial level. J Cell Biol. 2000;151:483–494. doi: 10.1083/jcb.151.3.483. [DOI] [PMC free article] [PubMed] [Google Scholar]