

Abstract
Huntington's disease (HD) is characterized by a progressive loss of neurons in the striatum and cerebral cortex and is caused by a CAG repeat expansion in the gene encoding huntingtin. Mice with the mutation inserted into their own huntingtin gene (knock-in mice) are, genetically, the best models of the human disease. Here we show for the first time that knock-in mice with 94 CAG repeats develop a robust and early motor phenotype at 2 months of age, characterized by increased rearing at night. This initial increase in repetitive movements was followed by decreased locomotion at 4 and 6 months, despite a normal life span. The decrease in striatal enkephalin mRNA that is known to occur at 4 months was not present at 2 months, when increased rearing was observed. Both the hyperactive and hypoactive phases of motor dysfunction preceded the detection of nuclear microaggregates of mutated huntingtin in striatal neurons. Nuclear microaggregates, defined as small huntingtin-positive punctas detected by light microscopy, were very rare at 4 months but became widely distributed in striatal neurons at 6 months. Nuclear inclusions did not appear until 18 months. When present, nuclear microaggregates predominated in the striosomal compartment of the striatum, providing a possible explanation for the different neuronal vulnerability of striatal compartments observed in humans. The early motor phenotype observed in the knock-in mouse is reminiscent of repetitive movements often observed in early HD and provides a novel opportunity to assess the ability of therapies to prevent the initial effects of the mutationin vivo.
Keywords: mouse, Huntington's disease, behavior, striosomes, microaggregates, nuclear inclusions, μ-opioid receptor, knock-in, striatum, immunohistochemistry, CAG repeats
The mutation causing Huntington's disease (HD) is a CAG repeat expansion in the gene encoding huntingtin (The Huntington's Disease Collaborative Research Group, 1993). Progress in understanding this disease depends critically on the generation of a wide range of mouse models because each model only partially reproduces disease characteristics (Chesselet and Levine, 2000; Menalled and Chesselet, 2001). In mice with an expanded CAG repeat inserted within the normal gene (“knock-in” mice), the mutation resides in the same genomic context as in patients (White et al., 1997; Levine et al., 1999; Shelbourne et al., 1999; Wheeler et al., 2000; Lin et al., 2001). Therefore, knock-in mice should provide an ideal model for understanding the progressive motor dysfunction and loss of striatal and cortical neurons characteristic of HD (Vonsattel et al., 1985; Harper, 1996).
Knock-in mice, however, have been much less studied than transgenics, primarily because they do not present any overt and robust motor anomalies at an early age (Shelbourne et al., 1999; Wheeler et al., 2000; Lin et al., 2001). Yet early cellular dysfunction exists in these mice, as indicated by molecular and electrophysiological alterations (Levine et al., 1999; Usdin et al., 1999; Menalled et al., 2000). A first goal of the present study was to examine in greater detail the behavior of knock-in mice with 94 CAG repeats inserted into the mouseHdh gene by homologous recombination (Levine et al., 1999). We report biphasic motor alterations in these mice, starting as early as 2 months of age, with an increase in repetitive movements followed at 4 and 6 months by decreased locomotion.
Because the progression of the disease is slow in HD knock-in mice, they are ideal for establishing the temporal relationship between various effects of the mutation. In particular, nuclear aggregates of huntingtin are a pathological hallmark of the disease both in mice and in humans (Davies at al. 1997; DiFiglia et al., 1997). Whether they are responsible for the anomalies caused by the mutation is unclear. A causal relationship was originally suggested because aggregates and abnormal behavior tended to appear at the same time in many mouse models. Results in postmortem human brain, as well as in some in vivo and in vitro models of HD and other CAG repeat diseases, have begun to challenge this hypothesis; however, the issue remains highly controversial (Klement et al., 1998; Saudou et al., 1998; Gutekunst et al., 1999; Kim et al., 1999; Kuemmerle et al., 1999).
A second goal of this study was to examine the regional and temporal distribution of huntingtin microaggregates in relation to behavioral and molecular alterations occurring over time in the same knock-in mice. The data show that nuclear aggregates can only be detected several months after the onset of other phenotypes in these mice, thus arguing against a primary role of protein aggregates in the earliest manifestations of the mutation. When they appear, the aggregates are more abundant in the striosomal compartment of the striatum, indicating a striking regional selectivity of the pathology in these knock-in mice.
MATERIALS AND METHODS
Animals
Animal care was in accordance with the United States Public Health Service Guide for the Care and Use of Laboratory Animals, and procedures were approved by the Institutional Animal Care and Use Committee at University of California Los Angeles (UCLA). The knock-in mice contained a chimeric mouse/human exon 1 with 94 CAG repeats interrupted by a CGG triplet in position 42 and inserted into the mouse gene by homologous targeting (Levine et al., 1999).
Knock-in mice homozygous for the 94 CAG repeat mutation were backcrossed once with C57BL/6 mice, and their progeny were then crossed to generate the homozygous and wild-type parents of the mice used in this study. Most of the mice in the study were F1 from this cross, and none were farther than F2. Homozygous and wild-type mice were matched for gender. Experiments were performed in 2-, 4-, and 6-month-old mice, and each group (n = 5–13) contained animals of both genders from three to five litters.
Open field testing
Knock-in and wild-type mice were subjected to the same handling and feeding schedules. Behavioral testing was performed from 6:30 until 10:30 P.M. (i.e., during the dark phase of the diurnal cycle) in the room in which they were normally housed. All measurements were made during the first 15 min after placement of the mice in a novel open field under red light (25 W). The open field consisted of a clear Plexiglas box (21 × 31 × 45 cm), with a floor divided into two rows of three squares (15 × 16.5 cm) each, that was placed in the center of a fume hood to avoid contamination (Lab Products Inc., Seaford, DE). Each mouse was initially placed in the middle square of the back row. Quantitative analysis was done simultaneously on three behaviors: the number of lines crossed by all four limbs (locomotor activity), the number of rears (lifting forelimbs off the ground and standing on hindlimbs regardless of occurrence on or off the walls), and the number of grooming episodes (licking, scratching, or cleaning any body part). The open field box was cleaned with Power-Cide Plus (Intercem Corporation, Anaheim, CA) before the testing of each mouse. Each mouse was tested in the open field for 15 min and filmed with an 8 mm Canon ES270 video camera (Canon Inc., Irvine, CA) for further quantification. Each 15 min of testing was analyzed as three periods of 5 min intervals to study the influence of novelty in the measured behavior. The animals were coded, and the researcher was blind to the genotype of the mice during testing and later during analysis of the tapes. Each mouse was tested only once in the course of the study to avoid the confounding effects of habituation to the open field.
Immunohistochemistry
For light microscopic studies, mice were perfused transcardially with 4% paraformaldehyde and 0.5% glutaraldehyde in 0.1 mphosphate buffer (PB), pH 7.2, except in the case of two mice (6-month-old knock-ins) that were perfused with paraformaldehyde alone. The brains were removed, postfixed in 4% paraformaldehyde in 0.1m PB for 6–8 hr at 4°C, cryoprotected in 30% sucrose, and cut into 35-μm-thick coronal sections on a Leica CM 1800 cryostat (Deerfield, IL). Sections were processed for immunostaining with EM48, an antibody that preferentially recognizes aggregated huntingtin (Gutekunst et al., 1999). After washes in 0.01 m PBS, the sections were treated with H2O2 (1%) in PBS containing 0.5% of Triton X-100 for 20 min to block endogenous peroxidase activity. Nonspecific sites were blocked by incubating the sections for 30 min at room temperature in PBS containing 3% bovine serum albumin and 2% normal goat serum (NGS). The sections were incubated with EM48 (1:300) in PBS containing 3% bovine serum albumin, 2% NGS, 0.08% sodium azide, and 0.2% Triton X-100 (incubation buffer) for 24 hr at room temperature. After rinses in PBS, the sections were incubated in biotinylated goat anti-rabbit antibody (1:200) (Vector ABC Elite, Burlingame, CA) in the incubation buffer for 2 hr at room temperature. After several rinses in PBS, the sections were incubated for 2 hr in avidin–biotin complex (Vector ABC Elite) in PBS containing 0.2% Triton X-100. Immunoreactivity was visualized by incubation in 0.03% 3–3′-diaminobenzidine tetrahydrochloride (Sigma, St. Louis, MO) and 0.0006% H2O2 in 0.05 mTris buffer, pH 7.6. After rinses in cold Tris buffer, the sections were dehydrated, defatted in xylene, and mounted with Eukitt (Calibrated Instruments, Hawthorne, NY). Some sections were counterstained with methyl green (Sigma) before mounting to identify the cell nuclei.
Immunostaining with a polyclonal antiserum against μ-opiate receptor (MOR-1) (kindly donated by Dr. R. P. Elde, University of Minnesota, Minneapolis, MN) was used to identify the striosomal compartment of the striatum. Immunostaining was performed according to a protocol from Harlan and Graybiel, slightly modified fromCanales and Graybiel (2000). Briefly, sections for MOR-1 immunostaining were washed with 0.01 m PBS containing 0.2% Triton X-100 (PBS-TX) and treated for 10 min with H2O2 (3%) in PBS-TX to block endogenous peroxidase. Nonspecific sites were blocked for 30 min at room temperature in PBS-TX containing 5% NGS. The sections were incubated with MOR-1 at a dilution of 1:50,000 in PBS containing 1% normal rat serum, 1% NGS, 0.1% sodium azide, and 1% Triton X-100 for 48 hr at room temperature. After rinses in PBS-TX, the sections were incubated in biotinylated goat anti-rabbit antibody (1:500) (Vector ABC Elite) in PBS-TX containing 1% NGS, for 1 hr at room temperature. After several rinses in PBS-TX, the sections were incubated for 20 min in biotinyl tyramide (0.1% of stock solution) (NEN Life Science Products, Boston, MA) in PBS-TX containing H2O2 (0.005%). After several washes in PBS-TX, the sections were incubated in avidin–biotin complex (Vector ABC Elite) in PBS-TX containing 1% NGS for 1 hr. Immunoreactivity was visualized by incubation in 0.02% 3–3′-diaminobenzidine tetrahydrochloride, 0.08% nickel ammonium sulfate, and 0.0066% H2O2in 0.1 m PB. All sections were dehydrated, defatted in xylene, and mounted with Eukitt (Calibrated Instruments).
For electronmicroscopic studies, immunogold labeling was performed as described previously (Li et al., 1999, 2000). Briefly, mice were perfused transcardially with 4% paraformaldehyde and 0.2% glutaraldehyde in 0.1 m PB, pH 7.2. The brains were removed, postfixed with 4% paraformaldehyde in 0.1 m PB for 6–8 hr at 4°C, and then sectioned with a vibratome. Brain sections were incubated with EM48 antibody in PBS containing 4% NGS for 24–60 hr at 4°C and then with Fab fragments of goat anti-rabbit secondary antibodies (1:50) conjugated to 1.4 nm gold particles (Nanoprobes Inc., Stony Brook, NY) in PBS with 4% NGS overnight at 4°C. After rinsing in PBS, sections were fixed again in 2% glutaraldehyde in PB for 1 hr, silver intensified using the IntenSEM kit (Amersham International, Buckinghamshire, UK), osmicated in 1% OsO4 in PB, and stained overnight in 2% aqueous uranyl acetate. Sections were dehydrated in ascending concentrations of ethanol and propylene oxide/Eponate 12 (1:1) and embedded in Eponate 12 (Ted Pella, Redding, CA). Ultrathin sections (60 nm) were cut with a Leica Ultracut S ultramicrotome. Thin sections were counterstained with 5% aqueous uranyl acetate for 5 min followed by Reynolds lead citrate for 5 min and examined with a Hitachi H-7500 electron microscope.
Data analysis
Sections processed for light microscopy were examined under bright-field illumination with a Zeiss Axioskop microscope (Göttingen, Germany). To analyze the distribution of groups of cells containing dense clusters of microaggregates within the striosome-matrix organization of the striatum, serially adjacent tissue sections (35 μm) were processed for EM48 and MOR-1, respectively, and were examined at low-power magnification (6.3×) on an Olympusmicroscope (Tokyo, Japan) equipped with a camera lucida. Two or three pairs of serially adjacent coronal sections taken at the level of the striatum were analyzed in each mouse. A chart of each section was drawn with the camera lucida. Morphological landmarks such as the corpus callosum, ventricle, and blood vessels were used to overlie each corresponding pair of charts. The clusters of cells intensely labeled with EM48 were sketched onto the drawings of the striosomes identified as areas of dense immunohistochemical staining for the μ-opioid receptor. By adjusting the intensity of the microscope light and the illumination of the page, the investigator was prevented from seeing the outlines of striosomes on the maps while sketching the clusters of intense labeling for EM48. Composite maps of each couple of sections were drawn by two independent investigators to confirm the results.
The composite maps were digitized with a Hewlett-Packard ScanJet 4 (Roseville, CA), and the images were visualized and analyzed with a Power Macintosh 9500 equipped with the public domain NIH program version 1.6 (Bethesda, MD). The surface area of each cluster of cells with intense EM48 immunostaining was measured and then added to calculate the total surface area (A) containing darkly labeled cells in each composite. The surface area of the clusters that did not overlap with the striosomes was also measured (B). The surface area that colocalized with the striosomes (C) was calculated as the subtractionA − B = C. The surface area of the striosomes (defined as areas of positive MOR-1 labeling) was measured and then added to obtain a total striosome area (D) on each composite. Finally, the total area of the striatum was measured (S), and the extrastriosomal matrix area (M) was obtained by subtractingS − D = M. The ratio of cluster areas with intense EM48 immunostaining within the striosomes (C) to the total area of clusters with intense EM48 immunostaining (A) was calculated byC/A. Values obtained in two or three pairs of sections were averaged for each animal, and the resulting numbers were used to calculate group means.
Filter trap assay
Cerebral hemisphere of knock-in (2, 7, and 11 month old) and wild-type (4 month old) mice were homogenized and fractionated according to Aronin et al., 1991. Briefly, 1 gm of tissue was homogenized with 0.25 m sucrose, 15 mmTris-HCl, pH 7.9, 60 mm KCl, 15 mm NaCl, 5 mm EDTA, and 1 mm EGTA, and the homogenate was spun at 2000 × g for 10 min. The pellet was resuspended and agitated in 10 mm HEPES, pH 7.9, 1.5 mm MgCl2 and 10 mm KCl for 10 min, and the extract was spun at 4000 × g for 10 min. The pellet was then resuspended in 0.5 HEPES, pH 7.9, 0.75 mmMgCl2, 0.5 mm EDTA, 0.5m KCl, and 12.5% glycerol, agitated for 30 min, and spun at 14,000 × g for 30 min. All the above steps were performed at 4°C. The supernatants of each spin contained soluble proteins. The final pellet was sonicated and heated at 95°C in 10% SDS solution to reach maximal suspension. Large insoluble debris was removed by a quick spin at 2000 × g. Proteins were quantified using the BCA Protein Assay Kit (Pierce, Rockford, IL). Identical amounts of protein were filtered through a cellulose acetate membrane with an Easy-Titer ELIFA System (Pierce) and fixed to the membrane with a solution of 0.1% glutaraldehyde in Tris-buffered saline (TBS). The membranes were washed with TBS, and SDS-insoluble aggregates retained in the filter were detected by incubation with anti-ubiquitin antibody (1:500) (Chemicon, Temecula, CA), followed by an anti-rabbit secondary antibody conjugated with horseradish peroxidase (1:5000) (Chemicon). The signal was developed with the Enhanced Chemoluminescence (ECL) system as described by the manufacturer (NEN Life Science Products).
In situ hybridization histochemistry
The cDNA for preproenkephalin was generously provided by Dr. S. L. Sabol (National Institute of Mental Health, Bethesda, MD). The plasmid containing the cDNA was linearized with SacI (Promega, Madison, WI), and an 35S-labeled RNA probe was generated (Chesselet et al., 1987). In situhybridization was performed on 10 μm fresh-frozen sections as described (Chesselet et al., 1987). All tissue sections were processed for film autoradiography with 3H-Hyperfilm (Amersham, Arlington Heights, IL). Films were digitized with a Hewlett-Packard ScanJet 4c, and the image was visualized and analyzed with a Power Macintosh 9500 equipped with the public domain NIH Image program version 1.6. Gray values were converted into optical densities with the help of a standard curve generated from Kodak autoradiographic standards (Kodak, Rochester, NY). Optical measurements were taken separately for the right and left dorsolateral striatum. These measurements were averaged to determine a single value per section. Values obtained in two sections were averaged for each animal, and the resulting number was used to calculate the group means.
Stereological studies
The optical dissector method with the following modifications was used to estimate the total number of neurons in the striatum of knock-in and wild-type mice (Oorschot, 1996). Briefly, mice were perfused through the heart with 4% paraformaldehyde and 0.5% glutaraldehyde in 0.1 m PB, pH 7.2. The brains were removed, postfixed in 4% paraformaldehyde in 0.1 m PB for 6–8 hr at 4°C, and cryoprotected in 30% sucrose. Sections through one cerebral hemisphere were cut in a coronal plane with a Leica CM 1800 cryostat at 40 μm. Sections were stored in cryoprotective solutions. The first sections containing the striatum were identified, and the first section to be analyzed was chosen at random from the first 10 sections containing the striatum. This section and every 10th section (sampling interval) thereafter were stained with 0.5% cresyl violet (Sigma), dehydrated, defatted in xylene, and mounted with Eukitt (Calibrated Instruments). Sections were digitized (Image-Pro Plus, Media Cybernetics, Silver Spring, MD) using a 2× lens (Olympus BX60, Ludl Electronic Products Ltd., Hawthorne, NY). A grid (squares of 198 × 198 μm) was superimposed on the image of the section, and the final image was printed out. The printout of each section was used as a guide. The striatum was outlined, and every sixth intersection of the grid (from the random starting point) was sampled when it was located within the outline of the striatum. The frames to be analyzed were localized in the section with an XYZ programmable motorized stage. For each sampled area, the number of nuclei (Q) that came into focus throughout 10 μm (h) within the thickness of the section and did not fall on the sampling frame borders was counted. The analysis was done with a 100× lens, and the frame had an area of 2730 μm (a). The printouts were digitized with a Hewlett-Packard ScanJet 4c, and the area of the striatum (A) was calculated with NIH Image version 1.6. The total volume of the striatum (Vref) was calculated with the Cavalieri method, whereas the neuronal density (NV) and finally the total number of neurons were calculated following the protocols described byOorschot (1996).
Statistical analysis
Behavioral experiments. Behavioral observations over the 15 min observation period were expressed as mean ± SEM. Data were analyzed by a three-way ANOVA considering the genotype (wild-type or knock-in), the age (2, 4, and 6 months of age), and the time (0–5, 5–10, and 10–15 min in the open field) as main factors. Note that separated cohorts of mice were studied at each age to prevent the confounding effect of habituation to the open field; therefore, no repeated measures were performed. When a significant interaction was found (p < 0.05), a post hoc Fisher PLSD test was performed for comparisons between groups.
In situ hybridization histochemistry and stereological results. Comparisons between the knock-in and wild-type mice were made with an unpaired two-tailed Student's t test.
Location of microaggregate-containing cells with respect to the striosomal compartments. The χ2goodness-of-fit test was used to determine whether the areas with intensely labeled cells were randomly distributed in the striatum.
Three-way ANOVAs were performed by the Mental Retardation Research Center Biostatistical Core at UCLA, and all other analyses were performed with the StatView Interactive Statistics and Graphics Package (version 5.0.1, SAS Institute Inc., Cary, NC). In all cases,p < 0.05 was considered significant.
RESULTS
Behavioral studies
Rearing
As we reported previously (Menalled et al., 2000), knock-in mice did not show any obvious abnormal behavior, such as clasping or tremor, up to 2 years of age. The earliest abnormal behavior observed in the knock-in mice was an increase in rearing activity during the dark phase of the diurnal cycle, which corresponds to the time of maximum motor activity in mice (Fig.1A). A rear was recorded when the animal lifted up both forelimbs, regardless of their contact with the wall of the open field. Analysis revealed an effect of age on the number of rears (ANOVA,F(2,67) = 3.82; p < 0.03) and a strong interaction between age and genotype (ANOVA, genotype × age, F(2,67) = 6.33;p = 0.003). In contrast to the marked increase in rearing observed at 2 months, knock-in mice showed significantly fewer rears than wild-type at 4 months (Fig. 1A). Furthermore, although wild-type mice displayed a significant increase in the number of rears at 4 and 6 months compared with 2 months of age, knock-in mice presented no significant changes in number of rears as they aged (Fig. 1A). Comparison of wild-type and knock-in mice during three successive periods of 5 min in the open field revealed that the increase in rears in 2-month-old mice was significant during the first 10 min of observation, indicating that the effects persisted beyond the initial period of exploration in a novel environment (Fig. 2).
Fig. 1.
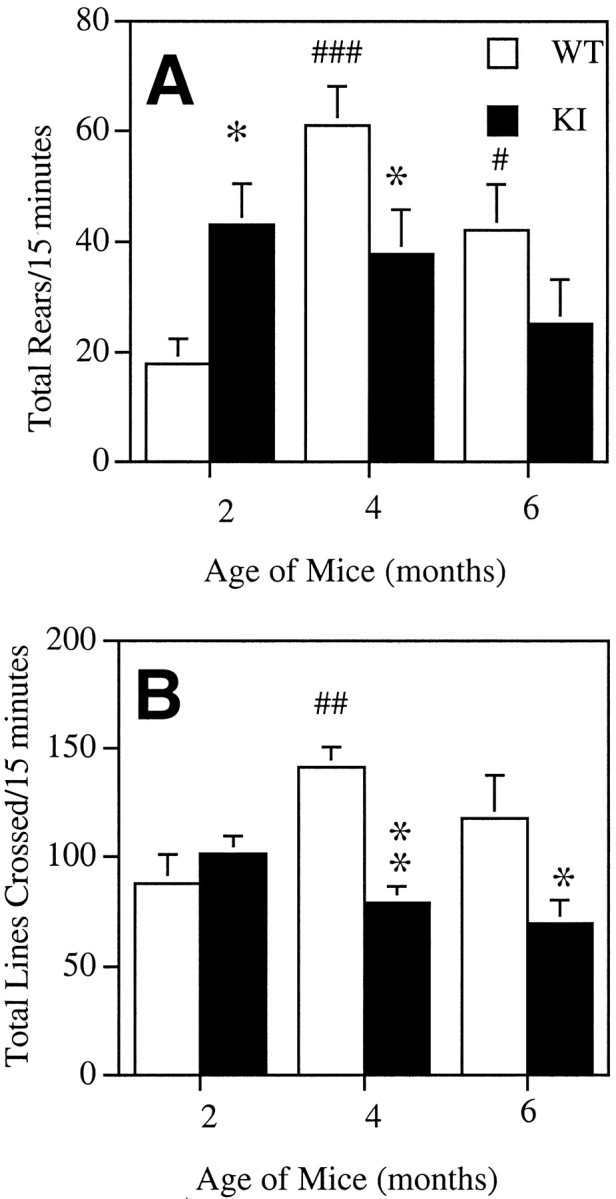
Number of rears (A) and locomotor activity (B) of 2-, 4-, and 6-month-old wild-type (open bars) and knock-in mice (filled bars). The data are the mean ± SEM of the total number of rears (A) and lines crossed (B) in 15 min (n = 12–13 per group). *p < 0.05, **p < 0.001 when compared with wild-type mice at the same age; #p < 0.05,##p < 0.005,###p < 0.0001 compared with 2-month-old wild-types (ANOVA followed by Fisher PLSD tests).
Fig. 2.

Number of rears (top panels) and locomotor activity (bottom panels) of 2-, 4-, and 6-month-old wild-type (○) and knock-in mice (●). Data are the mean ± SEM of the number of rears and lines crossed in three successive 5 min internals in the open field (n = 12–13 per group). *p < 0.05 when compared with corresponding wild-type mice during the same time period.
Locomotor activity
The locomotor activity of knock-in and wild-type mice was calculated by measuring the number of lines crossed during the first 15 min in the open field (Fig. 1B). Analysis revealed a strong effect of genotype (ANOVA, F(2,67) = 10.47; p < 0.002) and a strong interaction of genotype with age (ANOVA, genotype × age:F(2,67) = 5.44; p < 0.007). Although no difference was observed between 2-month-old knock-in and wild-type mice, a significant decrease in locomotor activity was detected in 4- and 6-month-old knock-in mice compared with age-matched wild types (Fig. 1B). As for rearing activity, wild types showed an increased locomotor activity with age, whereas knock-in mice did not (Fig. 1B). Importantly, the marked decrease in locomotor activity observed in 4-month-old knock-in mice persisted during the entire period of observation (Fig.2). At 6 months, the effect was more modest and significant only during the first 5 min (Fig. 2).
Grooming
A grooming episode was recorded every time the animal licked, scratched, or cleaned any body part. The mean number of grooms in 15 min ± SEM in knock-in versus wild-type 2-, 4- or 6-month-old mice was very similar (6.7 ± 1.1 vs 7.3 ± 1.1, 8 ± 1.5 vs 7.9 ± 1.8, 7 ± 1.2 vs 12.5 ± 2.2, respectively), and three-way ANOVA did not uncover any significant interactions of genotype, age, or time.
Levels of enkephalin mRNA in the striatum of knock-in mice
We have shown previously that striatal neurons of 4-month-old knock-in mice exhibited decreased levels (47%) of enkephalin mRNA without significant changes in the level of expression of mRNAs encoding substance P and glutamic acid decarboxylase (Mr 67,000, GAD67; Mr65,000, GAD65) (Menalled et al., 2000). To determine whether the early behavioral changes in rearing coincided with or preceded changes in enkephalin mRNA, the level of expression of enkephalin mRNA was examined in the striatum of 2-month-old mice, i.e., when early motor anomalies were detected. Sections were processed for in situhybridization histochemistry with a35S-labeled RNA probe complementary to preproenkephalin mRNA. Quantitative analysis of film autoradiograms did not reveal any significant differences in the level of enkephalin mRNA in the striatum of 2-month-old knock-in versus wild-type mice (wild-type mice 0.417 ± 0.016, knock-in mice 0.395 ± 0.026, mean optical density ± SEM; n = 7; unpaired Student's t test; p = 0.4769).
Stereological studies
Stereological methods were used to estimate the total number of medium spiny neurons in the striatum of 18- to 26-month-old knock-in and wild-type mice (Table 1). No significant differences were detected in the total number of these neurons. However, the total volume of the striatum of knock-in mice was significantly reduced compared with wild type. Consequently, the density of neurons was significantly increased in the striatum of knock-in mice.
Table 1.
Stereological parameters of the striatum of wild-type and knock-in mice
| Wild type (n = 5) | Knock-in (n = 6) | |
|---|---|---|
| Total neurons | 1,303,908 ± 72,030 | 1,346,885 ± 87,909 |
| Neuronal density | 333,158 ± 16,774 | 400,436 ± 14,320* |
| Volume (mm) | 3.914 ± 0.191 | 3.352 ± 0.1461-160 |
The data are presented as mean ± SEM;
p < 0.015 and
F1-160: p< 0.05.
Huntingtin microaggregates
To study the relationship between the appearance of abnormal behavior and the development of huntingtin microaggregates, brain sections from mice of different ages were stained with EM48, an antibody that selectively recognizes aggregated huntingtin (Gutekunst et al., 1999. Li et al., 2000). Only sections at the level of the striatum (including the overlying cortex) were examined in detail. Immunostaining with the EM48 antibody was absent, and huntingtin microaggregates were not detected in the striatum of wild-type mice at any of the ages examined in this study (Fig.3A). In knock-in mice, microaggregates were also absent at 2 month of age (Fig.3B). Only very weak cytoplasmic staining was observed in the striatum of two of six knock-in mice at this age (data not shown).
Fig. 3.
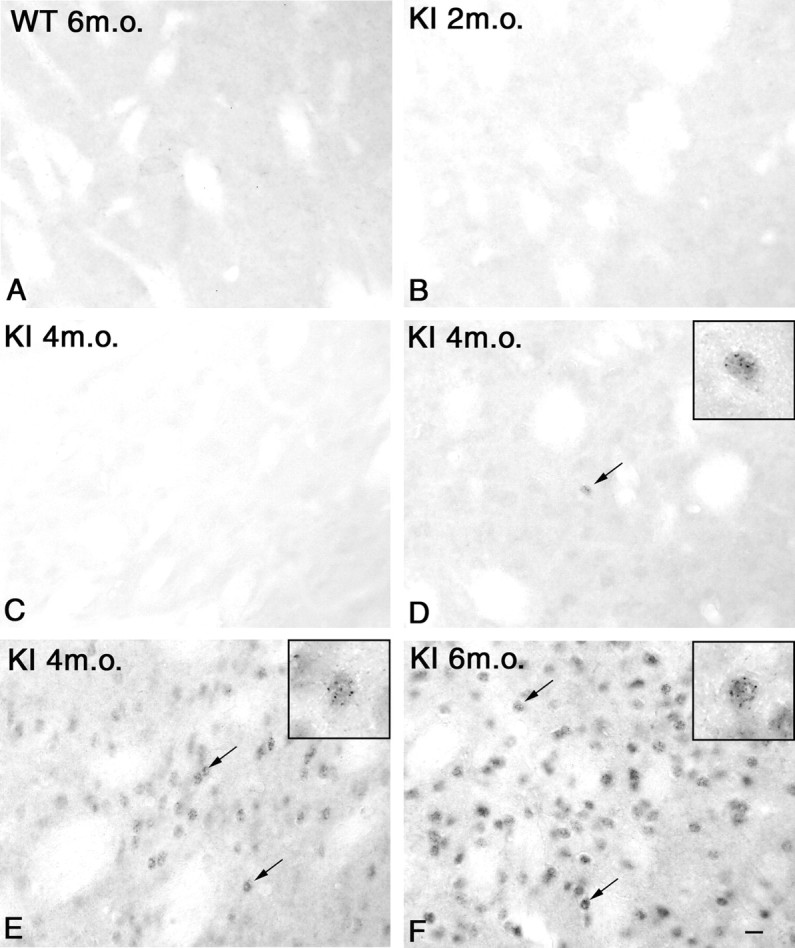
High-power photomicrographs of immunostaining with the EM48 antibody in the striatum of a 6-month-old (m.o.) wild-type (A) and 2- (B), 4- (C, D,E), and 6- (F) month old knock-in mice (KI). No or weak cytoplasmic staining was observed in the striatum of wild-type (A) and 2-month-old knock-in mice (B). Note the substantial inter-animal variability of huntingtin staining at 4 months of age, ranging from animals with no staining (C), with nuclear microaggregates in isolated cells (D) (arrow), or with numerous cells displaying nuclear microaggregates (E) (arrows). At 6 months of age, striatal cells contained numerous nuclear microaggregates (F) (arrows). Note that only the nucleus of striatal neurons is immunostained. Scale bar (shown inF): 10 μm. High-magnification microphotographinsets in D, E, andF show EM48 immunoreactivity in the nuclei of striatal cells (insets show 3× magnification of large photomicrograph).
At 4 months, great inter-individual variability was observed in the knock-in mice. Of the five knock-in mice studied, two had weak or no cytoplasmic staining in the striatum (Fig. 3C). Two mice displayed small huntingtin-positive punctas, in a few clustered or isolated striatal neurons, together with a very weak diffuse staining of other striatal cells (Fig. 3D). These punctas were designated “microaggregates” to distinguish them from the much larger inclusions described previously in transgenic mice (Davies et al., 1997). Only one 4-month-old knock-in mouse had many neurons with microaggregates in the striatum (Fig. 3E). In contrast, by 6 months of age, large clusters of striatal cells contained numerous nuclear microaggregates in all mice examined (Fig.3F). The presence of nuclear huntingtin aggregates in striatal neurons of the knock-in mice was confirmed by electronmicroscopy (Fig. 4). In contrast to microaggregates, single nuclear inclusions were not observed in the striatum of knock-in mice until 18 months of age, and even then they remained fairly small (data not shown) (Menalled et al., 2000).
Fig. 4.

Electron micrograph of EM48 immunogold labeling of the nucleus of a striatal neuron (n) from a 12-month-old knock-in mouse. Microaggregates are indicated by clusters of immunogold particles (arrows). Diffuse immunogold particles (arrowheads) are also observed. Scale bar, 0.5 μm.
No obvious cytoplasmic or neuropil aggregates were observed in the striatum of 2-, 4-, and 6-month-old knock-in mice. In the cerebral cortex overlying the striatum, the majority of neurons in both wild-type and knock-in mice showed weak diffuse cytoplasmic staining (Fig.5A,B). Aside from occasional microaggregates, similar to those sometimes observed in wild-type mice in this region, no conspicuous microaggregates were observed in the cerebral cortex of the 2- to 6-month-old knock-in mice. Similarly, no microaggregates were observed in the hippocampus at these ages (data not shown).
Fig. 5.
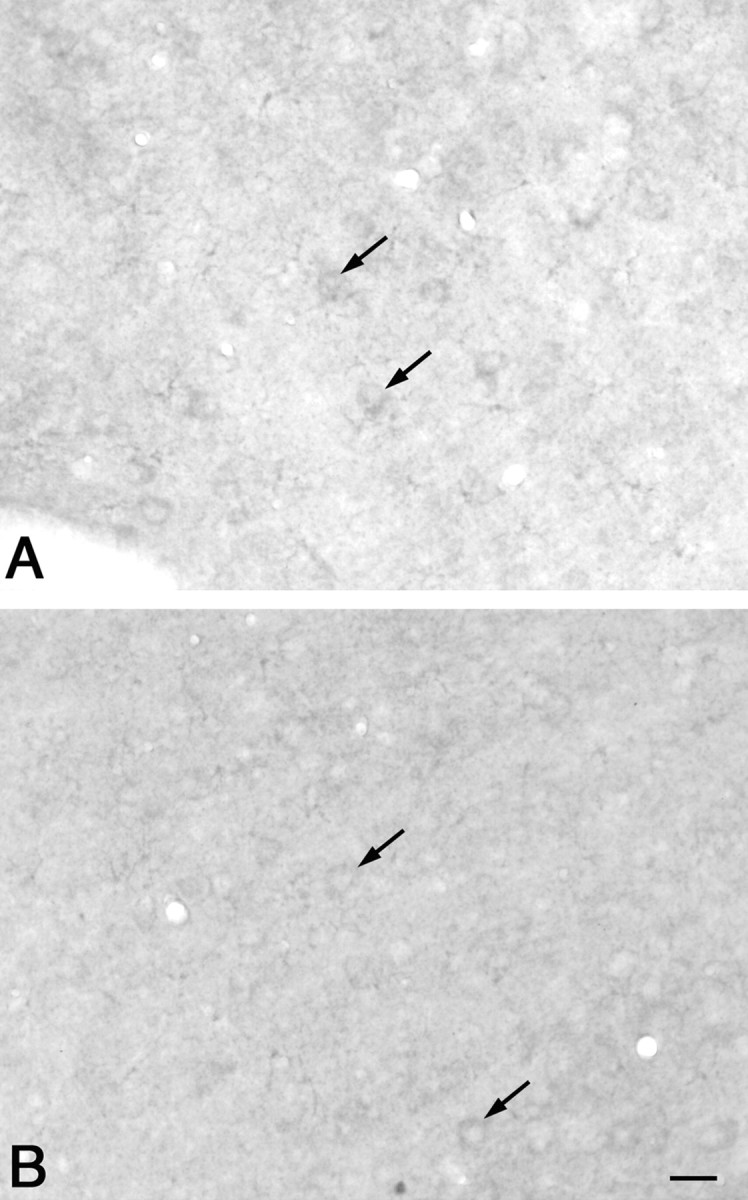
High-power photomicrographs of EM48 antibody immunostaining of the cortex of wild-type (A) and knock-in (B) mice. Note that only the cytoplasm showed weak staining (arrows) in both cases. Scale bar (shown in B): 20 μm.
It is likely that the microaggregates observed in the nuclei of striatal neurons correspond to protein aggregates containing huntingtin. To confirm the presence of insoluble protein aggregates in the brain of the knock-in mice at an age when microaggregates were observed microscopically, a filter assay was performed on brain tissue from wild-type and knock-in mice. Trapped proteins were detected with an anti-ubiquitin antibody because EM48 is not suitable for use on Western blots (Y. Wu and M.-F. Chesselet, unpublished observations). Protein aggregates retained by the filter assay were clearly detected with an anti-ubiquitin antibody in homogenates from the brain of 7-month-old knock-in mice (Fig. 6), i.e., when numerous EM 48-positive microaggregates were found in striatal neurons (Fig. 3F). Interestingly, a slightly higher signal was observed in brain tissue from 2-month-old knock-in mice compared with wild-types; however, further studies are necessary to determine whether this corresponds to a specific increase in ubiquinated protein aggregates in brain before the detection of huntingtin microaggregates by light microscopy in striatal neurons (Fig. 3B).
Fig. 6.

Detergent-resistant aggregates were detected in 7- and 11-month-old (mo) knock-in (KI) mice and not in wild-type (WT) or 2-month-old knock-in mice. Extracts of brain tissue were filtered through a cellulose acetate membrane, and the membranes were probed with an anti-ubiquitin antibody as described in Materials and Methods.
Striosome-matrix localization of microaggregate-containing neurons
Surprisingly, when nuclear microaggregates were present, they were usually not distributed uniformly within the striatum. In the single 4-month-old mouse with many aggregates and in six of the seven mice examined at 6 months, most of the aggregate-containing neurons were grouped in conspicuous clusters (Fig.7D), particularly in the dorsolateral part of the striatum. These clusters were surrounded by neurons with many fewer nuclear microaggregates (Fig. 7C), and only very few isolated neurons with many microaggregates were observed outside the clusters. Although individual microaggregates could not be distinguished at low-power magnification, it was possible to observe clear differences in the intensity of labeling that were proportional to the number of microaggregates seen at higher magnification. The clusters of densely labeled cells were reminiscent of the size, shape, and spatial distribution of the striosomal compartment of the striatum (Graybiel, 1995). To determine whether these intensely labeled clusters corresponded to striosomes, pairs of serially adjacent sections of the striatum were immunostained for MOR-1 as a marker of striosomes (Fig.7A,B) or for EM48 (Fig.7C,D). Neurons with numerous nuclear microaggregates (Fig. 7D) were usually found within the areas of dense MOR-1 staining (Fig. 7A, arrow,B), whereas areas devoid of MOR-1 staining (Fig.7A, asterisk) contained cells with very few if any nuclear microaggregates (Fig. 7C).
Fig. 7.
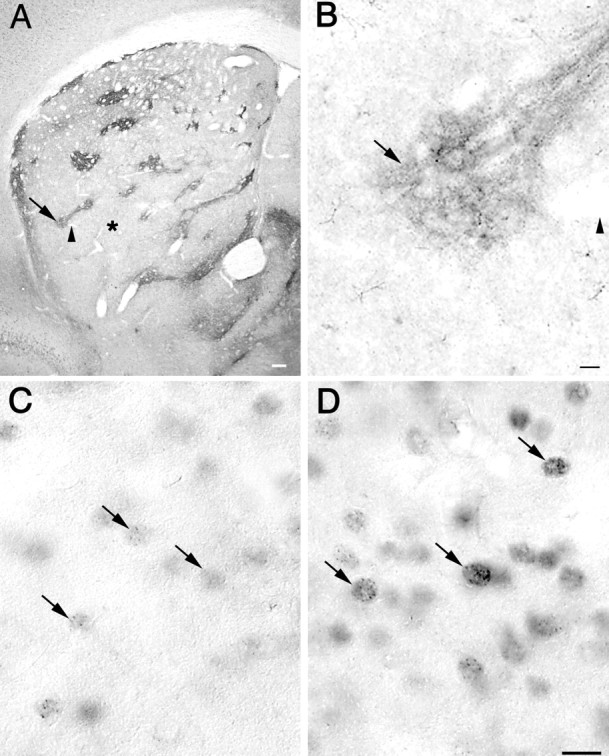
A, Low-power photomicrograph of immunostaining with an MOR-1 antibody in the striatum of a 6-month-old knock-in mouse. Areas positive for MOR-1 correspond to the striosomal compartment (arrow), whereas immunonegative areas correspond to the matrix compartment (asterisk). The arrowhead points to the same blood vessel indicated by the arrowhead in B. Scale bar, 100 μm. B, High-power photomicrograph of MOR-1 immunostaining showing in detail one of the striosomes (arrow) also seen in A atarrow. Scale bar, 10 μm. C,D, High-power photomicrographs of immunostaining with the EM48 antibody in a serially adjacent section to that shown inA and B. C, Striatal neurons with few nuclear microaggregates were found in the matrix compartment (area indicated by the asterisk in A).D, Striatal neurons with numerous microaggregates were located in the striosomal compartment shown in A andB at arrow. Note that only the nucleus of striatal neurons is immunostained. Scale bar (shown in Dfor C and D): 10 μm.
When areas of cells with intense EM48 staining (i.e., containing numerous microaggregates) and areas positive for MOR-1 were compared in serially adjacent sections, it was clear that the majority overlapped (Fig. 8). Although only 10.16% of the area of the striatum corresponded to striosomes, 57.90% of the total surface area of the clusters with densely labeled cells was found to overlap the striosomes (Table 2) (χ2 test; df = 5; χ2 = 223.9; p < 0.001). This indicates that the areas with cells containing numerous nuclear microaggregates were not distributed randomly in the striatum but were located preferentially in the striosomal compartment of the striatum.
Fig. 8.
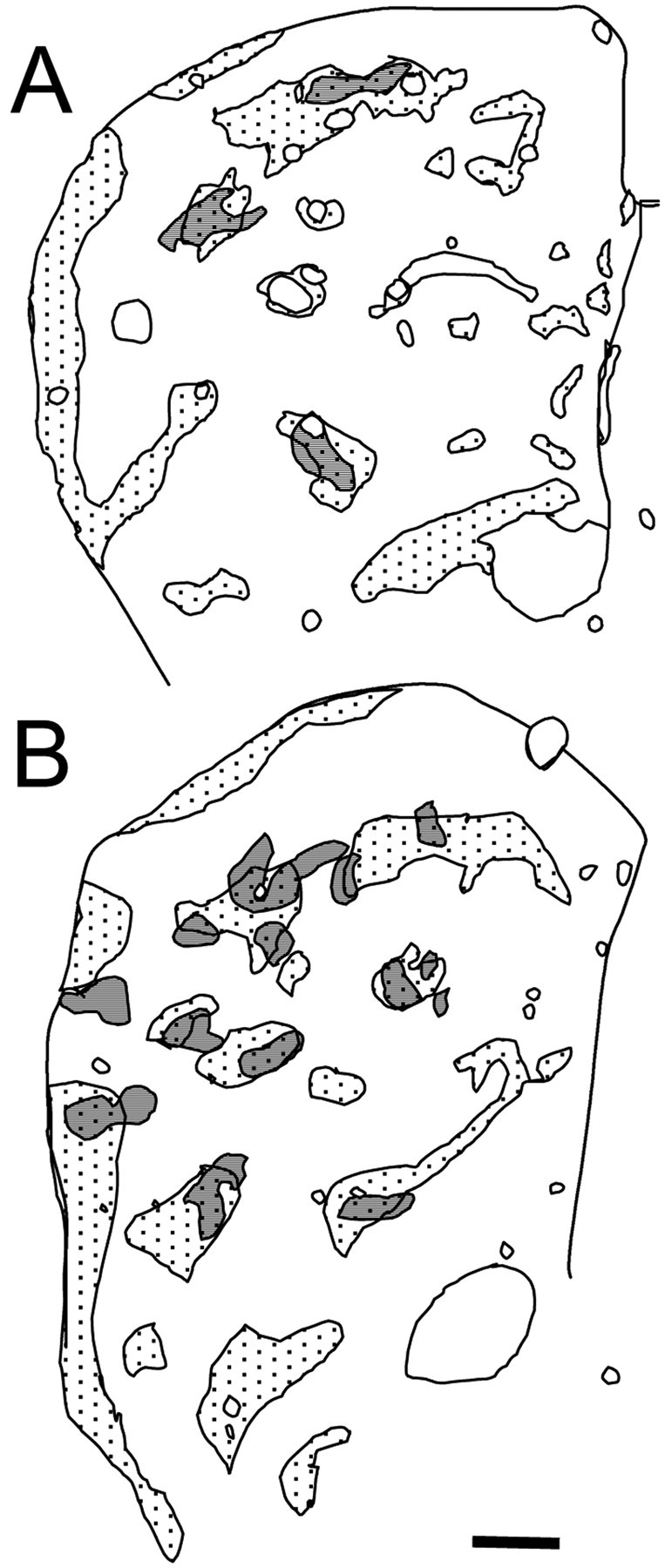
A, B, Maps showing MOR-1-positive areas (striosomes, represented as dotted areas) and cell clusters with intense EM48 immunostaining (represented as gray areas) in the striatum of 4-month-old (A) and 6-month-old (B) knock-in mice. Note the correspondence between the areas occupied by cells with intense EM48 immunostaining and the striosomes (represented as gray areas withdots). Scale bar, 0.2 mm.
Table 2.
Striosome-matrix localization of microaggregate-rich cell clusters in knock-in mice
| Area of striatum (S) (mm2) | 2.1346 ± 0.1156 |
| Area of striosomes (D) (mm2) | 0.2149 ± 0.0380 |
| Area of matrix (M = S − D) (mm2) | 1.9197 ± 0.1194 |
| % of striatum composed of striosomes | 10.16 ± 1.89 |
| Area of clusters cells with numerous microaggregates (A) (mm2) | 0.0987 ± 0.0149 |
| Area of Aoverlapping striosomes (C) (mm2) | 0.0546 ± 0.0081 |
| Area of A overlapping M (B = A − C) (mm2) | 0.0441 ± 0.0079 |
| % of the total area of clusters that overlapped the striosomes | 57.90 ± 3.22* |
The data are presented as mean ± SEM; n = 6.
p < 0.001; χ2 test, df = 5, χ2 = 223.7.
DISCUSSION
Genetically, knock-in mice provide a more faithful model of the human disease than transgenics because the mutation is expressed in the same genomic context as in humans. However, these models have received less attention than the transgenics because their phenotype is subtle. This study reports for the first time robust abnormal motor behaviors in a knock-in mouse model of HD as early as 2 months of age. The behavioral phenotype of the knock-in mice was biphasic, evolving from an increase in repetitive motor behavior to a decrease in locomotor activity. Nuclear aggregates of the mutated protein were either absent (2 months) or very rare (4 months) when abnormal behavior was first observed, indicating that they form after a prolonged phase of neuronal dysfunction. When they first appeared, nuclear staining and huntingtin aggregates were restricted to the striatum and predominated in the striosomal compartment, as defined by the presence of dense expression of μ-opioid receptors. Thus, knock-in models of Huntington's disease present a motor phenotype that precedes regionally specific neuropathology. The presence of early behavioral anomalies, the slow progression of symptoms, and similarities of the regional pathology to that in humans all suggest that these mice provide an important new model for studying HD and testing new therapies.
Abnormal motor behavior in knock-in mice
Abnormal motor behavior has been detected in gene carriers of the HD mutation up to 8 years before the presumed onset of overt symptoms (Smith et al., 2000). In addition, repetitive, uncontrolled movements are often observed before overt symptoms (S. Perlman, personal communication). Although the knock-in mice did not display obvious motor symptoms such as hyperactive turning, tremor, freezing, or clasping up to 2 years of age (Levine et al., 1999; Menalled et al., 2000), they showed an increase in repetitive movements at an early age (2 months). This initial increase in repetitive movements gave way to decreased locomotion at 4 and 6 months. It is unlikely that the decreased locomotor activity observed in the 4-month-old knock-in mice was caused solely by increased anxiety (File et al., 1998). Indeed it was present during the entire observation period and was not limited to the initial exploration phase during which the effect of anxiety is more likely to occur. The decreased locomotor activity that we have observed is reminiscent of the observation, in another line of mice, of a greater proportion of knock-in than wild-type mice that remain inactive when their cage is open (Lin et al., 2001).
A shift from hyperkinetic to hypokinetic behavior with age has also been reported in transgenic models of HD (Davies et al., 1997; Reddy et al., 1998; Luesse et al., 2001). It is interesting to note that patients with adult onset HD display dyskinetic movements during early stages but become akinetic later on (Harper, 1996). Furthermore, patients have reduced motor activity despite chorea, suggesting that hypokinesia is a major component of the movement disorder in HD (van Vugt et al., 2001). Thus, knock-in mice display a progressive behavioral phenotype with early onset and characteristics that are reminiscent of that observed in humans.
Microaggregates in knock-in mice
The antibody used to detect huntingtin in the present study, EM48, preferentially recognizes the aggregated form of the protein (Gutekunst et al., 1999). This offers a distinct advantage over antibodies that also detect normal huntingtin because very small aggregates are not obscured by staining for nonaggregated huntingtin. Accordingly, more aggregates are usually detected with EM48 than with other anti-huntingtin antibodies (Gutekunst et al., 1999). Despite the use of this highly sensitive antibody, microaggregates were detected with light microscopy only several months after the first behavioral symptoms. Because immunoblots revealed similar levels of huntingtin as in the wild type, the absence of microaggregates was not related to lack of protein (Wu and Chesselet, unpublished observation). Nuclear inclusions, defined as single large round aggregates similar to those originally described in transgenic R6/2 mice (Davies et al., 1997) and human brain (Roizin et al., 1979; DiFiglia et al., 1997), were present only in mice older than 18 months.
In cell culture, the toxicity of mutated huntingtin was enhanced when the formation of aggregates was prevented, suggesting that aggregate formation can be separated from the pathological effects of the mutation (Saudou et al., 1998). Similarly, SCA1 transgenic mice expressing the mutant ataxin-1 in which the self-association domain has been deleted develop an abnormal behavioral and cellular phenotype without ataxin-1 aggregates (Klement et al., 1998). Therefore, both huntingtin and ataxin-1 with expanded polyglutamine repeats can cause cellular dysfunction by other means than the formation of aggregates. This was further supported in our model by the absence of detectable aggregates in most 4-month-old knock-in mice, an age when we have observed decreased locomotion and a marked decrease in enkephalin mRNA (Menalled et al., 2000) as well as an increased response to the stimulation of NMDA receptors (Levine et al., 1999). Thus if abnormal protein aggregates form before behavioral symptoms in the brain of mice carrying a knock-in HD mutation, these must be very subtle because they were not detected with the sensitive EM48 antibody. This raises the issue of whether prevention of protein aggregation should be a primary target for treatment.
Despite these clear behavioral and cellular phenotypes, no cell loss was found in the knock-in mice. This supports the hypothesis that a prolonged phase of neuronal dysfunction precedes cell death in HD. Similar to observations in humans and R6/2 transgenic mice, striatal volume was decreased, which could be attributed to loss of neuropil (Vonsattel et al., 1985; Mangiarini et al., 1996). Indeed, although it is not yet known whether similar anomalies exist in knock-in mice, we have observed a decrease in spine density and extent of dendritic field and cross-sectional areas of striatal and cortical neurons in R6/2 mice (Klapstein et al., 2001).
Regional distribution of nuclear microaggregates
The knock-in mice showed a remarkable regional selectivity of neuropathology within the brain, with a selective presence of nuclear staining and huntingtin aggregates in the striatum, the area most affected in HD (Vonsattel et al., 1985). The absence of nuclear aggregates in cortex, in contrast to adult onset HD (Gutekunst et al., 1999), may be related to the relatively short (94) CAG repeat length. Indeed, cortical aggregates were observed in lines of knock-in mice carrying 140 (Menalled et al., 2001) and 150 CAG (Lin et al., 2001).
When nuclear microaggregates were present, they were not uniformly distributed in the striatum. Clusters of microaggregate-rich neurons fell within, or in close apposition to, patches of dense immunostaining for μ-opioid receptors identified in serially adjacent sections. This indicates that aggregate-rich neurons are found primarily in the striosomal compartment, a finding also reported in humans (Hedreen, 1998). The absence of a perfect match in 35-μm-thick serially adjacent sections is compatible with the shape of the striosomal network (Groves et al., 1988; Desban et al., 1993). It should be pointed out that our study is very different from a recent study in R6/2 transgenic mice that reported a preferential location of nuclear inclusions stained with ubiquitin in the matrix compartment (Morton et al., 2000). First, we focused on microaggregates of huntingtin, which are likely to represent an earlier form of protein aggregation than ubiquitinated inclusions (Morton et al., 2000). Second, in the transgenic mice, the striosomes were defined as areas of low-calbindin immunoreactivity that include a large area of the dorsolateral striatum, which contains both striosomes and matrix based on μ-opioid staining.
Results of normal huntingtin expression in striatal compartments differ not only between but also within species. Some studies in humans reported lower levels of huntingtin in striosomes than in matrix (Ferrante et al., 1997; Sapp et al., 1997), but opposite results were found in rats, monkeys, and also humans (Gutekunst et al., 1995;Kosinski et al., 1997). Moreover, similar levels of huntingtin were found in both compartments in rats (Fusco et al., 1999) as well as in mice (Bhide et al., 1996; L. B. Menalled and M.-F. Chesselet, unpublished observations). Because our studies were done in mice, it is unlikely that differences in huntingtin expression in the two compartments played a major role in the preferential formation of aggregates within striosomal neurons. Striosomal neurons are born earlier than those in the matrix, and both compartments have multiple molecular differences that could contribute to a greater effect of the mutation in striosomal neurons (Graybiel, 1995). It will be interesting to determine whether the instability of CAG repeat length detected in the striatum of knock-in mice (Kennedy and Shelbourne, 2000) occurs preferentially in the striosomes.
The preferential location of microaggregates in striosomes in both knock-in mice and humans (Hedreen, 1998) points to important similarities between this mouse model and the human disease. Although early neuronal loss has been reported to occur in the striosomes in postmortem human brain (Hedreen and Folstein, 1995), most studies have found a preservation of striosome areas with a decrease in matrix surface area and cell number (Ferrante et al., 1987; Seto-Ohshima et al., 1988). Thus the preferential location of aggregates in striosomes would further support a protective role of huntingtin aggregation in HD.
Footnotes
This work was supported by the Hereditary Disease Foundation's Cure HD Initiative. We thank L. Christian, A. Koppel, and E. Gruen for their assistance with the mouse colony, Armelle Leviel for her technical support, and M. H. Shomer and Dr. M. Romero-Ramos for helpful discussions. We are also grateful to P. Harlam, Dr. A. M. Graybiel (Massachusetts Institute of Technology), and Dr. C.-A. Gutekunst (Emory University) for their advice with immunohistochemistry, Dr. R. Elde (University of Minnesota) for the μ-opioid receptor antiserum, and Dr. S. L. Sabol (National Institutes of Health) for the preproenkephalin cDNA. We thank Dr. William Melega (UCLA) for the use of his stereology system, and Sharon Sampogna (Brain Research Institute Microscope Technique Core, UCLA) and Gwen Gordon (Mental Retardation Research Center Biostatistic Core at UCLA) for their assistance.
Correspondence should be addressed to Dr. Marie-Françoise Chesselet, Department of Neurology, University of California Los Angeles School of Medicine, 710 Westwood Plaza, Reed Neurological Research Center B114, Los Angeles, CA 90095. E-mail:MChesselet@mednet.ucla.edu.
REFERENCES
- 1.Aronin N, Chase K, Sagar SM, Sharp FR, DiFiglia M. N-methyl-d-aspartate receptor activation in the neostriatum increases c-fos and fos-related antigens selectively in medium-sized neurons. Neuroscience. 1991;44:409–420. doi: 10.1016/0306-4522(91)90065-v. [DOI] [PubMed] [Google Scholar]
- 2.Bhide PG, Day M, Sapp E, Schwarz C, Sheth A, Kim J, Young AB, Penney J, Golden J, Aronin N, DiFiglia M. Expression of normal and mutant huntingtin in the developing brain. J Neurosci. 1996;16:5523–5535. doi: 10.1523/JNEUROSCI.16-17-05523.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Canales JJ, Graybiel AM. A measure of striatal function predicts motor stereotypy. Nat Neurosci. 2000;3:377–383. doi: 10.1038/73949. [DOI] [PubMed] [Google Scholar]
- 4.Chesselet MF, Levine MS. Mouse modules of Huntington's disease. In: Chesselet MF, editor. Molecular mechanisms of neurodegenerative disease. Humana; Totowa, NJ: 2000. pp. 327–346. [Google Scholar]
- 5.Chesselet MF, Weiss L, Wuenschell C, Tobin AJ, Affolter HU. Comparative distribution of mRNAs for glutamic acid decarboxylase, tyrosine hydroxylase, and tachykinins in the basal ganglia: an in situ hybridization study in the rodent brain. J Comp Neurol. 1987;262:125–140. doi: 10.1002/cne.902620110. [DOI] [PubMed] [Google Scholar]
- 6.Davies SW, Turmaine M, Cozens BA, DiFiglia M, Sharp AH, Ross CA, Scherzinger E, Wanker EE, Mangiarini L, Bates GP. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90:537–548. doi: 10.1016/s0092-8674(00)80513-9. [DOI] [PubMed] [Google Scholar]
- 7.Desban M, Kemel ML, Glowinski J, Gauchy C. Spatial organization of patch and matrix compartments in the rat striatum. Neuroscience. 1993;57:661–671. doi: 10.1016/0306-4522(93)90013-6. [DOI] [PubMed] [Google Scholar]
- 8.DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- 9.Ferrante RJ, Kowall NW, Beal MF, Martin JB, Bird ED, Richardson EP., Jr Morphologic and histochemical characteristics of a spared subset of striatal neurons in Huntington's disease. J Neuropathol Exp Neurol. 1987;46:12–27. doi: 10.1097/00005072-198701000-00002. [DOI] [PubMed] [Google Scholar]
- 10.Ferrante RJ, Gutekunst CA, Persichetti F, McNeil SM, Kowall NW, Gusella JF, MacDonald ME, Beal MF, Hersch SM. Heterogeneous topographic and cellular distribution of huntingtin expression in the normal human neostriatum. J Neurosci. 1997;17:3052–3063. doi: 10.1523/JNEUROSCI.17-09-03052.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.File SE, Mahal A, Mangiarini L, Bates GP. Striking changes in anxiety in Huntington's disease transgenic mice. Brain Res. 1998;805:234–240. doi: 10.1016/s0006-8993(98)00736-7. [DOI] [PubMed] [Google Scholar]
- 12.Fusco FR, Chen Q, Lamoreaux WJ, Figueredo-Cardenas G, Jiao Y, Coffman JA, Surmeier DJ, Honig MG, Carlock LR, Reiner A. Cellular localization of huntingtin in striatal and cortical neurons in rats: lack of correlation with neuronal vulnerability in Huntington's disease. J Neurosci. 1999;19:1189–1202. doi: 10.1523/JNEUROSCI.19-04-01189.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Graybiel AM. The basal ganglia. Trends Neurosci. 1995;18:60–62. [PubMed] [Google Scholar]
- 14.Groves PM, Martone M, Young SJ, Armstrong DM. Three-dimensional pattern of enkephalin-like immunoreactivity in the caudate nucleus of the cat. J Neurosci. 1988;8:892–900. doi: 10.1523/JNEUROSCI.08-03-00892.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gutekunst CA, Levey AI, Heilman CJ, Whaley WL, Yi H, Nash NR, Rees HD, Madden JJ, Hersch SM. Identification and localization of huntingtin in brain and human lymphoblastoid cell lines with anti-fusion protein antibodies. Proc Natl Acad Sci USA. 1995;92:8710–8714. doi: 10.1073/pnas.92.19.8710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Harper PS. Huntington's disease. Saunders; Philadelphia: 1996. [Google Scholar]
- 18.Hedreen JC. Neuronal intranuclear inclusions in neostriatal striosomes and matrix in Huntington's disease. International Basal Ganglia Society Meeting VI, Brewster, MA, October; 1998. [Google Scholar]
- 19.Hedreen JC, Folstein SE. Early loss of neostriatal striosome neurons in Huntington's disease. J Neuropathol Exp Neurol. 1995;54:105–120. doi: 10.1097/00005072-199501000-00013. [DOI] [PubMed] [Google Scholar]
- 20.Kennedy L, Shelbourne PF. Dramatic mutation instability in HD mouse striatum: does polyglutamine load contribute to cell-specific vulnerability in Huntington's disease? Hum Mol Genet. 2000;9:2539–2544. doi: 10.1093/hmg/9.17.2539. [DOI] [PubMed] [Google Scholar]
- 21.Kim M, Lee HS, LaForet G, McIntyre C, Martin EJ, Chang P, Kim TW, Williams M, Reddy PH, Tagle D, Boyce FM, Won L, Heller A, Aronin N, DiFiglia M. Mutant huntingtin expression in clonal striatal cells: dissociation of inclusion formation and neuronal survival by caspase inhibition. J Neurosci. 1999;19:964–973. doi: 10.1523/JNEUROSCI.19-03-00964.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klapstein GJ, Fisher RS, Zanjani H, Cepeda C, Jokel ES, Chesselet MF, Levine MS. Electrophysiological and morphological changes in striatal spiny neurons in R6/2 Huntington's disease transgenic mice. J Neurophysiol. 2001;86:2667–2677. doi: 10.1152/jn.2001.86.6.2667. [DOI] [PubMed] [Google Scholar]
- 23.Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, Zoghbi HY, Orr HT. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41–53. doi: 10.1016/s0092-8674(00)81781-x. [DOI] [PubMed] [Google Scholar]
- 24.Kosinski CM, Cha JH, Young AB, Persichetti F, MacDonald M, Gusella JF, Penney JB, Jr, Standaert DG. Huntingtin immunoreactivity in the rat neostriatum: differential accumulation in projection and interneurons. Exp Neurol. 1997;144:239–247. doi: 10.1006/exnr.1997.6441. [DOI] [PubMed] [Google Scholar]
- 25.Kuemmerle S, Gutekunst CA, Klein AM, Li XJ, Li SH, Beal MF, Hersch SM, Ferrante RJ. Huntington aggregates may not predict neuronal death in Huntington's disease. Ann Neurol. 1999;46:842–849. [PubMed] [Google Scholar]
- 26.Levine MS, Klapstein GJ, Koppel A, Gruen E, Cepeda C, Vargas ME, Jokel ES, Carpenter EM, Zanjani H, Hurst RS, Efstratiadis A, Zeitlin S, Chesselet MF. Enhanced sensitivity to N-methyl-d-aspartate receptor activation in transgenic and knockin mouse models of Huntington's disease. J Neurosci Res. 1999;58:515–532. [PubMed] [Google Scholar]
- 27.Luesse HG, Schiefer J, Spruenken A, Puls C, Block F, Kosinski CM. Evaluation of R6/2 HD transgenic mice for therapeutic studies in Huntington's disease: behavioral testing and impact of diabetes mellitus. Behav Brain Res. 2001;126:185–195. doi: 10.1016/s0166-4328(01)00261-3. [DOI] [PubMed] [Google Scholar]
- 28.Li H, Li SH, Cheng AL, Mangiarini L, Bates GP, Li XJ. Ultrastructural localization and progressive formation of neuropil aggregates in Huntington's disease transgenic mice. Hum Mol Genet. 1999;8:1227–1236. doi: 10.1093/hmg/8.7.1227. [DOI] [PubMed] [Google Scholar]
- 29.Li H, Li SH, Johnston H, Shelbourne PF, Li XJ. Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat Genet. 2000;25:385–389. doi: 10.1038/78054. [DOI] [PubMed] [Google Scholar]
- 30.Lin CH, Tallaksen-Greene S, Chien WM, Cearley JA, Jackson WS, Crouse AB, Ren S, Li XJ, Albin RL, Detloff PJ. Neurological abnormalities in a knock-in mouse model of Huntington's disease. Hum Mol Genet. 2001;10:137–144. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- 31.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 32.Menalled L, Chesselet M-F. Mouse models of Huntington's disease. Trends Pharmacol Sci. 2001;23:32–39. doi: 10.1016/s0165-6147(00)01884-8. [DOI] [PubMed] [Google Scholar]
- 33.Menalled L, Zanjani H, MacKenzie L, Koppel A, Carpenter E, Zeitlin S, Chesselet MF. Decrease in striatal enkephalin mRNA in mouse models of Huntington's disease. Exp Neurol. 2000;162:328–342. doi: 10.1006/exnr.1999.7327. [DOI] [PubMed] [Google Scholar]
- 34.Menalled LB, Sison J, Zeitlin S, Li XJ, Chesselet MF. Temporal and regional development of huntingtin aggregates in a knock-in mouse model of Huntington's disease. Soc Neurosci Abstr. 2001;31:325.6. [Google Scholar]
- 35.Morton AJ, Lagan MA, Skepper JN, Dunnett SB. Progressive formation of inclusions in the striatum and hippocampus of mice transgenic for the human Huntington's disease mutation. J Neurocytol. 2000;29:679–702. doi: 10.1023/a:1010887421592. [DOI] [PubMed] [Google Scholar]
- 36.Oorschot DE. Total number of neurons in the neostriatal, pallidal, subthalamic, and substantia nigral nuclei of the rat basal ganglia: a stereological study using the Cavalieri and optical dissector methods. J Comp Neurol. 1996;366:580–599. doi: 10.1002/(SICI)1096-9861(19960318)366:4<580::AID-CNE3>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- 37.Reddy PH, Williams M, Charles V, Garrett L, Pike-Buchanan L, Whetsell WO, Jr, Miller G, Tagle DA. Behavioural abnormalities and selective neuronal loss in HD transgenic mice expressing mutated full-length HD cDNA. Nat Genet. 1998;20:198–202. doi: 10.1038/2510. [DOI] [PubMed] [Google Scholar]
- 38.Roizin L, Stellar S, Liu JD. Neuronal nuclear-cytoplasmic changes in Huntington chorea: electron microscope investigations. In: Chase TN, Wexler NS, Barbeau A, editors. Advances in neurology: Huntington's disease. Raven; New York: 1979. pp. 95–122. [Google Scholar]
- 39.Sapp E, Schwarz C, Chase K, Bhide PG, Young AB, Penney J, Vonsattel JP, Aronin N, DiFiglia M. Huntingtin localization in brains of normal and Huntington's disease patients. Ann Neurol. 1997;42:604–612. doi: 10.1002/ana.410420411. [DOI] [PubMed] [Google Scholar]
- 40.Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- 41.Seto-Ohshima A, Emson PC, Lawson E, Mountjoy CQ, Carrasco LH. Loss of matrix calcium-binding protein-containing neurons in Huntington's disease. Lancet. 1988;1:1252–1255. doi: 10.1016/s0140-6736(88)92073-9. [DOI] [PubMed] [Google Scholar]
- 42.Shelbourne PF, Killeen N, Hevner RF, Johnston HM, Tecott L, Lewandoski M, Ennis M, Ramirez L, Li Z, Iannicola C, Littman DR, Myers RM. A Huntington's disease CAG expansion at the murine Hdh locus is unstable and associated with behavioural abnormalities in mice. Hum Mol Genet. 1999;8:763–774. doi: 10.1093/hmg/8.5.763. [DOI] [PubMed] [Google Scholar]
- 43.Smith MA, Brandt J, Shadmehr R. Motor disorder in Huntington's disease begins as a dysfunction in error feedback control. Nature. 2000;403:544–549. doi: 10.1038/35000576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 45.Usdin MT, Shelbourne PF, Myers RM, Madison DV. Impaired synaptic plasticity in mice carrying the Huntington's disease mutation. Hum Mol Genet. 1999;8:839–846. doi: 10.1093/hmg/8.5.839. [DOI] [PubMed] [Google Scholar]
- 46.van Vugt JP, Siesling S, Piet KK, Zwinderman AH, Middelkoop HA, van Hilten JJ, Roos RA. Quantitative assessment of daytime motor activity provides a responsive measure of functional decline in patients with Huntington's disease. Mov Disord. 2001;16:481–488. doi: 10.1002/mds.1097. [DOI] [PubMed] [Google Scholar]
- 47.Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 48.Wheeler VC, White JK, Gutekunst CA, Vrbanac V, Weaver M, Li XJ, Li SH, Yi H, Vonsattel JP, Gusella JF, Hersch S, Auerbach W, Joyner AL, MacDonald ME. Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Hum Mol Genet. 2000;9:503–513. doi: 10.1093/hmg/9.4.503. [DOI] [PubMed] [Google Scholar]
- 49.White JK, Auerbach W, Duyao MP, Vonsattel JP, Gusella JF, Joyner AL, MacDonald ME. Huntingtin is required for neurogenesis and is not impaired by the Huntington's disease CAG expansion. Nat Genet. 1997;17:404–410. doi: 10.1038/ng1297-404. [DOI] [PubMed] [Google Scholar]