

Abstract
Huntington's disease (HD) results from polyglutamine expansion in huntingtin (htt), a protein with several consensus caspase cleavage sites. Despite the identification of htt fragments in the brain, it has not been shown conclusively that htt is cleaved by caspases in vivo. Furthermore, no study has addressed when htt cleavage occurs with respect to the onset of neurodegeneration. Using antibodies that detect only caspase-cleaved htt, we demonstrate that htt is cleaved in vivo specifically at the caspase consensus site at amino acid 552. We detect caspase-cleaved htt in control human brain as well as in HD brains with early grade neuropathology, including one homozygote. Cleaved htt is also seen in wild-type and HD transgenic mouse brains before the onset of neurodegeneration. These results suggest that caspase cleavage of htt may be a normal physiological event. However, in HD, cleavage of mutant htt would release N-terminal fragments with the potential for increased toxicity and accumulation caused by the presence of the expanded polyglutamine tract. Furthermore, htt fragments were detected most abundantly in cortical projection neurons, suggesting that accumulation of expanded htt fragments in these neurons may lead to corticostriatal dysfunction as an early event in the pathogenesis of HD.
Keywords: Huntington's disease, huntingtin, caspase cleavage, in vivo, corticostriatal pathway, neurodegeneration
N terminus of huntingtin (htt) (Hayden, 1981; Huntington's Disease Collaborative Research Group, 1993). Htt was the first neuronal protein shown to be a caspase substrate (Goldberg et al., 1996; Wellington et al., 1998, 2000), with defined sites for caspase-3 at amino acids 513 and 552, for caspase-2 at amino acid 552, and for caspase-6 at amino acid 586 (Wellington et al., 1998, 2000). Additionally, there are two caspase-3 consensus sites at amino acids 530 and 589 that appear to be silent (Wellington et al., 2000). We previously proposed the toxic fragment hypothesis, suggesting that caspase-mediated cleavage of expanded htt would generate toxic N-terminal fragments that induce neuronal death (Cooper et al., 1998;Hackam et al., 1998; Martindale et al., 1998; Saudou et al., 1998). We have shown that the toxicity of caspase-resistant expanded htt is markedly reduced in transfected cells (Wellington et al., 2000). Furthermore, full-length, wild-type htt has been shown recently to be neuroprotective in a variety of in vitro and in vivo models (Rigamonti et al., 2000; Leavitt et al., 2001; Zuccato et al., 2001). These observations suggest that inhibition of htt proteolysis would both reduce the load of toxic N-terminal fragments and maintain the neuroprotective function of intact htt.
Despite numerous in vitro and in vivo studies that support a correlation between htt length and toxicity, little is known about the proteolytic processing of htt in vivo. Several studies have identified htt fragments in control and HD brains (DiFiglia et al., 1995; Kim et al., 2001; Mende-Mueller et al., 2001), but several important questions remain unaddressed. For example, the identity of the proteases responsible for cleaving htt in vivo has not been demonstrated conclusively, in part because the site of htt cleavage has not been determined precisely in any in vivo study to date. Indeed, there is controversy in the literature about whether expanded htt is even susceptible to cleavage in vivo compared with htt with a normal polyglutamine tract (Dyer and McMurray, 2001). Furthermore, whether cleavage of htt is an early or late event in HD pathogenesis has not been specifically addressed. Finally, the cellular populations that may be particularly enriched in cleaved htt are not known.
To unequivocally detect caspase cleavage products of htt, we have developed novel antibodies specifically designed to detect N-terminal htt fragments generated by caspase cleavage at amino acid 513 or 552. Here, we provide direct evidence for caspase-mediated cleavage of htt. In early grade HD postmortem tissue as well as in young transgenic mice expressing expanded human htt, both normal and expanded htt are cleaved at amino acid 552 before the onset of gross striatal atrophy. Cleaved htt was also observed in control brain, suggesting that caspase cleavage of htt may be part of its normal regulated catabolism. The observation of htt cleavage most abundantly in cortical projection neurons suggests that disruption of the corticostriatal pathway may be an early event in the pathogenesis of HD.
MATERIALS AND METHODS
Human and murine tissues. Human HD and control brain postmortem tissues were obtained from the Canadian Brain Tissue Bank (Toronto, Canada) and the Harvard Brain Tissue Resource Center (Cambridge, MA). Tissue from a presymptomatic patient was obtained from Emory University as described previously (Gutekunst et al., 1999). All human tissues were obtained after ethical review. Yeast artificial chromosome (YAC) transgenic animals expressing human htt with 72 (CAG) repeats (Hodgson et al., 1999) were used as a source of murine tissues according to the University of British Columbia animal protocol A00-0254. Line 2511 contains one to two copies of the YAC and has been described previously (Hodgson et al., 1999). Line 44 was generated by independent microinjection and contains two to four copies of the YAC transgene.
Determination of CAG repeat number from brain tissue. The number of CAG repeats in htt from patient brain was determined by nonradioactive PCR using standards of known CAG size as described previously (Andrew et al., 1993; Bruland et al., 1999).
Antibodies. Antibodies specific for the C-terminal ends of htt caspase cleavage products ending at amino acid 513 or 552 were prepared using the immunizing peptides KLH-CHTLQADSVD and KLH-CSDPAMDLND, respectively. Antibodies were affinity purified as described previously (Gervais et al., 1999). The polyclonal N-terminal htt antibody BKP1 has been described previously (Kalchman et al., 1997). In addition to polyclonal BKP1, we have also generated a monoclonal version of BKP1 that recognizes the N-terminal 17 aa of htt. Polyclonal and monoclonal BKP1 were used interchangeably. Monoclonal antibodies specific for glyceraldehyde phosphate dehydrogenase (GAPDH) and expanded polyglutamine tracts (1C2) were purchased from Chemicon(Temecula, CA). EM48, an antibody that recognizes htt aggregates, has been described previously (Gutekunst et al., 1999; Li et al, 1999).
Cell culture and transfection. Human embryonic kidney (HEK) 293T cells were cultured and transfected by calcium phosphate as described previously, using the htt constructs pRcCMV-3949-15 and pRcCMV-3949-128 (Wellington et al., 2000). The constructs pRcCMV-3949-15-514X and pRcCMV-3949-128-514X were generated by inserting a translation terminal codon at amino acid 514. Apoptosis in cultured cells was induced by the addition of 35 μm tamoxifen as described previously (Hackam et al., 1998; Ellerby et al., 1999).
Western blotting. Murine and human tissues were homogenized in a buffer containing 0.25 m sucrose, 20 mm Tris HCl, pH 7.2, 1 mmMgCl2, 0.5 mm EDTA, complete protease inhibitor (Roche, Indianapolis, IN), and, unless otherwise indicated, 5–10 μmN-benzyloxycarbonyl-val-ala-asp-fluoromethyl ketone (zVAD-fmk). Lysates were sonicated for 10 sec and cleared of debris by centrifugation for 10 min at 9000 rpm at 4°C. To cleave htt in murine or human homogenates, 20 μg of protein prepared without zVAD-fmk was incubated in cleavage buffer alone [50 mm HEPES/KOH, pH 7.0, 10% (w/v) sucrose, 2 mm EDTA, 0.1% (w/v) 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, 5 mm DTT] or cleavage buffer with 2 nm purified recombinant caspase-3 for 60 min at 37°C as described previously (Wellington et al., 1998). As a control, replicate samples were pretreated with 50 nmacetyl-aspartyl-glutamyl-valyl-aspart-1-aldehyde (DEVD-CHO) for 15 min at 25°C before the addition of caspase-3. After the addition of 5× SDS loading buffer, heat-denatured samples were electrophoresed through 7.5% SDS-PAGE gels and transferred to a polyvinylidene difluoride membrane. Blots were probed with BKP1, Htt513, Htt552, or 1C2 overnight in 5% skim milk in PBS. After several washes in PBS with 0.5% Tween 20 (PBST), HRP-conjugated goat anti-rabbit or goat anti-mouse secondary antibodies were added for 1 hr at room temperature. After extensive washes in PBST, blots were developed using enhanced chemiluminescence (Amersham Biosciences, Arlington Heights, IL) according to recommended protocols.
Immunofluorescence. HEK 293T cells were processed for immunofluorescence exactly as described previously (Hackam et al., 1998) and stained with Htt513 and Htt552 as primary antibodies (1:5000) and goat anti-rabbit Alexa 594-conjugated secondary antibody (Molecular Probes, Eugene, OR; 1:800). Nuclei were stained with 4′,6′-diamidino-2-phenylindole (DAPI), and images were captured and processed using a CCD camera and Northern Eclipse software (Empix Imaging, Mississauga, Ontario, Canada) as described previously (Hackam et al., 1998).
Two-month-old wild-type and YAC transgenic mice (line 44, 72 CAG repeats) were killed by intraperitoneal injection of 0.8 ml of 2.5% avertin for immunohistochemical analysis of huntingtin cleavage in brain slices. The brains were snap frozen in optimal cutting temperature compound at −80°C, and 30 μm coronal sections were prepared on a cryostat. The sections were fixed with acetone for 10 min at room temperature and permeabilized with 0.1% Triton X-100. Sections were incubated in either Htt512 (1:500) or Htt552 (1:1000) in PBS for 48 hr at 4°C, washed in PBS, and incubated with goat anti-rabbit CY3-conjugated 2° antibody (1:200; Chemicon) for 2 hr at room temperature. Sections were then double labeled with neuronal-specific nuclear protein (NeuN) (1:50; Chemicon) in block (4% goat serum, 5% bovine serum albumin, and 0.1% Tween 20 in PBS) for 24 hr at 4°C, washed in PBS, and incubated in goat anti-mouse Alexa 488 (1:500; Molecular Probes) secondary antibody at room temperature for 2 hr. Omission of the primary antibodies was used as a negative control for immunohistochemical procedures. For a positive control, quinolinic acid was injected into the striatum to induce huntingtin cleavage. For this procedure, mice were anesthetized with inhaled isofluorine and placed in a stereotaxic apparatus, and a small craniotomy was performed at the injection site. Using a microinjector (World Precision Instruments, Sarasota, FL) and glass-pulled pipettes, 800 nl of 8 nmol of quinolinic acid (Sigma, St. Louis, MO) in PBS were slowly injected (10 nl/min) into the striatum (rostral 0.3 mm and lateral 2.3 mm to bregma, 4 mm deep). Five minutes after injection, the glass pipette was slowly withdrawn, and the scalp was closed with surgical sutures. The animal was given postoperative care in a heated chamber before returning to its home cage. The positive controls were killed and sectioned as described previously.
Immunohistochemistry. Immunocytochemistry on frozen brain tissue sections (50 μm) from one juvenile (grade 4) and two adult (presymptomatic and grade 4) HD cases and three normal individuals was performed as described previously (Gutekunst et al., 1999). To reduce nonspecific antibody binding, sections were incubated in 3% hydrogen peroxide and then PBS containing 4% normal goat serum (NGS) and 10 μg/ml avidin for 30 min followed by PBS rinses. Sections were then incubated in 2% NGS in PBS containing Htt552 and 50 μg/ml biotin for 20 hr, rinsed in PBS, and incubated for 12 hr at 4°C in biotinylated goat anti-rabbit secondary antibody (Jackson ImmunoResearch, West Grove, PA) in PBS containing 2% NGS. Sections were rinsed, incubated in avidin–biotin complex (ABC Elite; Vector Laboratories, Burlingame, CA) for 2 hr at 4°C, rinsed, and incubated in 0.05% DAB (Sigma) and 0.01% hydrogen peroxide in 50 mmTris buffer for 5–15 min. Sections were rinsed, mounted on glass slides, counterstained with thionin, dehydrated, and coverslipped for light microscopic examination using a Leica (Nussloch, Germany) DMRE.
Toluidine blue staining. Semithin sections (1.5 μm) were cut from striatum of 3- and 6-month-old YAC44 and wild-type littermate control mice. Sections were cut with a Leica Ultracut S ultramicrotome, counterstained with toluidine blue, differentiated in 95% alcohol, and coverslipped. Degeneration was quantified as described previously (Hodgson et al., 1999).
RESULTS
Validation of htt cleavage-specific antibodies in vitro and in vivo
To determine the specificity of Htt513 and Htt552 in tissues expressing endogenous htt, homogenates prepared from wild-type murine brain were either untreated or cleaved ex vivo by the addition of recombinant caspase-3. As a control, ex vivocaspase-3 cleavage was inhibited by preincubation of the homogenate with DEVD-CHO. Western blot analysis using the N-terminal antibody BKP1 identified both intact and cleaved htt (Fig.1A, left panel). Reprobing these blots demonstrates that Htt513 and Htt552 detect only the specific htt cleavage products ending at amino acid 513 or 552, respectively, with no detection of intact htt (Fig.1A, center and right panels).
Fig. 1.

Validation of cleavage-specific antibodies Htt513 and Htt552. A, Western blot of wild-type murine cortical tissue demonstrating detection of a single htt fragment ending at either amino acid 513 or 552 that is present only after ex vivo cleavage of endogenous htt with recombinant caspase-3.B, Immunofluorescence of HEK 293T cells transfected with htt constructs as indicated, demonstrating that the Htt513 or Htt552 antibodies detect htt only after tamoxifen (tam)-induced cleavage or, for Htt513, in the presence of constructs terminating at amino acid 513 (H-15X), in red. Nuclei are counterstained with DAPI (blue).
To demonstrate the ability of Htt513 and Htt552 to detect cleaved htt in its native conformation, HEK 293T cells were transfected with htt constructs with 15 or 138 glutamines ending at amino acid 1212 or 513, and apoptosis was induced by the addition of 35 μm tamoxifen (Wellington et al., 2000). Immunofluorescence was detected only after apoptotic induction or for Htt513, when the transfected construct was specifically engineered to end at amino acid 513 (Fig. 1B). These data confirm that both Htt513 and Htt552 specifically detect htt ending at amino acid 513 or 552, that neither antibody detects uncleaved htt, and that both antibodies recognize cleaved normal or expanded htt both by Western blot and by immunocytochemical methods. In vitro, Htt552 was able to detect cytoplasmic aggregates of cleaved expanded htt (Fig. 1B).
Htt is cleaved at amino acid 552 in HD brains with early grade neuropathology
Having demonstrated the specificity of the Htt513 and Htt552 antibodies, postmortem samples were analyzed from patients with HD and age-matched controls (Table 1). In each case, the neuropathological severity was noted either using the Vonsattel grade (Vonsattel et al., 1985) or by the detailed neuropathogical report (Table 1). Determination of the CAG sizes in these samples revealed that one of the patients was homozygous for the CAG expansion (HDB171), having 39 repeats on each allele (Fig.2). We also verified three heterozygous patients (HDB65, HDB173, and HDB174) with expansions of 41, 41, and 44 CAGs, respectively, and confirmed normal CAG sizes in four control brains (COB58, COB3, COB55, and COB56) (Fig. 2).
Table 1.
Summary of human disease and control brain tissue
| Brain | Neuropathology | CAG repeat number | Age at death (years) | Postmortem interval (hr) | Sex |
|---|---|---|---|---|---|
| HDB Emory | Grade 1/2 | 48/20 | 32 | 6 | F |
| HDB65 | No atrophy noted | 41/29 | 64 | 2 | M |
| HDB58 | Moderate atrophy | 42/21 | 63 | 3.5 | F |
| HDB171 | Grade 1 | 39/39 | 75 | 18.5 | M |
| HDB173 | Grade 3 | 41/24 | 76 | 7 | M |
| HDB174 | Grade 3 | 43/17 | 76 | 20 | F |
| COB3 | Control | 17/14 | 75 | 10 | F |
| COB55 | Control | 21/17 | 70 | 15 | M |
| COB56 | Control | 19/12 | 70 | 22.5 | F |
| COB58 | Control | 17/17 | 72 | 14.7 | F |
F, Female; M, male.
Fig. 2.
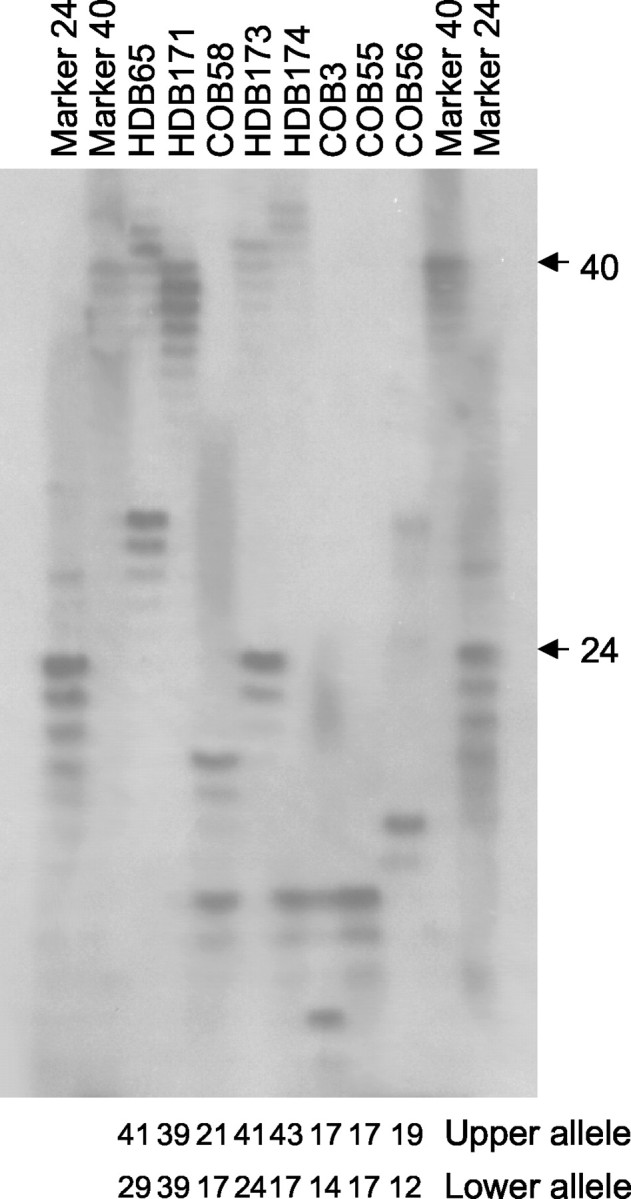
Determination of the CAG repeat length in HD brains (HDB) and control brains (COB). PCR products containing the htt CAG repeat were resolved on 0.5 mm sequencing gels alongside markers with a known CAG size. The total CAG size detected in the upper and lower alleles is listed below eachlane.
Homogenates were prepared from the frontal cortex from two patients with no gross striatal atrophy (HDB65 and HDB171), two patients with marked striatal atrophy (HDB173 and HDB174), and three controls of similar ages. Lysates were prepared in the presence of ≥5 μm zVAD-fmk to inhibit ex vivo caspase activation. Western blot analysis using BKP1 showed the presence of several htt cleavage fragments in both HD and control samples, including the presence of fragments that migrated at the expected size range for caspase cleavage products (Fig.3A, left panel). In HD patients, these caspase-generated fragments clearly showed a mobility shift induced by the expanded mutant allele compared with control brains. Importantly, lysates prepared from the patient homozygous for CAG expansion (HDB171) yielded a single band at the expected size for caspase-cleaved expanded htt (Fig.3A). These fragments from both HD and control cortex are generated by cleavage of htt at amino acid 552, as demonstrated by immunoreactivity with Htt552 (Fig. 3A, center panel). However, no immunoreactivity with the 90–95 kDa fragments was observed when these filters were reprobed with 1C2, an antibody reported to recognize expanded polyglutamine tracts (Trottier et al., 1995) (Fig. 3A, right panel). Under our conditions, 1C2 detected predominately full-length expanded htt in human brain homogenates.
Fig. 3.

Htt is cleaved at amino acid 552 in vivo. A, Parallel blots prepared from human cortex homogenized in the presence of zVAD-fmk were probed with BKP1, Htt552, and 1C2. Cleavage products showing CAG-dependent mobility are detected by BKP1 (left panel) in all samples except HD brain (HDB) 174 (in this exposure) and by Htt552 (center panel) but not 1C2 (right panel). B, Human postmortem tissue was homogenized in the absence of zVAD-fmk and incubated at 37°C with or without 2 μm recombinant caspase-3. Replicate blots were probed with BKP1 (left panel), Htt552 (center panel), or 1C2 (right panel). Caspase cleavage products arising from wild-type or expanded human htt in human brain lysates are detected with BKP1 and Htt552 but not 1C2. COB, Control brain.C, Whole murine brain from 6-month-old wild-type (WT) or YAC72 (line 2511) transgenic mice was homogenized in the absence of zVAD-fmk and incubated at 37°C with (+) or without (−) caspase-3 (C3). Replicate blots were probed with BKP1 (left panel), Htt552 (center panel), or 1C2 (right panel). Caspase cleavage products arising from wild-type murine (double open arrow) or expanded human htt (double asterisk) are distinguishable by the polyglutamine-mediated mobility shift. Full-length htt as well as normal and expanded fragments cleaved at amino acid 552 (upper band per allele) or amino acid 513 (lower band per allele) are detected with BKP1 (left panel). Htt552 recognizes only htt cleaved at amino acid 552 with normal and expanded polyglutamine (center panel). 1C2 detects both caspase cleavage products generated from the expanded human htt transgene in the context of murine brain. Faint immunoreactivity to intact expanded htt is also evident (left panel).
To test whether 1C2 can detect expanded htt cleavage fragments within brain homogenates, we prepared human or murine lysate in the absence of zVAD-fmk and then treated these samples with caspase-3 to generate maximal amounts of htt cleavage products. Under these conditions, 1C2 did not detect htt cleavage products in human lysates, although they were clearly detected by BKP1 and Htt552 (Fig.3B). In contrast, 1C2 detected htt cleavage products in the context of murine brain. Whole brains from YAC transgenic mice expressing low levels (0.5× endogenous) of human htt with 72 CAG repeats (Hodgson et al., 1999) were processed under identical conditions as human postmortem tissue, treated ex vivo with caspase-3, and probed with BKP1, Htt552, and 1C2. Htt fragments containing the polyglutamine expansion were clearly detected by 1C2, Htt552, and BKP1 (Fig. 3C). The lower level of mutant htt fragments detected by BKP1 reflects expression levels of the transgene and not decreased efficiency of cleavage of expanded htt, as detailed below. These findings suggest that detection of caspase cleavage products of expanded human htt may be possible with 1C2 in murine but not in human brain.
Western blot analysis showed that cleavage of htt in human caudate was present at low levels compared with frontal cortex in both control and HD brain (data not shown). In addition, no htt products were detected with Htt513 in frontal cortex, caudate, control, or HD brain (data not shown), suggesting that amino acid 552 is the preferred site of htt cleavage in vivo.
Caspase-cleaved htt is abundant in cortical projection neurons
To identify the specific cellular populations that contained Htt552-immunoreactive species, immunohistochemistry was performed on postmortem tissues from a 32-year-old presymptomatic individual with the HD gene mutation who died in an accident (Gutekunst et al., 1999), the grade I patient HDB171, and controls. The presymptomatic patient had CAG sizes of 48 and 20 (Table 1) and persons with 48 CAGs have a mean age of clinical onset of ∼35 years (Brinkman et al., 1997). Neuropathological analysis of this patient showed evidence of grade I/II neuropathology (data not shown), whereas a neurologic examination 6 weeks antemortem was completely normal.
Distinct cytoplasmic staining of many pyramidal neurons was noted in the frontal cortex of control and HD brains, with more intense labeling of HD neurons by Htt552 using immunohistochemical techniques (Fig.4). The omission of primary antibody resulted in no detectable staining (Fig. 4B). Nearly all Htt552 staining was cytoplasmic, suggesting that the majority of htt cleavage occurs in the cytoplasm. High-power imaging of Htt552-immunoreactive neurons in the grade I homozygote HDB171 revealed that staining was often punctate and associated with neurites (Fig.4C–H), although no costaining with EM48, an antibody that detects htt aggregates (Gutekunst et al., 1999; Li et al., 1999), was observed (data not shown). Frontal cortex from the 32-year-old presymptomatic patient (HDB Emory) contained Htt552-immunoreactive neurons in cortical layers II, III, V, and VI, with staining in layers II/III being less intense than in layers V/VI (Fig. 4, compare I with J). Striatal neurons from this presymptomatic patient also stained weakly with Htt552 (Fig. 4K), which may reflect the lower level of htt expression in striatal compared with cortical neurons (Fusco et al., 1999). These results confirmed that the htt cleavage product detected by Htt552 on Western blots primarily originated from cleavage of htt in cortical pyramidal neurons, at least some of which (layers III and V) project to the striatum. Additionally, detection of Htt552-positive neurons in the homozygote HDB171 confirms that expanded htt is indeed cleaved by caspases.
Fig. 4.

Immunohistochemical analysis of htt cleavage. A, Htt552 immunostaining of frontal cortex from control brain showing Htt552 immunoreactivity in many but not all pyramidal cortical neurons. B, Control panel for Htt552 immunostaining on the same control brain showing no immunoreactivity in the absence of primary antibody. Scale bar, 100 μm.C–H, High-power magnification of Htt552 immunostaining on frontal cortex from the grade I homozygote HDB171, showing the punctate, predominately cytoplasmic staining evident in cortical projection neurons. Scale bars: (in C) C–E,G, H, 20 μm; F, 10 μm.I, Htt552 immunostaining of frontal cortex from a 32-year-old presymptomatic patient (HDB Emory) showing the presence of cleaved htt in some neurons in layers II and III. J, Htt552 staining of frontal cortex from the same presymptomatic patient (HDB Emory) showing abundant immunoreactivity in pyramidal cortical neurons of layers V and VI. K, Htt552 immunostaining on striatum from the same presymptomatic patient, showing that some striatal neurons contain caspase-cleaved htt. Scale bar, 100 μm.
Similar to the Western blot results, the Htt513 antibody failed to detect specific cleavage products in any early grade HD or control brain examined (data not shown).
Caspase cleavage of htt precedes neurodegeneration in the YAC animal model of HD
Because brain tissue is not readily available for human HD patients before the onset of neurodegeneration, we examined the YAC transgenic animal model of HD (Hodgson et al., 1999) to specifically address when htt cleavage is first detectable relative to the onset of neurodegeneration. We examined tissue prepared from YAC72 mice expressing high levels (2 to 4× endogenous) of human htt at various time intervals by toluidine blue staining, Western blot, and immunohistochemistry. In these mice, degenerative changes in the striatum first become evident at ∼6 months of age based on toluidine blue staining of striatal neurons (Fig.5A). Although not a measure of neuronal loss, toluidine blue staining reflects highly dysfunctional neurons and represents a minimal estimate of neurodegeneration (Gallyas et al., 1993; Ribak and Baram, 1996; Kawamura et al., 1997; Toth et al., 1998). Cortical tissue from 2-, 4-, 6-, and 11-month-old YAC72 mice and wild-type littermate controls was then homogenized and blotted under conditions identical to those for processing of human samples. Immunoblotting with the N-terminal htt antibody BKP1 demonstrated the presence of intact htt and several smaller molecular weight species in both YAC72 and wild-type tissues at all ages examined (Fig.5B, left panel). Of note, specific caspase cleavage products are visible: a 100 kDa product observed only in YAC72 homogenates (cleaved mutant), suggesting that it is derived specifically from expanded htt, and a 70 kDa product present in both YAC72 and control tissue (cleaved wild-type) (Fig. 5B,left panel). The observation that endogenous htt is cleaved at early ages in both wild-type and transgenic mice suggests that cleavage of htt is a normal event. We also observed one additional product with an apparent mobility of ∼140 kDa that is present only in YAC72 homogenates, suggesting that it is also derived from expanded htt. There is no quantitative difference in htt expression at different ages when protein loading is normalized to GAPDH, and the relatively higher level of expanded cleavage products reflects the high level of transgene expression.
Fig. 5.

Degeneration and cleavage of htt in YAC transgenic mice with 72 CAG repeats. A, Semithin striatal sections from wild-type (WT) and YAC72 (line 44) transgenic mice stained with toluidine blue. Healthy neurons appear clear (white arrowhead), and degenerating neurons appear darkly stained (black arrows). B, Western blots of cortex from 2-, 4-, 6-, and 11-month-old YAC72 (line 44) transgenic mice or wild-type littermate controls. Blots were probed with BKP1 (top portion) and GAPDH (bottom portion) for a loading control (left panel), Htt552 (center panel), and 1C2 (right panel). Intact htt is indicated by anopen arrowhead and is detected by BKP1 (normal and expanded) and by 1C2 (expanded). The specific 100 and 70 kDa caspase cleavage products of expanded (cleaved mut) and normal (cleaved WT) alleles are indicated by theclosed and open arrows, respectively. Expanded cleaved htt is detected by all three antibodies, where as only BKP1 and Htt552 detect cleaved htt with a normal CAG repeat.
Immunoblotting with Htt552 confirmed that the 100 and 70 kDa products represent expanded and normal human htt cleaved by caspases at amino acid 552 (Fig. 5B, center panel). Interestingly, minor lower-molecular-weight species were also detected on murine blots using the Htt552 antibody that appear to be derived from additional proteolysis of the caspase cleavage products of normal and expanded htt (Fig. 5B, center panel). Immunoblotting with Htt513 failed to detect any cleavage product (data not shown), suggesting that the preferred site of cleavage in murine as well as human tissues is amino acid 552.
Finally, immunoblotting with 1C2 permitted the detection of intact htt as well as the 100 kDa product, confirming that the 100 kDa band represents cleavage of expanded htt (Fig. 5B, right panel). Overlaying the three blots also showed that 1C2 detected the 140 kDa species detected by BKP1 but not Htt552, suggesting that additional proteases may also cleave expanded httin vivo (Fig. 5B).
Fluorescent immunocytochemical staining of YAC72 and control brains confirmed that Htt552-positive neurons were present in the cortex at 2 months of age (Fig.6A), which is before the onset of detectable behavioral or neuropathological changes in these mice. Double staining with the neuronal marker NeuN confirmed that the Htt552 immunoreactivity was neuronal in origin. Htt552 staining in the YAC transgenic mice paralleled observations in the human samples. For example, murine Ht552 staining was punctate and largely cytoplasmic, similar to the staining pattern observed in human postmortem tissue. Compared with cortex, very few Htt552-positive neurons were identified in the striatum of either wild-type or YAC72 mice at 2 months of age (Fig. 6B), which may reflect the lower level of htt expression in the striatum compared with the cortex (Fusco et al., 1999). In addition, no Htt513-immunoreactive neurons were identified in YAC72 or control mice (data not shown).
Fig. 6.
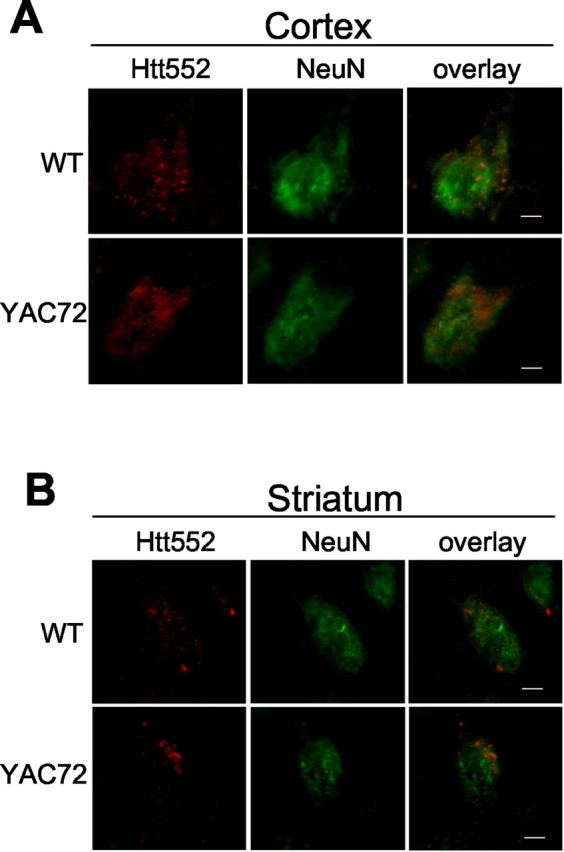
Immunofluorescence of 2-month-old YAC72 (line 44) and wild-type (WT) mice. A, In the cortex, Htt552 immunostaining (red) appears in a punctate pattern throughout the neuron, as confirmed by costaining with the neuronal marker NeuN (green).B, Immunofluorescence of the striatum from YAC72 (line 44) and wild-type mice at 2 months of age. Htt552 immunostaining (red) appears in a punctate pattern throughout the neuron, as confirmed by costaining with the neuronal marker NeuN (green). Htt552 staining was more abundant in the cortex than in the striatum. Scale bars, 2.5 μm.
Mutant and wild-type htt are cleaved with equal efficiency in brain lysates
Detection of caspase cleavage fragments from both mutant and wild-type htt suggests that caspases may cleave htt as part of its normal catabolism. To test for differences in susceptibility of cleavage between normal and expanded htt in brain tissue, we treated whole-brain homogenates prepared from YAC72 mice with high transgene expression with caspase-3 to control internally for all variables except for polyglutamine size (Fig.7A). Quantitation of cleavage over time showed that cleaved mutant and wild-type htt fragments accumulated at equivalent rates, as indicated by similar slopes in cleavage over time (Fig. 7B). These results show that mutant and wild-type htt are equally susceptible to caspase cleavage in brain lysates.
Fig. 7.
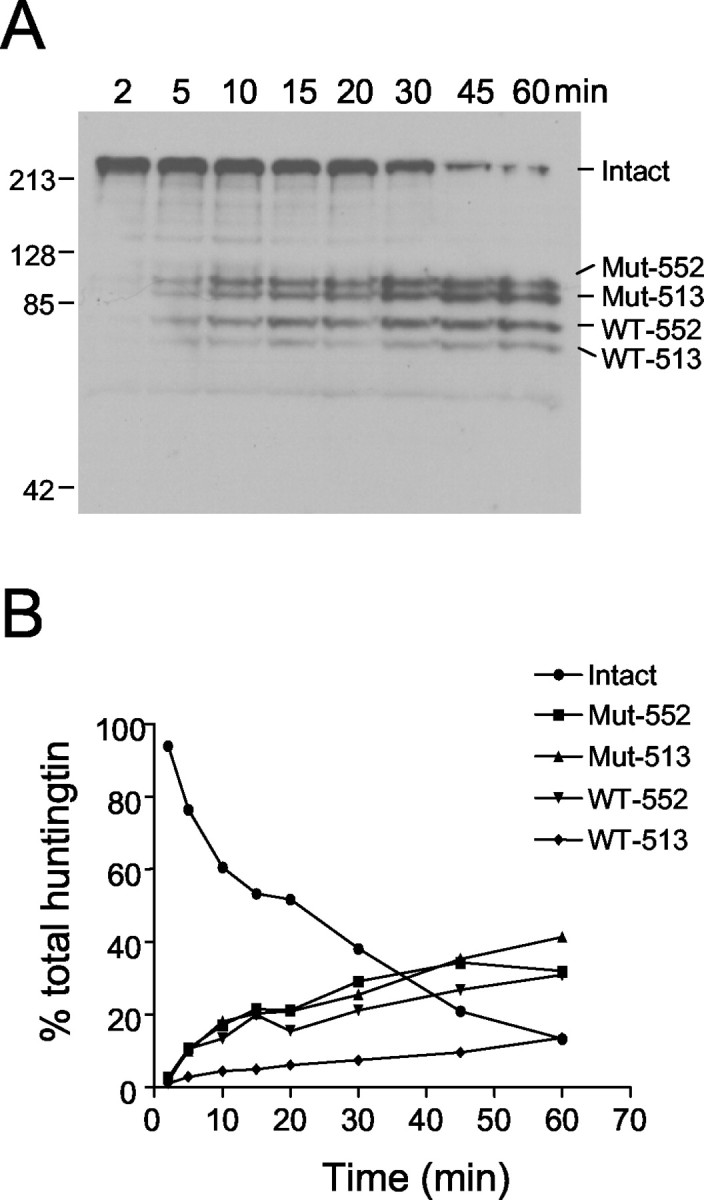
Kinetic analysis of htt cleavage in brain homogenates. A, YAC72 (high expressing) whole-brain homogenate prepared in the absence of zVAD-fmk was incubated for various times with a final concentration of 2 nm caspase-3, followed by immunodetection of cleavage fragments with BKP1. Over time, total intact htt (Intact) is converted to four major cleavage products, including mutant transgenic htt cleaved at amino acid 552 (Mut-552), mutant transgenic htt cleaved at amino acid 513 (Mut-513), wild-type endogenous htt cleaved at amino acid 552 (WT-552), and wild-type endogenous htt cleaved at amino acid 513 (WT-513).B, Quantitation of the percentage of reduction of intact total htt and percentage of accumulation of each of the htt cleavage products shows that mutant and wild-type cleavage products accumulate at equivalent rates.
DISCUSSION
In this study, we demonstrate caspase cleavage of htt in vivo using novel antibodies that specifically detect the C-terminal ends of caspase-cleaved htt. Htt cleavage was demonstrated in a patient homozygous for 39 CAG repeats using both immunohistochemistry and Western blotting, showing that caspase cleavage of htt clearly occurs in the presence of the polyglutamine expansion. In human postmortem patients, htt cleavage was particularly abundant in the cerebral cortex of patients with low-grade neuropathology in which striatal atrophy was still in its early stages (Vonsattel et al., 1985). Additionally, we demonstrate that htt cleavage precedes the onset of neurodegeneration in the YAC animal model of HD by the detection of cleaved expanded htt in YAC72 transgenic mice as early as 2 months of age. The demonstration of cleaved htt in HD brain with early neuropathology is consistent with the toxic fragment hypothesis, which suggests that the liberation of N-terminal htt fragments containing the expanded polyglutamine tract actively contributes to the pathogenesis of HD. Although htt cleavage products have been found in postmortem HD patients (Kim et al., 2001;Mende-Mueller et al., 2001) and development of a rapid neurological phenotype is well established in mice expressing truncated htt (Mangiarini et al., 1996; Bates et al., 1998), our study is the first to demonstrate that htt cleavage precedes the onset of the HD phenotypein vivo.
Cleavage of wild-type htt was also observed in control as well as HD brains, suggesting that caspase cleavage of htt may be a normal event. We detected no difference in the cleavage efficiency of expanded compared with wild-type htt, both during the natural history of the YAC transgenic model or after ex vivo treatment with caspase-3. This finding suggests that htt cleavage per se may not be solely a pathogenic event. Rather, the N-terminal cleavage fragments liberated in HD patients may lead to cellular dysfunction in HD through the toxic properties of the expanded polyglutamine tract within the truncated fragment. For example, it is possible that fragments generated from wild-type htt are efficiently cleared through the ubiquitin–proteasome pathway, whereas fragments containing an expanded polyglutamine tract accumulate and result in a number of downstream events that contribute to neuronal death. In addition, because full-length htt is neuroprotective, whereas truncated htt is toxic, cleavage of htt would presumably inactivate the neuroprotective function of intact normal htt. For htt with normal polyglutamine lengths, the protective role of intact htt is balanced with the proapoptotic role of the N-terminal fragment. However, expansion of the polyglutamine tract would be expected to disrupt this balance by the sustained presence of the toxic expanded cleavage product. In HD, loss of intact htt in addition to accumulation of htt fragments with an expanded polyglutamine may result in an overall shift toward a toxic state.
The vast majority of cleaved htt was detected in the cytoplasm, consistent with previous reports that cytoplasmic htt cleavage precedes nuclear uptake of N-terminal htt fragments (Hodgson et al., 1999; Li et al., 2000). Htt552 detected cytoplasmic aggregates generated in cultured HEK 293T cells but did not detect EM48-positive nuclear inclusions in HD brain, suggesting that additional cleavage events may trim the caspase-generated htt fragment to a smaller size, which then accumulates in the nucleus.
Cleaved htt was particularly abundant in pyramidal cortical projection neurons in layers II, III, V, and VI, which may reflect the overall abundance of htt in these neurons (Fusco et al., 1999). The presence of cleaved expanded htt in cortical layers III and V supports the hypothesis that cellular dysfunction of the corticostriatal pathway may contribute to the onset of HD. Interest in corticostriatal dysfunction has gained attention with the observation that expanded htt reduces expression of BDNF, which is made in cortical neurons and delivered to striatal neurons (Zuccato et al., 2001). Inhibition of BDNF transcription by expanded htt may result in insufficient trophic support for striatal neurons, leaving the striatum vulnerable to a variety of stressors. Cleaved htt can enter the nucleus, interact with the transcriptional coactivator cAMP response element-binding protein (CREB)-binding protein (CBP), and interfere with CBP-mediated gene transcription (Nucifora et al., 2001). Several genes, including BDNF, enkephalin, and c-jun, are regulated by CREB and/or CBP and are downregulated in HD tissue (Richfield et al., 1995; Luthi-Carter et al., 2000). Cortical changes, including the appearance of dysmorphic neurites and alterations in striatal NMDA receptor function, have been shown recently to precede behavioral deficits and neurodegeneration in animal models of HD (Levine et al., 1999; Laforet et al., 2001). Primary striatal neurons cultured from YAC72 mice have an increased susceptibility to excitotoxic death (Zeron et al., 2002), and mutant htt has been shown to enhance death of cells expressing NR1A/NR2B receptor subunits (Chen et al., 1999; Zeron et al., 2001), which are enriched in medium spiny striatal neurons. These results suggest that certain features of the pathogenesis of HD may originate in the cortex, and the resulting dysfunction of the corticostriatal pathway may enhance the selective vulnerability of medium spiny striatal neurons and ultimately lead to their degeneration. Although cleaved htt is known to be toxic in severalin vitro and in vivo models that express htt exon 1 (Mangiarini et al., 1996), until now it has not been possible to provide a molecular link between htt cleavage in particular neurons and toxicity. Our observations that cleaved htt exists in cortical neurons early in pathogenesis suggest that cortical dysfunction mediated through htt fragments may ultimately result in death of striatal neurons by several potential mechanisms.
Our analysis defines the preferred site of in vivo htt cleavage to be at amino acid 552, which is used in vitro by either caspase-3 or caspase-2 (Wellington et al., 1998, 2000). It is not yet possible to distinguish which caspase cleaves htt at amino acid 552 in vivo. Caspase-3 is a potent caspase with a high catalytic rate (Garcia-Calvo et al., 1999), is abundant in the developing and early postnatal brain (Shimohama et al., 1999), and is essential for normal developmental neuronal apoptosis (Kuida et al., 1996). In contrast, elimination of caspase-2 does not result in an overt neuronal phenotype during development (Bergeron et al., 1998), but the high basal level expression of caspase-2 in adult brain suggests that it may rather play a more prominent role in the adult CNS (Shimohama et al., 1999). Furthermore, caspase-2 but not caspase-3 has been suggested to mediate neuronal cell death induced by β-amyloid (Troy et al., 2000). Interestingly, caspase-2 but not caspase-3 coimmunoprecipitates with expanded full-length htt from brain (L. Ellerby, unpublished observations). Additionally, coexpression of dominant-negative caspase-2 but not caspase-3 was found to protect transfected primary striatal neurons from the toxicity of expanded htt, and expression of caspase-2 is upregulated in HD postmortem brains compared with age-matched controls (Ellerby, unpublished observations). Therefore, there are arguments in favor of either caspase-2 or -3 as playing a primary role in htt cleavage.
Although caspase activation is conventionally considered to be a late event in cell death from which cells do not recover, caspases can be active and cleave selected substrates without imminent cell death. Procaspase-3 has low-level catalytic activity and is capable of cleaving the same substrates as activated caspase-3 (Roy et al., 2001). In addition, activated caspase-3 has been identified in living hippocampal neurons, and inhibition of caspase activity interferes with the development of long-term spatial memory (Dash et al., 2000). Caspase activation is also suggested to play a functional role in synaptic plasticity (Chan and Mattson, 1999). It is possible that low levels of caspase activity that are normally present within neurons may generate sufficient htt cleavage products containing an expanded polyglutamine tract to initiate the pathogenesis of HD.
Recently, expanded htt was reported to be resistant to proteolysisin vivo (Dyer and McMurray, 2001), based primarily on the inability to detect cleavage products of expanded htt in human brain lysates with the 1C2 antibody. We show that htt cleavage fragments in HD patient brain homogenates can be detected on Western blots with BKP1 and Htt552 but not with 1C2. Furthermore, we have clear evidence of htt cleavage in a patient homozygous for 39 CAG repeats by immunohistochemistry as well as Western blot. Our results demonstrate that cleavage of expanded htt by caspases does indeed occur in vivo, a finding supported by two other reports (Kim et al., 2001;Mende-Mueller et al., 2001). Finally, we detected no difference in cleavage efficiency for normal compared with expanded htt when assayed in the same brain homogenate.
In this study, we demonstrate that caspase cleavage of both normal and expanded htt occurs in vivo. We define amino acid 552 as a preferred site of caspase cleavage in human HD, in YAC72 transgenic mice, and in control human and murine brain. Importantly, we have demonstrated caspase cleavage in a homozygote for HD, showing that cleavage is not restricted to the product of the normal allele. Cleaved htt is abundant in cortical projection neurons, suggesting that increased levels of N-terminal htt fragments with an expanded polyglutamine tract may mediate dysfunction of the corticostriatal pathway as an early event in HD. Corticostriatal dysfunction may lead to selective degeneration of the striatum through several mechanisms, including decreased trophic support and increased excitotoxicity. Finally, we show that cleavage fragments containing expanded htt are present in HD patient brains with early grade neuropathology and are present in YAC transgenic mice ∼4 months before the onset of detectable neurodegeneration or aggregates. Our results suggest that caspase cleavage of htt may be upstream of many adverse events correlated with htt fragments, including aggregate formation, nuclear localization of htt, altered transcription, and neurodegeneration. The observation that caspase cleavage of expanded htt is an early event in HD suggests that caspase inhibitors may be effective potential therapeutic agents for the treatment of HD. To be effective for HD, it is clear that the dose, route of administration, duration, and frequency of treatment will require detailed optimization to minimize undesirable side-effects. However, early and optimized treatment of HD patients with caspase inhibitors may reduce the load of toxic N-terminal htt fragments with expanded polyglutamine tracts in cortical neurons, which may delay or prevent the onset or progression of HD through preservation of functional corticostriatal connections.
Footnotes
This work was supported by National Institutes of Health (NIH) Grant NS40251 (L.M.E.), the Huntington's Disease Society of America, and the Multiple Dystrophy Association. C.-A.G. and D.B. are supported by the NIH. S.M.H. is supported by NIH Grants NS35255 and AT00613, the Hereditary Disease Foundation (HDF), and the Huntington Disease Society of America (HDSA). M.R.H. is supported by a Merck Frosst Grant to the Centre for Molecular Medicine and Therapeutics, the Canadian Institutes of Health Research, the Canadian Networks of Centres of Excellence and is a holder of a Canada Research Chair. We thank members of our respective laboratories for their support and critical evaluation of the data. We also thank the Canadian Brain Tissue Bank, the Harvard Brain Tissue Resource Center, and the HDF for generously sharing rare HD tissue. We thank the HDSA and the HDF for their generous support of this work.
∗C.L.W. and L.M.E. contributed equally to this work.
Correspondence should be addressed to Michael R. Hayden, Centre for Molecular Medicine and Therapeutics, 980 West 28th Avenue, Vancouver, British Columbia, Canada V5Z 4H4. E-mail: mrh@cmmt.ubc.ca.
REFERENCES
- 1.Andrew SE, Goldberg YP, Kremer B, Telenius H, Theilmann J, Adam S, Starr E, Squitieri F, Lin B, Kalchman MA. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington's disease. Nat Genet. 1993;4:398–403. doi: 10.1038/ng0893-398. [DOI] [PubMed] [Google Scholar]
- 2.Bates GP, Mangiarini L, Davies SW. Transgenic mice in the study of polyglutamine repeat expansion diseases. Brain Pathol. 1998;8:699–714. doi: 10.1111/j.1750-3639.1998.tb00196.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bergeron L, Perez GI, Macdonald G, Shi L, Sun Y, Jurisicova A, Varmuza S, Latham KE, Flaws JA, Salter JCM, Hara H, Moskowitz MA, Li E, Greenberg A, Tilly JL, Yuan J. Defects in regulation of apoptosis in caspase-2-deficient mice. Genes Dev. 1998;12:1304–1314. doi: 10.1101/gad.12.9.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brinkman RR, Mezei MM, Theilmann J, Almqvist E, Hayden MR. The likelihood of being affected with Huntington disease by a particular age, for a specific CAG size. Am J Hum Genet. 1997;60:1202–1210. [PMC free article] [PubMed] [Google Scholar]
- 5.Bruland O, Almqvist EW, Goldberg YP, Boman H, Hayden MR, Knappskog PM. Accurate determination of the number of CAG repeats in the Huntington disease gene using a sequence-specific internal DNA standard. Clin Genet. 1999;55:198–202. doi: 10.1034/j.1399-0004.1999.550308.x. [DOI] [PubMed] [Google Scholar]
- 6.Chan SL, Mattson MP. Caspase and calpain substrates: roles in synaptic plasticity and cell death. J Neurosci Res. 1999;58:167–190. [PubMed] [Google Scholar]
- 7.Chen N, Luo T, Wellington C, Metzler M, McCutcheon K, Hayden MR, Raymond LA. Subtype-specific enhancement of NMDA receptor currents by mutant huntingtin. J Neurochem. 1999;72:1890–1898. doi: 10.1046/j.1471-4159.1999.0721890.x. [DOI] [PubMed] [Google Scholar]
- 8.Cooper JK, Schilling G, Peters MF, Herring WJ, Sharp AH, Kaminsky Z, Masone J, Khan FA, Delanoy M, Borchelt DR, Dawson VL, Dawson TM, Ross CA. Truncated N-terminal fragments of huntingtin with expanded glutamine repeats form nuclear and cytoplasmic aggregates in cell culture. Hum Mol Genet. 1998;7:783–790. doi: 10.1093/hmg/7.5.783. [DOI] [PubMed] [Google Scholar]
- 9.Dash PK, Blum S, Moore AN. Caspase activity plays an essential role in long-term memory. NeuroReport. 2000;11:2811–2816. doi: 10.1097/00001756-200008210-00040. [DOI] [PubMed] [Google Scholar]
- 10.DiFiglia M, Sapp E, Chase K, Schwarz C, Meloni A, Young C, Martin E, Vonsattel JP, Carraway R, Reeves SA. Huntingtin is a cytoplasmic protein associated with vesicles in human and rat brain neurons. Neuron. 1995;14:1075–1081. doi: 10.1016/0896-6273(95)90346-1. [DOI] [PubMed] [Google Scholar]
- 11.Dyer RB, McMurray CT. Mutant protein in Huntington disease is resistant to proteolysis in affected brain. Nat Genet. 2001;29:270–278. doi: 10.1038/ng745. [DOI] [PubMed] [Google Scholar]
- 12.Ellerby LM, Andrusiak RL, Wellington CL, Hackam AS, Propp SS, Wood JD, Sharp AH, Margolis RL, Ross CA, Salvesen GS, Hayden MR, Bredesen DE. Cleavage of atrophin-1 at caspase site aspartic acid 109 modulates cytotoxicity. J Biol Chem. 1999;274:8730–8736. doi: 10.1074/jbc.274.13.8730. [DOI] [PubMed] [Google Scholar]
- 13.Fusco FR, Chen Q, Lamoreaux WJ, Figueredo-Cardenas G, Jiao Y, Coffman JA, Surmeier DJ, Honig MG, Carlock LR, Reiner A. Cellular localization of huntingtin in striatal and cortical neurons in rats: lack of correlation with neuronal vulnerability in Huntington's disease. J Neurosci. 1999;19:1189–1202. doi: 10.1523/JNEUROSCI.19-04-01189.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gallyas F, Zoltay G, Horvath Z, David K, Kelenyi L. An immediate morphopathologic response of neurons to electroshock; a reliable model for producing “dark” neurons in experimental neuropathology. Neurobiology. 1993;1:133–142. [PubMed] [Google Scholar]
- 15.Garcia-Calvo M, Peterson EP, Rasper DM, Vaillancourt JP, Zamboni R, Nicholson DW, Thornberry NA. Purification and catalytic properties of human caspase family members. Cell Death Differ. 1999;6:362–369. doi: 10.1038/sj.cdd.4400497. [DOI] [PubMed] [Google Scholar]
- 16.Gervais FG, Xu D, Robertson GS, Vaillancourt JP, Zhu Y, Huang J, LeBlanc A, Smith D, Rigby M, Shearman MS, Clarke EE, Zheng H, Van Der Ploeg LH, Ruffolo SC, Thornberry NA, Xanthoudakis S, Zamboni RJ, Roy S, Nicholson DW. Involvement of caspases in proteolytic cleavage of Alzheimer's amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell. 1999;97:395–406. doi: 10.1016/s0092-8674(00)80748-5. [DOI] [PubMed] [Google Scholar]
- 17.Goldberg YP, Nicholson DW, Rasper DM, Kalchman MA, Koide HB, Graham RK, Bromm M, Kazemi-Esfarjani P, Thornberry NA, Vaillancourt JP, Hayden MR. Cleavage of huntingtin by apopain, a proapoptotic cysteine protease, is modulated by the polyglutamine tract. Nat Genet. 1996;13:442–449. doi: 10.1038/ng0896-442. [DOI] [PubMed] [Google Scholar]
- 18.Gutekunst CA, Li SH, Yi H, Mulroy JS, Kuemmerle S, Jones R, Rye D, Ferrante RJ, Hersch SM, Li XJ. Nuclear and neuropil aggregates in Huntington's disease: relationship to neuropathology. J Neurosci. 1999;19:2522–2534. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hackam AS, Singaraja R, Wellington CL, Metzler M, McCutcheon K, Zhang T, Kalchman M, Hayden MR. The influence of huntingtin protein size on nuclear localization and cellular toxicity. J Cell Biol. 1998;141:1097–1105. doi: 10.1083/jcb.141.5.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hayden MR. Huntington's chorea. Springer; Berlin: 1981. [Google Scholar]
- 21.Hodgson JG, Agopyan N, Gutekunst CA, Leavitt BR, LePiane F, Singaraja R, Smith DJ, Bissada N, McCutcheon K, Nasir J, Jamot L, Li XJ, Stevens ME, Rosemond E, Roder JC, Phillips AG, Rubin EM, Hersch SM, Hayden MR. A YAC mouse model for Huntington's disease with full-length mutant huntingtin, cytoplasmic toxicity, and selective striatal neurodegeneration. Neuron. 1999;23:181–192. doi: 10.1016/s0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- 22.Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 23.Kalchman MA, Koide HB, McCutcheon K, Graham RK, Nichol K, Nishiyama K, Kazemi-Esfarjani P, Lynn FC, Wellington C, Metzler M, Goldberg YP, Kanazawa I, Gietz RD, Hayden MR. HIP1, a human homologue of S. cerevisiae Sla2p, interacts with membrane-associated huntingtin in the brain. Nat Genet. 1997;16:44–53. doi: 10.1038/ng0597-44. [DOI] [PubMed] [Google Scholar]
- 24.Kawamura T, Akira T, Watanabe M, Kagitani Y. Prostaglandin E1 prevents apoptotic cell death in superficial dorsal horn of rat spinal cord. Neuropharmacology. 1997;36:1023–1030. doi: 10.1016/s0028-3908(97)00096-8. [DOI] [PubMed] [Google Scholar]
- 25.Kim YJ, Yi Y, Sapp E, Wang Y, Cuiffo B, Kegel KB, Qin Z-H, Aronin N, DiFiglia M. Caspase 3-cleaved N-terminal fragments of wild-type and mutant huntingtin are present in normal and Huntington's disease brains, associate with membranes, and undergo calpain-dependent proteolysis. Proc Natl Acad Sci USA. 2001;98:12784–12789. doi: 10.1073/pnas.221451398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuida K, Zheng TS, Na S, Kuan C, Yang D, Karasuyama H, Rakic P, Flavell RA. Decreased apoptosis in the brain and premature lethality in CPP32-deficient mice. Nature. 1996;384:368–372. doi: 10.1038/384368a0. [DOI] [PubMed] [Google Scholar]
- 27.Laforet GA, Sapp E, Chase K, McIntyre C, Boyce FM, Campbell M, Cadigan BA, Warzecki L, Tagle DA, Reddy PH, Cepeda C, Calvert CR, Jokel ES, Klapstein GJ, Ariano MA, Levine MS, DiFiglia M, Aronin N. Changes in cortical and striatal neurons predict behavioral and electrophysiological abnormalities in a transgenic murine model of Huntington's disease. J Neurosci. 2001;21:9112–9123. doi: 10.1523/JNEUROSCI.21-23-09112.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Leavitt BR, Guttman JA, Hodgson JG, Kimel GH, Singaraja R, Vogl AW, Hayden MR. Wild-type huntingtin reduces the cellular toxicity of mutant huntingtin in vivo. Am J Hum Genet. 2001;68:313–324. doi: 10.1086/318207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Levine MS, Klapstein GJ, Koppel A, Gruen E, Cepeda C, Vargas ME, Jokel ES, Carpenter EM, Zanjani H, Hurst RS, Efstratiadis A, Zeitlin S, Chesselet MF. Enhanced sensitivity to N-methyl-d-aspartate receptor activation in transgenic and knockin mouse models of Huntington's disease. J Neurosci Res. 1999;58:515–532. [PubMed] [Google Scholar]
- 30.Li H, Li SH, Cheng AL, Mangiarini L, Bates GP, Li XJ. Ultrastructural localization and progressive formation of neuropil aggregates in Huntington's disease transgenic mice. Hum Mol Genet. 1999;8:1227–1236. doi: 10.1093/hmg/8.7.1227. [DOI] [PubMed] [Google Scholar]
- 31.Li H, Li S-H, Johnston H, Shelbourne PF, Li X-J. Amino-terminal fragments of mutant huntingtin show selective accumulation in striatal neurons and synaptic toxicity. Nat Genet. 2000;25:385–389. doi: 10.1038/78054. [DOI] [PubMed] [Google Scholar]
- 32.Luthi-Carter R, Strand A, Peters NL, Solano SM, Hollingsworth ZR, Menon AS, Frey AS, Spektor BS, Penney EB, Schilling G, Ross CA, Borchelt DR, Tapscott SJ, Young AB, Cha J-H, Olson JM. Decreased expression of striatal signaling genes in a mouse model of Huntington's disease. Hum Mol Genet. 2000;9:1259–1271. doi: 10.1093/hmg/9.9.1259. [DOI] [PubMed] [Google Scholar]
- 33.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 34.Martindale D, Hackam A, Wieczorek A, Ellerby L, Wellington C, McCutcheon K, Singaraja R, Kazemi-Esfarjani P, Devon R, Kim SU, Bredesen DE, Tufaro F, Hayden MR. Length of huntingtin and its polyglutamine tract influences localization and frequency of intracellular aggregates. Nat Genet. 1998;18:150–154. doi: 10.1038/ng0298-150. [DOI] [PubMed] [Google Scholar]
- 35.Mende-Mueller LM, Toneff T, Hwang S-R, Chesselet M-F, Hook VYH. Tissue-specific proteolysis of Huntingtin (htt) in human brain: evidence of enhanced levels of N- and C-terminal htt fragments in Huntington's disease striatum. J Neurosci. 2001;21:1830–1837. doi: 10.1523/JNEUROSCI.21-06-01830.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nucifora FC, Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Ross CA. Interference by huntingtin and atrophin-1 with CBP-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- 37.Ribak CE, Baram TZ. Selective death of hippocampal CA3 pyramidal cells with mossy fiber afferents after CRH-induced status epilepticus in infant rats. Brain Res Dev Brain Res. 1996;91:245–251. doi: 10.1016/0165-3806(95)00183-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Richfield EK, Maguire-Zeiss KA, Cox C, Gilmore J, Voorn P. Reduced expression of preproenkephalin in striatal neurons from Huntington's disease patients. Ann Neurol. 1995;37:335–343. doi: 10.1002/ana.410370309. [DOI] [PubMed] [Google Scholar]
- 39.Rigamonti D, Bauer JH, De Fraja C, Conti L, Sipione S, Sciorati C, Clementi E, Hackam A, Hayden MR, Li Y, Cooper JK, Ross CA, Govoni S, Vincenz C, Cattaneo E. Wild-type huntingtin protects from apoptosis upstream of caspase-3. J Neurosci. 2000;20:3705–3713. doi: 10.1523/JNEUROSCI.20-10-03705.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roy S, Bayly CI, Gareau Y, Houtzager VM, Kargman S, Keen SLC, Rowland K, Seiden IM, Thornberry NA, Nicholson DW. Maintenance of caspase-3 proenzyme dormancy by an intrinsic “safety catch” regulatory tripeptide. Proc Natl Acad Sci USA. 2001;98:6132–6137. doi: 10.1073/pnas.111085198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saudou F, Finkbeiner S, Devys D, Greenberg ME. Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell. 1998;95:55–66. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- 42.Shimohama S, Tanino H, Fujimoto S. Changes in caspase expression in Alzheimer's disease: comparison with development and aging. Biochem Biophys Res Commun. 1999;256:381–384. doi: 10.1006/bbrc.1999.0344. [DOI] [PubMed] [Google Scholar]
- 43.Toth Z, Yan XX, Haftoglou S, Ribak CE, Baram TZ. Seizure-induced neuronal injury: vulnerability to febrile seizures in an immature rat model. J Neurosci. 1998;18:4285–4294. doi: 10.1523/JNEUROSCI.18-11-04285.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Trottier Y, Lutz Y, Stevanin G, Imbert G, Devys D, Cancel G, Saudou F, Weber C, David G, Tora L. Polyglutamine expansion as a pathological epitope in Huntington's disease and four dominant cerebellar ataxias. Nature. 1995;378:403–406. doi: 10.1038/378403a0. [DOI] [PubMed] [Google Scholar]
- 45.Troy CM, Rabacchi SA, Friedman WJ, Frappier TF, Brown K, Shelanski ML. Caspase-2 mediates neuronal cell death induced by β-amyloid. J Neurosci. 2000;20:1386–1392. doi: 10.1523/JNEUROSCI.20-04-01386.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington's disease. J Neuropathol Exp Neurol. 1985;44:559–577. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- 47.Wellington CL, Ellerby LM, Hackam AS, Margolis RL, Trifiro MA, Singaraja R, McCutcheon K, Salvesen GS, Propp SS, Bromm M, Rowland KJ, Zhang T, Rasper D, Roy S, Thornberry N, Pinsky L, Kakizuka A, Ross CA, Nicholson DW, Bredesen DE, Hayden MR. Caspase cleavage of gene products associated with triplet expansion disorders generates truncated fragments containing the polyglutamine tract. J Biol Chem. 1998;273:9158–9167. doi: 10.1074/jbc.273.15.9158. [DOI] [PubMed] [Google Scholar]
- 48.Wellington CL, Singaraja R, Ellerby L, Savill J, Roy S, Leavitt B, Cattaneo E, Hackam A, Sharp A, Thornberry N, Nicholson DW, Bredesen DE, Hayden MR. Inhibiting caspase cleavage of huntingtin reduces toxicity and aggregate formation in neuronal and nonneuronal cells. J Biol Chem. 2000;275:19831–19838. doi: 10.1074/jbc.M001475200. [DOI] [PubMed] [Google Scholar]
- 49.Zeron MM, Chen N, Moshaver A, Lee AT, Wellington CL, Hayden MR, Raymond LA. Mutant huntingtin enhances excitotoxic cell death. Mol Cell Neurosci. 2001;17:41–53. doi: 10.1006/mcne.2000.0909. [DOI] [PubMed] [Google Scholar]
- 50.Zeron MM, Hansson O, Chen N, Wellington CL, Leavitt BR, Brundin P, Hayden MR, Raymond LA. Increased sensitivity to N-methyl-d-aspartate receptor-mediated excitotoxicity in a mouse model of Huntington's disease. Neuron. 2002;14:849–860. doi: 10.1016/s0896-6273(02)00615-3. [DOI] [PubMed] [Google Scholar]
- 51.Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]