

Abstract
Dysregulation of dopamine transmission is thought to contribute to schizophrenic psychosis and drug dependence. Dopamine release is regulated by D2 dopamine autoreceptors, and D2receptor ligands are used to treat psychosis and addiction. To elucidate the long-term effects of D2 autoreceptor activity on dopamine signaling, dopamine overflow evoked by single or paired-pulse stimulation was compared in striatal slices from D2-null mutant and wild-type mice. Quinpirole, a D2/D3 receptor agonist, had no effect on evoked dopamine release in D2 mutant mice, indicating that D2 receptors are the only release-regulating receptors at the axon terminal. Dopamine release inhibition by GABABreceptor activation was unchanged in D2 mutant mice, suggesting that other G-protein-coupled pathways remained normal in the absence of D2 autoreceptors. Paired-pulse stimulation revealed that autoinhibition of dopamine release was maximal 500 msec after stimulation and lasted <5 sec. In D2-null mutants, dopamine overflow in response to single stimuli was severely decreased. Experiments with the uptake inhibitor nomifensine indicated that this was caused by enhanced dopamine uptake rather than reduced release. Analysis of dopamine overflow kinetics using a simulation model suggested that the enhanced uptake was caused by an increase in the maximal velocity of uptake, Vmax. These results from D2-null mutant mice support the suggestion that D2 autoreceptors and dopamine transporters interact to regulate the amplitude and timing of dopamine signals.
Keywords: dopamine, D2 receptor, autoreceptor, dopamine transporter, release, paired-pulse depression, striatum
Dopamine (DA) release and uptake are severely affected by drugs of abuse, such as cocaine and amphetamine, and alterations in DA levels have also been postulated to occur in mental illnesses, including schizophrenia. The D2-like DA receptors, main targets of antipsychotic drugs, are expressed as postsynaptic receptors and as presynaptic autoreceptors. As autoreceptors, they regulate extracellular DA levels.
It is well established that activation of DA autoreceptors located at the soma decreases the firing rate of DAergic neurons (Bunney et al., 1973) and that axon-terminal autoreceptors inhibit DA release (Cubeddu and Hoffmann, 1982). However, a wide range of the duration of DA release autoinhibition has been reported in in vivo andin vitro studies, with estimates ranging from milliseconds up to 30 sec (Mayer et al., 1988; Limberger et al., 1991; Kennedy et al., 1992; Benoit-Marand et al., 2001). Moreover, D2 receptor activity may have long-term effects on DA release by inhibiting DA synthesis (Kehr et al., 1972; Haubrich and Pflueger, 1982; O'Hara et al., 1996; Pothos et al., 1998; Lindgren et al., 2001).
In addition to regulating firing rates, DA synthesis, and DA release, an interaction between D2 autoreceptors and the plasmalemmal DA transporter (DAT) has been proposed by several studies (Meiergerd et al., 1993; Cass and Gerhardt, 1994; Batchelor and Schenk, 1998), possibly including long-term effects on DAT expression (Kimmel et al., 2001; Mayfield and Zahniser, 2001).
Recently, the introduction of D2 receptor mutant mice has made it possible to study the long-term effects of the absence of D2 activity on presynaptic aspects of DAergic transmission. Three mouse lines have been generated: two with null mutations (Baik et al., 1995; Jung et al., 1999) and one with a deletion mutation (Kelly et al., 1997). Studies on the Baik et al. (1995) null-mutant mice indicated that the D2receptor is the only release-regulating (L'hirondel et al., 1998) and the only firing rate-regulating (Mercuri et al., 1997) autoreceptor. In the same mouse line, immunolabeling with anti-DAT antibodies revealed that the axonal innervation of the striatum was denser than in wild-type (WT) mice (Parish et al., 2001). These findings suggest that long-term adaptations in DA release and uptake occur in response to the lack of D2 activity. More direct evidence for changes in release and uptake was provided by two recent in vivo studies. Dickinson et al. (1999) reported that D2-null mice (D2−/−) have normal extracellular DA levels but decreased DA uptake, andBenoit-Marand et al. (2001) demonstrated a lack of DA release autoinhibition in D2−/− versus WT mice.
To isolate axon-terminal D2 autoreceptor effects on DA release and uptake, we have used electrochemical recordings of evoked DA overflow in striatal slice preparations. Comparison of WT with D2−/− mice (Jung et al., 1999) revealed that the absence of D2 autoreceptor activity alters the kinetics of DA signaling via effects on both release and uptake.
MATERIALS AND METHODS
Animals and striatal slice preparation. We used mice lacking the D2 receptor (D2−/−) and their WT littermates (Jung et al., 1999). These animals had either a C57BL/6 × 129Sv mixed genetic or a congenic C57BL/6 background. There were no differences with respect to DA release and reuptake between the two different genetic backgrounds (data not shown). All mice were between 8 and 16 weeks of age.
Mice were anesthetized with ketamine/xylazine and decapitated. Striatal brain slices were cut on a vibratome at 300 μm thickness. Recordings were obtained from the second to fourth frontal slice of caudate putamen (bregma +1.54 mm to + 0.62 mm) (Franklin and Paxinos, 1997). Slices were allowed to recover for 1 hr in a holding chamber in oxygenated artificial CSF (ACSF) at room temperature; they were then placed in a recording chamber and superfused (1 ml/min) with ACSF (in mm): 125 NaCl, 2.5 KCl, 26 NaHCO3, 2.4 CaCl2, 1.3 MgSO4, 0.3 KH2PO4, and 10 glucose at 36°C. Nomifensine, (−)sulpiride, quinpirole, andR(+)-baclofen were obtained from Sigma (St. Louis, MO).
Electrochemical recordings. Disk carbon fiber electrodes of 5 μm in diameter with a freshly cut surface (Kawagoe et al., 1993) were placed in the dorsal striatum ∼50 μm into the slice. For cyclic voltammetry (CV), a triangular voltage wave (−400 to +1000 mV at 300 V/sec vs Ag/AgCl) was applied to the electrode every 100 msec using a waveform generator (Model 39; Wavetek, Ltd., Norwich, Norfolk, UK). Current was recorded with an Axopatch 200B amplifier (Axon Instruments, Foster City, CA), with a low-pass Bessel Filter setting at 10 kHz, digitized at 25 kHz (Instrunet board; GW Instruments, Somerville, MA), and acquired with the Superscope II program (GW Instruments). For amperometry, a constant voltage of +400 mV was applied via the Axopatch 200B. Amperometric traces were filtered with a digital hamming filter (125 Hz cutoff frequency). Striatal slices were electrically stimulated with a bipolar stimulating electrode placed ∼100 μm from the recording electrode using an Iso-Flex stimulus isolator triggered by a Master-8 pulse generator (A.M.P.I., Jerusalem, Israel). Background-subtracted cyclic voltammograms served to calibrate the electrodes and to identify the released substance.
Simulation model. To estimate DA release and uptake parameters from CV recordings of evoked DA overflow, we used a one-dimensional random walk/finite difference model of diffusion (Berg, 1983; Sulzer and Pothos, 2000), which incorporated a function for DA uptake according to Michaelis–Menten kinetics. A detailed description of the simulation has been described by Schmitz et al. (2001). Examples of spreadsheets that can be used to run this simulation model can be downloaded from our laboratory web site (http://www.columbia.edu/∼ds43/download.html). To find the best fit to an actual recording trace, we used the Mini Analysis Program (Synaptosoft, Decatur, GA), which contains a subroutine with our simulation model and uses a simplex algorithm to perform nonlinear regression.
HPLC analysis of catecholamine content in striatal slices.Corticostriatal slices from D2−/− and WT mice were prepared as described above. The striatum was dissected and homogenized in 300 μl of 2% perchloric acid either immediately or after an incubation period in ACSF. Samples were sonicated, frozen, thawed, and again sonicated to ensure disruption of membranes and finally centrifuged at 15,000 × g at 4°C. The pellet was used to determine protein concentrations with a protein assay kit (Bio-Rad, Hercules, CA). The supernatant was kept frozen at −80°C, and catecholamine content was measured the next day by HPLC. The mobile phase (adjusted to a pH 4.6 of with glacial acetic acid) contained 10% methanol and (in mm): 50 sodium acetate, 0.05 EDTA, and 0.7 heptanesulfonic acid. The HPLC system consisted of an ESA (Chelmsford, MA) Coulochem 5100A with a 5011 analytical cell and a BAS Biophase ODS column (250 × 4.6 mm; 5 μm).
Statistics. The two-tailed Student's t test was used for pairs of data, whereas ANOVA with post hoccomparison (Newman–Keul test) was used to analyze groups of data (GB Stat software, Silver Spring, MD). Significance levels ofp < 0.05 (*) or p < 0.01(**) are indicated in the figures. The best fit of simulations to DA overflow recordings was found by nonlinear regression, and the goodness of fit is reported by R2 values (Schmitz et al., 2001).
RESULTS
Inhibition of DA release by G-protein-coupled receptors
DA overflow was recorded with carbon fiber disk electrodes in the dorsal striatum of corticostriatal slices using fast CV. DA overflow was evoked by single-pulse stimulation (1 msec, 400 μA) with a bipolar stimulation electrode placed ∼100 μm from the recording electrode (Fig. 1a). A typical response is shown in Figure 1c, and background-subtracted voltammograms for the peak DA overflow and for electrode calibration in a 5 μm DA solution are shown in Figure1b.
Fig. 1.

DA overflow recorded in coronal striatal mouse slices with CV. a, DA overflow was stimulated with a bipolar electrode placed on the surface of the slice and recorded with a carbon fiber electrode (CFE) placed ∼100 μm away from both poles of the stimulating electrode and inserted ∼50 μm deep into the slice. CP, Caudate putamen;aca, anterior commissure anterior. b, CV subtraction voltammograms for a calibration in 5 μm DA (top trace) and for the peak of the DA signal recording in c (bottom trace). Calibration of the electrode before and after the recording provided identification of the measured substance and conversion of current into DA concentration.c, Example of DA overflow in response to a single-pulse stimulus (400 μA, 1 msec) recorded with CV. A triangular voltage wave is applied every 100 msec to the carbon fiber electrode, and the recorded current trace is sampled at the voltage that elicits maximal DA oxidation. Solid line indicates the best fit found by a random walk simulation of DA diffusion corrected for uptake with Michaelis–Menten kinetics (see Materials and Methods).
Previous studies suggested that in addition to the D2 receptor, other members of the D2-like receptor family may act as autoreceptors (Tepper et al., 1997; Diaz et al., 2000). Therefore, we compared the effects of the D2/D3agonist quinpirole on stimulated DA release in slices derived from D2−/− and WT mice. DA overflow was stimulated once per 2 min. When a stable response was obtained, superfusion of the slice was switched to medium containing quinpirole (0.5 μm). Three recordings of evoked DA overflow before the switch to quinpirole-containing medium were averaged to normalize the data. Figure 2a plots the normalized maximal DA overflow during 10 min of superfusion with quinpirole, and subsequent washout with control medium in slices derived from WT and D2−/− mice. In WT mice, DA overflow was inhibited by 75% after 10 min of superfusion and recovered only slowly and partially within 20 min after the switch to control medium. In contrast, quinpirole had no effect on stimulated release in slices from D2−/− mice. Thus, D3 receptors do not appear to play a role in inhibiting evoked DA release in the dorsal striatum.
Fig. 2.
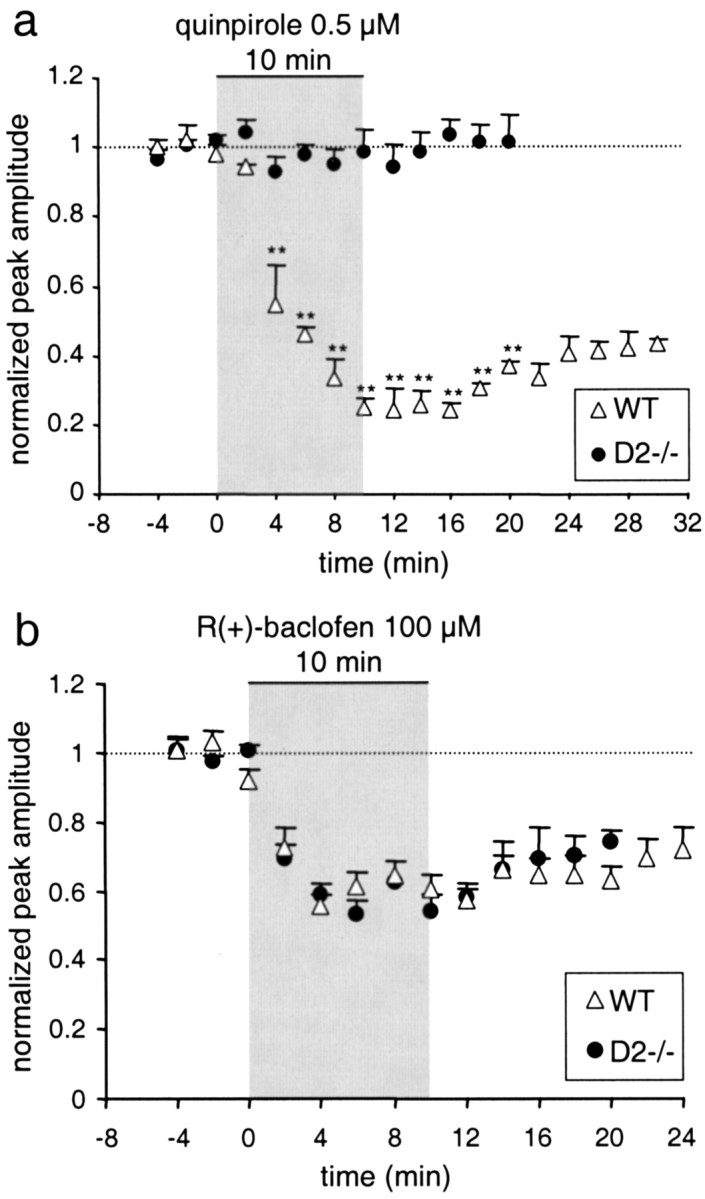
Autoreceptor and heteroreceptor inhibition of DA release. a, DA overflow was evoked once per 2 min in slices of WT (▵, n = 3) and D2−/− (●, n = 5) mice. After stable responses were obtained, slices were superfused for 10 min with the D2/D3 receptor agonist quinpirole (0.5 μm). The peak amplitude of evoked DA overflow, normalized to DA overflow evoked by three stimuli before the switch to quinpirole, is plotted versus time (means ± SEM; double asterisks denote a statistically significant difference from WT with p < 0.01). The superfusion with quinpirole is indicated by the gray area. b, Effects of the GABAB agonist R(+)-baclofen (100 μm) on evoked DA overflow in slices from D2−/− mice (n = 6) and WT mice (n = 8). As in a, the normalized peak response is plotted versus time. The gray areaindicates the 10 min superfusion with R(+)-baclofen. There was no statistically significant difference between WT and D2−/− mice (p > 0.05).Dotted lines indicate normalized amplitude = 1.
It is possible that in D2−/− mice, inhibition of DA release by G-protein-coupled heteroreceptors is altered as a result of compensatory adaptation. At somata of midbrain DAergic neurons, GABAB receptors, which are coupled to the inhibitory G-proteins Gi and Go (Kerr and Ong, 1995), affect the same inwardly rectifying potassium currents and voltage-gated calcium currents as the D2 autoreceptor (Lacey et al., 1988; Cardozo and Bean, 1995). Therefore, we tested the effect of the GABAB agonist R(+)-baclofen (100 μm) on evoked DA release in slices from WT and D2−/− mice. Superfusion with R(+)-baclofen reduced stimulated DA overflow by 40% (within 4 min) in both WT and D2−/− mice alike (Fig. 2b).
In summary, these results indicate that the axon-terminal D2 receptor is the only release-regulating autoreceptor. Because the response to GABABagonists was unaffected, G-protein-mediated release inhibition by heteroreceptors appears to remain functional in D2−/− mice.
Evoked DA release and reuptake in D2−/− mice
We subsequently examined how the absence of D2 autoreceptors affects axon-terminal DA release and uptake parameters. We found that in D2−/− mice, DA overflow in response to a single electrical stimulus (1 msec, 400 μA) reached only 54% of the amplitude of evoked DA overflow recorded in slices from WT mice (Fig.3a,b).
Fig. 3.
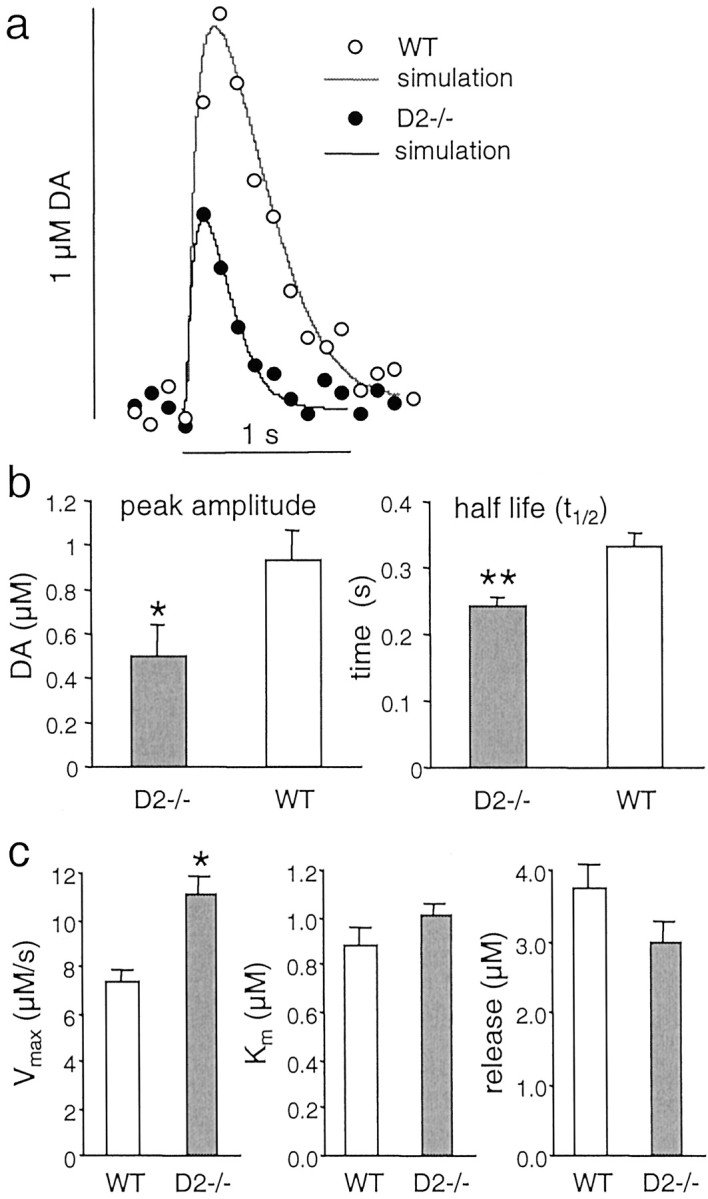
DA overflow in response to single-pulse stimulation in D2−/− and WT mice. a, Examples of evoked DA overflow from WT (○) and D2−/− mice (●) are shown with their corresponding best fit simulations (solid lines). The simulation parameters for the WT trace are Vmax = 6 μm/sec, Km = 1 μm, initial DA release = 2.7 μm, and dead radius (r) = 6.5 μm (R2 = 0.92). For the D2−/− trace, the parameters areVmax = 9.5 μm/sec,Km = 0.9 μm, DA = 2.3 μm, and r = 7 μm (R2 = 0.96). b, Average peak amplitude and half life (mean ± SEM;asterisk denotes statistical difference from WT withp < 0.05) of evoked DA overflow in slices from D2−/− (n = 7 mice for peak amplitude and n = 11 mice for half life) and WT mice (n = 8 mice for peak amplitude andn = 13 mice for half life).c, Histograms showing the average parameters (mean ± SEM; one asterisk denotes p < 0.05; two asterisks denote p < 0.01) obtained by fitting the simulation to the data (averageR2 = 0.97 for WT and 0.95 for D2−/−).
Because evoked DA overflow is determined by both DA release and reuptake, the reduced overflow in D2−/− mice may be a result of either decreased release or increased uptake. To differentiate between the two, we recently developed a simulation model to estimate release and uptake parameters, which consists of a random walk/finite difference simulation of diffusion of DA in brain tissue corrected for DA uptake according to Michaelis–Menten kinetics (Schmitz et al., 2001). The simulation is fit to recordings of DA overflow using a simplex algorithm to perform nonlinear regression by varying four parameters: the initial DA concentration elicited by the stimulation, the maximal DAT uptake velocity (Vmax), the apparent affinity (Km), and the diffusion distance to the electrode (“dead volume”).
The solid lines in Figure 3a are the best fit simulations for the respective recording traces. The most striking difference in DA overflow parameters between D2−/− and WT mice was an increased Vmax of 11.1 μm/sec in D2−/− mice, compared with 7.3 μm/sec in WT mice (+51%;p < 0.05). The initial DA concentration was slightly decreased in D2−/− mice (3 μm compared with 3.8 μmin WT mice, −20%, not significant).Km (0.9 μm in WT and 1 μm in D2 −/−) and the estimated diffusion distance (7.3 ± 0.4 μm in WT and 8.2 ± 0.6 μm in D2−/−) were not significantly different. These results suggested that the reduced amplitude of DA overflow in D2−/− mice was primarily caused by enhanced uptake attributable to an increasedVmax.
Effects of uptake blockade
To further test the possibility that the difference in DA overflow between D2−/− and WT mice was primarily caused by enhanced uptake in the mutants, we recorded DA overflow in slices from D2−/− and WT mice in the presence of the competitive uptake inhibitor nomifensine (Meiergerd and Schenk, 1994). In the dorsal striatum, nomifensine (5–10 μm) causes an increase in Km, estimated to lie between 11 and 20 μm (Jones et al., 1995;Schmitz et al., 2001; Wu et al., 2001), resulting in DA overflow that is primarily determined by DA release and diffusion. Thus, if the difference between genotypes was attributable to reduced DA release in the D2−/− mutant, DA overflow in the presence of nomifensine would remain lower in D2−/− mice than in WT mice. In contrast, if the decrease in stimulated DA overflow in D2−/− mice was caused by increased DA uptake, nomifensine would be expected to eliminate the difference in DA overflow between D2−/− mice and WT mice.
DA overflow was stimulated once per minute, and slices were superfused for 20 min with 5 μm nomifensine, a concentration well above its estimated Ki of 0.09–0.49 μm (Meiergerd and Schenk, 1994; Jones et al., 1995). Figure 4a shows an example of evoked DA overflow recorded before and after 20 min of nomifensine superfusion in a D2−/− and WT striatal slice. The plot of normalized amplitudes of DA overflow in Figure 4b shows that nomifensine caused a twofold increase in DA overflow in slices from WT mice and a fivefold increase in slices from D2−/− mice. This relatively greater enhancement of DA overflow in D2−/− mice resulted in identical absolute amplitudes of evoked DA overflow in the two genotypes (Fig. 4c; 2.41 μm in WT and 2.44 μm in D2−/−). These results thus confirm that DA release was nearly unaltered in D2−/− mice, whereas DA uptake was enhanced. The enhanced uptake could be attributable either to an increase inVmax or to a decrease inKm. As shown above (Fig.3c), the simulation of evoked DA overflow suggested an enhanced Vmax in D2−/− mice.
Fig. 4.
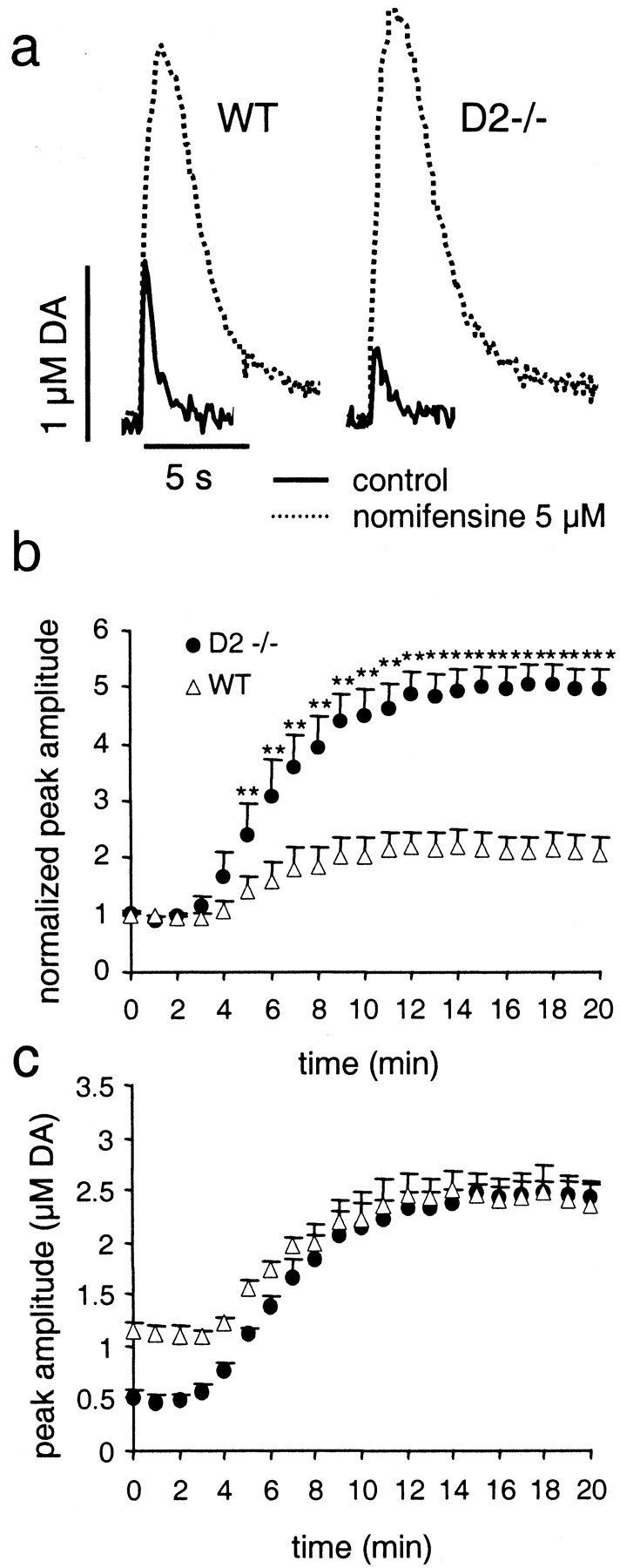
Effects of uptake inhibition by nomifensine on stimulated DA overflow in D2−/− and WT mice.a, Examples of evoked DA overflow in slices from WT and D2−/− mice before (solid lines) and after (dashed lines) a 20 min superfusion with 5 μm nomifensine. b, Increase in peak DA overflow over time in slices superfused with 5 μmnomifensine, normalized to overflow recorded in control medium (mean ± SEM; asterisks denote statistical difference from WT with p < 0.01), in WT mice (▵, n = 5), and D2−/− mice (●,n = 7). c, The data shown inb are plotted as absolute values of peak DA overflow. Peak DA overflow in D2−/− mice was smaller in control medium but reached the same level as DA overflow in WT mice after a 10 min superfusion with 5 μm nomifensine.
Striatal content of DA and metabolites
To further test the possibility that altered DA release might contribute to the observed reduction of DA overflow in D2−/− mice, we measured overall DA tissue content in striatal slices immediately after sectioning and after 30 and 120 min of incubation in the holding chamber in ACSF at room temperature. The tissue content of DA and its metabolites 3,4-dihydroxyphenylacetic acid (DOPAC) and homovanilic acid (HVA) was determined in homogenates of striatal slices by HPLC analysis. Figure5a shows that DA levels were not different in D2−/− and WT mice. There was a slight drop in DA levels after incubation for 30 min and a recovery to the original level after 2 hr of incubation for both genotypes. Although absolute DA levels were similar in both genotypes, the ratio of DA to DOPAC and HVA was slightly but not significantly elevated in D2−/− mice (Fig. 5b). We conclude that the observed reduced DA overflow in D2−/− mice was not caused by reduced tissue levels of DA.
Fig. 5.
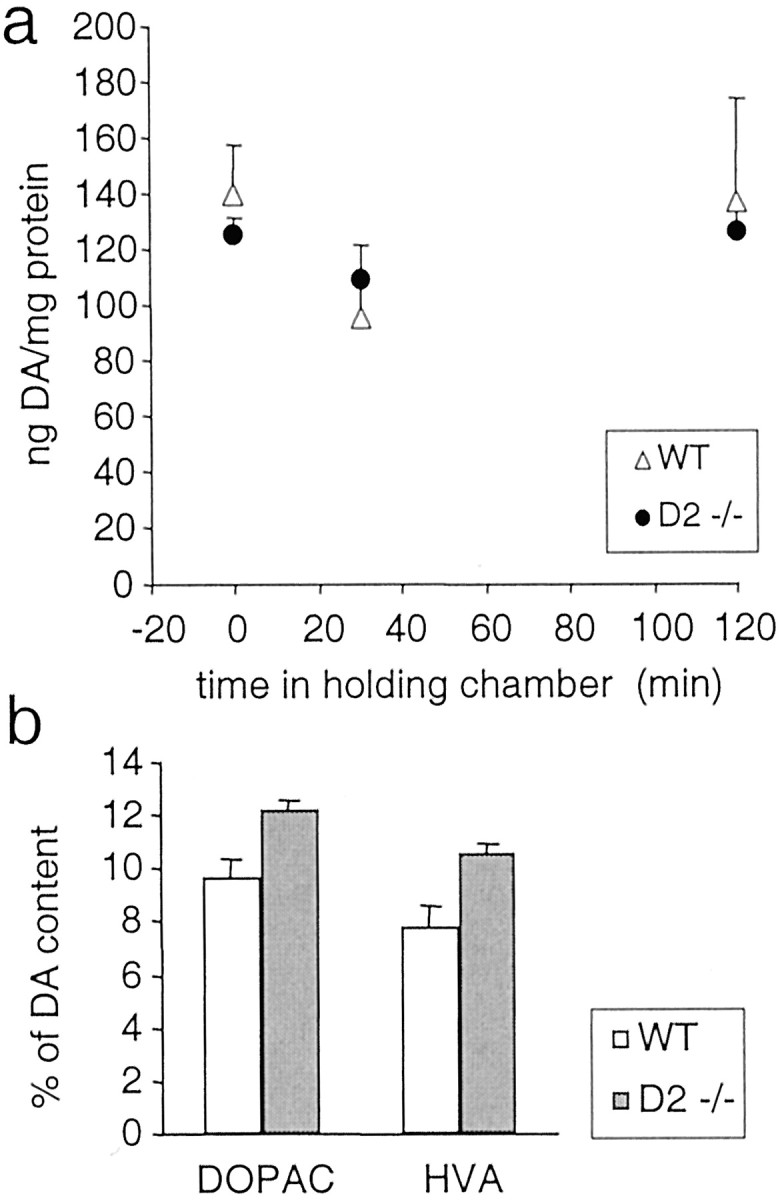
HPLC analysis of tissue levels of DA and its metabolites DOPAC and HVA in striatal slices from WT and D2−/− mice. a, DA content of striatal slices (3 per animal) from WT mice (▵, n = 6 for time 0 and n = 3 for 30 min and 2 hr) and D2−/− mice (●, n = 4 for time 0 andn = 2 for 30 min and 2 hr) was determined immediately after sectioning and after a 30 min and 2 hr recovery in the holding chamber at room temperature. There was no difference between WT and D2−/− mice (p> 0.05). b, Tissue content of the metabolites DOPAC and HVA determined in slices from WT mice (n = 6) and D2−/− mice (n = 4) immediately after sectioning, expressed as a percentage of DA content. There was no difference between WT and D2−/− mice (p > 0.05).
Time course of DA release autoinhibition
Although the inhibition of DA release by D2receptors has been studied extensively using receptor agonists, there are few data on the autoinhibition by endogenous DA release itself. Furthermore, estimates of the duration of DA release autoinhibition have been especially divergent (see the introductory remarks). In rat striatal slices, D2 receptor antagonists were shown to affect paired-pulse depression (PPD) as long as 30 sec (Kennedy et al., 1992). We repeated this experiment with slices from WT mice and compared PPD before and after superfusion with the D2 receptor antagonist sulpiride (10 μm), but found no significant differences (n = 5, data not shown). Similarly, we compared PPD in slices from WT and D2−/− mice. Paired pulses were applied at intervals of 5, 10, 20, 30, and 60 sec (examples for 60, 10, and 5 sec in Fig. 6a). In Figure 6a (inset), the ratio of the maximal response amplitudes for the second and first stimulus (PPD) is plotted versus interpulse intervals. Consistent with the lack of effect of sulpiride, there was no significant difference in the response between the genotypes. The data were fit by two double exponentials with the time constants τslow = 20 sec (67% in D2−/− and 62% in WT mice) and τfast = 3 sec (33 and 38%, respectively).
Fig. 6.
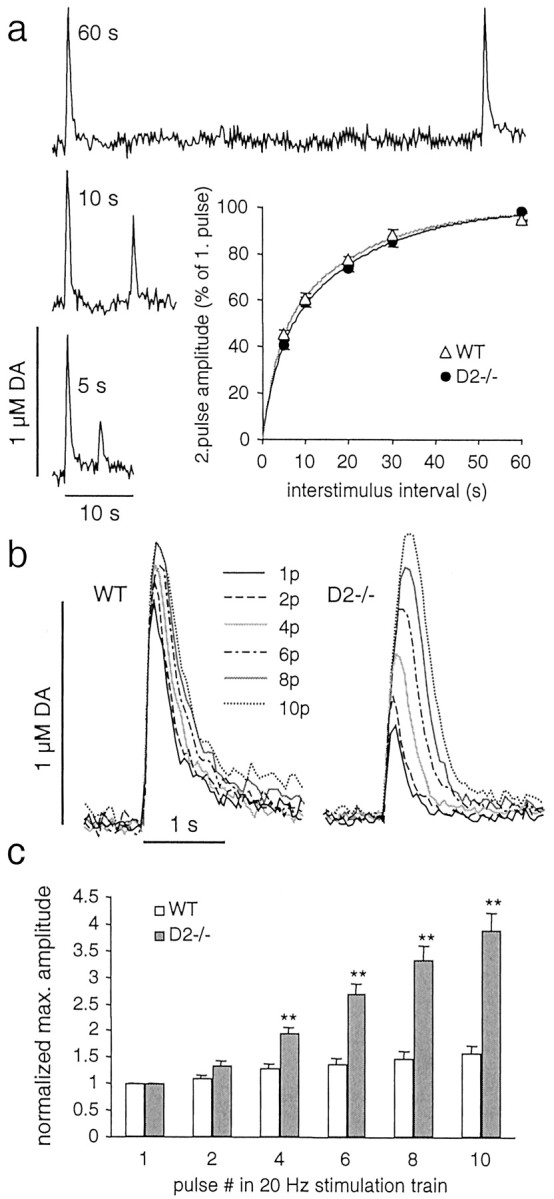
DA overflow in response to paired-pulse stimulation and train stimulation at 20 Hz in slices from D2−/− and WT mice. a, DA overflow recorded in slices from D2−/− mice (n = 5) and WT mice (n = 4) was evoked by paired-pulse stimulation with interpulse intervals of 5, 10, 20, 30, and 60 sec. Examples of recording traces are shown for a WT slice for 5, 10, and 60 sec interpulse intervals. The inset is a plot of PD expressed as maximal DA overflow evoked by the second stimulus in percentage of maximal DA overflow evoked by the first stimulus versus interpulse interval. The solid lines are double exponential fits to the data with the time constants τslow = 20 sec (62% in WT and 67% in D2−/−) and τfast = 3 sec (38% in WT and 33% in D2−/−). There was no difference between WT and D2−/− mice (p > 0.05).b, Examples of DA overflow, recorded in a slice from a WT and a D2−/− mouse, in response to stimulation trains of increasing pulse (p) number (1, 2, 4, 6, 8, and 10) with a frequency of 20 Hz. c, Average maximal amplitudes (mean ± SEM; double asterisksdenote statistical difference from WT with p < 0.01) obtained by stimulation trains as inb, normalized to the peak amplitude evoked by a single-pulse stimulation for D2−/− (n = 7) and WT (n = 7) mice.
Because there was no difference in PPD between the genotypes for intervals in the second range, we subsequently tested the response to stimulation trains in the subsecond range. Figure 6b shows examples of CV recordings of DA overflow in response to 20 Hz train stimulations with increasing pulse number (1–10). The averaged normalized amplitude of DA overflow plotted versus pulse number in the stimulation train is shown below (Fig. 6c). In WT mice, there was only a slight increase in DA overflow with each additional pulse, whereas in D2−/− mice, DA overflow increased substantially with each pulse. A slight increase was apparent already for a two pulse stimulation, indicating that the onset of D2 autoreceptor effects on DA release occurred within 50 msec. We also observed an increase in DA overflow in response to a 10 pulse train at 20 Hz in slices from WT mice superfused with the D2 receptor antagonist sulpiride (10 μm). The peak amplitude for a 10 pulse train was 190% of that for a single-pulse stimulation (n = 4, data not shown).
To further explore the time course of the D2autoreceptor effect on DA release, amperometric recordings were used that have faster kinetics. As a result, recordings of DA signals evoked with shorter interpulse intervals do not overlap (Dugast et al., 1994;Schmitz et al., 2001). Striatal slices from both genotypes were stimulated with trains of five pulses with interpulse intervals of 1, 0.5, or 0.2 sec, respectively. An amperometric recording trace of DA overflow in response to stimulation with 0.5 sec intervals is shown in Figure 7a. The averaged response to stimulus 2–5 is plotted as the fraction of the first response (Fig. 7b). For all three intervals, a significant difference between D2−/− and WT mice was found (p < 0.01). A summary of the difference in PPD between WT and D2−/− mice for all interpulse intervals used is shown in Figure 8. The autoinhibition of DA release mediated by D2receptors had an onset of ∼50 msec, was maximal at 500 msec, and terminated between 1 and 5 sec.
Fig. 7.
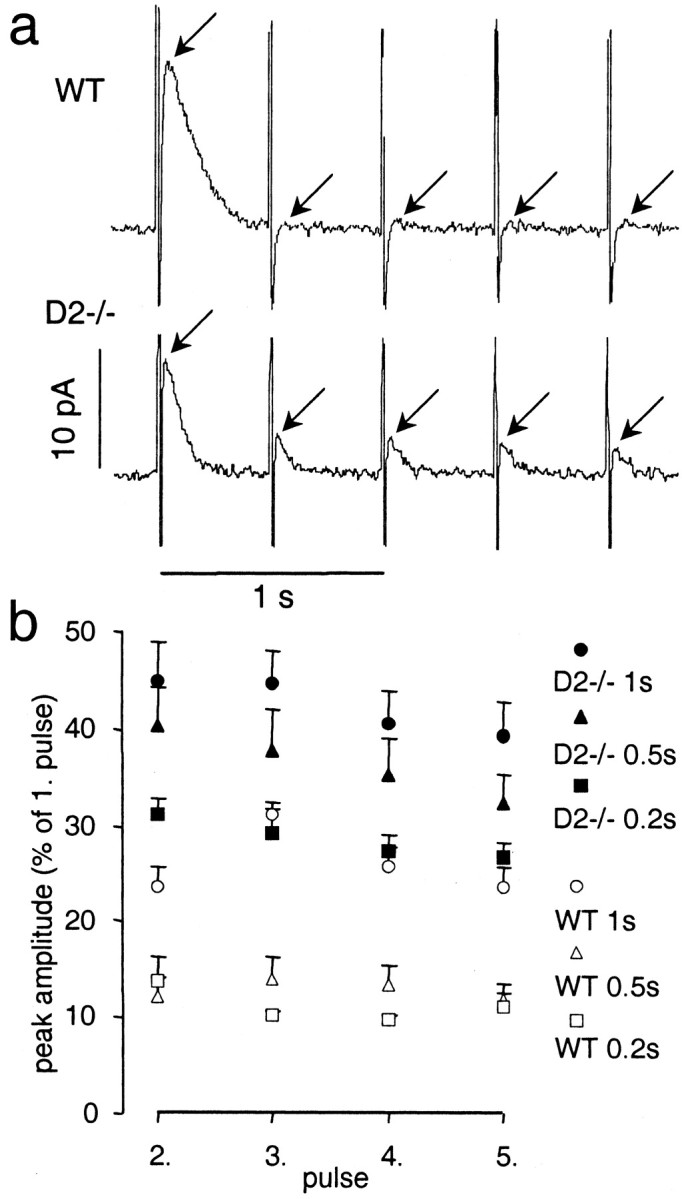
DA overflow recorded with amperometry in response to stimulation trains of five pulses with intervals of 0.2, 0.5, and 1 sec. a, Examples of amperometric recordings of DA overflow in response to stimulation trains of five pulses at 2 Hz in a slice from a WT and a D2−/− mouse (arrowsindicate peak of evoked DA overflow; vertical lines are stimulation artifacts). b, Average response (mean ± SEM) to pulse numbers 2–5 expressed as a fraction of the maximal response to the first pulse for stimulation trains with intervals of 1, 0.5, and 0.2 sec for WT mice (n = 5 for 1 and 0.5 sec intervals; n = 3 for 0.2 sec interval) and D2−/− mice (n = 8 for 1 and 0.5 sec intervals; n = 5 for 0.2 sec interval). Responses to all three stimulation trains were significantly different from WT (p < 0.01).
Fig. 8.

Summary diagram of the difference in PPD between WT and D2−/− mice for interpulse intervals ranging from 50 msec to 5 sec. The maximal PPD occurred ∼500 msec after the stimulation and terminated between 1 and 5 sec.
DISCUSSION
We studied long-term adaptation of DA axon terminals to the lack of D2 autoreceptors by comparing striatal DA release in D2−/− and WT mice. There was no compensatory expression of other release-regulating autoreceptors of the D2 receptor family. Thus, the D2 receptor appears to be the only release-regulating autoreceptor on nigrostriatal terminals. Heteroreceptor-mediated DA release inhibition was unaltered in D2−/− mice, because GABABreceptor activation suppressed DA release to the same extent as in WT mice. Comparison of paired-pulse stimulation of DA overflow in slices from D2−/− and WT mice revealed that D2-mediated autoinhibition of DA release was maximal at ∼500 msec after stimulation and lasted for <5 sec. PPD observed for longer interstimulus intervals (5–30 sec) was not mediated by autoreceptor activation. DA overflow evoked by a single stimulus was reduced in amplitude and duration in D2−/− mice compared with WT mice. Our results suggest that this reduced DA overflow was caused by an increase in the maximal velocity of uptake. This finding points to an interaction between the two principal regulators of extracellular DA levels, the D2 autoreceptor and DAT.
Autoreceptor and heteroreceptor-mediated DA release inhibition
Although mesencephalic dopamine neurons are known to express D3 receptors (Diaz et al., 2000), our experiments using the D2/D3 receptor agonist quinpirole confirmed conclusions of previous studies (Rubinstein et al., 1997; Koeltzow et al., 1998; L'hirondel et al., 1998) that the D2 receptor is the only release-inhibiting DA receptor on nigrostriatal terminals. We tested whether the lack of D2 receptors would result in a loss of inhibitory G-proteins or an increased response to G-protein-coupled heteroreceptors. GABAB and D2 receptors affect the same potassium and calcium currents in DA somata (Lacey et al., 1988; Cardozo and Bean, 1995) and may use the same subset of inhibitory G-proteins (Kerr and Ong, 1995; Huff et al., 1998). However, the response to GABAB receptor agonists was unchanged in D2−/− mice, indicating an intact set of inhibitory G-proteins and no compensatory changes in the response to heteroreceptor activation.
Autoinhibition of DA release
The inhibition of DA release by autoreceptors at axon terminals (Cubeddu and Hoffmann, 1982; Mayer et al., 1988; Limberger et al., 1991; Kennedy et al., 1992) and somata of DA neurons (Cragg and Greenfield, 1997) is well established. However, as emphasized in a recent publication by Benoit-Marand et al. (2001), the reported time course of the autoreceptor effect has been quite variable, with estimates of the duration ranging from milliseconds to several seconds. A discrepancy appears to exist, especially between in vivoand in vitro studies. In vitro, in rat striatal slices using a paired-pulse stimulation paradigm, autoreceptor effects blocked by sulpiride were found to last as long as 30 sec (Kennedy et al., 1992), whereas in vivo, comparison of WT and D2−/− mice indicated a duration of maximally 600 msec (Benoit-Marand et al., 2001). This discrepancy may be attributable to the different behavior of stimulated DA releasein vivo and in vitro. DA release in response to a single stimulus in vitro elicits ∼10-fold more DA release than in vivo (Michael and Wightman, 1999). Furthermore, evoked DA release exhibits a marked PPD for interpulse intervals between 5 and 30 sec in vitro (Kennedy et al., 1992) that is not found in vivo (Chergui et al., 1994). These differences in response are not well understood (Michael and Wightman, 1999). Possible explanations include continuous spontaneous activity in vivo versus no activity in vitro, different release probabilities because of the different stimulation conditions, and/or release of an unknown inhibitory factor in vitro. Nevertheless, our estimates of the time course of autoreceptor effectsin vitro, derived from experiments using the D2 receptor antagonist sulpiride in WT mice and from comparing D2−/− and WT mice, are in agreement with the results obtained in vivo (Benoit-Marand et al., 2001). In vivo, autoinhibition was maximal between 150 and 300 msec after stimulation and lasted for ∼600 msec. In vitro, we found that the maximal effect occurred at 500 msec and lasted <5 sec. This slightly prolonged time course could be attributable to the larger amplitude and duration of DA overflowin vitro.
The time course of maximal autoinhibition appears to be suitable to enhance the DA signal in response to “meaningful” burst firing (Schultz, 1986), as opposed to baseline firing of substantia nigra neurons. In the rat, burst firing consists of two to six spikes at 15 Hz. Thus, all spikes occur before maximal release inhibition is reached. In contrast, tonic activity consists of single spikes with interpulse intervals (250 msec) that allow maximal autoinhibition (Grace and Bunney, 1984a,b). Beyond the physiological role of release-regulating D2 autoreceptors, this activity is likely to play an important role in situations when DA transmission is disturbed, as for instance in the short- and long-term response to psychostimulant drugs (Pierce et al., 1995; Schmitz et al., 2001).
Compensatory interaction between DAT and D2 autoreceptors
We found a striking reduction of DA overflow in response to a single-pulse stimulus in D2−/− mice. The simulation model indicated that this reduction was caused by an increased maximal velocity of uptake rather than by decreased DA release. Experiments with nomifensine confirmed this finding, because DA overflow was nearly identical in WT and D2−/− mice in the presence of the uptake inhibitor.
In contrast to our findings, in mice with a deletion mutation of the D2 receptor, the in vivo DAT activity was found to be decreased, as determined by the clearance rate of DA injections into the striatum (Dickinson et al., 1999). High potassium-evoked DA release and basal DA levels assessed by microdialysis were unaltered in these mice. The basis of these findings remains to be elucidated, because decreased DA uptake alone would result in higher basal DA levels. A possibility is that there are changes in DA release in this mutant that cannot be observed by microdialysis with its limited time resolution.
In another mouse line carrying a null mutation for the D2 receptor, Benoit-Marand et al. (2001) reported no change in DA overflow in response to three pulses at 100 Hz in vivo, suggesting at first glance that DA release and uptake are unaltered in these animals. However, in contrast to in vitroconditions, the baseline activity of DA neurons in vivo may result in a tonic activation of D2 autoreceptors. Accordingly, Benoit-Marand et al. (2001) reported that haloperidol increased the half life of DA overflow in vivo, suggesting that the basal level of autoreceptor activation in vivostimulates DA uptake. Similar findings have been reported previouslyin vivo and in striatal homogenates in vitro(Meiergerd et al., 1993; Cass and Gerhardt, 1994; Batchelor and Schenk, 1998).
Therefore, it appears that in WT mice, DA uptake is enhanced by basal D2 activity, whereas, according to our results, DA uptake is enhanced because of the long-term absence of D2 activity in D2−/− mice. The short-term regulation of DAT by D2receptors may involve DAT phosphorylation and/or trafficking (Batchelor and Schenk, 1998; Zahniser and Doolen, 2001), whereas long-term regulation may involve changes in DAT protein expression. The results of our simulation suggest that the increased DA uptake in D2−/− mice is because of increasedVmax (i.e., increased plasmalemmal DAT expression). This is in agreement with a recent immunocytochemistry study that found an increased staining for DAT in the striatum of D2−/− mice (Parish et al., 2001). Several studies have reported changes in DAT expression in response to D2 activity. An oocyte expression study suggested D2 receptor-induced upregulation of DAT expression by a voltage-dependent mechanism (Mayfield and Zahniser, 2001). An in vivo study found that D2receptor agonists decreased DAT expression in the caudate putamen and increased DAT expression in the nucleus accumbens (Kimmel et al., 2001). From these studies, it appears that D2activity may change DAT expression in either direction via mechanisms not yet elucidated.
However, DAT expression can also affect D2autoreceptor function, because D2 autoreceptor activity is virtually absent in DAT mutant mice. Moreover, the tissue content of DA is severely reduced in these mutants, whereas DA metabolism is elevated (Giros et al., 1996; Jones et al., 1999). In D2−/− mice, however, we found no change in tissue levels of DA and only a slight increase in metabolite levels, also reported previously for these mice (Jung et al., 1999). We conclude that the absence of D2 autoreceptors results in only minor changes in DA synthesis and metabolism, whereas autoinhibition of DA release and reuptake are strongly affected.
In summary, several studies have provided evidence for an interaction between D2 autoreceptors and DAT activity and expression. Our results support such an interaction by demonstrating a compensatory regulation of DA uptake in D2−/− mice. This mutual regulation appears to ensure that DA signals are transmitted with the appropriate amplitude and timing. Short- and long-term effects of psychostimulant and antipsychotic drugs are therefore likely to include changes in both D2autoreceptors and DAT.
Note added in proof. During the course of the publication of this manuscript, an in vitro study on wild-type mice was published that confirms the time course of D2 autoreceptor effects on DA release reported here (Phillips et al., 2002).
Footnotes
This work was supported by the National Association for Research on Schizophrenia and Depression, the National Institute on Drug Abuse, the National Institute of Mental Health, the National Science Foundation, the Lowenstein Foundation, and the Parkinson's Disease Foundation. We thank Marianne Benoit-Marand and François Gonon for helpful discussion. We also thank Meisheng Jiang and Lutz Birnbaumer for generously providing Go protein mutant mice for preliminary experiments.
Correspondence should be addressed to Dr. David Sulzer, Columbia University, Department of Neurology, 650 West 168th Street, New York, NY 10032. E-mail: ds43@columbia.edu.
REFERENCES
- 1.Baik JH, Picetti R, Saiardi A, Thiriet G, Dierich A, Depaulis A, Le Meur M, Borrelli E. Parkinsonian-like locomotor impairment in mice lacking dopamine D2 receptors. Nature. 1995;377:424–428. doi: 10.1038/377424a0. [DOI] [PubMed] [Google Scholar]
- 2.Batchelor M, Schenk JO. Protein kinase A activity may kinetically upregulate the striatal transporter for dopamine. J Neurosci. 1998;18:10304–10309. doi: 10.1523/JNEUROSCI.18-24-10304.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Benoit-Marand M, Borrelli E, Gonon F. Inhibition of dopamine release via presynaptic D2 receptors: time course and functional characteristics in vivo. J Neurosci. 2001;21:9134–9141. doi: 10.1523/JNEUROSCI.21-23-09134.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berg HC. Random walks in biology. Princeton UP; Princeton, NJ: 1983. [Google Scholar]
- 5.Bunney BS, Walters JR, Roth RH, Aghajanian GK. Dopaminergic neurons: effect of antipsychotic drugs and amphetamine on single cell activity. J Pharmacol Exp Ther. 1973;185:560–571. [PubMed] [Google Scholar]
- 6.Cardozo DL, Bean BP. Voltage-dependent calcium channels in rat midbrain dopamine neurons: modulation by dopamine and GABAB receptors. J Neurophysiol. 1995;74:1137–1148. doi: 10.1152/jn.1995.74.3.1137. [DOI] [PubMed] [Google Scholar]
- 7.Cass WA, Gerhardt GA. Direct in vivo evidence that D2 dopamine receptors can modulate dopamine uptake. Neurosci Lett. 1994;176:259–263. doi: 10.1016/0304-3940(94)90096-5. [DOI] [PubMed] [Google Scholar]
- 8.Chergui K, Suaud-Chagny MF, Gonon F. Nonlinear relationship between impulse flow, dopamine release and dopamine elimination in the rat brain in vivo. Neuroscience. 1994;62:641–645. doi: 10.1016/0306-4522(94)90465-0. [DOI] [PubMed] [Google Scholar]
- 9.Cragg SJ, Greenfield SA. Differential autoreceptor control of somatodendritic and axon terminal dopamine release in substantia nigra, ventral tegmental area, and striatum. J Neurosci. 1997;17:5738–5746. doi: 10.1523/JNEUROSCI.17-15-05738.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cubeddu LX, Hoffmann IS. Operational characteristics of the inhibitory feedback mechanism for regulation of dopamine release via presynaptic receptors. J Pharmacol Exp Ther. 1982;223:497–501. [PubMed] [Google Scholar]
- 11.Diaz J, Pilon C, Le Foll B, Gros C, Triller A, Schwartz JC, Sokoloff P. Dopamine D3 receptors expressed by all mesencephalic dopamine neurons. J Neurosci. 2000;20:8677–8684. doi: 10.1523/JNEUROSCI.20-23-08677.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dickinson SD, Sabeti J, Larson GA, Giardina K, Rubinstein M, Kelly MA, Grandy DK, Low MJ, Gerhardt GA, Zahniser NR. Dopamine D2 receptor-deficient mice exhibit decreased dopamine transporter function but no changes in dopamine release in dorsal striatum. J Neurochem. 1999;72:148–156. doi: 10.1046/j.1471-4159.1999.0720148.x. [DOI] [PubMed] [Google Scholar]
- 13.Dugast C, Suaud-Chagny MF, Gonon F. Continuous in vivo monitoring of evoked dopamine release in the rat nucleus accumbens by amperometry. Neuroscience. 1994;62:647–654. doi: 10.1016/0306-4522(94)90466-9. [DOI] [PubMed] [Google Scholar]
- 14.Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Academic; San Diego: 1997. [Google Scholar]
- 15.Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- 16.Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: single spike firing. J Neurosci. 1984a;4:2866–2876. doi: 10.1523/JNEUROSCI.04-11-02866.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: burst firing. J Neurosci. 1984b;4:2877–2890. doi: 10.1523/JNEUROSCI.04-11-02877.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Haubrich DR, Pflueger AB. The autoreceptor control of dopamine synthesis: an in vitro and in vivo comparison of dopamine agonists. Mol Pharmacol. 1982;21:114–120. [PubMed] [Google Scholar]
- 19.Huff RM, Chio CL, Lajiness ME, Goodman LV. Signal transduction pathways modulated by D2-like dopamine receptors. Adv Pharmacol. 1998;42:454–457. doi: 10.1016/s1054-3589(08)60786-3. [DOI] [PubMed] [Google Scholar]
- 20.Jones SR, Garris PA, Wightman RM. Different effects of cocaine and nomifensine on dopamine uptake in the caudate-putamen and nucleus accumbens. J Pharmacol Exp Ther. 1995;274:396–403. [PubMed] [Google Scholar]
- 21.Jones SR, Gainetdinov RR, Hu XT, Cooper DC, Wightman RM, White FJ, Caron MG. Loss of autoreceptor functions in mice lacking the dopamine transporter. Nat Neurosci. 1999;2:649–655. doi: 10.1038/10204. [DOI] [PubMed] [Google Scholar]
- 22.Jung MY, Skryabin BV, Arai M, Abbondanzo S, Fu D, Brosius J, Robakis NK, Polites HG, Pintar JE, Schmauss C. Potentiation of the D2 mutant motor phenotype in mice lacking dopamine D2 and D3 receptors. Neuroscience. 1999;91:911–924. doi: 10.1016/s0306-4522(98)00705-2. [DOI] [PubMed] [Google Scholar]
- 23.Kawagoe KT, Zimmerman JB, Wightman RM. Principles of voltammetry and microelectrode surface states. J Neurosci Methods. 1993;48:225–240. doi: 10.1016/0165-0270(93)90094-8. [DOI] [PubMed] [Google Scholar]
- 24.Kehr W, Carlsson A, Lindqvist M, Magnusson T, Atack C. Evidence for a receptor-mediated feedback control of striatal tyrosine hydroxylase activity. J Pharm Pharmacol. 1972;24:744–747. doi: 10.1111/j.2042-7158.1972.tb09104.x. [DOI] [PubMed] [Google Scholar]
- 25.Kelly MA, Rubinstein M, Asa SL, Zhang G, Saez C, Bunzow JR, Allen RG, Hnasko R, Ben-Jonathan N, Grandy DK, Low MJ. Pituitary lactotroph hyperplasia and chronic hyperprolactinemia in dopamine D2 receptor-deficient mice. Neuron. 1997;19:103–113. doi: 10.1016/s0896-6273(00)80351-7. [DOI] [PubMed] [Google Scholar]
- 26.Kennedy RT, Jones SR, Wightman RM. Dynamic observation of dopamine autoreceptor effects in rat striatal slices. J Neurochem. 1992;59:449–455. doi: 10.1111/j.1471-4159.1992.tb09391.x. [DOI] [PubMed] [Google Scholar]
- 27.Kerr DI, Ong J. GABAB receptors. Pharmacol Ther. 1995;67:187–246. doi: 10.1016/0163-7258(95)00016-a. [DOI] [PubMed] [Google Scholar]
- 28.Kimmel HL, Joyce AR, Carroll FI, Kuhar MJ. Dopamine D1 and D2 receptors influence dopamine transporter synthesis and degradation in the rat. J Pharmacol Exp Ther. 2001;298:129–140. [PubMed] [Google Scholar]
- 29.Koeltzow TE, Xu M, Cooper DC, Hu XT, Tonegawa S, Wolf ME, White FJ. Alterations in dopamine release but not dopamine autoreceptor function in dopamine D3 receptor mutant mice. J Neurosci. 1998;18:2231–2238. doi: 10.1523/JNEUROSCI.18-06-02231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lacey MG, Mercuri NB, North RA. On the potassium conductance increase activated by GABAB and dopamine D2 receptors in rat substantia nigra neurones. J Physiol (Lond) 1988;401:437–453. doi: 10.1113/jphysiol.1988.sp017171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.L'hirondel M, Cheramy A, Godeheu G, Artaud F, Saiardi A, Borrelli E, Glowinski J. Lack of autoreceptor-mediated inhibitory control of dopamine release in striatal synaptosomes of D2 receptor-deficient mice. Brain Res. 1998;792:253–262. doi: 10.1016/s0006-8993(98)00146-2. [DOI] [PubMed] [Google Scholar]
- 32.Limberger N, Trout SJ, Kruk ZL, Starke K. “Real time” measurement of endogenous dopamine release during short trains of pulses in slices of rat neostriatum and nucleus accumbens: role of autoinhibition. Naunyn Schmiedebergs Arch Pharmacol. 1991;344:623–629. doi: 10.1007/BF00174745. [DOI] [PubMed] [Google Scholar]
- 33.Lindgren N, Xu ZQ, Herrera-Marschitz M, Haycock J, Hokfelt T, Fisone G. Dopamine D2 receptors regulate tyrosine hydroxylase activity and phosphorylation at Ser40 in rat striatum. Eur J Neurosci. 2001;13:773–780. doi: 10.1046/j.0953-816x.2000.01443.x. [DOI] [PubMed] [Google Scholar]
- 34.Mayer A, Limberger N, Starke K. Transmitter release patterns of noradrenergic, dopaminergic and cholinergic axons in rabbit brain slices during short pulse trains, and the operation of presynaptic autoreceptors. Naunyn Schmiedebergs Arch Pharmacol. 1988;338:632–643. doi: 10.1007/BF00165627. [DOI] [PubMed] [Google Scholar]
- 35.Mayfield RD, Zahniser NR. Dopamine D2 receptor regulation of the dopamine transporter expressed in Xenopus laevis oocytes is voltage-independent. Mol Pharmacol. 2001;59:113–121. doi: 10.1124/mol.59.1.113. [DOI] [PubMed] [Google Scholar]
- 36.Meiergerd SM, Schenk JO. Kinetic evaluation of the commonality between the site(s) of action of cocaine and some other structurally similar and dissimilar inhibitors of the striatal transporter for dopamine. J Neurochem. 1994;63:1683–1692. doi: 10.1046/j.1471-4159.1994.63051683.x. [DOI] [PubMed] [Google Scholar]
- 37.Meiergerd SM, Patterson TA, Schenk JO. D2 receptors may modulate the function of the striatal transporter for dopamine: kinetic evidence from studies in vitro and in vivo. J Neurochem. 1993;61:764–767. doi: 10.1111/j.1471-4159.1993.tb02185.x. [DOI] [PubMed] [Google Scholar]
- 38.Mercuri NB, Saiardi A, Bonci A, Picetti R, Calabresi P, Bernardi G, Borrelli E. Loss of autoreceptor function in dopaminergic neurons from dopamine D2 receptor deficient mice. Neuroscience. 1997;79:323–327. doi: 10.1016/s0306-4522(97)00135-8. [DOI] [PubMed] [Google Scholar]
- 39.Michael DJ, Wightman RM. Electrochemical monitoring of biogenic amine neurotransmission in real time. J Pharm Biomed Anal. 1999;19:33–46. doi: 10.1016/s0731-7085(98)00145-9. [DOI] [PubMed] [Google Scholar]
- 40.O'Hara CM, Uhland-Smith A, O'Malley KL, Todd RD. Inhibition of dopamine synthesis by dopamine D2 and D3 but not D4 receptors. J Pharmacol Exp Ther. 1996;277:186–192. [PubMed] [Google Scholar]
- 41.Parish CL, Finkelstein DI, Drago J, Borrelli E, Horne MK. The role of dopamine receptors in regulating the size of axonal arbors. J Neurosci. 2001;21:5147–5157. doi: 10.1523/JNEUROSCI.21-14-05147.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Phillips PE, Hancock PJ, Stamford JA. Time window of autoreceptor-mediated inhibition of limbic and striatal dopamine release. Synapse. 2002;44:15–22. doi: 10.1002/syn.10049. [DOI] [PubMed] [Google Scholar]
- 43.Pierce RC, Duffy P, Kalivas PW. Sensitization to cocaine and dopamine autoreceptor subsensitivity in the nucleus accumbens. Synapse. 1995;20:33–36. doi: 10.1002/syn.890200106. [DOI] [PubMed] [Google Scholar]
- 44.Pothos EN, Przedborski S, Davila V, Schmitz Y, Sulzer D. D2-Like dopamine autoreceptor activation reduces quantal size in PC12 cells. J Neurosci. 1998;18:5575–5585. doi: 10.1523/JNEUROSCI.18-15-05575.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rubinstein M, Phillips TJ, Bunzow JR, Falzone TL, Dziewczapolski G, Zhang G, Fang Y, Larson JL, McDougall JA, Chester JA, Saez C, Pugsley TA, Gershanik O, Low MJ, Grandy DK. Mice lacking dopamine D4 receptors are supersensitive to ethanol, cocaine, and methamphetamine. Cell. 1997;90:991–1001. doi: 10.1016/s0092-8674(00)80365-7. [DOI] [PubMed] [Google Scholar]
- 46.Schmitz Y, Lee CJ, Schmauss C, Gonon F, Sulzer D. Amphetamine distorts stimulation-dependent dopamine overflow: effects on D2 autoreceptors, transporters, and synaptic vesicle stores. J Neurosci. 2001;21:5916–5924. doi: 10.1523/JNEUROSCI.21-16-05916.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schultz W. Responses of midbrain dopamine neurons to behavioral trigger stimuli in the monkey. J Neurophysiol. 1986;56:1439–1461. doi: 10.1152/jn.1986.56.5.1439. [DOI] [PubMed] [Google Scholar]
- 48.Sulzer D, Pothos EN. Regulation of quantal size by presynaptic mechanisms. Rev Neurosci. 2000;11:159–212. doi: 10.1515/revneuro.2000.11.2-3.159. [DOI] [PubMed] [Google Scholar]
- 49.Tepper JM, Sun BC, Martin LP, Creese I. Functional roles of dopamine D2 and D3 autoreceptors on nigrostriatal neurons analyzed by antisense knockdown in vivo. J Neurosci. 1997;17:2519–2530. doi: 10.1523/JNEUROSCI.17-07-02519.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu Q, Reith ME, Kuhar MJ, Carroll FI, Garris PA. Preferential increases in nucleus accumbens dopamine after systemic cocaine administration are caused by unique characteristics of dopamine neurotransmission. J Neurosci. 2001;21:6338–6347. doi: 10.1523/JNEUROSCI.21-16-06338.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zahniser NR, Doolen S. Chronic and acute regulation of Na(+)/Cl(−)-dependent neurotransmitter transporters: drugs, substrates, presynaptic receptors, and signaling systems. Pharmacol Ther. 2001;92:21–55. doi: 10.1016/s0163-7258(01)00158-9. [DOI] [PubMed] [Google Scholar]