

Abstract
The “primitive” neurons of the peripheral nervous system (PNS) have the remarkable ability to regenerate new fibers. This regenerative process requires a sequence of gene activation and repression that is poorly understood. One gene that is almost exclusively expressed in neurons of the PNS and is activated after nerve injury is the peripherin intermediate filament gene, but little is known about the genomic elements that control either its restricted expression or its response to nerve injury in adult mice. Previous studies suggested that both 5′ flanking sequence and intragenic regions were required for cell type-specific and injury-specific expression. To determine which intragenic regions were critical, mice were generated that expressed peripherin transgenes lacking different introns. Analyses of these mice revealed that deletion of introns 2–8 had no effect on either the cell type-specific or injury-specific expression of the peripherin gene; however, the remaining intron, intron 1, differentially bound Sp1 transcription-related proteins/protein complexes in extracts fromperipherin-expressing and nonexpressing tissues. Furthermore, a transgene that lacked intron 1 was not expressed in many neurons that contain endogenous peripherin but was activated after injury. Thus, accurate cell type-specific peripherin gene expression in the PNS depends on elements within intron 1, but other sequences, most likely in the 5′flanking region, are required for activating the peripherin gene in response to nerve injury.
Keywords: peripherin, gene regulation, injury-induced expression, cell type-specific expression, intermediate filament, intron, nerve injury, Sp1, NFI
In humans, recovery from nerve injury is ineffective and incomplete. Those nerve fibers that eventually regenerate are successful at sensing injury, increasing transcription of needed components, and navigating to appropriate targets. One strategy for improving recovery is to identify, analyze, and ultimately enhance the molecular mechanisms that mediate these processes. To decipher the transcriptional pathways controlling nerve regeneration, specific genomic sequences and DNA-binding proteins that mediate the injury response must be identified.
We are focusing on understanding these transcriptional responses by analyzing expression of the peripherin intermediate filament (IF) gene, one of a group of cytoskeletal genes that is transcriptionally activated after nerve injury (Oblinger et al., 1989; for review, seeSkene, 1989; Bisby and Tetzlaff, 1992; Tanabe et al., 1999). These genes may play key roles in recovery from injury, but little is known about the identity or hierarchy of “injury-response elements,” sequences within the genes that mediate induction after injury.
Previous studies have partially localized elements that regulate peripherin gene cell type-specific expression and activation after nerve injury. A transgene consisting of 5.8 kb of 5′ flanking sequence linked to all peripherin exons and introns was accurately expressed in a cell type-specific manner in adult mice and was injury responsive (Belecky-Adams et al., 1993). Trangenes that includeperipherin 5′ flanking sequences linked to bacterial reporter genes, however, generally have not reproduced expression of the endogenous peripherin gene. In vitro, the 5′ peripherin flanking region activated bacterial reporter gene expression (Desmarais et al., 1992; Thompson et al., 1992), but it was not cell appropriate, leading Thompson et al. (1992) to suggest that intragenic sequences may be required. Similarly, peripherin 5′ flanking sequence linked to lacZ restricted expression to neurons in transgenic mice but did not promote accurate cell type-specific expression in either embryos (Leconte et al., 1996) or adults (Belecky-Adams et al., 1993). Addition of intragenic sequences behind aperipherin/lacZ transgene did permit accurate expression in mouse embryos (Leconte et al., 1996) but not in adult mice (B. E. Kramer, T. Belecky-Adams, and L. M. Parysek, unpublished observations). Taken together, these findings suggest that intragenic sequences are required for cell type-specific expression in adult mice and that the position adjacent to the 5′ flanking region may be important. The location of injury-response elements was not reliably determined in any of the lacZ-containing mouse lines because the transgenes were expressed in insufficient numbers of injury-responsive neurons in the adult.
We have undertaken studies, therefore, using a myc-tagged rat peripherin gene as reporter, to define specific regions of the gene critical for cell type-specific expression and for injury responsiveness in adult mice. We found that accurate cell type-specific expression of the rat peripherin gene in adult mice requires intron 1. Through in vitro footprint and binding assays on intron 1, we identified two Sp1 family member-binding sites that flank a Nuclear Factor I (NFI) family member protein-binding site. None of the intronic sequences, however, were required for activation of the peripherin gene after nerve injury; in this case, 5′ flanking, 3′ flanking, and coding sequences were sufficient.
MATERIALS AND METHODS
Transgene construction. Transgenes were constructed by replacing regions of the ratPeriph/periph-myc transgene (Belecky-Adams et al., 1993) with the corresponding region of a myc-tagged rat peripherin cDNA. Convenient restriction sites were used to replace all peripherin genomic sequences that included introns 3–8 to makePeriph/Δi3–8-myc. A similar strategy was used to makePeriph/Δi2–8-myc and Periph/intronless-myc. As with the Periph/periph-myc transgene, each transgene contained 5.8 kb of flanking sequence, a human c-myc tag in exon 9, and the same 600 bp of 3′ flanking region (see Fig. 1). Expression of all transgenes was tested in PC12 cells before injection into mice.
Fig. 1.

Rat peripherin transgenes. The open boxes represent 5.8 kb of 5′ flanking sequence (to scale), thesolid shaded boxes represent peripherin exons (to scale), and solid lines represent intronic sequences (intron numbers below) and 3′ flanking sequence (to scale). A human c-myc tag (v) was placed in-frame at the 3′ end of the coding region in exon 9 to facilitate detection of protein derived from each transgene.
Transgenic mice. Transgenic mice were generated and genotyped as described in Belecky-Adams et al. (1993). All mice tested were F1 or later generation SJL-C57BL/6 hybrids.
Immunohistochemistry. Tissues were dissected from mice of 4–8 weeks of age, embedded in TBS freeze medium (Fisher Scientific, Pittsburgh, PA), and frozen in a dry ice–ethanol bath. The tissue was stored at −70°C until sectioning. Seven micrometer cryostat sections were fixed in dehydrated methanol for 4 min at −20°C and sequentially double labeled. Monoclonal antibody labeling was performed using the M.O.M. kit (Vector Laboratories Inc., Burlingame, CA), according to the kit protocol. After monoclonal labeling, polyclonal labeling was performed as described in Foley et al. (1991). The sections were mounted using Biomedia aqueous mounting medium (Fisher Scientific). Primary antibodies were a rabbit polyclonal c-myc antibody (1:100; Upstate Biotechnology, Waltham, MA), a monoclonal peripherin antibody (1:100; Chemicon International, Temecula, CA), and a monoclonal antibody to neurofilament-M and -H, 15G1 [neat culture supernatant; Brody et al. (1989)]. Secondary antibodies were goat anti-rabbit Alexa-488 (1:300; Molecular Probes, Eugene, OR) and streptavidin conjugated to Alexa-594 (1:200; Molecular Probes) for detection of the monoclonal antibody after M.O.M kit labeling. Labeling was imaged using a Zeiss LSM510 confocal laser-scanning microscope. The nerve crushes were performed as described in Belecky-Adams et al. (1993), except that tissues were placed in TBS freeze medium and analyzed as described above.
Nuclear extracts. Isolation of nuclear extracts was performed as described in Dusing et al. (2001), except that KCl replaced NaCl in buffer C, andN-acetyl-l-cysteine was not used for extract preparation from the brain and liver. For each nuclear extract isolation procedure, the entire mouse small intestine, brain, or liver was used from each of eight mice.
Whole-cell extracts. All solutions were used at 4°C and contained 5 mm PMSF (Sigma, St. Louis, MO), 1 mm leupeptin (Sigma), and 1 mm pepstatin A (Sigma). All dorsal root ganglia (DRGs) down to L4 (∼40) were dissected from each of four mice and placed into 1 ml of PBS. They were washed once in PBS and then treated with 0.5% collagenase (Worthington, Lakewood, NJ) for 30 min at 37°C. The cells were washed in PBS and pelleted. The cells were then resuspended in lysis buffer (20 mm HEPES-KOH, pH 7.9, 450 mm NaCl, 0.4 mmEDTA, 0.5 mm DTT, 25% glycerol). Cells were placed three times in a dry ice–ethanol bath for 2 min and then put at 37°C for 2 min. Subsequently, they were spun for 10 min at 4°C. The supernatant was removed and frozen at −80°C (Dent and Latchman, 1993).
Electrophoretic mobility shift assay. Gel-purified double-stranded oligonucleotides at 2.25 nm were labeled at one end by use of a Klenow-mediated (NEB, Beverly, MA) fill-in reaction with dATP α-32P (3000 Ci/mmol) at a 5′ overhang. Labeled oligonucleotide was purified on a nick column (Amersham Biosciences, Piscataway, NJ). Binding reactions of 25 μl contained 5 μl of 5× buffer (125 mmTris, pH 8.0, 32.5 mmMgCl2, 2.5 mm DTT, 2.5 mm EDTA, 250 mm KCl, and 0.6 μg/μl BSA), 0.5 μl of 4 μg/μl of poly I/poly C (Sigma), 0.5 μl of 5 m NaCl, 10 μl of nuclear extract or whole-cell extract diluted with Buffer D [20 mm HEPES-KOH, pH 7.9, 20% glycerol, 0.1m KCl, 0.2 mm EDTA, 5 mm PMSF (Sigma), 1 mmleupeptin (Sigma), and 1 mm pepstatin A (Sigma) and 0.5 mm DTT], and 100× molar excess of competitor oligonucleotide (in reactions containing inhibitors) plus water to a final volume of 24 μl. One microliter of labeled oligonucleotide (15,000–30,000 cpm) was added and incubated for 30 min on ice. One microliter of loading dye was added, and the entire reaction was run on a 5.7% nondenaturing polyacrylamide gel at 30 mA in 1× TBE. Gels were dried under vacuum and exposed to x-ray film overnight. Each experiment was repeated with extracts from different nuclear protein isolations.
DNA footprint. Overlapping PCR fragments spanning the length of intron 1 were labeled at one end by use of a Klenow-mediated fill-in reaction with dATP α-32P (3000 Ci/mmol) at a 5′ overhang created by restriction digest. The labeled fragments were purified on a nick column (Amersham Biosciences). Twenty-five microliters of nuclear extract or whole-cell extract in Buffer D [formulated as for electrophoretic mobility shift assay (EMSA)] were incubated on ice for 30 min with 10 μl of 5× buffer (125 mm HEPES-KOH, pH 7.9, 25 mmMgCl2, 5 mm DTT, and 0.5 μg/μl of BSA), 1 μl of 1 μg/μl of poly I/poly C, 0.5 μl of 5 m NaCl, and 13.5 μl of water to a final volume of 50 μl. Control reactions were incubated with BSA in the place of nuclear extract. Labeled probe was added (0.5–1 ng; 100,000–150,000 cpm) and incubated on ice for 1 hr. DNase I, at a concentration of 0.05 Kunitz units, was added to each reaction mixture and incubated for 1 min at 25°C. One hundred fifty microliters of ice-cold stop solution (15 mm EDTA, 0.2% SDS, 40 μg/ml of salmon sperm DNA, and 20 mg/ml of proteinase K) were added. The tubes were then incubated at 50°C for 20 min. Each reaction was phenol–chloroform extracted three times and ethanol precipitated. The samples were resuspended in 2 μl of water and 4.5 μl of sequencing stop dye (95% formamide, 10 mm EDTA, 0.05% bromophenol blue, and 0.1% xylene cyanol). The products were denatured and run on a 6% polyacrylamide gel (Sequagel 6, National Diagnostic, Atlanta, GA). The gel was dried and placed on x-ray film overnight. Each experiment was repeated with extracts from different nuclear protein isolations. Size standards were obtained by Sequenase reactions (USB, Cleveland OH) prepared by the provided protocol.
RESULTS
To define regions of the peripherin gene that are critical for cell type-specific expression and activation in response to nerve injury in adult mice, we deleted sequential segments from a correctly expressed full-length rat peripherin genomic construct (Fig.1, Periph/periph-myc). We decided against the use of lacZ reporter-based constructs because our unpublished studies indicated that a lacZ reporter flanked by intragenic sequences was not expressed reliably in adult mice (Kramer, Belecky-Adams, and Parysek, unpublished observations). The structure of the first construct for our current studies was guided by previous observations that only introns 1 and 2 contain significant sequences identical in rat, mouse, and human (Foley et al., 1994). Thus, the first construct contained 5.8 kb of rat peripherin 5′ flanking sequence driving a rat peripherin gene that lacked introns 3–8 (Fig. 1, Periph/Δi3–8-myc). A second construct,Periph/Δi2–8-myc, was identical to the first but lacked intron 2 (Fig. 1). A third transgene contained the entireperipherin coding region but no introns (Fig. 1,Periph/intronless-myc). To facilitate detection of the transgene-derived protein, an in-frame myc tag was placed at the 3′ end of these transgenes.
Introns 3–8 are not required for accurate cell type-specific expression of peripherin
We tested the effects on cell type-specific expression in adult mice of deleting introns 3–8 (Periph/Δi3–8-myc) from a full-length rat peripherin transgene. This transgene was introduced into the genome of six lines of mice, five of which expressed myc-tagged peripherin protein. Double immunofluorescence revealed that distribution of myc-tagged transgenic protein was identical to that of endogenous peripherin protein in sensory neurons of the DRG, enteric nervous system, and lower motor neurons, all areas that exhibit distinctive patterns of peripherin expression (Portier et al., 1984; Parysek and Goldman, 1988). In DRG neurons, myc labeling was seen in small-diameter neurons but was not detectable in the large-diameter neurons, matching the distribution pattern of endogenous protein (data not shown). In the small intestine, both transgenic and endogenous proteins were observed in the enteric ganglia of the myenteric and submucosal plexuses, and in the spinal cord, allperipherin-expressing lower motor neurons coexpressed transgenic protein (data not shown). In addition, in regions of the brain and brainstem examined, the transgene was expressed in cells that express endogenous peripherin, and not in any other neurons (data not shown). These expression patterns were essentially identical to that seen with the full-length peripherin transgene (Belecky-Adams et al., 1993) and indicated that introns 3–8 are not required for accurate cell type-specific expression of peripherin. Because intragenic sequences were required for regulation of the peripherin gene in adult mice (Belecky-Adams et al., 1993), further experiments focused on the remaining introns, 1 and 2.
Intron 2 is not required for cell type-specificperipherin expression
To test the role of intron 2, a transgene lacking introns 2–8 (Periph/Δi2–8-myc) was analyzed. Six lines expressed the myc-tagged transgenic protein of nine lines that incorporated the transgene. As with the Periph/Δi3–8-myc transgenic lines, expression of the Periph/Δi2–8-myc transgene was identical to that of endogenous peripherin in these six lines. The distribution of transgenic protein and endogenous protein was virtually identical in the sensory neurons of the DRG (Fig.2A,B), the enteric nervous system (Fig.3A,B), and the lower motor neurons of the cervical spinal cord (Fig.4A,B). These data indicate that intron 2 does not contain sequences critical for precise cell type-specific expression of the peripherin gene, suggesting that these control elements are likely contained within intron 1.
Fig. 2.

Intron 1 is required for cell type-specific expression of peripherin transgenes in the DRG. Cross sections of adult DRGs from Periph/Δi2–8-myc (A,B) and Periph/intronless-myc(C, D) transgenic mice were double labeled with a polyclonal myc antibody (A,C) and a monoclonal peripherin antibody (B, D) to determine whether the transgenic protein pattern matched the endogenous pattern. In DRG sections from intron 1-containing mice, essentially perfect overlap was observed between the transgenic protein and endogenousperipherin (A, B), but DRGs from the intron 1-deleted transgenic mice revealed that many neurons expressed endogenous peripherin (D) but did not express transgenic peripherin (C). Scale bar, 20 μm.
Fig. 3.
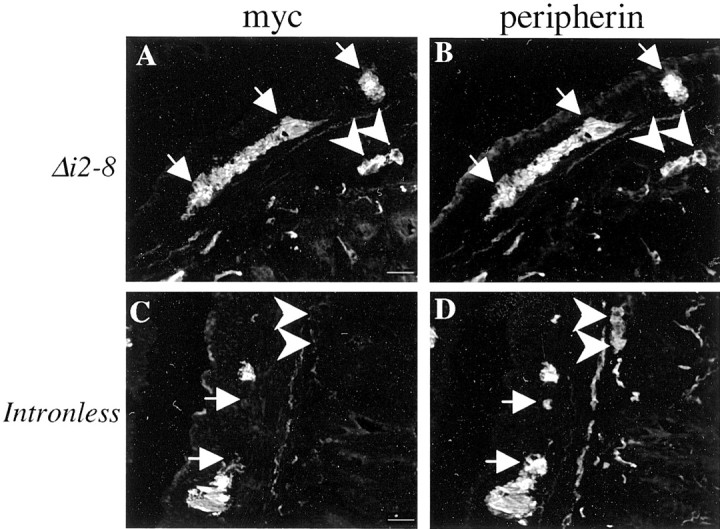
Intron 1 is required for cell type-specific expression of peripherin transgenes in the enteric nervous system. Cross sections of adult small intestine were double labeled with polyclonal myc antibody (A, C) and monoclonal peripherin antibody (B, D) to determine whether transgenes were expressed in endogenousperipherin-expressing neurons in the myenteric (white arrows) and submucosal (white arrowheads) plexuses. Expression of the intron 1-containing transgene,Periph/Δi2–8-myc, perfectly matched endogenousperipherin expression (A, B), but the Periph/intronless transgene (C,D) was expressed in <50% of the peripherin-positive structures in both the myenteric and submucosal plexuses in a majority of the lines. Scale bar, 20 μm.
Fig. 4.
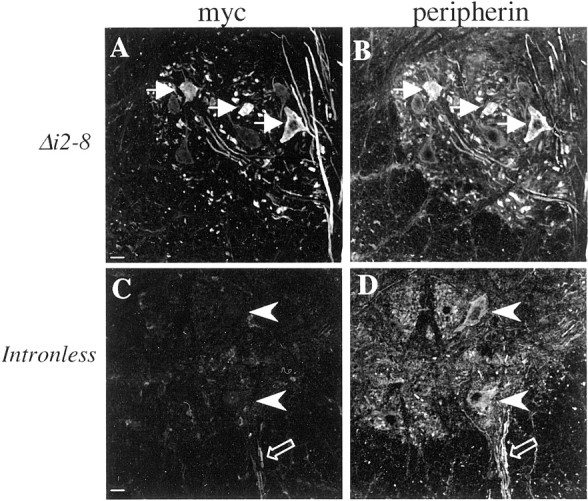
Cell type-specific expression ofperipherin in lower motor neurons requires intron 1. Cross sections of adult mouse cervical spinal cord were double labeled with a polyclonal myc antibody (A, C) and a monoclonal peripherin antibody (B, D) to determine the degree of overlap between transgenic and endogenous protein. In the intron-1 containing Periph/Δi2–8-myctransgenic mice (A, B, white arrows), transgene expression mimics that of endogenousperipherin, but in most of thePeriph/intronless transgenic mice, the only evidence of transgene expression in the lower motor neurons (C,D, white arrowheads) was an occasional labeled fiber in the ventral root (C,D, open arrows). Scale bar, 20 μm.
Intron 1 contains elements required for accurate cell type-specific expression of peripherin
To determine directly whether intron 1 is required for peripherin gene regulation in adult mice, expression of a transgene lacking all introns (Periph/intronless-myc) was analyzed. Because genomic elements surrounding transgene integration sites can unduly limit the basal expression of transgenes lacking introns (Choi et al., 1991; Webster et al., 1997), we examined an exceptionally large number of lines containing the intronless peripherin transgene. Seventeen lines of mice were generated that carried the transgene, and 11 of these lines (64.7%) were found to express transgenic protein. Approximately the same percentage of lines expressed the intron 1-containing transgene (Periph/Δi2–8-myc) (66.7%), suggesting that absence of an intron in the transgene did not have a significant impact on its basal expression.
In striking contrast to transgenic lines that contained intron 1, expression of transgenic protein varied significantly among the 11 expressing lines of intronless mice. No line had an overall distribution pattern identical to that of endogenous peripherin, although in some cases, expression among neurons of the DRG was close to normal. In DRG sensory neurons, myc-tagged protein was detected in each of the 11 expressing lines of mice, but only 3 lines showed reasonably good correspondence (75–85%) to endogenous expression, and in each case, myc-negative cells belonged to the medium-sized neuron size class. In these same three lines, however, the transgene was not expressed at all in enteric neurons. Four other lines expressed myc-tagged protein in ∼50% of the peripherin-positive DRG neurons (Fig. 2C,D), and the remaining four lines expressed transgenic protein in <50% of peripherin-positive neurons. These data are consistent with the idea that intron 1 contains an enhancer required for complete peripherin expression in the DRG. In some lines, sequences surrounding the integration site may compensate, to a degree, for the loss of the enhancer, but expression identical to endogenous peripherin was obtained only when intron 1 was present.
In the enteric nervous system, dramatic differences were observed between expression of the intronless transgene and endogenous peripherin gene. Of the 11 expressing lines, 8 lines expressed the transgene in the gut, but none of these lines expressed the transgene in all peripherin-positive neurons. In four lines, transgene was expressed in >50% of peripherin-positive structures in the myenteric and submucosal plexuses. In the other four lines, the transgene was expressed in >50% of myenteric structures, but in much less than 50% of submucosal structures, leading to an overall expression level of <50% of peripherin-positive structures in the enteric nervous system (Fig. 3C,D). Thus, the expression pattern of the intronless transgene indicated the presence of regulatory sequences within intron 1 critical for correct peripherin gene expression in the enteric nervous system.
Differences also were observed between expression of the endogenous peripherin gene and the intronless transgene in lower motor neurons of the cervical spinal cord, indicating that sequences in intron 1 play a critical role in regulating peripherin gene expression in these cells as well. Myc-tagged protein was observed in the cervical cord in all 11 intronless lines, but in no more than 50% of motor neurons in four lines and only in rare neurons in the remaining seven lines, indicated by the presence of one or two myc-labeled fibers in the ventral root (Fig. 4C,D). Examination of other areas of the CNS, parts of the brain and brainstem, revealed similar findings as in the PNS; that is, loss of intronlesstransgene expression in some peripherin-positive neurons and no ectopic expression.
In summary, these data revealed that deletion of intron 1, but not the other seven peripherin gene introns, results in loss of transgene expression in some cells that express endogenous peripherin. Intron 1, therefore, must contain elements critical for full expression of the peripherin gene.
Which regions of intron 1 are responsible for cell type-specific regulation?
To identify specific sequences in intron 1 that may be responsible for regulating cell type-specific expression of peripherin, DNase I footprint analyses were performed. Addition of nuclear extracts from the small intestine, brain, or liver or whole-cell extracts from the DRG inhibited DNase I degradation of five regions within intron 1 (Figs. 5,6, 7). These regions were located at rat gene [(GenBank Accession no. M26232;Thompson and Ziff (1989)] nucleotides 1483–1493 [Footprint (FP) 1], 1528–1537 (FP2), 1546–1555 (FP3), 1678–1686 (FP4), and 1697–1707 (FP5) (Fig. 5, top). To confirm that the footprints represented specific protein–DNA interactions, EMSAs were performed using oligonucleotides of appropriate length (>25 bp) for the assays.
Fig. 5.

Extracts from tissues that express peripherin contain different intron 1-binding Sp1 or Sp1-related proteins than nonexpressing tissues. A region of intron 1, from 1360–1664 of the rat peripherin gene, was radiolabeled and digested with DNase I in the presence of BSA (Control), whole-cell extract from the DRG, or nuclear extract from the small intestine (SI), brain, or liver. Nuclear extracts from all tested tissues protected nucleotides 1483–1493, footprint 1 (FP1) (A). EMSA of Oligo 1, corresponding to FP1 and surrounding sequences, illustrated specific DNA–protein complexes with nuclear extract from the SI (left, arrowhead) and liver (right). These complexes were competed off by unlabeled Oligo 1 (Specific), a short oligonucleotide (Oligo 1B) to the protected sequence, and by an oligonucleotide containing an Sp1 consensus site. The formation of these complexes was not inhibited by a nonspecific oligonucleotide or by an oligonucleotide containing a mutant Sp1 consensus site (B) (Oligo sequences are shown in Table 1).
Fig. 6.

Intron 1 interacts with different NFI- or NFI-related proteins, depending on whether the tissue extract of origin contains neurons. Sequence from 1475–1594 of the rat peripherin gene was radiolabeled and digested with DNase I in the presence of BSA (Control), whole-cell extract from the DRG, or nuclear extract from the small intestine (SI), brain, or liver. Nucleotides 1528–1537 (FP2) and 1546–1555 (FP3) were protected by extracts from all tissues (A). Oligo 2, containing FP2 and FP3 and surrounding nucleotides, formed specific DNA–protein complexes with extracts from the DRGs (B). These complexes were competed off by unlabeled Oligo 2 (Specific), an oligonucleotide (Oligo 2B) containing sequence between FP2 and FP3, and by an oligonucleotide containing an NFI consensus site. These complexes were not inhibited by a nonspecific oligonucleotide, Oligo 2A, Oligo 2C, Oligo 2D, or an oligonucleotide containing a mutant NFI consensus site.
Fig. 7.

A second site in intron 1 binds Sp1 or Sp1-related proteins that correlate with the status of peripherin expression in the tissue from which the extract was derived. A region spanning 1665–1800 of the rat gene was radiolabeled and digested with DNase I in the presence of BSA (Control), whole-cell extract from the DRG, or nuclear extract from the SI, brain, or liver. There was protection of nucleotides 1678–1686 (FP4) and 1697–1707 (FP5) by extracts from all tissues (A). An oligonucleotide containing FP4, FP5, and surrounding sequences formed specific DNA–protein complexes with whole-cell extract from the DRG (B,arrowhead) and nuclear extract from the liver (C). These complexes were competed off by unlabeled Oligo 3 (Specific) and by an oligonucleotide containing an Sp1 consensus site. The liver nuclear extract formed one complex that was reduced, but not completely competed off, by the Sp1 oligonucleotide. All complexes were not inhibited by a nonspecific oligonucleotide or by an oligonucleotide containing a mutant Sp1 consensus site (B, C).
The first experiment focused on FP1 (Fig. 5A) and used an oligonucleotide (Oligo 1) that contained FP1 and adjacent nucleotides (Table 1; see Sequence). The mobility of Oligo 1 was reduced when incubated with whole-cell extract from mouse DRG (data not shown) or nuclear extracts from small intestine (Fig.5B, left), liver (Fig. 5B,right), or brain (data not shown). To show that the DNA–protein interactions were specific, EMSAs using competitor oligonucleotides were performed. These experiments showed that the DNA–protein complexes were specific because a specific inhibitor, excess unlabeled Oligo 1, blocked the formation of the DNA–protein complexes, and excess nonspecific inhibitor had no effect on complex formation. Extracts from DRGs or small intestine formed a single high-mobility complex (Fig. 5B, left,arrowhead). In contrast, nuclear extracts from liver (Fig.5B, right) or brain (data not shown) resulted in several complexes of lower mobility, suggesting that these tissues contain multiple proteins interacting at this site. To define the exact location of protein interaction, short Oligos 1A, 1B (corresponds to FP1), and 1C (Table 1; see Sequence) were used in competitive binding experiments. These assays showed that only Oligo 1B competed for binding, indicating that the regions of protein binding in Oligo 1 resided completely within the region protected in DNA footprint analysis. Using the Transcription Element Search System (TESS) sequence analysis database, computer analysis of FP1 revealed the presence of a potential Sp1 binding site. To determine whether an Sp1 family member does interact with this sequence, an oligonucleotide containing an Sp1 consensus binding site and an identical oligonucleotide containing a mutant Sp1 binding site (Table 1; see Sequence) were tested for their ability to compete with labeled Oligo 1 for binding to nuclear protein. Excess Sp1 oligonucleotide greatly reduced or eliminated binding of protein from all tissues to labeled Oligo 1, whereas excess mutant Sp1 oligonucleotide did not, signifying that members of the Sp1 family of transcription factors interact with this sequence.
Table 1.
Composition of oligonucleotides and competitive inhibitors used for EMSA
| Oligo | Sequence | Region1-a |
|---|---|---|
| Oligo 1 | TCTAACAACCATAGGGCGTGGTCGGGCTGGC | 1474–1505 |
| Oligo 1A | TCTAACAACCAT | 1474–1486 |
| Oligo 1B | ATAGGGCGTGGTCG | 1484–1498 |
| Oligo 1C | GTCGGGCTGGC | 1495–1505 |
| Oligo 2 | TGCAGCATCCTAGCGGGCCCTCTGCCAGCACTTTGCTCCGAG | 1525–1565 |
| Oligo 2A | TGCAGCATCCTAGCG | 1525–1538 |
| Oligo 2B | GGCCCTCTGCCAG | 1539–1551 |
| Oligo 2C | CACTTTGCTCCGAG | 1552–1565 |
| Oligo 2D | CCAGCACTTTGCTCCGAG | 1548–1565 |
| Oligo 3 | GCTGCAGAAGTAGATGGGGGCGGGGTGCTGGATGTAGCACGAGGTCTGG | 1665–1713 |
| Non-Specific | CAGTCATGACTTCGACTAGCACTTCATCAG | |
| Sp1 | CCCTTGGTGGGGGCGGGGCCTAAGCTGCG | |
| Mutant Sp1 | CCCTTGGTGGGTTGGGGGCCTAAGCTGCG1-b | |
| NFI | GGCACCTGTTTCAATTTGGCACGGAGCCAACAG | |
| Mutant NFI | GGCACCTGTTTCAATTTGTTACGGATTCAACAG1-b |
Region of the rat peripherin gene, numbered according to National Center for Biotechnology Information accession M26232.
Bold nucleotides are changes to the consensus binding site.
A second oligonucleotide, Oligo 2 (Table 1; see Sequence), spanning footprinted regions 2 (FP2) and 3 (FP3) (Fig. 6A) and adjacent nucleotides, showed an identical high-mobility shift when incubated with whole-cell extract from mouse DRGs (Fig.6B) or nuclear extract from the small intestine or brain (data not shown). Nuclear extract from a non-neuronal tissue, liver, produced multiple lower-mobility protein–DNA complexes (data not shown). Formation of complexes from all tissue extracts was abolished by addition of excess unlabeled Oligo 2 (specific inhibitor) but not by excess nonspecific oligonucleotide. Short competitor Oligos 2A, 2B, 2C, and 2D (Table 1; see Sequence), spanning the length of Oligo 2, indicated protein bound to nucleotides 1539–1551 (Oligo 2B), which is located between FP2 and FP3. Computer analysis of nucleotides 1539–1551, using the TESS sequence analysis program, indicated a potential NFI binding site. To determine whether NFI protein family members in the tissue extracts interacted with Oligo 2, competitive oligonucleotides containing either an NFI consensus site or a mutant NFI consensus site (Table 1; see Sequence) were tested for their ability to inhibit binding of the extract to labeled Oligo 2. The NFI consensus oligonucleotide inhibited binding of all proteins from DRGs (Fig. 6B), small intestine, brain, and liver (data not shown), but the mutant NFI oligonucleotide did not interfere with these interactions (Fig. 6B). These data indicate that NFI family member proteins bind to the 1539–1551 region of the rat peripherin gene. Although the EMSAs localized the protein–DNA interaction to an NFI consensus site, the footprint results suggest an area of protein binding localized outside of this area. Because these assays use the same extracts but are done under very different conditions, it is likely that an NFI or a related protein is a critical member of a multiple protein complex that interacts with the entire region between nucleotides 1528 and 1555 of the rat peripherin gene.
A single high-mobility protein–DNA complex also was formed when Oligo 3 (Table 1; see Sequence), corresponding to footprinted regions 4 (FP4) and 5 (FP5) (Fig. 7A) and adjacent nucleotides, was incubated with whole-cell extract from mouse DRGs (Fig. 7B,arrowhead) or nuclear extract from the small intestine (data not shown). This interaction was specific, because unlabeled Oligo 3 (specific inhibitor) abolished the interaction, whereas nonspecific inhibitor had little effect. Nuclear extracts from liver (Fig.7C) or brain (data not shown) formed multiple low-mobility DNA–protein complexes. These interactions also were specific because they were eliminated by unlabeled Oligo 3 (data not shown) but not by a nonspecific inhibitor. Sequence analysis showed that Oligo 3 contains a perfect Sp1 motif. To confirm binding of an Sp1 family member protein to this site, Sp1 and mutant Sp1 oligonucleotides were tested to determine their efficiency in competing for the interaction between the protein and labeled Oligo 3. The Sp1 oligonucleotide completely inhibited all interactions in all tissue extracts (Fig. 7B), except for one partially inhibited protein–DNA complex formed by liver nuclear extract (Fig. 7C). The mutant Sp1, however, did not inhibit any interactions, confirming that Sp1 family member proteins interact with this region of intron 1 (Fig.7B,C).
Taken together, the data indicate that intron 1 is required for regulating accurate cell type-specific expression and that known transcription factor family proteins bind to intron 1.
Intragenic sequences are not required for activation ofperipherin after nerve injury
To determine whether intron 1 was required for activation of the peripherin gene after nerve injury, transgene expression was studied after sciatic nerve crush in the intron 1-containingPeriph/Δi2–8-myc lines and in the intron 1-deletedPeriph/intronless-myc lines that expressed the peripherin transgene in a near-perfect manner in the DRGs. Seven days after injury, expression of the transgene was assessed by myc-immunofluorescence. Expression of endogenous peripherinwas assayed as a positive control. In each experiment, sham-operated mice were used as a negative control.
Analyses of nerve crush experiments performed on thePeriph/Δi2–8-myc transgenic line of mice revealed an increase in peripherin transgene expression that paralleled the normal increase in endogenous peripherin gene expression (data not shown) in large-diameter sensory neurons of the DRG after injury. After the sham operation, the large-diameter neurons expressed minimal or no peripherin (data not shown), as is normal for the uninjured DRG. These initial studies revealed that introns 2–8 were not required for injury-induced peripherin gene activation.
Similarly, in the Periph/intronless-myc transgenic mouse lines that expressed transgene-derived peripherin/myc in 75–85% of the peripherin-positive neurons, myc-tagged protein was expressed robustly in large-diameter neurons after nerve crush (Fig.8A, arrows), paralleling the increase in endogenous peripherin expression (Fig.8B, arrows). No increase was observed in sham-operated animals (Fig. 8C,D,arrows). This increase in myc-tagged protein was observed in 85% of the large-diameter neurons in this ganglion, as determined by comparing neurofilament and myc labeling (data not shown). Importantly, the percentage of large-diameter neurons expressing transgenic protein was identical to the percentage of cells expressing endogenous peripherin protein, indicating that the intronless transgene was activated in the appropriate neurons. These data indicate that injury response elements are not located within the introns but are most likely located in the 5′ flanking sequence, although the possibility that they are located in the 3′ untranslated region or exons of the peripherin gene cannot be excluded.
Fig. 8.
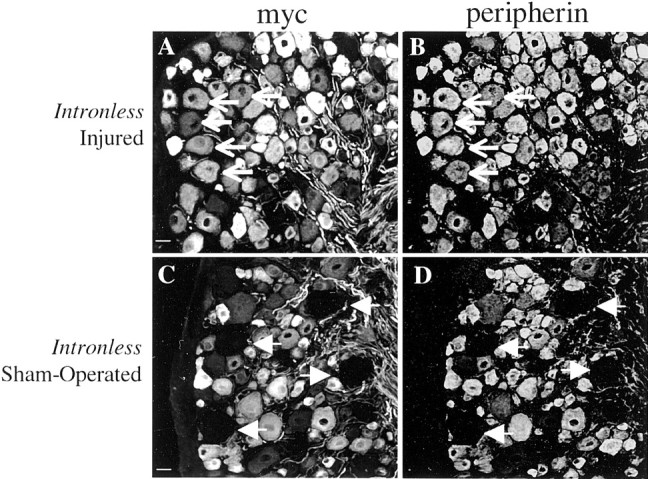
Intron 1 is not required to activateperipherin expression after nerve injury.Periph/intronless-myc transgenic mice that expressed the transgene in 85% of endogenous peripherin-expressing neurons underwent either sciatic nerve crush (A, B) or surgery alone (sham operated) (C, D). Cross sections of the L5 DRG from these mice, which were double labeled with polyclonal myc antibody (A, C) and monoclonal peripherin antibody (B, D), revealed that the Periph/intronless-myc transgene was upregulated after injury in a manner identical to that of endogenousperipherin. Both transgenic and endogenous peripherin protein were expressed in the large-diameter sensory neurons of injured mice (A, B, arrows) but not in these neurons in the uninjured DRGs (C,D, arrows). Scale bar, 20 μm.
DISCUSSION
To understand the transcriptional pathways leading to activation of the peripherin gene after nerve injury, we identified genomic segments required for cell type-specific expression and induction after nerve injury. We found that 5.8 kb of 5′ peripherin flanking sequence was sufficient for injury-induced activation of an intronless peripherin gene that contained peripherin coding and 600 bp 3′ flanking sequence, but was insufficient to direct transgene expression to all peripherin-expressing neurons. Only transgenes that also contained intron 1 were expressed in all of the same neurons that express endogenous peripherin. Thus, injury-inducible elements are likely to lie in the 5′ flanking region but also could be located in the 3′ untranslated region or coding region, whereas critical cell-specific elements lie in intron 1.
The intron 1-dependent loss of transgene expression from some cells that express endogenous peripherin suggests that intron 1 contains one or more enhancer elements that are not of the general enhancer type. Loss of a general enhancer would be expected to reduce expression levels overall and result in an apparent preferential loss of transgene-derived peripherin in neurons that normally express low levels of endogenous peripherin. This is not what we observed. Instead, in mice bearing the intronless transgene, cells that appeared to be labeled similarly by peripherin antibody [such as the small-sized DRG cells (Fig. 8D, top) or the myenteric elements (Fig. 3D, bottom)] were dissimilarly labeled by myc antibody (corresponding regions of Figs.8C and 3C, respectively), indicating that transgene expression was lost only from certain cells. This enhancer appeared to be especially important for expression in medium-sized DRG neurons, the submucosal enteric neurons, and lower motor neurons of the spinal cord, suggesting that the element is more in the nature of a cell type-specific, rather than general, enhancer.
Two additional conclusions can be drawn about the role of intron 1. First, it is clear that it plays no role in restricting expression to peripherin-positive neurons. Whether intron 1 was present or not, we never observed ectopic expression of any of the peripherin transgenes, even in the brain (data not shown). Second, intron 1 does not contain an “all-or-none” enhancer because deletion of intron 1 impaired expression in every 1 of 17 mouse lines with distinct transgene integration sites, yet did not abolish all peripherin expression. Thus, intron 1 is critical, but it is likely to be aided in executing its enhancer function by the 5′ flanking sequence, exons, or the 3′ untranslated region.
EMSA analysis identified candidate sequences within intron 1 that are likely to play roles in the cell-type-specific expression ofperipherin. Two Sp1 consensus sites bound distinct proteins or protein complexes that correlated with peripherin expression in the tissue extract of origin. Extracts from tissues that expressed robust amounts of peripherin (DRGs, small intestine) formed a single, high-mobility complex with each of the two Sp1 sites. It is most likely that this high-mobility complex contained an uncommon, perhaps tissue-specific, Sp1 family member or unknown protein because binding of Sp1 itself or a closely related protein to DNA produces multiple complexes with lower mobility shifts (Andrew et al., 2000; Teunissen et al., 2002) such as we observed with extracts derived from tissues that expressed little or no peripherin (liver and brain). Similarly, extracts from neuron-enriched tissues (brain, small intestine, and DRGs) formed a single complex with the NFI site in intron 1 that was very different from the multiple complexes formed by proteins from liver. As yet, we have no direct evidence that this Sp1/NFI/Sp1 motif is required for cell-type-specific expression of peripherin, but the close correlation between the type of complex formed and the origin of the tissue extract strongly supports this conclusion.
Previous work on the mouse peripherin gene by Lecomte et al. (1999)also supports the importance of these Sp1 sites in peripherin gene regulation. Mouse neuroblastoma cell nuclear extract bound to a core Sp1 motif in intron 1 of the mouse peripherin gene that is 90% identical in sequence to our first Sp1 binding site (1483–1493 of the rat gene). The binding protein was identified as “AP-2-like” but was not characterized. Lecomte et al. (1999) also observed binding of a member of the Sp1 family to a region of the mouse gene that is identical in sequence to our second Sp1 site (1678–1686). TESS sequence analyses indicate that both Sp1 sites are conserved in the human gene as well, suggesting that Sp1 or related proteins that interact with this region of intron 1 in rat and mouse also interact with the same sites in the human gene. There are several theories about the role of Sp1 (for review, see Fry and Farnham, 1999) and NFI (for review, see Gronostajski, 2000) or related proteins in regulating gene transcription when present in the promoter, but how they may function together when present within an intron remains unknown. The role of the Sp1 and NFI sites in intron 1 of the peripherin gene will be determined by testing the effect of mutating these sites on peripherinexpression in transgenic mice.
Our finding that intron 1 of the peripherin gene contains elements required for cell-type-specific expression adds to previous studies showing the importance of intron 1 for precise cell type-specific and temporal expression of other IF genes, including those encoding glial fibrillary acidic protein (Sarkar and Cowan, 1991), keratin 18 (Rhodes and Oshima, 1998), neurofilament-L (Charron et al., 1995), and nestin (Zimmerman et al., 1994). Given the common evolutionary origin of the IF genes, some consistency in location of key regulatory elements might be expected. Interestingly, perfect Sp1 and NFI consensus sites are found in intron 1 of the neurofilament-L gene (Charron et al., 1995) in an arrangement similar to that in the peripherin gene. These observations raise the possibility that primitive Sp1 and NFI recognition sequences present in the evolutionary antecedent of the IF genes may have been duplicated along with the structural genes and then evolved to mediate distinct patterns of cell-type-specific expression through interaction with different Sp1 and NFI family members.
Intron 1 is critical for full expression of peripherin transgenes inperipherin-expressing neurons, but the observation that an intronless transgene was activated by nerve injury suggests that required injury-response elements are not present in intragenic regions. This finding was somewhat unexpected because our previous studies (Belecky-Adams et al., 1993) indicated that 5′ flanking sequence of peripherin was insufficient to activate a lacZ reporter gene after nerve injury. In that study, however, the lacZ reporter construct was either not expressed at all or poorly expressed in injury-responsive neurons (Belecky-Adams et al., 1993). The intronless myc-tagged peripherin transgene used in the present studies was expressed more robustly in mice, and three lines expressed this transgene at high levels in DRG neurons, allowing a more reliable test of injury responsiveness. The observation that this intronless transgene was activated after nerve injury suggests that the injury-response elements are located within the 5′ region, although the coding sequences or 3′ flanking sequences of the gene cannot yet be completely excluded. Indeed, injury-mediated repression of mRNA encoding neurofilament-L occurs via elements in the 3′ untranslated region (Schwartz et al., 1995). Both 5′ flanking sequence and a minimum of 11 kb of intron 1 are needed for accurate cell type-specific regulation and activation after injury of the GAP-43 gene (Vanselow et al., 1994), which, like peripherin, shows an injury-mediated increase in mRNA (Woolf et al., 1990). The precise location of theGAP-43 injury-response elements, however, remains unknown.
The identification of intron 1 as critical for full cell-type specific expression facilitates the design of transgenes for determining 5′ flanking sequences required for activation after injury. Several potential regulatory sequences, including NGF-inducible and IL-6-binding elements, have been identified within the 5′ flanking sequence of peripherin via in vitro studies (Thompson et al., 1992; Lecomte et al., 1998). NGF activator domains lie both distal and proximal to the TATA box (Thompson et al., 1992;Desmarais and Royal, 1996), and NGF is believed to be a primary activator of the peripherin gene (Leonard et al., 1987), but this has not been verified in vivo. Significantly, functional recovery after nerve regeneration is an NGF-independent process (Diamond et al., 1992). Thus, activation of the peripherin gene after nerve crush might not require NGF. We believe that the IL-6 element is a particularly good candidate for an injury-response element. IL-6 activates the peripherin gene (Sterneck et al., 1996; Lecomte et al., 1998), it is induced in Schwann cells within 12 hr after nerve injury (Bolin et al., 1995), making it readily available during regeneration, and it increases the rate of regeneration of peripherin-positive neurons (Hirota et al., 1996; Shuto et al., 2001). Furthermore, the location and sequence of the binding site for LIF/IL-6 are highly conserved in the mouse and human peripherin genes. The role of the IL-6 site and other potential sites in injury responsiveness can now be tested through further deletion analyses. Once such elements in the peripherin gene have been defined, it will be important to determine whether they are shared among other nerve injury-activated genes.
Footnotes
This work was supported by Grant 35313 from the National Institute of Neurological Disorders and Stroke. We thank Dr. Dan Wiginton for his advice and guidance with the DNase I footprinting and EMSA experiments, Molly McFarland for her help with construction of the deletion transgenes, and Dr. Robert Brackenbury for helpful comments on this manuscript.
Correspondence should be addressed to Dr. Linda M. Parysek, Department of Cell Biology, Neurobiology and Anatomy, University of Cincinnati, 3125 Eden Avenue, Cincinnati, OH 45267-0521. E-mail:Linda.Parysek@uc.edu.
REFERENCES
- 1.Andrew SD, Delhanty PJ, Mulligan LM, Robinson BG. Sp1 and Sp3 transactivate the RET proto-oncogene promoter. Gene. 2000;256:283–291. doi: 10.1016/s0378-1119(00)00302-4. [DOI] [PubMed] [Google Scholar]
- 2.Belecky-Adams T, Wight DC, Kopchick JJ, Parysek LM. Intragenic sequences are required for cell type-specific and injury-induced expression of the rat peripherin gene. J Neurosci. 1993;13:5056–5065. doi: 10.1523/JNEUROSCI.13-12-05056.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bisby MA, Tetzlaff W. Changes in cytoskeletal protein synthesis following axon injury and during axon regeneration. Mol Neurobiol. 1992;6:107–123. doi: 10.1007/BF02780547. [DOI] [PubMed] [Google Scholar]
- 4.Bolin LM, Verity AN, Silver JE, Shooter EM, Abrams JS. Interleukin-6 production by Schwann cells and induction in sciatic nerve injury. J Neurochem. 1995;64:850–858. doi: 10.1046/j.1471-4159.1995.64020850.x. [DOI] [PubMed] [Google Scholar]
- 5.Brody BA, Ley CA, Parysek LM. Selective distribution of the 57 kDa neural intermediate filament protein in the rat CNS. J Neurosci. 1989;9:2391–2401. doi: 10.1523/JNEUROSCI.09-07-02391.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Charron G, Guy LG, Bazinet M, Julien JP. Multiple neuron-specific enhancers in the gene coding for the human neurofilament light chain. J Biol Chem. 1995;270:30604–30610. doi: 10.1074/jbc.270.51.30604. [DOI] [PubMed] [Google Scholar]
- 7.Choi T, Huang M, Gorman C, Jaenisch R. A generic intron increases gene expression in transgenic mice. Mol Cell Biol. 1991;11:3070–3074. doi: 10.1128/mcb.11.6.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dent CL, Latchman DS. The DNA mobility shift assay. In: Latchman DS, editor. Transcription factors: a practical approach. Oxford UP; New York: 1993. pp. 1–26. [Google Scholar]
- 9.Desmarais D, Royal A. The TATA motif is a target for efficient transcriptional activation and nerve growth factor induction of the peripherin gene. J Biol Chem. 1996;271:24976–24981. doi: 10.1074/jbc.271.40.24976. [DOI] [PubMed] [Google Scholar]
- 10.Desmarais D, Filion M, Lapointe L, Royal A. Cell-specific transcription of the peripherin gene in neuronal cell lines involves a cis-acting element surrounding the TATA box. EMBO J. 1992;11:2971–2980. doi: 10.1002/j.1460-2075.1992.tb05367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diamond J, Foerster A, Holmes M, Coughlin M. Sensory nerves in adult rats regenerate and restore sensory function to the skin independently of endogenous NGF. J Neurosci. 1992;12:1467–1476. doi: 10.1523/JNEUROSCI.12-04-01467.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dusing MR, Florence EA, Wiginton DA. Pdx-1 is required for activation in vivo from a duodenum-specific enhancer. J Biol Chem. 2001;276:14434–14444. doi: 10.1074/jbc.M009249200. [DOI] [PubMed] [Google Scholar]
- 13.Foley J, Cohn SL, Salwen HR, Chagnovich D, Cowan J, Mason KL, Parysek LM. Differential expression of N-myc in phenotypically distinct subclones of a human neuroblastoma cell line. Cancer Res. 1991;51:6338–6345. [PubMed] [Google Scholar]
- 14.Foley J, Ley CA, Parysek LM. The structure of the human peripherin gene (PRPH) and identification of potential regulatory elements. Genomics. 1994;22:456–461. doi: 10.1006/geno.1994.1410. [DOI] [PubMed] [Google Scholar]
- 15.Fry CJ, Farnham PJ. Context-dependent transcriptional regulation. J Biol Chem. 1999;274:29583–29586. doi: 10.1074/jbc.274.42.29583. [DOI] [PubMed] [Google Scholar]
- 16.Gronostajski RM. Roles of the NFI/CTF gene family in transcription and development. Gene. 2000;249:31–45. doi: 10.1016/s0378-1119(00)00140-2. [DOI] [PubMed] [Google Scholar]
- 17.Hirota H, Kiyama H, Kishimoto T, Taga T. Accelerated nerve regeneration in mice by upregulated expression of interleukin (IL) 6 and IL-6 receptor after trauma. J Exp Med. 1996;183:2417–2419. doi: 10.1084/jem.183.6.2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lecomte MJ, Basseville M, Landon F, Karpov V, Fauquet M. Transcriptional activation of the mouse peripherin gene by leukemia inhibitory factor: involvement of STAT proteins. J Neurochem. 1998;70:971–982. [PubMed] [Google Scholar]
- 19.Lecomte MJ, Basseville M, Fauquet M. Involvement of intronic sequences in cell-specific expression of the peripherin gene. J Neurochem. 1999;73:1806–1815. [PubMed] [Google Scholar]
- 20.Leconte L, Santha M, Fort C, Poujeol C, Portier M-M, Simonneau M. Cell type-specific expression of the mouse peripherin gene requires both upstream and intragenic sequences in transgenic mouse embryos. Dev Brain Res. 1996;92:1–9. doi: 10.1016/0165-3806(95)00182-4. [DOI] [PubMed] [Google Scholar]
- 21.Leonard DG, Ziff EB, Greene LA. Identifcation and characterization of mRNAs regulated by nerve growth factor in PC12 cells. Mol Cell Bio. 1987;7:3156–3167. doi: 10.1128/mcb.7.9.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oblinger MM, Wong J, Parysek LM. Axotomy-induced changes in the expression of a type III neuronal intermediate filament gene. J Neurosci. 1989;9:3766–3775. doi: 10.1523/JNEUROSCI.09-11-03766.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parysek LM, Goldman RD. Distribution of a novel 57 kDa intermediate filament (IF) protein in the nervous system. J Neurosci. 1988;8:555–563. doi: 10.1523/JNEUROSCI.08-02-00555.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Portier MM, de Nechaud B, Gros F. Peripherin, a new member of the intermediate filament protein family. Dev Neurosci. 1984;6:335–344. doi: 10.1159/000112360. [DOI] [PubMed] [Google Scholar]
- 25.Rhodes K, Oshima RG. A regulatory element of the human keratin 18 gene with AP-1-dependent promoter activity. J Biol Chem. 1998;273:26534–26542. doi: 10.1074/jbc.273.41.26534. [DOI] [PubMed] [Google Scholar]
- 26.Sarkar S, Cowan NJ. Intragenic sequences affect the expression of the gene encoding glial fibrillary acidic protein. J Neurochem. 1991;57:675–684. doi: 10.1111/j.1471-4159.1991.tb03799.x. [DOI] [PubMed] [Google Scholar]
- 27.Schwartz ML, Bruce J, Shneidman PS, Schlaepfer WW. Deletion of 3′-untranslated region alters the level of mRNA expression of a neurofilament light subunit transgene. J Biol Chem. 1995;270:26364–26369. doi: 10.1074/jbc.270.44.26364. [DOI] [PubMed] [Google Scholar]
- 28.Shuto T, Horie H, Hikawa N, Sang K, Tokashiki A, Murata H, Yamamoto I, Ishikawa Y. IL-6 up-regulates CNTF mRNA expression and enhances neurite regeneration. NeuroReport. 2001;12:1081–1085. doi: 10.1097/00001756-200104170-00043. [DOI] [PubMed] [Google Scholar]
- 29.Skene JH. Axonal growth-associated proteins. Annu Rev Neurosci. 1989;12:127–156. doi: 10.1146/annurev.ne.12.030189.001015. [DOI] [PubMed] [Google Scholar]
- 30.Sterneck E, Kaplan DR, Johnson PF. Interleukin-6 induces expression of peripherin and cooperates with Trk receptor signaling to promote neuronal differentiation in PC12 cells. J Neurochem. 1996;67:1365–1374. doi: 10.1046/j.1471-4159.1996.67041365.x. [DOI] [PubMed] [Google Scholar]
- 31.Tanabe K, Nakagomi S, Kiryu-Seo S, Namikawa K, Imai Y, Ochi T, Tohyama M, Kiyama H. Expressed-sequence-tag approach to identify differentially expressed genes following peripheral nerve axotomy. Mol Brain Res. 1999;64:34–40. doi: 10.1016/s0169-328x(98)00302-7. [DOI] [PubMed] [Google Scholar]
- 32.Teunissen BE, van Amersfoorth SC, Opthof T, Jongsma HJ, Bierhuizen MF. Sp1 and Sp3 activate the rat connexin40 proximal promoter. Biochem Biophys Res Commun. 2002;292:71–78. doi: 10.1006/bbrc.2002.6621. [DOI] [PubMed] [Google Scholar]
- 33.Thompson MA, Ziff EB. Structure of the gene encoding peripherin, an NGF-regulated neuronal-specific type III intermediate filament protein. Neuron. 1989;2:1043–1053. doi: 10.1016/0896-6273(89)90228-6. [DOI] [PubMed] [Google Scholar]
- 34.Thompson MA, Lee E, Lawe D, Gizang-Ginsberg E, Ziff EB. Nerve growth factor-induced derepression of peripherin gene expression is associated with alterations in proteins binding to a negative regulatory element. Mol Cell Biol. 1992;12:2501–2513. doi: 10.1128/mcb.12.6.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vanselow J, Grabczyk E, Ping J, Baetscher M, Teng S, Fishman MC. GAP-43 transgenic mice: dispersed genomic sequences confer a GAP-43-like expression pattern during development and regeneration. J Neurosci. 1994;14:499–510. doi: 10.1523/JNEUROSCI.14-02-00499.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Webster J, Donofrio G, Wallace R, Clark AJ, Whitelaw CB. Intronic sequences modulate the sensitivity of β-lactoglobulin transgenes to position effects. Gene. 1997;193:239–243. doi: 10.1016/s0378-1119(97)00128-5. [DOI] [PubMed] [Google Scholar]
- 37.Woolf CJ, Reynolds ML, Molander C, O'Brien C, Lindsay RM, Benowitz LI. The growth-associated protein GAP-43 appears in dorsal root ganglion cells and in the dorsal horn of the rat spinal cord following peripheral nerve injury. Neuroscience. 1990;34:465–478. doi: 10.1016/0306-4522(90)90155-w. [DOI] [PubMed] [Google Scholar]
- 38.Zimmerman L, Parr B, Lendahl U, Cunningham M, McKay R, Gavin B, Mann J, Vassileva G, McMahon A. Independent regulatory elements in the nestin gene direct transgene expression to neural stem cells or muscle precursors. Neuron. 1994;12:11–24. doi: 10.1016/0896-6273(94)90148-1. [DOI] [PubMed] [Google Scholar]