

Abstract
Nitric oxide (NO) suppressed the transcription of the mouse κ opioid receptor (KOR) gene, mediated by a rapid downregulation ofc-myc gene expression. KOR was constitutively expressed in postnatal day 19 (P19) embryonal carcinoma stem cells and is suppressed by NO donors [sodium nitroprusside (SNP), 3-morpholinosydnonimine-1, and S-nitrosoglutathione] in P19 stem cells within 4 hr. The suppression was reversed by 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide, an NO scavenger, but could not be blocked by dithiothreitol, ruling outS-nitrosylation as the underlying mechanism. The suppressive effect of NO on KOR occurred at the level of gene transcription, mediated by E boxes located in promoters I and II of this gene. Protein–DNA complexes that formed on these E boxes contained c-myc; c-myc expression was suppressed by NO in P19 stem cells within 2 hr of treatment. Furthermore, chromatin immunoprecipitation demonstrated reducedc-myc binding to the E boxes and hypoacetylation of histone H3 on the chromatin of endogenous KOR promoters in P19 stem cells treated with SNP. It is proposed that NO regulates KOR at the level of gene transcription, mediated by a rapid suppression ofc-myc gene expression and its binding to KOR promoters, and followed by chromatin hypoacetylation of and reduced transcription from KOR promoters in P19 stem cells. A novel pathway mediating the potential interplay between NO and opioid systems is discussed.
Keywords: nitric oxide, κ opioid receptor, c-Myc, gene regulation, transcription, P19
Opioids exert various pharmacological effects, including pain relief through their receptors, which are primarily detected in the brain and spinal cord (Slowe et al., 1999; Rosin et al., 2000). Three types of opioid receptors are present in animals, including the μ opioid receptor, δ opioid receptor, and κ opioid receptor (KOR); each receptor is encoded by a different gene (Goldstein and Naidu, 1989; Kieffer, 1995; Wei and Loh, 1996). The mouse KOR gene is constitutively expressed in postnatal day 19 (P19) embryonal carcinoma stem cells; the level of KOR expression changes as cells undergo differentiation (Chen et al., 1999; Bi et al., 2001). In addition to P19 carcinoma cells, various cancer cell lines also express KOR, including lung cancer (Maneckjee and Minna 1990; Kim et al., 1997), CNE2 human epithelial tumor (Diao et al., 2000), and human breast cancer (Hatzoglou et al., 1996).
The mouse KOR gene contains two promoters, promoter I (PI) and PII. PII is located in intron I of this gene. Its changing pattern of expression in differentiating P19 cultures provided a model to address the regulatory mechanisms controlling KOR gene expression. Recently, we demonstrated a negative pathway that controlled KOR gene transcription in both P19 cells and mouse embryos, which involved vitamin A hormones. In differentiating cultures treated for the long term, retinoic acid (RA), the active ingredient of vitamin A hormone, downregulated KOR transcription by inducing Ikaros that recruited histone deacetylases to an Ikaros-binding site located in the second promoter (overlapping with intron I) of this gene (Hu et al., 2001). Interestingly, we observed a rapid and dramatically suppressive effect of nitric oxide (NO) on KOR expression in this cell-culture model.
It has been reported that long-term administration of either morphine (Bhargava et al., 1998) or cocaine (Bhargava and Kumar, 1997) increased neuronal NO synthase activity in certain brain regions. Furthermore, opioid alkaloid activation resulted in the release of NO in neutrophils, monocytes, and endothelial cells (Magazine et al., 1996;Kowalski, 1998; Stefano et al., 2000). Despite the demonstration of potential cross talks between the opioid and the NO systems as shown in these pharmacological studies, it remains unclear how the NO system interacts with the opioid system at the molecular level.
NO is a well defined signaling molecule that is involved in pathophysiological processes such as inflammation, apoptosis, regulation of enzyme activity, and gene expression. The actions of NO in gene expression were reported to involve, primarily,S-nitrosylation of proteins with subsequent alteration of transcription factors binding to the target promoter site (Lee et al., 2001; Marshall and Stamler, 2001), as well as the activation of soluble guanylate cyclase that stimulated the cGMP–protein kinase G pathway (Morris, 1995). Cadet et al. (2001) reported recently that NO downregulated the expression of the μ opioid receptor gene and suggested that the suppression possibly involved the apoptotic pathway mediated by peroxynitrite.
In this report, we demonstrate the identification of a novel signaling pathway of NO in the opioid receptor system, mediated by the immediate suppression of c-myc expression. As a result, occupancy ofc-myc binding sites on both PI and PII of the endogenous KOR gene became reduced, chromatin histone on the two promoters became hypoacetylated, and KOR expression was rapidly suppressed in P19 stem cells.
MATERIALS AND METHODS
Plasmid construction. A series of reporter constructs with luciferase inserted into the cloning site of pGL3B (Promega, Madison, WI) were as described by Hu et al. (2001). All of the deletion mutants were constructed in pGL3B. Deletions of PI include Kd36 (−1319 to −743), Kd37 (−1319 to −994), Kd38 (−1044 to −743), Kd39 (−1019 to −883), and Kd40 (−903 to −743). Deletions of PII include Kd50 (−404 to −210), Kd53 (−209 to −15), Kd54 (−163 to −15), Kd56 (−73 to −15), and Kd57 (−43 to −15).
Cell culture and transient transfection. P19 cells were maintained in α-MEM (Invitrogen Corporation, Grand Island, NY) supplemented with 2.5% FBS and 7.5% calf serum in 5% CO2 at 37°C (Lu et al., 1997). COS-1 cells were grown in α-DMEM supplemented with 10% FBS. Transient transfection and reporter assays were as described previously (Lee et al., 1998). For cotransfection assays, the human c-myc expression vector (a gift from Dr. J. M. Bishop, University of California, San Francisco, CA) (Felsher et al., 2000) was used to express c-mycectopically in P19 and COS-1 cells.
Reverse transcription-PCR of endogenous KOR and c-Myc genes.RNA was isolated and analyzed by reverse transcription-PCR (RT-PCR) as described previously (Wei et al., 2000). Primers specific to total KOR mRNA are 5′-ATCAGGGCTGAACAGCTA-3′ and 5′-GCAAGGAGCATTCAATGAC-3′. Primers for c-myc mRNA are 5′-CCATATG-CCCCTCAACGTGAAC-3′ and 5′-GGGATCCTTATGCACCAGAGTTT-3′, which span a 1350 bp coding region.
Electrophoretic mobility shift assay. Two putative E boxes were found in the promoter fragments of both Kd40 and Kd57 constructs by computer alignment. P1-E and P2-E oligos correspond to PI (Kd40) and PII (Kd57), respectively. The electrophoretic mobility shift assay (EMSA) was conducted as described previously (von Knethen et al., 1999), with 10 μg of nuclear extract incubated in a 10 μl final reaction volume containing 2 ng of labeled DNA at 4°C for 30 min.
Chromatin immunoprecipitation assay. Control P19 cells or those treated with sodium nitroprusside (SNP) for 6 hr were cross-linked with formaldehyde as described previously (Hu et al., 2001). Cell lysates were subjected to immunoprecipitation overnight at 4°C using 2 μg of anti-c-myc (Santa Cruz Biotechnology, Santa Cruz, CA), anti-acetylated-histone H3 (Upstate Biotechnology, Lake Placid, NY), or preimmune rabbit sera (Pierce, Rockford, IL). After reversed cross-linking, DNA was precipitated and detected by PCR with pairs of primers specific to the KOR promoter regions containing an E box (i.e., 5′-GATGCACAGTAGCTTTCC-3′ and 5′-GCAAGGAAGCAAGTGGTA-3′ for PI and 5′-TCCTTCCTTGGGATG-3′ and 5′-CTGGAAAGCGAGAAGGTG-3′ for PII).
Southwestern blot analysis. Nuclear extracts and whole-cell lysates (Sommer et al., 1998) were isolated, separated by SDS-PAGE, and transferred to a polyvinylidene difluoride (PVDF) membrane. The membrane was immediately neutralized and hybridized with an α-32P-labeled probe for 5 hr at room temperature. The blot was extensively washed and exposed to a PhosphoImager screen (Amersham Biosciences, Piscataway, NJ). The same membrane was reprobed with anti-c-Myc (Park et al., 2001).
RESULTS
Suppression of endogenous KOR gene activity by NO in P19 cells
To explore factors that might have an immediate effect on KOR gene expression in the P19 cell differentiation model, various agents were used to treat P19 stem cells, followed by the examination of the endogenous KOR mRNA expression. It was interesting to observe a rapid and dramatically suppressive effect of NO on KOR expression in P19 stem cells. As shown in Figure1A, the steady-state KOR mRNA expression in P19 cells was dramatically suppressed after the addition of SNP, an NO donor, within 4 hr of treatment. The suppression continued for up to 24 hr (data not shown), suggesting a prolonged, suppressive effect of NO. Furthermore, the suppressive effect of SNP was dose dependent (Fig. 1B), supporting a specific effect of NO on the expression of KOR.
Fig. 1.

Suppression of endogenous KOR by NO. KOR mRNA isolated from SNP-treated P19 cells was analyzed by RT-PCR using primer pairs specific to KOR and actin. A, Different time points. B, Concentrations of SNP.
Suppression of KOR promoters by NO in P19 cells
Because the effect of SNP on KOR gene expression persisted over an extended period, we then determined whether this suppressive effect was mediated by the regulatory region of the KOR gene. We took advantage of a previously engineered KOR-luc reporter, which contained the regulatory region of both PI and PII of the mouse KOR gene, K45. As shown in Figure 2A, all of the NO donors tested, including SNP, 3-morpholinosydnonimine-1 (SIN-1), and S-nitrosoglutathione (GSNO), were equally effective in suppressing this reporter, suggesting that NO indeed was able to suppress KOR gene expression in P19 cells, and that the suppression was mediated by the regulatory region of the KOR gene. To substantiate the specificity of NO on KOR transcriptional regulation further, we used an NO scavenger, 2-phenyl-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (PTIO), to block the effect of SNP. As shown in Figure 2B, PTIO effectively blocked the suppressive effect of SNP; PTIO alone had no significant effect. To gain an insight into the cell-type specificity of SNP in relation to suppression of the KOR gene, the responses of the same reporter K45 to SNP in P19 cells and COS-1 cells were compared (Fig. 2C). It appeared that SNP had no effect on this reporter in the COS-1 background, suggesting the involvement of specific cellular factors of P19 in mediating the suppressive effect of NO on KOR transcription.
Fig. 2.
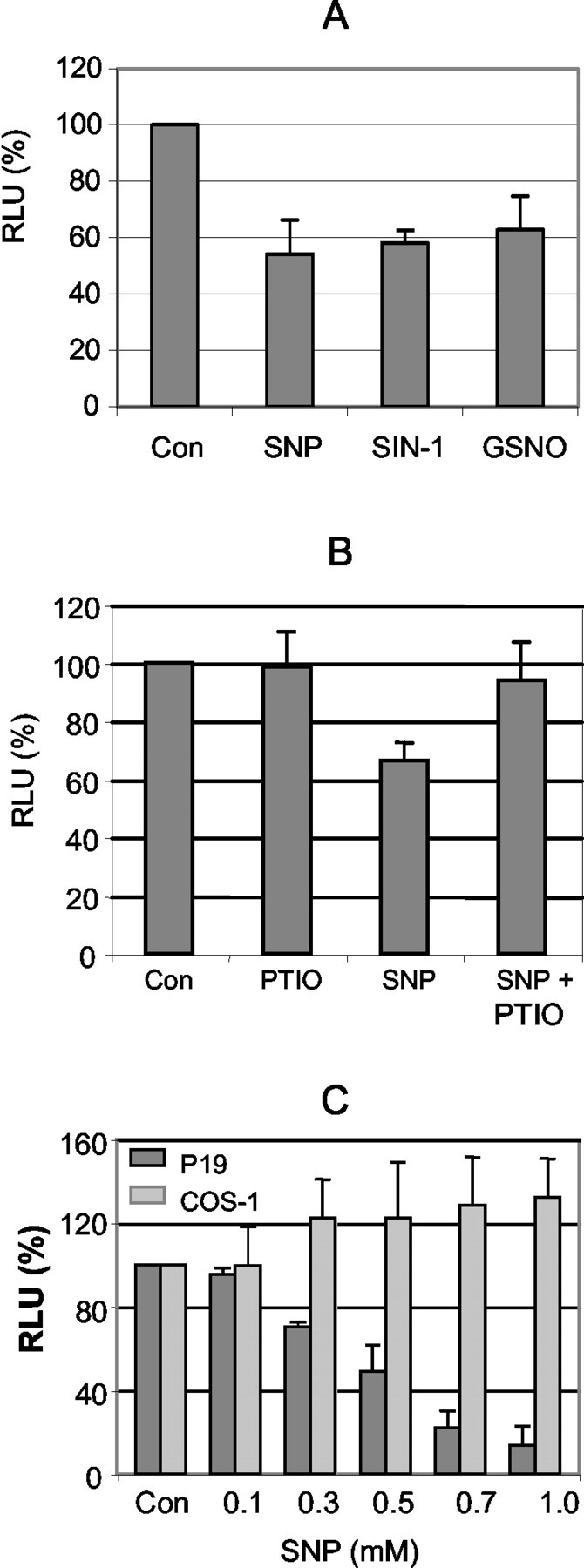
Effect of NO on the KOR promoter. P19 cells were transiently transfected with K45 and lacZ internal control. After 24 hr of transfection, cells were treated with SNP, SIN-1, and GSNO (A, each 0.5 mm) and with 50 μm PTIO (B) in the presence or absence of SNP for 6 hr at 37°C. The relative luciferase units (RLU) were shown as the averages ± SD of three independent experiments performed in triplicate.C, P19 and COS-1 cells transfected with K45 were exposed to various concentrations of SNP for 6 hr. The relative luciferase units are the averages ± SD of three experiments.Con, Controls.
Suppression of KOR expression by NO through the E-box elements in PI and PII
The suppressive effect of SNP on both the endogenous KOR mRNA (Fig. 1) and the KOR reporter (Fig. 2) suggested a physiological relevance of NO on transcriptional regulation of the KOR gene. To determine the genetic elements that mediated this suppressive effect, transient transfection assays were conducted using serial deletion mutants of the K45 reporter. Because the KOR gene could use two promoters, PI and PII, we first determined which promoter(s) mediated the effect of SNP. It was interesting that both promoters were suppressed by SNP; therefore, we used two sets of deletion mutants to determine the responsible DNA elements. Figure3A shows the results using various deletions of PI (K19). Reporters K19, Kd36, Kd38, and Kd40 were all suppressed by SNP, whereas Kd37 and Kd39 were not affected, suggesting that the DNA fragment (−903 to −743) used in the smallest SNP-responsive construct, Kd40, contained the responding sequence. Figure 3B shows the results of deletion constructs from PII (K18). It appeared that K18, Kd53, Kd54, Kd56, and Kd57 were also suppressed by SNP, but not Kd50, which was deleted in aPstI fragment adjacent to the initiation site. The analysis of possible common sequences present in all of the SNP-responsive constructs revealed a putative c-myc binding site, an E box (CACGTG), in both promoters. The putative c-myc binding sites were located at −882 to −877 (CACTTG) and at −39 to −34 (CAAGTG) of PI and PII, respectively. These results suggest that NO might suppress KOR gene transcription through a putativec-myc binding site.
Fig. 3.

NO suppresses KOR PI and PII through E boxes. P19 cells were transiently transfected with various deletion constructs of PI (A) or PII (B) as indicated. The cells were untreated (Con) or treated with 0.5 or 1 mm SNP for 6 hr. The results presented at the right were shown as the averages ± SD of two independent experiments performed in triplicate. The E boxes present in the smallest genomic fragments Kd40 and Kd57 are indicated. Luc, Luciferase;RLU, relative luciferase units.
Suppression of protein–DNA complex formation on the E-box elements by NO
To first determine whether the E boxes were indeed the functional binding sites for nuclear factors of P19, presumably thec-myc transcription factor, we used EMSA with α-32P-labeled DNA fragments spanning the putative c-myc sites as the probe. One major band was detected in EMSA using nuclear extracts prepared from untreated P19 cells, for both PI (P1-E) and PII (P2-E). Furthermore, this band could be efficiently competed out by cold DNA fragments for both P1-E and P2-E (Fig. 4A). Interestingly, as the SNP concentration increased, the intensity of the retarded band decreased (lanes 4–6, 9–11), suggesting decreased protein–DNA interaction on these E boxes as a result of SNP treatment. A stronger reduction in the binding intensity of P2-E than that of P1-E by SNP suggests that P2-E responds to SNP more effectively. Furthermore, the specifically shifted band, which could be competed out by cold DNA fragments (Fig. 4B,lane 2), disappeared only in the presence of thec-myc antibody (lane 3), suggesting that protein complexes formed on E boxes indeed contained c-myc, which was specifically blocked by this antibody. The formation of this complex was not affected by the preimmune serum (lane 4). In addition to EMSA, Southwestern blotting showed binding of multiple proteins to the E box of PII for both P19 and COS-1 nuclear extracts; one major band was competed specifically with the cold probe (Fig. 4C), which was confirmed as thec-myc protein by probing the same membrane with ac-myc antibody (Fig. 4D). The identity ofc-myc binding to these E boxes was also confirmed on the endogenous KOR gene promoter regions using the chromatin immunoprecipitation (ChIP) assay (see Fig. 6).
Fig. 4.

c-myc binds to the E boxes of KOR promoters. A, EMSA. P19 cells were incubated alone (lanes 1, 4, and 9) or with 0.5 SNP for 6 hr (lanes 5 and 10) or 1 mmSNP for 6 hr (lanes 6 and 11). DTT (1 or 5 mm) was added to P19 cells for 30 min, followed by treatment with 1 mm SNP (lanes 7 and8) for 6 hr at 37°C. Nuclear extracts (10 μg) were incubated with α-32P-labeled probes of P1-E (lanes 1–8) in the presence of excess cold probes (lanes 2 and 3) or P2-E (lanes 9–11) for 30 min at 4°C. B, Antibody interference assay. Nuclear extracts were incubated with 100× excess of cold probe (lane 2), c-myc antibody (lane 3), or preimmune serum (lane 4) for 30 min at room temperature and then with α-32P-labeled P2-E for 30 min at 4°C. C, Southwestern (SW) and Western (WB) blots. Nuclear extracts (75 μg) of P19 cells and COS-1 cells were separated by 12% SDS-PAGE, transferred to PVDF membranes, and hybridized with α-32P-labeled P2-E in the absence (lanes 1and 2) or presence (lanes 3 and4) of 100× excess of cold probe. The same membranes were reprobed with c-myc antibody as shown on the Western blot. comp, Competition.Arrowheads indicate c-myc complexes.
Fig. 6.

NO downregulates in vivo binding ofc-myc and renders histone hypoacetylation on KOR promoters. +, SNP treatment for 6 hr; −, untreated cultures. ChIP assays were performed as described in Materials and Methods. Left, Schematic presentation of E boxes in PI and PII and the location of respective primer sequences for PCR analysis. Thenumbers below the promoters indicate the position of the PCR primers relative to the ATG codon. Right, Comparison of in vivo binding of c-myc and acetylated histone H3 (AcH3) between control and SNP-treated cultures. The relative intensities of the signals of SNP-treated samples to controls are presented below as the averages of two experiments. The input was the PCR product of total chromatin. A rabbit preimmune serum was used as a nonspecific control. Con, Control.
The reduced binding of the c-myc complex in SNP-treated cultures suggested two possibilities. NO might induce post-translational modification of c-myc, such asS-nitrosylation, widely known as the major modification affecting protein–DNA interactions (Marshall and Stamler, 2001). Alternatively, NO might downregulate c-myc expression in P19 cells. To test these two possibilities, DTT, a compound that inhibitsS-nitrosylation, was used to treat the nuclear extract of P19 in EMSA, as shown in lanes 7 and 8 in Figure4A. Interestingly, DTT could not recover protein binding to the c-myc binding site, ruling outS-nitrosylation as a cause for this reduced c-mycbinding to DNA.
To examine the alternative possibility that c-myc expression was suppressed by SNP, c-myc mRNA and protein levels were examined with RT-PCR and Western blotting, respectively. It appeared that SNP treatment for 2 hr began to suppress the steady-state level ofc-myc mRNA in P19 cells; the suppressive effect was even more dramatic at 4–8 hr (Fig.5A). Furthermore, suppression of c-myc expression by SNP was also concentration dependent (Fig. 5B). The level of c-myc protein was examined by Western blot analyses as shown in Figure 5C. SNP also suppressed c-myc protein levels in a dose-dependent manner (lanes 1–4). The suppression ofc-myc by SNP was recovered by treatment with an NO scavenger, PTIO (Fig. 5D), consistent with the results shown in Figure 2B. To confirm the effects ofc-myc on KOR regulation, a c-myc expression vector was used to overexpress c-myc. As shown in Figure5E, c-myc expression indeed induced KOR reporter activity approximately ninefold in both P19 and COS-1 cells, and SNP blocked the induction in P19 cells as predicted (Fig.5E). Consistent with the result showing that NO failed to suppress KOR reporter expression in COS-1 cells (Fig. 2C), SNP has no effect either on the expression of c-myc protein in COS-1 cells (Fig. 5C, lanes 5–7) or on the elevation of KOR reporter activity by c-myc expression (Fig. 5E), supporting the cell-type specificity of NO-mediated suppression of the KOR gene in P19 cells, which was attributable to the suppression of c-myc mRNA and protein expression.
Fig. 5.

NO suppresses c-mycexpression. A, B, c-myc mRNA was reverse-transcribed and amplified by PCR. The amounts ofc-myc mRNA were analyzed against the time (A) or the dose (B) of SNP added. C, Western blot. Whole-cell lysate of P19 cells (50 μg; lanes 1–4) or COS-1 cells (100 μg;lanes 5–7) was immunoblotted withc-myc antibody. D, Western blot analysis, with anti-c-myc, of the whole-cell lysate (50 μg) of P19 controls (lane 1), cells treated with PTIO (lane 2), or cells treated with 1 mm SNP in the absence (lane 3) or presence (lane 4) of 50 μm PTIO for 6 hr. E, Induction of KOR reporter by c-myc expression in P19 and COS-1 cells. Cells were cotransfected with K45, a c-myc expression vector, and an internal lacZ control. Cells were then treated with vehicle or 0.5 mm SNP for 6 hr. The relative luciferase units (RLU) of two independent experiments are presented.
Suppression of c-myc binding and histone acetylation on endogenous KOR promoters by NO
It was shown that c-myc was able to recruit histone acetyltransferases (HATs), enzymes involved in acetylating histone proteins and inducing an open chromatin conformation for gene activation (McMahon et al., 2000). To examine whether the histone acetylation status of KOR promoters and the binding of endogenousc-myc to these KOR regulatory sequences in P19 cells were altered by NO treatment, ChIP assays were performed to detect the acetylation status of PI and PII and the binding of c-myc to the endogenous KOR promoters. As shown in Figure6, SNP decreased c-myc binding to the endogenous E-box sequences of the KOR gene for both PI and PII (lanes 5 and 6). Acetylation of these DNA regions of the endogenous KOR gene was also consistently reduced (lanes 7 and 8). Furthermore, PII appears to be more sensitive to the SNP-triggered reduction of c-mycbinding, consistent with the results of the gel-shift data shown in Figure 4. This provided the evidence that NO indeed reducedc-myc occupancy on the regulatory regions of the endogenous KOR gene, which resulted in diminished histone acetylation on KOR promoter regions. Therefore, it was concluded that NO rapidly suppressed KOR gene transcription in P19 cells by a novel signaling pathway that involved immediate repression of c-mycexpression, formation of c-myc–DNA complexes, and ultimately hypoacetylation of chromatin on the endogenous KOR promoter regions.
DISCUSSION
We have shown previously that the mouse KOR gene was constitutively expressed in P19 stem cells and exhibited a unique pattern of expression in developing embryos, primarily in the CNS (Chen et al., 1999; Hu et al., 1999). Using the P19 culture model, we tried to explore potential regulatory pathways underlying the specific expression of the KOR gene. Vitamin A was found to be a negative factor for KOR expression in differentiating P19 cells, which was mediated by the induction of Ikaros that recruited HATs to silence the KOR gene in differentiating cells (Bi et al., 2001; Hu et al., 2001). This study showed that NO could rapidly and dramatically suppress KOR expression in stem cells by reducing c-mycexpression, revealing a novel pathway of NO signaling and an interesting interplay between the NO and KOR systems at the gene level in stem-cell populations.
All of the NO donors tested herein showed a similar suppressive effect on the KOR gene, which was reversed by an NO scavenger. NO was shown to affect the expression of a number of genes, such as intercellular adhesion molecule-1, gonadotropin-releasing hormone, MAP kinase phosphatase-3, and inducible NO synthase (Toyoshima et al., 1999;Belsham and Mellon, 2000; Rossig et al., 2000; Lee et al., 2001). However, in all of these studies, the action of NO was shown primarily to involve S-nitrosylation of Oct-1 (Lee et al., 2001), nuclear factor κB (Marshall and Stamler, 2001), and c-Myb (Brendeford et al., 1998) transcription factors. Our finding that NO was able to repress the expression of c-myc transcription would be the first example of a novel signaling pathway of NO that regulated the expression level of a transcription factor. Because this effect is cell-type-specific, it would be interesting to identify the specific cellular factors or transcription machinery that are responsible for this rapid suppression of the c-myc gene by NO in stem cells. In addition to NO, we have observed thatc-myc gene expression was suppressed by RA in P19 cells (data not shown). This finding is in agreement with the suppressive effect of RA on KOR expression (Bi et al., 2001).
A transient transfection assay with various deletions of PI and PII demonstrated that two E boxes [−882 to −877 (PI) and −39 to −34 (PII)] functioned as the NO-responsive element. As reviewed previously (Dang, 1999; Amati et al., 2001), the E box is a binding site ofc-myc transcription factor. The action of c-mycinvolved its interaction with other transcription factors as well as enzymes for modifying histone proteins, the HATs (Xu et al., 2001). In other words, c-myc formed complexes with many other transcription factors, such as Max, to act on gene promoters (Blackwood and Eisenman, 1991), and recruited the transformation–transactivation domain-associated protein coactivator complex (McMahon et al., 1998; Bouchard et al., 2001), which contained the HAT component hGCN5 (McMahon et al., 2000). The recruitment of the HAT complex ultimately leads to histone acetylation of histone H3 and H4 of the chromatin, consequently activating gene transcription (Amati et al., 2001; Xu et al., 2001). Our Southwestern blot results indeed suggested that protein complex binding to the E box of the KOR promoter contained proteins other than the confirmed c-myc. It would be interesting to identify specific protein factors that are associated with c-myc in protein complexes formed on the E box of KOR promoters. The results of the ChIP assay (Fig. 6) revealed hypoacetylation of histone proteins on the chromatin of KOR promoter regions, confirming that both the binding of c-myc complexes to E boxes and the acetylation of histone on KOR promoters were reduced in P19 stem cells after the cells were exposed to NO. This would provide a mechanistic explanation for the action of NO that resulted in a more prolonged effect on the KOR gene attributable to changes in its chromatin modification in specific cell types.
It has been demonstrated that many transcription factors are modulated by NO-induced S-nitrosylation of the proteins (Marshall et al., 2000; Marshall and Stamler, 2001). However, in the regulation of the KOR gene, DTT, a compound inhibiting S-nitrosylation (Brendeford et al., 1998), had no effect on either the DNA-binding ability of nuclear extracts from the cells treated with SNP or the reporter activity in the transient transfection assay. This result allowed us to rule out S-nitrosylation as the underlying mechanism. Alternatively, the transcription factors Oct-1 and CCAAT/enhancer-binding protein-β have been known to be involved in the NO/cGMP-mediated repression of gonadotropin-releasing hormone gene expression (Belsham and Mellon, 2000). However, the potent cGMP analog 8-bromoadenosine 3′,5′-cGMP had no effects either in the transient transfection assay using KOR promoter constructs or onc-myc protein expression in cells treated with SNP (data not shown). This result precluded the involvement of the cGMP–protein kinase G pathway in the regulation of the KOR gene by NO. Instead, NO reduced the expression of c-myc in P19 (Fig. 5). Furthermore, a detailed comparison of the kinetics of the suppression of c-myc and KOR in P19 cells treated with SNP also supported the sequential actions of NO (0 hr) followed byc-myc (2 hr) on the expression of the KOR gene (4 hr). More convincingly, SNP did not affect c-myc expression in COS-1 cells, in which SNP had no effect on KOR expression. Therefore, this novel signaling pathway of NO is cell-type-specific and involves certain cellular factors that affect the expression ofc-myc; these factors remain to be identified.
The action of NO can occur at the processes of cellular apoptosis and/or differentiation (Brune et al., 1998; Chung et al., 2001).Peunova et al. (2001) suggested that NO might be an essential negative regulator of neuron precursor proliferation during brain development in vertebrates. Our finding that NO suppressed KOR expression in P19 stem cells is consistent with this idea. However, it remains to be determined whether the effect of NO on KOR expression is associated with P19 apoptosis or differentiation, because this effect occurred within a very short period in stem cells. The physiological implications of a rapid suppression of KOR by NO in stem cells remain to be determined experimentally.
Footnotes
This work was supported by National Institutes of Health Grants DA11190, DK54733, and DK60521 (L.N.W.) and Grants DA11806 and DA00564 (H.H.L.). We thank Dr. J. Bi for help with RT-PCR and Southern blotting.
Correspondence should be addressed to Dr. Li-Na Wei, Department of Pharmacology, University of Minnesota Medical School, 6-120 Jackson Hall, 321 Church Street Southeast, Minneapolis, MN 55455. E-mail:weixx009@tc.umn.edu.
REFERENCES
- 1.Amati B, Frank SR, Donjerkovic D, Taubert S. Function of the c-myc oncoprotein in chromatin remodeling and transcription. Biochim Biophys Acta. 2001;1471:M135–M145. doi: 10.1016/s0304-419x(01)00020-8. [DOI] [PubMed] [Google Scholar]
- 2.Belsham DD, Mellon PL. Transcription factors Oct-1 and C/EBPβ (CCAAT/enhancer-binding protein-β) are involved in the glutamate/nitric oxide/cyclic-guanosine 5′-monophosphate-mediated repression of gonadotropin-releasing hormone gene expression. Mol Endocrinol. 2000;14:212–228. doi: 10.1210/mend.14.2.0418. [DOI] [PubMed] [Google Scholar]
- 3.Bhargava HN, Kumar S. Effect of multiple injections of U-50,488H, a κ-opioid receptor agonist, on the activity of nitric oxide synthase in brain regions and spinal cord of mice. Gen Pharmacol. 1997;29:397–399. doi: 10.1016/s0306-3623(96)00480-6. [DOI] [PubMed] [Google Scholar]
- 4.Bhargava HN, Kumar S, Barjavel MJ. Kinetic properties of nitric oxide synthase in cerebral cortex and cerebellum of morphine tolerant mice. Pharmacology. 1998;56:252–256. doi: 10.1159/000028205. [DOI] [PubMed] [Google Scholar]
- 5.Bi J, Hu X, Loh HH, Wei L-N. Regulation of mouse κ opioid receptor gene expression by retinoids. J Neurosci. 2001;21:1590–1599. doi: 10.1523/JNEUROSCI.21-05-01590.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blackwood EM, Eisenman RN. Max: a helix-loop-helix zipper protein that forms a sequence-specific DNA-binding complex with Myc. Science. 1991;251:1211–1217. doi: 10.1126/science.2006410. [DOI] [PubMed] [Google Scholar]
- 7.Bouchard C, Dittrich O, Kiermaier A, Dohmann K, Menkel A, Eilers M, Luscher B. Regulation of cyclin D2 gene expression by the Myc/Max/Mad network: myc-dependent TRRAP recruitment and histone acetylation at the cyclin D2 promoter. Genes Dev. 2001;15:2042–2047. doi: 10.1101/gad.907901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brendeford EM, Andersson KB, Gabrielsen OS. Nitric oxide (NO) disrupts specific DNA binding of the transcription factor c-Myb in vitro. FEBS Lett. 1998;425:52–56. doi: 10.1016/s0014-5793(98)00196-3. [DOI] [PubMed] [Google Scholar]
- 9.Brune B, von Knethen A, Sandau KB. Nitric oxide and its role in apoptosis. Eur J Pharmacol. 1998;351:261–272. doi: 10.1016/s0014-2999(98)00274-x. [DOI] [PubMed] [Google Scholar]
- 10.Cadet P, Mantione K, Bilfinger TV, Stefano GB. Real-time RT-PCR measurement of the modulation of μ opiate receptor expression by nitric oxide in human mononuclear cells. Med Sci Monit. 2001;7:1123–1128. [PubMed] [Google Scholar]
- 11.Chen HC, Wei L-N, Loh HH. Expression of μ-, κ-, and δ-opioid receptors in P19 mouse embryonal carcinoma cells. Neuroscience. 1999;92:1143–1155. doi: 10.1016/s0306-4522(99)00030-5. [DOI] [PubMed] [Google Scholar]
- 12.Chung HT, Pae HO, Choi BM, Billiar TR, Kim YM. Nitric oxide as a bioregulator of apoptosis. Biochem Biophys Res Commun. 2001;282:1075–1079. doi: 10.1006/bbrc.2001.4670. [DOI] [PubMed] [Google Scholar]
- 13.Dang CV. c-myc target genes involved in cell growth, apoptosis, and metabolism. Mol Cell Biol. 1999;19:1–11. doi: 10.1128/mcb.19.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diao CT, Li L, Lau SY, Wong TM, Wong NS. κ-Opioid receptor potentiates apoptosis via a phospholipase C pathway in the CNE2 human epithelial tumor cell line. Biochim Biophys Acta. 2000;1499:49–62. doi: 10.1016/s0167-4889(00)00107-5. [DOI] [PubMed] [Google Scholar]
- 15.Felsher DW, Zetterberg A, Zhu J, Tisty T, Bishop JM. Overexpression of MYC causes p53-dependent G2 arrest of normal fibroblasts. Proc Natl Acad Sci USA. 2000;97:10544–10548. doi: 10.1073/pnas.190327097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Goldstein A, Naidu A. Multiple opioid receptors: ligand selectivity profiles and binding site signatures. Mol Pharmacol. 1989;36:265–272. [PubMed] [Google Scholar]
- 17.Hatzoglou A, Bakogeorgou E, Castanas E. The antiproliferative effect of opioid receptor agonists on the T47D human breast cancer cell line, is partially mediated through opioid receptors. Eur J Pharmacol. 1996;296:199–207. doi: 10.1016/0014-2999(95)00703-2. [DOI] [PubMed] [Google Scholar]
- 18.Hu X, Cao S, Loh HH, Wei L-N. Promoter activity of mouse κ opioid receptor gene in transgenic mouse. Brain Res Mol Brain Res. 1999;69:35–43. doi: 10.1016/s0169-328x(99)00077-7. [DOI] [PubMed] [Google Scholar]
- 19.Hu X, Bi J, Loh HH, Wei L-N. An intronic Ikaros-binding element mediates retinoic acid suppression of the κ opioid receptor gene, accompanied by histone deacetylation on the promoters. J Biol Chem. 2001;276:4597–4603. doi: 10.1074/jbc.M005477200. [DOI] [PubMed] [Google Scholar]
- 20.Kieffer BL. Recent advances in molecular recognition and signal transduction of active peptides: receptors for opioid peptides. Cell Mol Neurobiol. 1995;15:615–635. doi: 10.1007/BF02071128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim JK, Summer SN, Schrier RW. Effect of κ opioid agonist RU 51599 on osmotic and non-osmotic stimulated arginine vasopressin release and gene regulation in small cell lung carcinoma cells. Neuropeptides. 1997;31:423–429. doi: 10.1016/s0143-4179(97)90035-4. [DOI] [PubMed] [Google Scholar]
- 22.Kowalski J. Augmenting effect of opioids on nitrite production by stimulated murine macrophages. Neuropeptides. 1998;32:287–291. doi: 10.1016/s0143-4179(98)90050-6. [DOI] [PubMed] [Google Scholar]
- 23.Lee BS, Kim YM, Kang HS, Kim HM, Pyun KH, Choi I. Octamer binding protein-1 is involved in inhibition of inducible nitric oxide synthase expression by exogenous nitric oxide in murine liver cells. J Biochem (Tokyo) 2001;129:77–86. doi: 10.1093/oxfordjournals.jbchem.a002839. [DOI] [PubMed] [Google Scholar]
- 24.Lee C-H, Chinpaisal C, Wei L-N. Cloning and characterization of mouse RIP140, a corepressor for nuclear orphan receptor TR2. Mol Cell Biol. 1998;18:6745–6755. doi: 10.1128/mcb.18.11.6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lu S, Loh HH, Wei L-N. Studies of dual promoters of mouse κ-opioid receptor gene. Mol Pharmacol. 1997;52:415–420. doi: 10.1124/mol.52.3.415. [DOI] [PubMed] [Google Scholar]
- 26.Magazine HI, Liu Y, Bilfinger TV, Fricchione GL, Stefano GB. Morphine-induced conformational changes in human monocytes, granulocytes, and endothelial cells and in invertebrate immunocytes and microglia are mediated by nitric oxide. J Immunol. 1996;156:4845–4850. [PubMed] [Google Scholar]
- 27.Maneckjee R, Minna JD. Opioid and nicotine receptors affect growth regulation of human lung cancer cell lines. Proc Natl Acad Sci USA. 1990;87:3294–3298. doi: 10.1073/pnas.87.9.3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marshall HE, Stamler JS. Inhibition of NF-κB by S-nitrosylation. Biochemistry. 2001;40:1688–1693. doi: 10.1021/bi002239y. [DOI] [PubMed] [Google Scholar]
- 29.Marshall HE, Merchant K, Stamler JS. Nitrosation and oxidation in the regulation of gene expression. FASEB J. 2000;14:1889–1900. doi: 10.1096/fj.00.011rev. [DOI] [PubMed] [Google Scholar]
- 30.McMahon SB, Van Buskirk HA, Dugan KA, Copeland TD, Cole MD. The novel ATM-related protein TRRAP is an essential cofactor for the c-myc and E2F oncoproteins. Cell. 1998;94:363–374. doi: 10.1016/s0092-8674(00)81479-8. [DOI] [PubMed] [Google Scholar]
- 31.McMahon SB, Wood MA, Cole MD. The essential cofactor TRRAP recruits the histone acetyltransferase hGCN5 to c-myc. Mol Cell Biol. 2000;20:556–562. doi: 10.1128/mcb.20.2.556-562.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Morris BJ. Stimulation of immediate early gene expression in striatal neurons by nitric oxide. J Biol Chem. 1995;270:24740–24744. [PubMed] [Google Scholar]
- 33.Park SW, Choi K, Kim IC, Lee HH, Hooper NM, Park HS. Endogenous glycosylphosphatidylinositol-specific phospholipase C releases renal dipeptidase from kidney proximal tubules in vitro. Biochem J. 2001;353:339–344. doi: 10.1042/0264-6021:3530339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peunova N, Scheinker V, Cline H, Enikolopov G. Nitric oxide is an essential negative regulator of cell proliferation in Xenopus brain. J Neurosci. 2001;21:8809–8818. doi: 10.1523/JNEUROSCI.21-22-08809.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosin A, van der Ploeg I, Georgieva J. Basal and cocaine-induced opioid receptor gene expression in the rat CNS analyzed by competitive reverse transcription PCR. Brain Res. 2000;872:102–109. doi: 10.1016/s0006-8993(00)02481-1. [DOI] [PubMed] [Google Scholar]
- 36.Rossig L, Haendeler J, Hermann C, Malchow P, Urbich C, Zeiher AM, Dimmeler S. Nitric oxide down-regulates MKP-3 mRNA levels: involvement in endothelial cell protection from apoptosis. J Biol Chem. 2000;275:25502–25507. doi: 10.1074/jbc.M002283200. [DOI] [PubMed] [Google Scholar]
- 37.Slowe SJ, Simonin F, Kieffer B, Kitchen I. Quantitative autoradiography of μ-, δ-, and κ1-opioid receptors in κ-opioid receptor knockout mice. Brain Res. 1999;818:335–345. doi: 10.1016/s0006-8993(98)01201-3. [DOI] [PubMed] [Google Scholar]
- 38.Sommer A, Bousset K, Kremmer E, Austen M, Luscher B. Identification and characterization of specific DNA-binding complexes containing members of the Myc/Max/Mad network of transcriptional regulators. J Biol Chem. 1998;273:6632–6642. doi: 10.1074/jbc.273.12.6632. [DOI] [PubMed] [Google Scholar]
- 39.Stefano GB, Goumon Y, Casares F, Cadet P, Fricchione GL, Rialas C, Peter D, Sonetti D, Guarna M, Welters ID, Bianchi E. Endogenous morphine. Trends Neurosci. 2000;9:436–442. doi: 10.1016/s0166-2236(00)01611-8. [DOI] [PubMed] [Google Scholar]
- 40.Toyoshima T, Kamijo R, Takizawa K, Sumitani K, Hatori M, Nagumo M. Nitric oxide up-regulates the expression of intercellular adhesion molecule-1 on cancer cells. Biochem Biophys Res Commun. 1999;257:395–399. doi: 10.1006/bbrc.1999.0476. [DOI] [PubMed] [Google Scholar]
- 41.von Knethen A, Callsen D, Brune B. NF-κB and AP-1 activation by nitric oxide attenuated apoptotic cell death in RAW 264.7 macrophages. Mol Biol Cell. 1999;10:361–372. doi: 10.1091/mbc.10.2.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wei L-N, Loh HH. Molecular biology of opioid receptors and associate proteins. In: Blum K, Noble EP, editors. Handbook of psychiatric genetics. CRC; New York: 1996. pp. 77–88. [Google Scholar]
- 43.Wei L-N, Hu X, Bi J, Loh HH. Post-transcriptional regulation of mouse κ-opioid receptor expression. Mol Pharmacol. 2000;57:401–408. [PubMed] [Google Scholar]
- 44.Xu D, Popov N, Hou M, Wang Q, Bjorkholm M, Gruber A, Menkel AR, Henriksson M. Switch from Myc/Max to Mad1/Max binding and decrease in histone acetylation at the telomerase reverse transcriptase promoter during differentiation of HL60 cells. Proc Natl Acad Sci USA. 2001;98:3826–3831. doi: 10.1073/pnas.071043198. [DOI] [PMC free article] [PubMed] [Google Scholar]