

Abstract
Recent studies have revealed that activation of extracellular signal-regulated kinase (ERK) may contribute to apoptosis, a cell death process involved in oxidative stress. We examined phosphorylation of ERK1/2 and oxidative stress after transient focal cerebral ischemia (FCI) using transgenic (Tg) mice that overexpress copper/zinc superoxide dismutase (SOD1). The mice were subjected to 60 min of middle cerebral artery (MCA) occlusion by intraluminal suture blockade followed by 1, 4, and 24 hr of reperfusion. Immunohistochemistry and Western blot analysis showed that phospho-ERK1 was markedly increased in the cortex within the MCA territory at 1 hr of reperfusion (p < 0.01), followed by a decrease at 24 hr in wild-type mice. Double staining with phospho-ERK1/2 and neuron-specific nuclear protein showed that phospho-ERK1/2 was primarily expressed in neurons. In SOD1 Tg mice, phospho-ERK1/2 was prominently reduced compared with nonischemic controls, shown by immunohistochemistry. Western blot analysis confirmed a significant decrease in phospho-ERK1/2 1 hr after FCI in the ischemic cortex (p < 0.005). Apoptotic-related DNA fragmentation was reduced in the ischemic cortex of SOD1 Tg mice compared with wild-type mice using a cell death assay. These results suggest that phosphorylation of ERK1/2 may be involved in apoptosis or cell death after transient FCI and that SOD1 may attenuate apoptotic cell death mediated by the mitogen-activated protein kinase/ERK pathway.
Keywords: cerebral ischemia, extracellular signal-regulated kinase, mitogen-activated protein kinase, copper/zinc-superoxide dismutase, apoptosis, reactive oxygen species
Members of the mitogen-activated protein kinase (MAPK) family, which include extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 MAPK, are known to play a critical role in the regulation of cell growth and differentiation (Boulton et al., 1991;Segal and Greenberg, 1996) and the control of cellular responses to cytokines and stress (Rouse et al., 1994; Raingeaud et al., 1995). They are activated by phosphorylation on threonine and tyrosine residues (Davis, 1993; Seger and Krebs, 1995). After activation by phosphorylation, MAPK phosphorylates other intracellular enzymes and transcription factors. ERK has two isoforms (ERK1/2), which are constitutively expressed in the normal brain (Boulton et al., 1991), and they are activated by MAPK/ERK kinase 1/2 (MEK1/2). Upstream of the MEK/ERK pathway, Raf-1 is the main effector recruited by Ras to activate MEK1/2 (Avruch et al., 1994). After activation, ERK1/2 phosphorylates several downstream elements that relate to transcription, such as Elk-1 (Sgambato et al., 1998a) or 90 kDa ribosomal S6 kinases (Frodin and Gammeltoft, 1999). ERK1/2 can be activated by various stimuli, such as growth factors (Boulton et al., 1991), oxidative stress (Aikawa et al., 1997), intracellular calcium influx (Rosen et al., 1994), or stimulation of glutamate receptors (Fiore et al., 1993; Kurino et al., 1995; Sgambato et al., 1998b). However, whether activation of ERK is protective or detrimental to neurons is controversial. Although an in vitro study suggests neuroprotective effects of ERK activation after ischemia (Hetman et al., 1999), recent in vivo studies reveal that inhibition of ERK could reduce infarct volume after focal cerebral ischemia (FCI), suggesting a deleterious effect of ERK activation (Alessandrini et al., 1999; Namura et al., 2001).
Apoptosis after FCI has been reported in a number of studies. The antioxidant enzyme is one of the major mechanisms by which cells counteract the deleterious effects of reactive oxygen species (ROS), and recent studies have revealed a protective effect of the antioxidant enzyme against apoptosis after cerebral ischemia and reperfusion. We have shown that superoxide dismutase (SOD) plays a protective role against FCI (Kinouchi et al., 1991; Chan, 1996; Kondo et al., 1997;Murakami et al., 1998; Fujimura et al., 1999a, 2000) as well as global ischemia (Murakami et al., 1997; Chan et al., 1998). We have also demonstrated that overexpression of cytosolic copper/zinc SOD (SOD1) in transgenic (Tg) mice could attenuate cytochrome c release from mitochondria (Fujimura et al., 2000) and subsequent DNA fragmentation after FCI (Kondo et al., 1997; Fujimura et al., 2000). These reports show that SOD1 is involved in cellular damage after ischemia and reperfusion. The MEK/ERK pathway is known to be activated by ROS and reactive nitrogen species (Baas and Berk, 1995; Guyton et al., 1996; Yun et al., 1998). However, whether an antioxidant like SOD1 can affect the MEK/ERK pathway after FCI has not yet been demonstrated. In the present study, we address this issue by studying MEK and ERK expression before and after transient FCI, using both wild-type and SOD1 Tg mice.
MATERIALS AND METHODS
SOD1 Tg mice. Heterozygous SOD1 Tg mice of the SOD1 TGHS/SF-218-3 strain with a CD-1 background, carrying human SOD1 genes with a threefold increase in SOD1, were derived from the founder stock described previously (Epstein et al., 1987). They were further bred with CD-1 wild-type mice to generate heterozygous mice. The SOD1 Tg mice were identified by quantitative demonstration of SOD1 using nondenaturing gel electrophoresis, followed by nitroblue tetrazolium staining (Epstein et al., 1987). There were no differences in the phenotypes, including the anatomy of the circle of Willis, between the SOD1 Tg mice and their wild-type littermates (Yang et al., 1994). There was no difference in the regional cerebral blood flow before and after FCI between the SOD1 Tg and wild-type mice (Chan et al., 1993).
Focal cerebral ischemia. Adult male SOD1 Tg mice and non-Tg littermates (3-month-old males; 35–40 gm) were subjected to transient FCI by intraluminal middle cerebral artery (MCA) blockade with a nylon suture as described previously (Yang et al., 1994). The mice were anesthetized with 1.5% isoflurane in 30% oxygen and 70% nitrous oxide using a face mask. The rectal temperature was controlled at 37°C with a homeothermic blanket. Cannulation of a femoral artery allowed the monitoring of blood pressure and arterial blood gases, with samples for analysis being taken immediately after cannulation, 10 min after occlusion, and 10 min after reperfusion. Blood gas was analyzed with a pH/Blood Gas Analyzer (Chiron Diagnostics, Ltd., Essex, UK). After a midline skin incision, the left external carotid artery was exposed, and its branches were electrocoagulated. An 11 mm 5-0 surgical monofilament nylon suture, blunted at the end, was introduced into the left internal carotid artery through the external carotid artery stump. After 60 min of MCA occlusion, blood flow was restored by withdrawal of the nylon suture. To examine the role of the MEK/ERK pathway after FCI, injection of a MEK1/2 inhibitor was performed. The MEK1/2 inhibitor 1,4-diamino-2,3-dicyano-1,4-bis(2-aminophenylthio) butadiene (U0126) was purchased from Cell Signaling (Beverly, MA) and dissolved in dimethylsulfoxide (DMSO) and PBS. The dose of the drug was decided by referring to a previous intravenous injection study (Namura et al., 2001). The scalp was incised on the midline, and the skull was exposed. U0126 (20 nmol in 20% DMSO in PBS) and the vehicle (20% DMSO in PBS) were injected intracerebroventricularly (2 μl, bregma; 1 mm lateral, 0.2 mm posterior, and 3.1 mm deep). U0126 and the vehicle were injected 1 hr before MCA occlusion.
Immunohistochemistry. Anesthetized animals were perfused with 10 U/ml heparin and subsequently with 4% paraformaldehyde in 0.1m PBS (pH 7.4) at 1, 4, and 24 hr of reperfusion, as well as normal controls (n = 3 each). The brains were removed, postfixed for 12 hr, sectioned at 50 μm on a vibratome, and processed for immunohistochemistry. The sections were incubated with blocking solution and reacted with rabbit polyclonal anti-phospho-ERK1/2 antibody (Cell Signaling) at a dilution of 1:100. Immunohistochemistry was performed using the avidin–biotin technique, and then the nuclei were counterstained with methyl green solution for 5 min. For histological assessment, alternate slices from each brain section were stained with cresyl violet.
Immunofluorescent double labeling with phospho-ERK1/2 and neuron-specific nuclear protein. To evaluate neuronal expression of phospho-ERK1/2, we performed double immunofluorescent staining for phospho-ERK1/2 and neuron-specific nuclear protein (NeuN). The sections fixed by 4% paraformaldehyde were immunostained with the phospho-ERK1/2 antibody as described above, with biotinylated goat anti-rabbit IgG (Vector Laboratories, Burlingame, CA), followed by fluorescein avidin DCS (Vector Laboratories). Next, the sections were incubated with blocking solution and reacted with mouse monoclonal anti-NeuN antibody (Chemicon International, Temecula, CA) at a dilution of 1:200, followed by Texas Red-conjugated donkey anti-mouse IgG antibody (Jackson ImmunoResearch, West Grove, PA) at a dilution of 1:500. Subsequently, the slides were covered with Vectashield mounting medium with 4′,6′-diamidino-2-phenylindole (DAPI; Vector Laboratories). Fluorescence of fluorescein was observed at excitation (Ex) of 495 nm and emission (Em) of >515 nm, and fluorescence of Texas Red was observed at Ex of 510 nm and Em of >580 nm. Fluorescence of DAPI was also observed at Ex of 360 nm and Em of >460 nm.
Western blot analysis. Whole-cell protein extraction was performed. Samples were obtained from the MCA territory cortex on the ischemic sides and from nonischemic controls (n = 4 each). Fresh brain tissue was cut into pieces after 1, 4, and 24 hr (n = 4 each) of reperfusion and homogenized in 7 vol of cold suspension buffer (in mm): 20 HEPES-KOH, pH 7.5, 250 sucrose, 10 KCl, 1.5 MgCl2, 1 EDTA, 1 EGTA, 0.7% protease, and phosphatase inhibitor cocktails (Sigma, St. Louis, MO). The homogenate was centrifuged at 10,000 ×g for 20 min at 4°C, and the supernatant was used for the analysis. After adding the same volume of Tris-glycine SDS sample buffer (Invitrogen, Carlsbad, CA) to the supernatant, equal amounts of the samples were loaded per lane. The primary antibodies were a 1:1000 dilution of rabbit polyclonal antibody against phospho-MEK1/2 (Ser217/221), phospho-stress-activated protein kinase/JNK (Thr183/Tyr185), MEK, and ERK1/2 (Cell Signaling); a 1:1000 dilution of mouse monoclonal phospho-ERK1/2 (Thr202/Tyr204) E10 monoclonal antibody (Cell Signaling); or a 1:10,000 dilution of anti-β-actin monoclonal antibody (Sigma). Western blots were performed with horseradish peroxidase-conjugated anti-rabbit IgG (Cell Signaling) or anti-mouse IgG (Chemicon International) using enhanced chemiluminescence Western blotting detection reagents (Amersham Biosciences, Buckinghamshire, UK). The film was scanned with a GS-700 imaging densitometer (Bio-Rad, Hercules, CA), and the results were quantified using Multi-Analyst software (Bio-Rad).
In situ detection of superoxide anion production. The early production of superoxide anion (O2−) in cerebral ischemia was investigated using hydroethidine (HEt) by a method described previously (Murakami et al., 1998). HEt is diffusible into the CNS parenchyma after an intravenous injection and is selectively oxidized to ethidium by O2− but not by other ROS, such as hydrogen peroxide, hydroxyl radical, or peroxynitrite (Bindokas et al., 1996; Murakami et al., 1998). HEt solution (200 μl; 1 mg/ml in PBS) was administered intravenously 15 min before induction of ischemia as described previously (Murakami et al., 1998). In the brains of animals intravenously injected with HEt, fluorescence was assessed microscopically at Ex of 355 nm and Em of >415 nm for HEt detection or at Ex of 510–550 nm and Em of >580 nm for ethidium detection. Animals were killed 1 hr after transient FCI by transcardial perfusion as described. After fixation with 4% paraformaldehyde for 12 hr, the brains were sectioned at 50 μm on a vibratome. These sections were observed with a microscope under fluorescent light. Immunofluorescent staining with phospho-ERK1/2 was also performed on some of these sections as described above.
Cell death assay. For quantification of apoptotic-related DNA fragmentation, we used a commercial enzyme immunoassay to determine cytoplasmic histone-associated DNA fragments (Roche Molecular Biochemicals, Mannheim, Germany); this assay detects apoptotic but not necrotic cell death (Leist et al., 1998). Samples were obtained from the entire MCA territory or the MCA territory cortex on the ischemic sides and from nonischemic controls (n = 4 each). Fresh brain tissue was cut into pieces after 1, 4, and 24 hr of reperfusion, homogenized with a Teflon homogenizer in 5 vol of ice-cold buffer (in mm: 50 KH2PO4 and 0.1 EDTA, pH 7.8), and spun for 10 min at 750 × g. The supernatant was collected and spun for 20 min at 10,000 × g. The supernatant was further centrifuged at 100,000 × g for 60 min at 4°C. The resulting supernatant was collected, and the protein concentration was determined. A cytosolic volume containing 20 μg of protein was used for the ELISA according to the manufacturer's protocol.
Quantification and statistical analysis. The data are expressed as mean ± SD. Comparisons among multiple groups were performed using an ANOVA (Fisher's protected least-significant difference test), whereas comparisons between two groups were achieved using Student's t test. p < 0.05 was considered statistically significant.
RESULTS
Physiological data and cerebral infarction
Physiological parameters showed no significant differences in mean arterial blood pressure and arterial blood gas analysis between the groups. The preischemic physiological values were as follows (wild-type and SOD1 Tg mice, respectively) (in mmHg): 89 ± 8.8 and 91 ± 10.7 mean arterial blood pressure, 151 ± 31 and 141 ± 19 PaO2, 33 ± 3.2 and 33 ± 5 PaCO2, pH of 7.3 ± 0.07 and 7.3 ± 0.05 (values are mean ± SD; n = 4). There was no deviation from these values over the period of assessment. An ischemic lesion of the core of the caudate putamen was visible as a pale, slightly stained area in the ischemic hemisphere as early as 1 hr after reperfusion and extended to the entire MCA territory at 4 hr by cresyl violet staining (data not shown). The time-dependent increase in infarction in mouse brain using the intraluminal suture blockade is consistent with previous reports that used the same focal stroke model in mice (Yang et al., 1994; Kondo et al., 1997).
Neuronal expression of phospho-ERK1/2 is temporally increased in the ischemic cortex after transient FCI
Phospho-ERK1/2 was predominantly expressed in the surface layer of the normal mouse brain cortex (Fig.1A). One hour after transient FCI, expression of phospho-ERK1/2 was markedly increased in the ischemic cortex of the MCA territory (Fig. 1B,C) but not in the caudate putamen (ischemic core). At 24 hr of reperfusion, the expression of phospho-ERK1/2 was reduced in the ischemic cortex and was primarily accumulated around nuclei (Fig.1D). In the caudate putamen, phospho-ERK1/2 was hardly seen after 1–24 hr of transient FCI (data not shown). Phospho-ERK1/2 expression in the contralateral hemisphere after reperfusion was not different from that in the normal brain (data not shown). Double immunofluorescence for phospho-ERK1/2 (Fig.2A) and NeuN (Fig.2B) demonstrated that phospho-ERK1/2 expression colocalized with neurons in the ischemic cortex 1 hr after reperfusion (Fig. 2C, overlapped image). The results suggest that phospho-ERK1/2 is expressed primarily in neurons of the ischemic cortex after FCI.
Fig. 1.
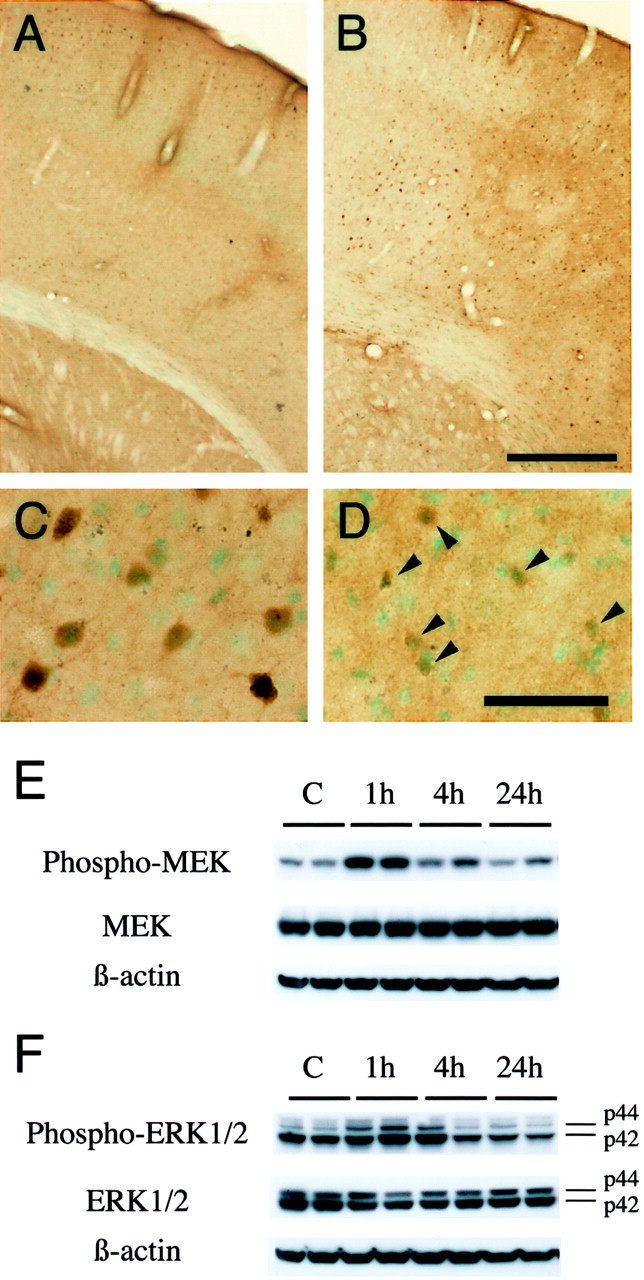
A–D, Phospho-ERK1/2 immunostaining with methyl green counterstaining in coronal brain sections from wild-type mice after transient FCI. Phospho-ERK1/2 was predominantly expressed in the surface layer of the normal mouse brain cortex (A) and was prominently increased 1 hr after reperfusion in the ischemic cortex of the MCA territory (B) but not in the caudate putamen (ischemic core). C, D, Representative photomicrographs of higher magnification in the MCA territory cortex after ischemia. In the ischemic cortex of the MCA territory, homogeneous cytoplasmic and nuclear immunoreactivity of phospho-ERK1/2 was markedly increased 1 hr after reperfusion (C). At 24 hr of reperfusion, the expression of phospho-ERK1/2 was reduced in the ischemic cortex and was primarily accumulated around nuclei (D,arrowheads). In the caudate putamen, phospho-ERK1/2 was hardly seen after 1–24 hr of transient FCI (data not shown). Phospho-ERK1/2 expression in the contralateral hemisphere after reperfusion was not different from that in the normal brain (data not shown). Scale bars: A, B, 400 μm; C, D, 50 μm. E, F, Western blot analysis of phospho-MEK, MEK, phospho-ERK1/2, and ERK1/2 after transient FCI. E, Phospho-MEK, MEK, and β-actin from the whole-cell samples in the nonischemic control brains (lane C) and ischemic brains (lanes 1h–24h). Phospho-MEK and MEK immunoreactivity were evident as a band with a molecular mass of 45 kDa in the whole-cell fraction from the MCA territory cortex of the mouse brains. Phospho- MEK and MEK were expressed constitutively in the nonischemic control brain (lane C). One hour after reperfusion, phospho-MEK was increased significantly compared with the nonischemic controls (top row, lane 1h), whereas it was decreased by 4 hr (top row, lane 4h). MEK showed no prominent increase or decrease after reperfusion in the ischemic cortex (middle row). The results of the β-actin analysis are shown as an internal control (bottom row). F, The bands of phospho-ERK1/2 and ERK1/2 were observed at 44 kDa (ERK1) and 42 kDa (ERK2) in the whole-cell fraction from the MCA territory cortex of the mouse brains. Phospho-ERK1 was increased significantly 1 hr after transient FCI (top row, p44, lane 1h), whereas it returned to the control level by 4 hr (top row, p44, lane 4h). In contrast, phospho-ERK2 was decreased after reperfusion (top row, p42). ERK1/2 did not show prominent changes before or after FCI (middle row). β-actin was used as an internal control, and no difference was observed between the samples (bottom row). Densitometric analysis showed that phospho-MEK was increased significantly 1 hr after reperfusion compared with the nonischemic controls (p < 0.005) and that phospho-ERK1 was increased significantly 1 hr after reperfusion compared with the nonischemic controls (p < 0.005). Phospho-ERK2 was decreased after ischemia, and the decrease was significant at 24 hr compared with the nonischemic control (p < 0.01).
Fig. 2.
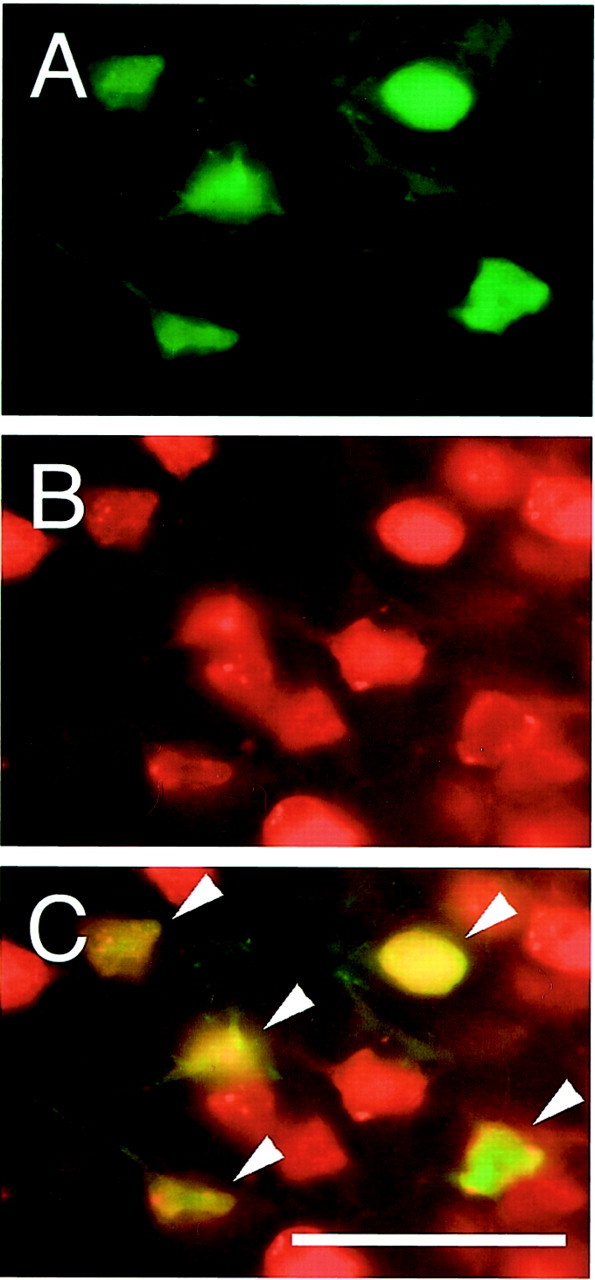
Double immunofluorescent staining for phospho-ERK1/2 and NeuN 1 hr after transient FCI. A, Expression of phospho-ERK1/2 was observed in the ischemic cortex 1 hr after reperfusion. B, NeuN immunoreactivity shows the distribution of neurons in the same view. C, Overlapped image of A and B demonstrates that phospho-ERK1/2 expression colocalized completely with neurons in the ischemic cortex 1 hr after reperfusion (arrowheads). The results suggest that phospho-ERK1/2 is expressed primarily in neurons of the ischemic cortex after FCI. Scale bar, 50 μm.
Western blot analysis demonstrates significant increase in phospho-MEK and phospho-ERK1 after transient FCI
As shown in Figure 1E, phospho-MEK and MEK immunoreactivity were evident as a band with a molecular mass of 45 kDa in the whole-cell fraction from the MCA territory cortex in the mouse brains. Phospho-MEK and MEK were constitutively expressed in the nonischemic control brains. One hour after reperfusion, phospho-MEK was significantly increased compared with the nonischemic controls (p < 0.005), whereas it was decreased by 4 hr. There was no prominent MEK increase or decrease after reperfusion in the ischemic cortex. The bands of phospho-ERK1/2 and ERK1/2 were observed at 44 kDa (ERK1) and 42 kDa (ERK2) in the whole-cell fraction from the MCA territory cortex (Fig. 1F). Phospho-ERK1 was significantly increased 1 hr after transient FCI (p < 0.005), whereas it returned to the control level by 4 hr. In contrast, phospho-ERK2 was decreased after FCI, and the decrease was significant at 24 hr (p < 0.01). ERK1/2 did not show prominent changes before or after FCI. β-actin was used as an internal control, and no difference was observed between the samples.
SOD production was not affected by the MEK1/2 inhibitor
As shown in Figure 3A, Western blot revealed that the injection of U0126 actually reduced phospho-ERK1/2 expression 1 hr after reperfusion compared with the vehicle injection. However, phosphorylation of JNK was not inhibited by U0126. The results show the specificity of U0126 on inhibition of ERK1/2 phosphorylation but not JNK phosphorylation. To confirm whether free radical induction is inhibited by U0126, we examined O2− production using an injection of U0126 or the vehicle 1 hr after reperfusion. Production of O2− was determined using HEt, a fluorescent dye selectively oxidized to ethidium O2−, at 1 hr of reperfusion as described previously (Murakami et al., 1998). O2− production was shown by oxidized HEt (red) as vesicular signals in the ischemic cortex of the vehicle-treated animals (Fig. 3B) or U0126-treated animals (Fig. 3C), and no significant difference was observed between those two groups. The data show that U0126 does not affect O2− production after transient FCI, suggesting that ROS may be upstream of ERK1/2 activation.
Fig. 3.

A, Western blot analysis of phospho-ERK1/2 and phospho-stress-activated protein kinase/JNK after transient FCI in U0126- or vehicle-injected mice. U0126 injection actually reduced phospho-ERK1/2 expression 1 hr after reperfusion compared with vehicle injection (V, vehicle;U, U0126). In contrast, phosphorylation of JNK was not inhibited by U0126. The results show the specificity of U0126 after inhibition for ERK1/2 phosphorylation but not JNK phosphorylation.B, C, Representative photomicrographs show O2− production 1 hr after transient FCI in U0126- or vehicle-injected mice. O2−production was shown by oxidized HEt (red) as vesicular signals in the ischemic cortex of vehicle-treated animals (B) or U0126-treated animals (C), and no significant difference was observed between these two groups. The data show that U0126 does not affect O2− production after transient FCI, suggesting that ROS may be upstream of ERK1/2 activation.
Production of O2− after FCI is decreased by SOD1
O2− production was shown by oxidized HEt signals (red) in the cytosol of the contralateral cortex in both the wild-type (Fig.4A) and SOD1 Tg mice (Fig. 4B); however, no conspicuous difference was observed between them. In the ischemic cortex, enhanced vesicular signals were observed in the wild-type mice (Fig. 4C). These vesicular signals were markedly decreased in the SOD1 Tg mice (Fig.4D) compared with the wild-type mice, suggesting inhibition of O2−production by SOD1. Nuclear staining (blue) revealed enhanced vesicular signals that meant O2−production (red) was located in the perinuclear area (Fig.4C).
Fig. 4.

Representative photomicrographs show O2− production 1 hr after transient FCI. O2− production was shown by oxidized HEt signals (red), which were observed constitutively in the cytosol of the contralateral cortex in both the wild-type mice (A) and SOD1 Tg mice (B). No conspicuous difference was observed between them. C, In the ischemic cortex, enhanced vesicular signals were observed in the wild-type mice.D, These vesicular signals were markedly decreased in the SOD1 Tg mice compared with the wild-type mice, suggesting inhibition of O2− production by SOD1. Nuclear staining (blue) revealed enhanced vesicular signals, which means that O2−production (red) was located in the perinuclear area (C). Scale bar, 20 μm.
Phosphorylation of ERK1/2 after transient FCI is markedly diminished in SOD1 Tg mice
To investigate the relationship between ERK1/2 phosphorylation and oxidative stress, immunofluorescent staining with phospho-ERK1/2 was performed after oxidized HEt signal detection. One hour after transient FCI, phospho-ERK1/2 was strongly expressed in the ischemic cortex (Fig.5A). Oxidized HEt signals were also observed in the same area at this time point (Fig. 5B). Most phospho-ERK1/2-positive cells were highly colocalized with oxidized HEt signals (Fig. 5C), suggesting that oxidative stress might be involved in phosphorylation of ERK1/2 after ischemia. To examine the effect of SOD1 on ERK1/2 phosphorylation, phospho-ERK1/2 expression after transient FCI was analyzed by immunohistochemistry in both the wild-type and SOD1 Tg mice. Phospho-ERK1/2 was remarkably expressed in the ischemic cortex at 1 hr of reperfusion in the wild-type animals (Fig.6A), whereas the immunoreactivity of phospho-ERK1/2 was markedly decreased in the SOD1 Tg mice (Fig. 6B). Western blot analysis showed that phosphorylation of MEK decreased 1 hr after reperfusion in the SOD1 Tg mice compared with the wild-type mice, but the decrease was not significant (Fig. 6C,E; p = 0.219). Regarding phosphorylation of ERK1/2 (Fig. 6D), both phospho-ERK1 and phospho-ERK2 were significantly decreased 1 hr after reperfusion in the SOD1 Tg mice compared with the wild-type mice (Fig.6F, phospho-ERK1, p < 0.005; Fig.6G, phospho-ERK2, p < 0.005). No difference was observed in MEK and ERK1/2 levels between the SOD1 Tg mice and the wild-type mice 1 hr after transient FCI. These results demonstrate that ERK1/2 phosphorylation is inhibited by SOD1, suggesting the protective effect of SOD1 on MEK/ERK pathway activation.
Fig. 5.

Representative photomicrographs show immunofluorescent staining for phospho-ERK1/2 (green) on oxidized HEt signal detection (red). A, Phospho-ERK1/2 was strongly expressed in the ischemic cortex 1 hr after transient FCI.B, Oxidized HEt signals were also observed in the same area 1 hr after transient FCI. C, Overlapped image ofA and B shows that most phospho-ERK1/2-positive cells (green) highly colocalized with oxidized HEt signals (red), suggesting that oxidative stress might be involved in phosphorylation of ERK1/2 after ischemia. D, Nuclear staining (blue) with oxidized HEt signals in the same view. Scale bar, 20 μm.
Fig. 6.

Immunohistochemistry and Western blot analysis of phospho-ERK1/2 after transient FCI in SOD1 Tg mice.A–D, To examine the effect of SOD1 on ERK1/2 phosphorylation, phospho-ERK1/2 expression after transient FCI was analyzed by immunohistochemistry in both the wild-type (WT) and SOD1 Tg mice. Phospho-ERK1/2 was expressed strongly in the ischemic cortex at 1 hr of reperfusion in the wild-type mice (A), whereas the immunoreactivity of phospho-ERK1/2 was markedly decreased in the SOD1 Tg mice (B). Dashed lines indicate the border of the lesion. C, D, Western blot analysis of phospho-MEK and phospho-ERK1/2 in the ischemic cortex 1 hr after transient FCI in both the wild-type and SOD1 Tg mice. C, Phospho-MEK (top row) was decreased 1 hr after reperfusion in the SOD1 Tg mice compared with the wild-type mice; however, the decrease was not significant (E;p = 0.219). D, Regarding phosphorylation of ERK1/2, both phospho-ERK1 and phospho-ERK2 (top row) were significantly decreased 1 hr after reperfusion in the SOD1 Tg mice compared with the wild-type mice (F, phospho-ERK1, ∗p < 0.005;G, phospho-ERK2, ∗p < 0.005). No difference was observed in MEK (C, middle row) and ERK1/2 (D, middle row) levels between the SOD1 Tg and the wild-type mice 1 hr after transient FCI. β-actin was used as an internal control, and no difference was observed between the samples (bottom rows). These results demonstrate that ERK1/2 phosphorylation is inhibited by SOD1, suggesting a protective effect of SOD1 on MEK/ERK pathway activation. Scale bar, 400 μm.
DNA fragmentation after transient FCI is significantly decreased in the ischemic cortex of SOD1 Tg mice compared with wild-type mice
Apoptotic-related DNA fragmentation after ischemia was analyzed with a commercial cell death detection kit. As shown in Figure7A, DNA fragmentation was significantly increased in the entire MCA territory lesion 24 hr after reperfusion compared with the nonischemic brain (p < 0.0001). Hence, we examined DNA fragmentation 24 hr after reperfusion in both the SOD1 Tg mice and the wild-type mice to assess the protective effect of SOD1 on DNA damage. In a preliminary study, no significant difference was observed in DNA fragmentation in the entire MCA territory region, including the caudate putamen and the cortex, between the SOD1 Tg and wild-type mice. As shown in Figure 7B, DNA fragmentation at 24 hr of reperfusion was significantly reduced in the MCA territory cortex of the SOD1 Tg mice compared with the wild-type mice at the same time point (p < 0.005). These results suggest that SOD1 plays a role in inhibiting DNA fragmentation in the ischemic cortex after transient FCI.
Fig. 7.

Apoptotic-related DNA fragmentation after ischemia was analyzed with a commercial cell death detection kit.A, DNA fragmentation was increased significantly in the entire MCA territory lesion 24 hr after reperfusion compared with the nonischemic brain (∗p < 0.0001).B, DNA fragmentation was increased significantly in the ischemic cortex 24 hr after reperfusion compared with the nonischemic brain (∗p < 0.0001). In the SOD1 Tg mice, DNA fragmentation at 24 hr of reperfusion was reduced significantly in the ischemic cortex compared with the wild-type (WT) mice at the same time point (∗∗p < 0.005). These results suggest that SOD1 plays a role in inhibiting DNA fragmentation in the ischemic cortex after transient FCI.C, Nonischemic control brain.
DISCUSSION
The current study demonstrates that phosphorylation of MEK and ERK1 was temporally enhanced in the ischemic cortex 1 hr after reperfusion, suggesting that MEK and ERK1 were activated during the early period after transient FCI (Fig. 1). In contrast, phosphorylation of ERK2 was not increased after FCI and was significantly decreased 24 hr after reperfusion. The dissociation of phosphorylation between ERK1 and ERK2 after the insult is also reported in vitro, suggesting that ERK1 and ERK2 play differential roles in cell survival and cell death, respectively (Namura et al., 2001). In that study, the authors surmised that a difference in stability to insult may exist between phospho-ERK1 and phospho-ERK2 in cortical neurons against oxygen deprivation. In the present study, we do not mention the exact role of each ERK after FCI. Further study is required to clarify the difference in function between phospho-ERK1 and phospho-ERK2.
Because activation of MEK and ERK was followed by DNA fragmentation at 24 hr of reperfusion (Fig. 7), it might be possible that the early enhancement of phosphorylation in these kinases is a forewarning of apoptosis. Previous studies have revealed that ERK1/2 was phosphorylated after FCI (Alessandrini et al., 1999; Namura et al., 2001), whereas the current study demonstrated MEK phosphorylation after FCI, as well as ERK1/2 phosphorylation. Because ERK1/2 is activated by phospho-MEK, inhibition of MEK activation can be effective to prevent ERK1/2 activation. A recent study using a MEK inhibitor showed that ERK1/2 activation was regulated by MEK after FCI (Namura et al., 2001). Furthermore, ERK1/2 activation seems to have a deleterious effect on FCI, because ERK1/2 inhibition by the MEK inhibitor reduced infarct volume (Alessandrini et al., 1999; Namura et al., 2001), which is consistent with our results (data not shown).
Immunohistochemistry showed that although phospho-ERK1/2 was decreased in the ischemic cortex at 24 hr of reperfusion, it was still expressed in and primarily accumulated around nuclei (Fig. 1D). The mechanism of nuclear accumulation of phospho-ERK1/2 after FCI is unclear; however, it is conceivable that activated ERK1/2 translocated from the cytoplasm to nuclei after FCI because phospho-ERK1/2 is known to activate transcription factors, such as Elk-1, in nuclei (Sgambato et al., 1998a). In fact, an in vitro study has shown the nuclear translocation of active ERK1/2 from the cytoplasm (Chen et al., 1992). Regarding spatial distribution of phospho-ERK1/2, immunohistochemistry showed a prominent increase in phospho-ERK1/2 in the MCA territory cortex but not in the ischemic core (Fig. 1). The distribution of phospho-ERK1/2 after FCI was consistent with previous studies (Alessandrini et al., 1999; Irving et al., 2000). However, the subcellular population of phospho-ERK1/2 expression has not yet been elucidated. Although a study showed that phospho-ERK1/2 was expressed in the cortex, where neurons were still preserved after FCI (Irving et al., 2000), neuronal expression of phospho-ERK1/2 was not directly confirmed. In the present study, we revealed the colocalized expression of phospho-ERK1/2 and NeuN by a double immunofluorescent method (Fig.2), suggesting neuronal expression of phospho-ERK1/2.
Our previous studies revealed that SOD1 has protective effects on ischemic damage (Kinouchi et al., 1991; Chan, 1996; Kondo et al., 1997;Murakami et al., 1997; Chan et al., 1998; Fujimura et al., 1999b,2000). In the present study, we focused on the role of the MEK/ERK pathway in the neuroprotective effect of SOD1. As seen in Figure 4, oxidized HEt signals were markedly decreased in the SOD1 Tg mice compared with the wild-type mice, showing inhibition of O2− production by SOD1. Moreover, overexpressed SOD1 significantly decreased ERK1/2 phosphorylation after FCI (Fig. 6), suggesting that ERK1/2 activation is mediated by O2−production after FCI. It is also reported that ROS can induce MEK/ERK pathway activation in vitro (Baas and Berk, 1995; Guyton et al., 1996; Seo et al., 2001). The conjecture that ERK activation after FCI is mediated by ROS is also supported by the fact that phospho-ERK1/2 was expressed in the cells that produced O2− (Fig. 5). Ras and Raf-1 are upstream of the MEK/ERK pathway (Marshall, 1995; Pritchard and McMahon, 1997; Downward, 1998; McNeill and Downward, 1999). The expression of dominant-negative Ras or Raf mutants was shown to decrease the ERK activity induced by hydrogen peroxide in cultured cells, suggesting that oxidative stress induces ERK activation through Ras or Raf-1 activation (Aikawa et al., 1997). Moreover, antioxidants prevented both ERK activation and cell death induced by Zn2+, which generated ROS (Seo et al., 2001). In that study, both inhibition of ERK with PD98059, a synthetic inhibitor of MEK1/2, and the expression of the dominant-negative Ras mutant significantly prevented cell death, suggesting that ERK activation mediates cell death induced by ROS. Ras is also known to be activated by nitric oxide induced by the NMDA receptor (Yun et al., 1998). Although Ras and Raf-1 activation has not been reported after FCI, it is conceivable that oxidative stress induces MEK and ERK activation through Ras and Raf-1 activation. In the present study, overexpressed SOD1 could have significantly reduced ERK1/2 phosphorylation after transient FCI (Fig. 6F,G;p < 0.005), whereas the decrease in phospho-MEK was not significant in the SOD1 Tg mice compared with the wild-type mice (Fig. 6E; p = 0.219). How oxidative stress contributes to activation of the MEK/ERK pathway after FCI is still unclear; however, it is obvious that oxidative stress plays a crucial role in ERK1/2 activation after transient FCI.
Because the MEK/ERK pathway interacts with other elements that determine cell survival or cell death, it is useful to investigate the change in such elements after ischemia to understand the role of oxidative stress on the MEK/ERK pathway. Akt is known to play a critical role in controlling the balance between survival and apoptosis (Burgering and Coffer, 1995; Franke et al., 1995, 1997). One study shows that inhibition of Akt induces phosphorylation of Raf at serine-259 and inactivates Raf, which results in ERK1/2 activation in cultured cells stimulated with insulin-like growth factor (Zimmermann and Moelling, 1999). Another study shows that phosphatidylinositol 3-kinase inhibitor, which inhibits Akt activation, prevented nuclear translocation of PKCζ, resulting in ERK1/2 activation after ischemic hypoxia and reoxygenation in cultured cells (Mizukami et al., 2000). Furthermore, we have shown that Akt is activated in the ischemic cortex after transient FCI (Noshita et al., 2001). All of this evidence together suggests that Akt activation after transient FCI may interact with activation of the MEK/ERK pathway. A future study is required to clarify the relationship between Akt activation and ERK1/2 activation after FCI.
Our results imply that ERK1/2 is activated in neurons of the ischemic cortex after transient FCI and that ROS production may be critical to activation of the MEK/ERK pathway. SOD1 contributes to the inhibition of apoptosis induced by FCI by reducing the early formation of superoxide radicals and preventing the phosphorylation of ERK1/2, showing the critical role of SOD1 on the MEK/ERK pathway.
Footnotes
This work was supported by National Institutes of Health Grants NS14543, NS25372, NS36147, and NS38653. P.H.C. is a recipient of the Jacob Javits Neuroscience Investigator Award. We thank Dr. Charles J. Epstein (Department of Pediatrics, University of California, San Francisco, School of Medicine, San Francisco, CA) for the breeding pairs of SOD1 transgenic mice. We also thank Cheryl Christensen for editorial assistance and Liza Reola and Bernard Calagui for technical assistance.
Correspondence should be addressed to Dr. Pak H. Chan, Neurosurgical Laboratories, Stanford University, 1201 Welch Road, MSLS #P314, Stanford, CA 94305-5487. E-mail: phchan@leland.stanford.edu.
REFERENCES
- 1.Aikawa R, Komuro I, Yamazaki T, Zou Y, Kudoh S, Tanaka M, Shiojima I, Hiroi Y, Yazaki Y. Oxidative stress activates extracellular signal-regulated kinases through Src and Ras in cultured cardiac myocytes of neonatal rats. J Clin Invest. 1997;100:1813–1821. doi: 10.1172/JCI119709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alessandrini A, Namura S, Moskowitz MA, Bonventre JV. MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc Natl Acad Sci USA. 1999;96:12866–12869. doi: 10.1073/pnas.96.22.12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Avruch J, Zhang XF, Kyriakis JM. Raf meets Ras: completing the framework of a signal transduction pathway. Trends Biochem Sci. 1994;19:279–283. doi: 10.1016/0968-0004(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 4.Baas AS, Berk BC. Differential activation of mitogen-activated protein kinases by H2O2 and O2− in vascular smooth muscle cells. Circ Res. 1995;77:29–36. doi: 10.1161/01.res.77.1.29. [DOI] [PubMed] [Google Scholar]
- 5.Bindokas VP, Jordán J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci. 1996;16:1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boulton TG, Nye SH, Robbins DJ, Ip NY, Radziejewska E, Morgenbesser SD, DePinho RA, Panayotatos N, Cobb MH, Yancopoulos GD. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell. 1991;65:663–675. doi: 10.1016/0092-8674(91)90098-j. [DOI] [PubMed] [Google Scholar]
- 7.Burgering BM, Coffer PJ. Protein kinase B (c-Akt) in phosphatidylinositol-3-OH kinase signal transduction. Nature. 1995;376:599–602. doi: 10.1038/376599a0. [DOI] [PubMed] [Google Scholar]
- 8.Chan PH. Role of oxidants in ischemic brain damage. Stroke. 1996;27:1124–1129. doi: 10.1161/01.str.27.6.1124. [DOI] [PubMed] [Google Scholar]
- 9.Chan PH, Kamii H, Yang G, Gafni J, Epstein CJ, Carlson E, Reola L. Brain infarction is not reduced in SOD-1 transgenic mice after a permanent focal cerebral ischemia. NeuroReport. 1993;5:293–296. doi: 10.1097/00001756-199312000-00028. [DOI] [PubMed] [Google Scholar]
- 10.Chan PH, Kawase M, Murakami K, Chen SF, Li Y, Calagui B, Reola L, Carlson E, Epstein CJ. Overexpression of SOD1 in transgenic rats protects vulnerable neurons against ischemic damage after global cerebral ischemia and reperfusion. J Neurosci. 1998;18:8292–8299. doi: 10.1523/JNEUROSCI.18-20-08292.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen RH, Sarnecki C, Blenis J. Nuclear localization and regulation of erk- and rsk-encoded protein kinases. Mol Cell Biol. 1992;12:915–927. doi: 10.1128/mcb.12.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis RJ. The mitogen-activated protein kinase signal transduction pathway. J Biol Chem. 1993;268:14553–14556. [PubMed] [Google Scholar]
- 13.Downward J. Ras signalling and apoptosis. Curr Opin Genet Dev. 1998;8:49–54. doi: 10.1016/s0959-437x(98)80061-0. [DOI] [PubMed] [Google Scholar]
- 14.Epstein CJ, Avraham KB, Lovett M, Smith S, Elroy-Stein O, Rotman G, Bry C, Groner Y. Transgenic mice with increased Cu/Zn-superoxide dismutase activity: animal model of dosage effects in Down syndrome. Proc Natl Acad Sci USA. 1987;84:8044–8048. doi: 10.1073/pnas.84.22.8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fiore RS, Murphy TH, Sanghera JS, Pelech SL, Baraban JM. Activation of p42 mitogen-activated protein kinase by glutamate receptor stimulation in rat primary cortical cultures. J Neurochem. 1993;61:1626–1633. doi: 10.1111/j.1471-4159.1993.tb09796.x. [DOI] [PubMed] [Google Scholar]
- 16.Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- 17.Franke TF, Kaplan DR, Cantley LC. PI3K: downstream AKTion blocks apoptosis. Cell. 1997;88:435–437. doi: 10.1016/s0092-8674(00)81883-8. [DOI] [PubMed] [Google Scholar]
- 18.Frodin M, Gammeltoft S. Role and regulation of 90 kDa ribosomal S6 kinase (RSK) in signal transduction. Mol Cell Endocrinol. 1999;151:65–77. doi: 10.1016/s0303-7207(99)00061-1. [DOI] [PubMed] [Google Scholar]
- 19.Fujimura M, Morita-Fujimura Y, Kawase M, Copin J-C, Calagui B, Epstein CJ, Chan PH. Manganese superoxide dismutase mediates the early release of mitochondrial cytochrome c and subsequent DNA fragmentation after permanent focal cerebral ischemia in mice. J Neurosci. 1999a;19:3414–3422. doi: 10.1523/JNEUROSCI.19-09-03414.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujimura M, Morita-Fujimura Y, Narasimhan P, Copin J-C, Kawase M, Chan PH. Copper-zinc superoxide dismutase prevents the early decrease of apurinic/apyrimidinic endonuclease and subsequent DNA fragmentation after transient focal cerebral ischemia in mice. Stroke. 1999b;30:2408–2415. doi: 10.1161/01.str.30.11.2408. [DOI] [PubMed] [Google Scholar]
- 21.Fujimura M, Morita-Fujimura Y, Noshita N, Sugawara T, Kawase M, Chan PH. The cytosolic antioxidant copper/zinc-superoxide dismutase prevents the early release of mitochondrial cytochrome c in ischemic brain after transient focal cerebral ischemia in mice. J Neurosci. 2000;20:2817–2824. doi: 10.1523/JNEUROSCI.20-08-02817.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ. Activation of mitogen-activated protein kinase by H2O2. Role in cell survival following oxidant injury. J Biol Chem. 1996;271:4138–4142. doi: 10.1074/jbc.271.8.4138. [DOI] [PubMed] [Google Scholar]
- 23.Hetman M, Kanning K, Cavanaugh JE, Xia Z. Neuroprotection by brain-derived neurotrophic factor is mediated by extracellular signal-regulated kinase and phosphatidylinositol 3-kinase. J Biol Chem. 1999;274:22569–22580. doi: 10.1074/jbc.274.32.22569. [DOI] [PubMed] [Google Scholar]
- 24.Irving EA, Barone FC, Reith AD, Hadingham SJ, Parsons AA. Differential activation of MAPK/ERK and p38/SAPK in neurones and glia following focal cerebral ischaemia in the rat. Brain Res Mol Brain Res. 2000;77:65–75. doi: 10.1016/s0169-328x(00)00043-7. [DOI] [PubMed] [Google Scholar]
- 25.Kinouchi H, Epstein CJ, Mizui T, Carlson E, Chen SF, Chan PH. Attenuation of focal cerebral ischemic injury in transgenic mice overexpressing CuZn superoxide dismutase. Proc Natl Acad Sci USA. 1991;88:11158–11162. doi: 10.1073/pnas.88.24.11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kondo T, Reaume AG, Huang T-T, Carlson E, Murakami K, Chen SF, Hoffman EK, Scott RW, Epstein CJ, Chan PH. Reduction of CuZn-superoxide dismutase activity exacerbates neuronal cell injury and edema formation after transient focal cerebral ischemia. J Neurosci. 1997;17:4180–4189. doi: 10.1523/JNEUROSCI.17-11-04180.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kurino M, Fukunaga K, Ushio Y, Miyamoto E. Activation of mitogen-activated protein kinase in cultured rat hippocampal neurons by stimulation of glutamate receptors. J Neurochem. 1995;65:1282–1289. doi: 10.1046/j.1471-4159.1995.65031282.x. [DOI] [PubMed] [Google Scholar]
- 28.Leist M, Kühnle S, Single B, Nicotera P. Difference between apoptotic and necrotic cell death by means of the BM cell death detection ELISA or annexin V staining. Biochemica. 1998;2:25–28. [Google Scholar]
- 29.Marshall CJ. Specificity of receptor tyrosine kinase signaling: transient versus sustained extracellular signal-regulated kinase activation. Cell. 1995;80:179–185. doi: 10.1016/0092-8674(95)90401-8. [DOI] [PubMed] [Google Scholar]
- 30.McNeill H, Downward J. Apoptosis: Ras to the rescue in the fly eye. Curr Biol. 1999;9:R176–R179. doi: 10.1016/s0960-9822(99)80107-9. [DOI] [PubMed] [Google Scholar]
- 31.Mizukami Y, Kobayashi S, Uberall F, Hellbert K, Kobayashi N, Yoshida K. Nuclear mitogen-activated protein kinase activation by protein kinase cζ during reoxygenation after ischemic hypoxia. J Biol Chem. 2000;275:19921–19927. doi: 10.1074/jbc.M907901199. [DOI] [PubMed] [Google Scholar]
- 32.Murakami K, Kondo T, Epstein CJ, Chan PH. Overexpression of CuZn-superoxide dismutase reduces hippocampal injury after global ischemia in transgenic mice. Stroke. 1997;28:1797–1804. doi: 10.1161/01.str.28.9.1797. [DOI] [PubMed] [Google Scholar]
- 33.Murakami K, Kondo T, Kawase M, Li Y, Sato S, Chen SF, Chan PH. Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J Neurosci. 1998;18:205–213. doi: 10.1523/JNEUROSCI.18-01-00205.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Namura S, Iihara K, Takami S, Nagata I, Kikuchi H, Matsushita K, Moskowitz MA, Bonventre JV, Alessandrini A. Intravenous administration of MEK inhibitor U0126 affords brain protection against forebrain ischemia and focal cerebral ischemia. Proc Natl Acad Sci USA. 2001;98:11569–11574. doi: 10.1073/pnas.181213498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Noshita N, Lewén A, Sugawara T, Chan PH. Evidence of phosphorylation of Akt and neuronal survival after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2001;21:1442–1450. doi: 10.1097/00004647-200112000-00009. [DOI] [PubMed] [Google Scholar]
- 36.Pritchard C, McMahon M. Raf revealed in life-or-death decisions. Nat Genet. 1997;16:214–215. doi: 10.1038/ng0797-214. [DOI] [PubMed] [Google Scholar]
- 37.Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J Biol Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- 38.Rosen LB, Ginty DD, Weber MJ, Greenberg ME. Membrane depolarization and calcium influx stimulate MEK and MAP kinase via activation of Ras. Neuron. 1994;12:1207–1221. doi: 10.1016/0896-6273(94)90438-3. [DOI] [PubMed] [Google Scholar]
- 39.Rouse J, Cohen P, Trigon S, Morange M, Alonso-Llamazares A, Zamanillo D, Hunt T, Nebreda AR. A novel kinase cascade triggered by stress and heat shock that stimulates MAPKAP kinase-2 and phosphorylation of the small heat shock proteins. Cell. 1994;78:1027–1037. doi: 10.1016/0092-8674(94)90277-1. [DOI] [PubMed] [Google Scholar]
- 40.Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- 41.Seger R, Krebs EG. The MAPK signaling cascade. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- 42.Seo SR, Chong SA, Lee S-I, Sung JY, Ahn YS, Chung KC, Seo JT. Zn2+-induced ERK activation mediated by reactive oxygen species causes cell death in differentiated PC12 cells. J Neurochem. 2001;78:600–610. doi: 10.1046/j.1471-4159.2001.00438.x. [DOI] [PubMed] [Google Scholar]
- 43.Sgambato V, Vanhoutte P, Pages C, Rogard M, Hipskind R, Besson MJ, Caboche J. In vivo expression and regulation of Elk-1, a target of the extracellular-regulated kinase signaling pathway, in the adult rat brain. J Neurosci. 1998a;18:214–226. doi: 10.1523/JNEUROSCI.18-01-00214.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sgambato V, Pages C, Rogard M, Besson MJ, Caboche J. Extracellular signal-regulated kinase (ERK) controls immediate early gene induction on corticostriatal stimulation. J Neurosci. 1998b;18:8814–8825. doi: 10.1523/JNEUROSCI.18-21-08814.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang G, Chan PH, Chen J, Carlson E, Chen SF, Weinstein P, Epstein CJ, Kamii H. Human copper-zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke. 1994;25:165–170. doi: 10.1161/01.str.25.1.165. [DOI] [PubMed] [Google Scholar]
- 46.Yun HY, Gonzalez-Zulueta M, Dawson VL, Dawson TM. Nitric oxide mediates N-methyl-d-aspartate receptor-induced activation of p21ras. Proc Natl Acad Sci USA. 1998;95:5773–5778. doi: 10.1073/pnas.95.10.5773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zimmermann S, Moelling K. Phosphorylation and regulation of Raf by Akt (protein kinase B). Science. 1999;286:1741–1744. doi: 10.1126/science.286.5445.1741. [DOI] [PubMed] [Google Scholar]