

Abstract
Prolonged exposure of most fast neurotransmitter-operated ion channels to agonist drives the receptors into a nonfunctional, or desensitized, state. Despite extensive investigation, desensitization remains a thoroughly characterized, yet poorly understood, process. Part of the difficulty in elucidating the mechanism of desensitization has been an inability to resolve the kinetics of both agonist binding and functional desensitization in the same set of operable receptors. To overcome this limitation, we applied single oocyte3H-ligand binding and two-electrode voltage clamp to oocytes expressing recombinant α1β2γ2 GABA receptors. Using this approach, we report several observations fundamental to the mechanism of desensitization. First, we confirm that desensitization reversibly shifts GABA receptors into a high-affinity state. For [3H]GABA binding, the half-maximal binding of the desensitized state was ∼0.040 μm. Second, we show that, upon agonist removal, this high-affinity state disappears with a time constant of 127 ± 12 sec (n = 4), similar to the time constant for functional recovery from desensitization of 124 ± 26 sec (n = 5). [3H]GABA, however, dissociates fourfold faster (τ = 30 ± 2 sec; n = 3) than functional recovery, indicating that desensitized receptors need not be bound by GABA. These data provide direct evidence for a cyclical model of receptor desensitization.
Keywords: GABAA receptor, desensitization, binding, kinetics, affinity, oocyte
Early studies of endplate nicotinic acetylcholine (nACh) receptors demonstrated that prolonged exposure to agonist drove the receptors into a refractory, or desensitized, state (Katz and Thesleff, 1957). Recovery from desensitization was a time-dependent process that first required removal of the agonist. It is now clear that desensitization is a general feature of most ligand-activated ion channels (Changeux and Edelstein, 1998). In GABA receptors, it has been suggested that desensitization may play an important role in shaping synaptic inhibition (Jones and Westbrook, 1995; Overstreet et al., 2000), and desensitization has also been implicated in the mechanism by which allosteric modulators exert their effects (Birnir et al., 1997; Zhu and Vicini, 1997). Still, despite extensive investigation, desensitization remains a thoroughly characterized, yet poorly understood, process.
In their pioneering studies, Katz and Thesleff (1957) proposed the following

cyclical model for desensitization: where R is the activatable receptor, A is the agonist molecule, and D is the desensitized receptor. They postulated that, if the affinity for the agonist in the desensitized state was greater than that of the activatable state, then this scheme could account for the following: (1) the profound desensitization in the absence of significant activation and (2) the slow rate of onset of desensitization compared with the rate of recovery when agonist was removed. Although evidence has accumulated that suggests the desensitized state has a higher affinity for agonist than that of the nondesensitized closed state (Rang and Ritter, 1970; Weber et al., 1975; Weiland et al., 1975; Quast et al., 1978; Neubig et al., 1982;Heidmann et al., 1983), the issue is far from resolved. Scheme 1 also predicts that agonist can dissociate directly from the desensitized state. In this scenario, a receptor may no longer be bound by agonist but yet still functionally desensitized. Again, direct evidence for this hypothesis is lacking.
We developed previously a technique that allows one to perform repeated radioactive ligand binding measurements in single intact oocytes expressing recombinant GABA receptors (Chang and Weiss, 1999a). One can then submit these oocytes to electrophysiological recording, allowing a direct correlation between binding and function in the same set of operable surface receptors. Here, we use this approach to investigate the desensitization of recombinant GABA receptors comprising rat α1, β2, and γ2 subunits.
MATERIALS AND METHODS
cDNA and cRNA preparation. The wild-type cDNAs of rat α1, β2, and γ2 subunits were subcloned into the vector pGEMHE for high expression (Liman et al., 1992). The plasmids were linearized byNheI, which left a >200 base pair tail for cRNA stability. RNase-free cDNA templates were prepared by treating linearized cDNA with proteinase K. Capped cRNAs were then transcribed by T7 RNA polymerase. After degradation of the DNA template by RNase-free DNase I, the cRNAs were purified and resuspended in diethyl pyrocarbonate (DEPC)-treated water. cRNA yield and integrity were examined on a 1% agarose gel.
Oocyte preparation and cRNA injection. FemaleXenopus laevis (Xenopus I, Ann Arbor, MI) were anesthetized by 0.2% MS-222. The ovarian lobes were surgically removed from the frog and placed in calcium-free OR2 incubation solution consisting of 92.5 mm NaCl, 2.5 mm KCl, 1 mmMgCl2, 1 mmNa2HPO4, 5 mm HEPES, 50 U/ml penicillin, and 50 μg/ml streptomycin, pH 7.5. The lobes were cut into small pieces and digested with 0.3% Collagenase A (Boehringer Mannheim, Indianapolis, IN) with constant stirring at room temperature for 1.5–2 hr. The dispersed oocytes were thoroughly rinsed with the above solution containing 1 mm CaCl2. Stage VI oocytes were selected, and the follicular layer, if still present, was manually removed with fine forceps. The oocytes were incubated at 18°C.
Micropipettes for cRNA injection were pulled from borosilicate glass (Drummond Scientific, Broomall, PA) on a Sutter Instruments (Novato, CA) P87 horizontal puller, and the tips were cut with scissors to an ∼40 μm outer diameter The cRNA with dilution in DEPC-treated water (for voltage clamp) or without dilution (for binding) was drawn up into the micropipette and injected into oocytes with a Nanoject microinjection system (Drummond Scientific) at a total injection volume of 20–60 nl.
Electrophysiology. One to 2 d after injection, the oocyte expressing α1β2γ2 GABA receptors was voltage clamped at −70 mV. Dose–response relationships were determined by measuring the current induced by a range of agonist concentrations. The EC50 and Hill coefficient were determined by fitting the data to the Hill equation of the following form:
| Equation 1 |
where I is the current amplitude for that particular agonist concentration ([A]), Imax is the maximum current amplitude, EC50 is the agonist concentration that induces a 50% maximal response, andn is the Hill coefficient.
Single oocyte binding. Two to 3 d after injection, the expression level of the α1β2γ2 GABA receptors was examined by two-electrode voltage clamp at −70 mV. Oocytes with a current in response to 10 μm GABA of >3000 nA were selected for the binding assay. Most of the oocytes tested had a current amplitude in the 4000–6000 nA range. The single oocyte binding was performed as described previously (Chang and Weiss, 1999a). Briefly, the oocyte expressing α1β2γ2 GABA receptors was held by gentle suction to the end of a sequencing gel loading pipette tip. The oocyte was then incubated in OR2 containing [3H]GABA or [3H]muscimol for 30 sec at room temperature, rinsed for 5 sec in a 0°C OR2 bath with constant stirring (to remove unbound 3H-ligand), and finally placed in 250 μl of OR2 at room temperature for 85 sec to let the bound 3H-ligand dissociate. The 250 μl of OR2 was then thoroughly mixed with 4 ml of scintillation fluid, and the radioactivity (in counts per minute) was determined in a liquid scintillation counter. This binding paradigm was followed by an accompanying run with the initial incubation in the same concentration of 3H-ligand plus 300 μm nonradiolabeled gabazine (SR95531) or bicuculline, antagonists of the GABAA receptor (Research Biochemicals, Natick MA). In this case, the counts per minute released is a measure of the nonspecific3H-ligand binding and, when subtracted from the total binding (counts per minute measured in the absence of the antagonist), provided a measure of the specific binding. Except for the experiments examining the dose dependence of3H-ligand binding, 1 μm3H-ligand was typically used to assess changes in the amount of bound ligand. Based on our estimate of the apparent KD(0.04 μm) and thekoff (τ = 30 sec) and the relationship kobs =kon * [GABA] +koff, we determined an association time constant for 1 μm GABA of 1.2 sec. Thus, our typical 30 sec incubation in 1 μm GABA is more than sufficient to reach equilibrium for binding to receptors already in the high-affinity state. However, during the incubation in3H-agonist, more receptors enter the desensitized state. In this case, the measurements were not at steady state. In addition, we demonstrate that GABA dissociates from the high-affinity state with a time constant of ∼30 sec at room temperature. Thus, we lose <15% of the bound [3H]GABA in the 5 sec cold rinse. In all binding paradigms, calculations confirmed that there would be no significant depletion of the3H-agonist.
For the measurement of the apparent binding affinity, a concentration range of 3H-ligand was examined. The relationship between the concentration of3H-ligand and the specific binding (B) were fit with the following form of the Hill equation:
| Equation 2 |
in which KD is the concentration of ligand (A) required for half-maximal binding,Bmax is the maximum binding of3H-agonist, and n is the Hill coefficient. We will use the term apparentKD (half-maximal binding) because this experiment does not necessarily measure the receptor affinity because the agonist–receptor interaction is not a simple bimolecular process (Colquhoun, 1998; Chang and Weiss, 1999a). The rate of dissociation was determined using the same binding protocol as described above, except that the final release of 3H-ligand was into sequential 250 μl wells of OR2. The dissociation data were least-squares fit by an exponential decay function.
The equilibrium dose–response relationship for [3H]GABA was determined by placing the oocyte in a 96 V-shaped well plate. The OR2 in the well was removed and replaced with 12 μl of OR2 with the desired [3H]GABA concentration. After a 30 min incubation, the solution was removed, and the oocyte was rinsed five times with 300 μl of cold OR2 over a 10 sec time period. Finally, 250 μl of room temperature OR2 was added to the well, and 10 min was allowed for the release of [3H]GABA. The OR2 was then collected, and radioactivity was determined in a liquid scintillation counter. The process was repeated but with the 30 min [3H]GABA incubation in the presence of 300 μm SR95531 to determine the nonspecific binding in the same oocyte.
Predictions of the proposed kinetic scheme. The prediction of the EC50 for functional desensitization by Scheme 2 (presented in Discussion) was determined from the following relationship:
| Equation 3 |
where D is the fraction of receptors in the desensitized state for a given GABA concentration, KR andKD are the agonist binding affinities of the resting and desensitized receptor, respectively, M is [R]/[D] or the ratio of the equilibrium occupancies of the resting and desensitized forms of the unbound receptor, and n is the maximum number of GABA molecules that can bind to the receptor, two in our case (Edelstein and Changeux, 1996).
RESULTS
Detection of specific binding in single oocytes expressing α1β2γ2 GABA receptors
Oocytes were injected with cRNA encoding for α1β2γ2 GABA receptors. Figure 1Ademonstrates that we can obtain specific binding of [3H]GABA or [3H]muscimol to individual oocytes expressing recombinant α1β2γ2 GABA receptors. We previously used this single oocyte radioactive ligand binding technique to study ρ1 GABA receptors in which, because of the lack of desensitization and extremely slow dissociation of GABA from its binding site, we were able to examine agonist association and dissociation to and from nondesensitized open and closed states (Chang and Weiss, 1999a). In contrast, the GABA-activated chloride currents from α1β2γ2 GABA receptors decay much faster than ρ1 receptors when agonist is removed (Amin and Weiss, 1994). Figure 1B shows a sample current response at 8°C in which one small drop (∼10 μl) of 20 μm GABA was added to an oocyte in a rapidly flowing 100 μl bath. The current decay was well fit by a single exponential function with a time constant of 0.56 ± 0.03 and 0.85 ± 0.08 sec at 23 and 8°C, respectively (n= 8). This 1.5-fold difference in the time constant of decay at the two temperatures was partly attributable to the measured 1.2-fold slower perfusion rate at 8°C compared with 23°C and was likely attributable to a slight shrinkage of the diameter of the tubing at the lower temperature. Recombinant α1β2γ2 GABA receptors expressed in HEK293 cells and assayed with the patch-clamp technique (allowing a faster solution exchange than oocyte recording) exhibit a biexponential deactivation with the slowest time constant on the order of 180 msec (Tia et al., 1996). Thus, the current decay we observed in oocytes expressing α1β2γ2 GABA receptors likely represents, in large part, the rate of agonist removal from the bath rather than the underlying receptor kinetics.
Fig. 1.

Specific binding to individual oocytes expressing recombinant GABAA receptors. A, Oocytes were incubated in 1 μm [3H]GABA or 2 μm [3H]muscimol for 30 sec as described in Materials and Methods. Nonspecific binding was determined in the presence of 300 μm SR95531. Specific binding would be the difference between the total and nonspecific binding (in counts per minute). B, An oocyte expressing GABAAreceptors was voltage clamped at −70 mV at 8°C, and 10 μl of 20 μm GABA was added in a rapidly flowing 100 μl bath. The current decay was well fit by a single exponential function with a time constant of 0.85 ± 0.08 sec (n = 8). Thearrow indicates 5 sec, the duration of the cold rinse in the binding experiments. Note that, by 5 sec, the current returned to baseline.
In the binding assay, the initial incubation in3H-ligand was followed by a 5 sec 0°C rinse to wash away the free 3H-ligand from the surface of the oocyte. The rinse time was approximately sixfold longer than the observed time constant of current decay (Fig.1B, arrow), indicating that ∼99.7% of the receptors would have closed by the end of the rinse. (This percentage is likely to be a lower limit given that the observed current decay reflects agonist removal in the perfusion chamber rather than agonist dissociation, and, in addition, the rinse with 150 ml of OR2 in a beaker with constant stirring should have a faster exchange rate around the oocyte surface than that of the recording chamber.) So, if channels are closing and agonist is dissociating within 5 sec, what is the source of the specific binding in Figure 1A? One possible explanation is that the3H-ligand is binding to a state of the receptor with higher affinity than that of the state(s) along the activation pathway. The data in Figure 1Bdemonstrates that the binding is clearly not to the presumed high-affinity open state (Chang and Weiss, 1999b). Although we cannot completely rule out that a small fraction of the binding in Figure1A is to nondesensitized states (closed or open), evidence will be provided that the majority of the binding is to the high-affinity desensitized state.
Increased binding in desensitized receptors
Figure 2A shows a current in response to a 150 sec application of 500 μm GABA in an oocyte expressing α1β2γ2 GABA receptors. Note that, by the end of the prolonged GABA application, the current decayed toward a steady-state level that was ∼5% of the peak current. Thus, a majority of the receptors were functionally desensitized. This current decay was not attributable to a change in the chloride gradient because we compared the reversal potentials during a maintained application of 2 μm GABA (−20.7 ± 1.7; n= 3), which shows little decline in current, and at the end of the prolonged application of 500 μm GABA (−23.4 ± 2.0; n = 3). This represents a 5.6 ± 1.4% decrease in the driving force at the end of the 500 μm GABA application, which would only account for a small fraction of the >95% decrease in current amplitude. Thefilled bar in Figure 2B plots the specific binding in single oocytes expressing α1β2γ2 GABA receptors that were first exposed to 500 μm nonradiolabeled GABA for 150 sec, rinsed for 30 sec, and then tested for specific binding of 1 μm[3H]GABA using the same protocol as in Figure 1A. The open bar plots the specific binding from the same oocytes but before the 150 sec 500 μm GABA incubation. Desensitization of the receptors increased the specific binding from 163 ± 25 to 1253 ± 309 cpm (n = 8), a 7.7-fold increase. In the case of the desensitized receptors, a fraction of the binding sites would still be bound by unlabeled GABA at the end of the 30 sec rinse. Thus, we would be measuring binding in the fraction of receptors from which GABA has dissociated. The observation that the specific binding increased with the 500 μm GABA preincubation suggests that, although the receptors released GABA, they are still desensitized. In a subsequent section, a more direct demonstration of this will be provided. Nevertheless, an interpretation of this increased binding is that the desensitized receptors have a higher affinity for agonist.
Fig. 2.

Increased binding of 1 μm[3H]GABA after receptor desensitization.A, Application of 500 μm GABA for 150 sec desensitized the GABA receptors. B, Specific binding of 1 μm [3H]GABA was determined in an oocyte, and then the oocyte was incubated in 500 μm GABA for 150 sec and rinsed for 30 sec, and the specific binding of 1 μm [3H]GABA was determined again in the same oocyte. Desensitization increased the specific binding from 163 ± 25 to 1253 ± 309 cpm (n = 8), a 7.7-fold increase.
Relationship between the concentration dependence of the increased binding and functional desensitization
If the increase in binding in Figure 2B were to the desensitized state of the receptor, then the concentration dependence of the increased binding should be similar to the concentration dependence of functional desensitization. Figure3A shows our method for assessing the concentration dependence of functional desensitization. First, we measured the current in response to 1 μm GABA. Next, a variable concentration of GABA was applied for 150 sec to desensitize the receptors. Thirty seconds from the termination of the GABA application, the current in response to 1 μm GABA was tested again. The level of functional desensitization was the ratio of the 1 μm GABA applications after and before desensitization. This ratio is normalized and plotted in Figure3B (open circles). The solid line is a fit of Equation 1 to these data and yielded an EC50 for functional desensitization of 9.8 ± 3.4 μm. We used the same protocol (agonist concentrations and incubation times) to examine the concentration dependence of the increased binding. The filled circles in Figure 3B plot the increase in binding of 1 μm [3H]GABA (B) normalized to the maximum binding increase (Bmax) as a function of the concentration of nonlabeled GABA used to desensitize the receptors. Thecontinuous line is a least-squares fit of Equation 2 to these data and yielded an apparent KDof 10.1 ± 1.2 μm (n = 4). Equilibrium was likely not achieved at the lower GABA concentrations in either the functional or binding experiments of Figure 3. Thus, the EC50s and apparentKD values are not accurate determinations of the GABA concentration required to desensitize half the receptors or the GABA concentration required for a half-maximal increase in binding. Nevertheless, the goal was to correlate the concentration dependence of functional desensitization and [3H]GABA binding. Because the relationships are remarkably similar, these data further support the notion that the measured binding is to the desensitized state of the receptor.
Fig. 3.

Correlation of the concentration dependence of the increased binding and functional desensitization. A, Oocytes were first tested with 1 μm GABA and desensitized for 150 sec with 1, 3.16, 10, 31.6, and 100 μm GABA, and then retested with 1 μm GABA to assess the fraction of functionally desensitized receptors. Three representativetraces from an oocyte desensitized with 1, 3.16, and 10 μm GABA are superimposed. B, Theopen circles plot the fraction of desensitized receptors as a function of the concentration of GABA used in the desensitizing application. The same protocol was used to assess the concentration dependence of increased binding (filled circles). Both functional desensitization and increased binding show the same concentration dependence with an EC50 of 9.8 ± 3.4 μm (n = 3) and an apparentKD of 10.1 ± 3.2 μm(n = 4), respectively.
Relationship between the time course of the disappearance of the increased binding and the time course of functional recovery
If the increased binding is to desensitized receptors, then the time course of the disappearance of the increased binding should be similar to the time course of functional recovery from desensitization. Figure 4A shows the paradigm for assessing the time course of recovery from the functionally desensitized state. After a stable response to 1 μm GABA was obtained, the receptors were desensitized for 150 sec in 500 μm GABA. At the end of this prolonged GABA application and a 30 sec rinse, 30 sec pulses of 1 μm GABA were applied at 2 min intervals. The filled circles in Figure 4Aplot the fractional recovery of the current versus time. The recovery was well described by a single exponential function with a time constant of 124 ± 26 sec (n = 5). A similar protocol (30 sec incubation, 5 sec rinse, 85 sec release) was used to assess the disappearance of the increased binding after a 150 sec incubation of 500 μm GABA and 30 sec rinse (Fig. 4B). The decay of the increased binding was dominated by an exponential function with a time constant of 127 ± 12 sec (n = 4). Note that, after 600 sec, the binding was still slightly elevated over the original baseline, suggesting the presence of an additional slower component. Nevertheless, the similarity of the time constants that dominate the recoveries further supports the hypothesis that the observed3H-ligand binding reflects the desensitized state of the receptor.
Fig. 4.

Correlation of the time course of the recovery of function and increased binding after receptor desensitization.A, After a stable response to 1 μm GABA was obtained, 500 μm GABA was applied for 150 sec, followed by a 30 sec rinse, and a 1 μm 30 sec test pulse of GABA was applied in 2 min intervals. The filled circles plot the recovery, and the solid line is from a fit of a single exponential function yielding the indicated time constant. B, The same protocol was performed to assess the decay of the increased binding, which also decreased as a single exponential function (solid line) with a time constant very similar to that observed for functional recovery.
Comparison of muscimol activation and binding
The EC50 for α1β2γ2 GABA receptors is ∼46 μm for GABA compared with ∼14 μmfor muscimol (Amin and Weiss, 1993). Furthermore, the available stock concentrations of [3H]GABA is 11 μm compared with 51 μm for [3H]muscimol. Thus, we can examine [3H]muscimol binding to the receptor across a wider activation range than for [3H]GABA. The open circles in Figure 5A plot the normalized current amplitude as a function of muscimol concentration in oocytes expressing α1β2γ2 GABA receptors. These data were fit with a Hill equation (continuous line) yielding an EC50 of 5.3 ± 1.0 μm and a Hill coefficient of 1.60 ± 0.10 (n = 5). We next conducted binding studies over a comparable range of [3H]muscimol concentrations. The filled circles in Figure 5Aplot the specific binding as a function of the [3H]muscimol concentration. Fitting a Hill equation to the mean of the binding data yielded a dominant apparent KD of 6.3 μm (n = 6), which agreed well with that determined in the electrophysiological analysis of muscimol-mediated activation.
Fig. 5.
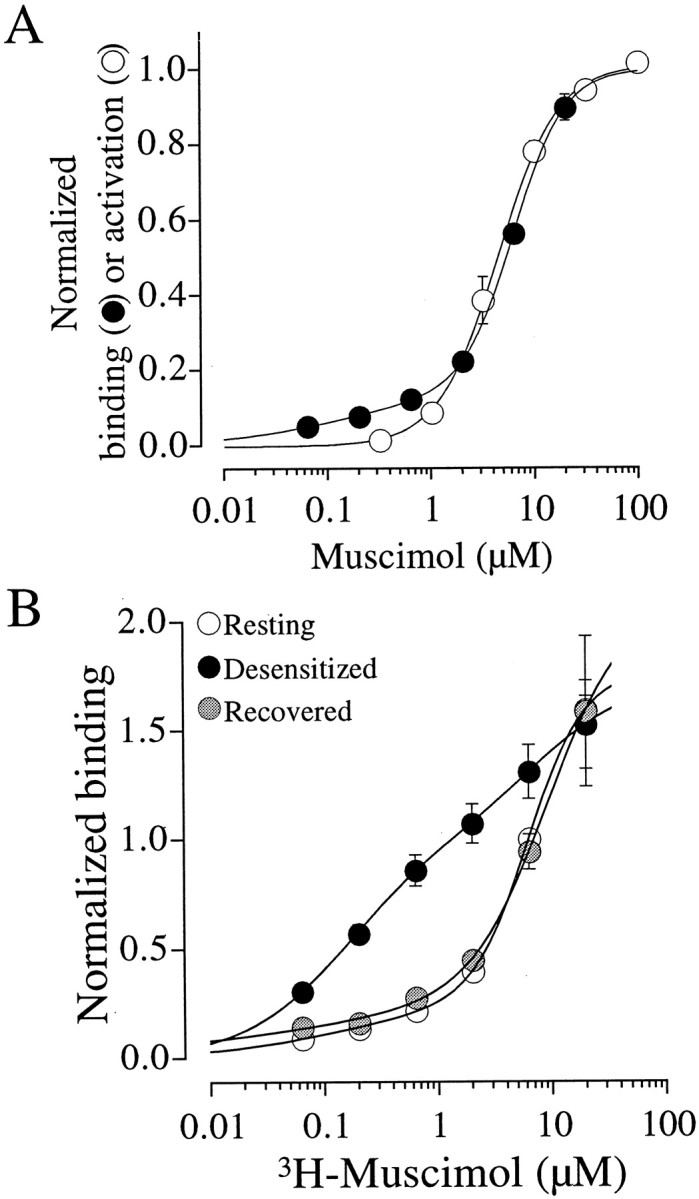
Muscimol binding and activation. A, Correlation of the concentration dependence of activation and binding by muscimol. The open circles plot the dose–response relationship for muscimol. The solid line is a fit of the Hill equation yielding an EC50 of 5.3 ± 1.0 μm and a Hill coefficient of 1.60 ± 0.10 (n = 5). The specific binding for [3H]muscimol (filled symbols) demonstrated a dominant apparentKD of 6.3 μm and a Hill coefficient of 1.76. The reported binding parameters represent fits to the mean rather than the mean of the fits to individual oocytes. For both binding and activation, the data were normalized to the maximum predicted from a least-squares fit to the data points.B, Desensitization shifts the receptors into a high-affinity state. Specific binding is plotted as a function of [3H]muscimol. For the resting (open symbols) and recovered (shaded symbols) states, oocytes were incubated with the indicated concentrations of [3H]muscimol and specific binding was determined as in Figure 1A. In the desensitized case (filled symbols), the oocyte was incubated in 500 μm GABA for 150 sec and rinsed for 30 sec before each test concentration. For all three relationships, the data were well described by the sum of two Hill equations with EC50 values and Hill coefficients provided in Table 1. For each oocyte, the data were normalized to the value at 6.3 μm in the resting state.
If the desensitized state is of higher affinity than the resting closed state, the question arises as to why the EC50 for activation and the apparent KD for binding in Figure 5A are in excellent agreement. In the binding studies, prolonged exposure to muscimol drives the receptors to the high-affinity desensitized state to which we can measure bound [3H]muscimol. In the electrophysiological studies, the receptors must be driven through the same set of states on the pathway to channel opening. Thus, a possible interpretation of the data in Figure 5A is that the concentration dependence is similar for both activation and desensitization because they share common agonist concentration-dependent binding steps.
Direct demonstration that desensitization increases the apparent affinity of the receptor
The data thus far suggests that the desensitized state has an increased agonist affinity. To directly test this hypothesis, we examined binding as a function of [3H]muscimol concentration as in Figure5A, except each test concentration of [3H]muscimol was preceded by a 150 sec preincubation in 500 μm GABA, followed by a 30 sec rinse. The open circles in Figure 5B plot the binding as a function of [3H]muscimol concentration without predesensitization (resting). The filled circles plot the binding as a function of [3H]muscimol with predesensitization before each test concentration of [3H]muscimol (desensitized). Note that the dose–binding relationship shifted to the left, indicating a higher apparent affinity. The receptors were then allowed to recover, and the binding as a function of [3H]muscimol (without predesensitization) was reexamined (Fig. 5B,shaded circles, Recovered). The dose–binding relationship returned to the control, resting level.
For all three conditions in Figure 5B (Resting,Desensitized, and Recovered), the sum of two Hill equations was required to fit the binding curves, suggesting a minimum of two apparent affinities. Fitting a single component gave an inadequate description of the data. Table1 provides the parameters for the two components. These data indicate a high-affinity component of <200 nm and a low-affinity component in the 6–9 μm range (Table 1). In the resting and recovered curves, the fraction of the high-affinity component was 0.15 in both cases, whereas the fraction of the high-affinity component after desensitization was 0.58. Because of the small amplitude of the high-affinity component in the resting and recovered states, as well as the low number of data points, the fitting failed to converge to a solution. To estimate the parameters, we fixed the apparentKD of the high-affinity component to the value we determined in the desensitized state. Regardless of the specific values of the parameters, we interpret the two components as follows. Without predesensitization, a small population of receptors may already exist in the desensitized state (see Discussion). Thus, we measure a minor high-affinity component and a dominant low-affinity component. The same is true for the recovered state. During desensitization, however, receptors shift to a high-affinity state, and, therefore, the fraction of the high-affinity component was increased. In the desensitized situation, the low-affinity component was still significant because recovery is occurring during both the 30 sec rinse after the 500 μm GABA desensitization and the 30 sec incubation in [3H]muscimol.
Table 1.
Parameters from the fit of the sum of two Hill equations to the relationship between the concentration dependence of [3H]muscimol binding
| High-affinity component | Low-affinity component | ||||
|---|---|---|---|---|---|
| EC50 1 (μm) | Hill 1 | Fraction 1 | EC50 2 (μm) | Hill 2 | |
| Resting | 0.16 | 0.72 | 0.15 | 6.3 | 1.76 |
| Desensitized | 0.16 | 0.95 | 0.15 | 7.8 | 1.02 |
| Recovered | 0.16 | 0.41 | 0.58 | 9.4 | 1.29 |
The data represent the average of six oocytes for the resting and desensitized states. Only two oocytes survived through the recovery. The parameters are a fit to the mean, and therefore no SEs are provided.
Rate of dissociation of [3H]GABA from desensitized receptors
As demonstrated in Figure 2, desensitized receptors show an enhanced binding of [3H]GABA. Unless new binding sites are being uncovered by desensitization (an unlikely scenario), these data suggest that GABA must dissociate from the desensitized receptor faster than the disappearance of the high-affinity state (Fig. 4B). To quantify the rate of dissociation, we examined the time course of [3H]GABA dissociation from the desensitized receptors. Oocytes were exposed to 500 μm nonradiolabeled GABA for 150 sec, rinsed for 30 sec, and then incubated in 1 μm[3H]GABA for 30 sec. Figure6 shows the time course of dissociation of [3H]GABA. The continuous line is the best fit of a single exponential component, which yielded a time constant of 30 ± 2 sec (n = 3). There is evidence of a faster component, but we do not have the temporal resolution to establish its kinetics. Nevertheless, these data, coupled with the data in Figure 4 demonstrating a functional recovery with a time constant of 124 sec, indicate that receptors can remain in a desensitized state long after agonist has dissociated from its binding site.
Fig. 6.

Dissociation rate of [3H]GABA from desensitized receptors. Receptors were first desensitized with 500 μm GABA and then bound with 1 μm[3H]GABA. The filled symbols plot the [3H]GABA released in 6 sec intervals. The dissociation was well described by a single exponential component (solid line) yielding the indicated time constant. The data were normalized to the first point of the control run before desensitization of the receptors. The plotted value is actually the absolute value of the derivative of dissociation. However, the derivative of an exponential function has the same time constant, so the reported τ is the same as that for the dissociation.
In a previous study using single oocyte binding and concomitant two-electrode voltage clamp on oocytes expressing recombinant ρ1 GABA receptors, we identified an ∼10-fold excess in the number of receptors determined by binding compared with electrophysiology (Chang and Weiss, 1999a). This comparison was possible because of the slow agonist dissociation and lack of desensitization of the receptor. Unfortunately, because of the rapid activation and desensitization kinetics of the α1β2γ2 receptor and because desensitized receptors do not conduct current, we were unable to make a similar comparison. Although we cannot rule out excess binding, the high correlation between the concentration and time dependence of desensitization as assessed by binding and electrophysiology (Figs. 3,4) gives us confidence that we are probing the same process(es) with the two measurements.
DISCUSSION
Part of the difficulty in elucidating the mechanism of desensitization has been an inability to resolve the kinetics of both agonist binding and functional desensitization in the same set of operable receptors. To this end, we used the single oocyte binding technique (Chang and Weiss, 1999a), along with the two-electrode voltage clamp, to investigate the mechanism of GABA receptor desensitization.
We were able to measure specific binding of [3H]GABA and [3H]muscimol in individual oocytes expressing recombinant α1β2γ2 GABA receptors. We concluded that the observed specific binding was to the desensitized state of the receptor based on the following observations. First, the specific binding increased dramatically (approximately eightfold) after the receptors were desensitized. Second, the concentration dependence of functional desensitization (EC50 of 9.8 μm) and increased binding (apparentKD of 10.1 μm) were similar. And third, the increase in binding after desensitization recovered with a time constant of 127 sec, similar to the 124 sec time constant of recovery from functional desensitization. Having established that the observed binding was to the functionally desensitized state, we addressed two fundamental issues regarding the mechanism of GABA receptor desensitization: (1) the dissociation rate of agonist and its relationship to functional recovery and (2) the affinity of the desensitized state.
Relationship between the dissociation rate of agonist and the lifetime of the functionally desensitized state
A cyclical model of receptor desensitization (Scheme 1) (Katz and Thesleff, 1957) implies that agonist can dissociate directly from the desensitized receptor. Thus, a receptor may have released agonist yet still be functionally desensitized. Previous evidence in support of this notion is that the recovery rate of desensitized nACh receptors does not depend on the particular agonist (Rang and Ritter, 1970). The basis for this argument is that, if agonist remained bound, the affinities of the different agonists would determine their rates of recovery. Additional evidence for the cyclical model is that the observed rate of agonist dissociation from desensitized receptors in membrane preparations was faster than the rate of functional recovery (Quast et al., 1978; Boyd and Cohen, 1980). The extent to which this holds true for operable surface receptors was unclear. This is an important consideration because membrane isolation can alter the apparent receptor affinity (Fenster et al., 1999).
In this study, we were able to directly correlate dissociation and recovery in intact surface receptors. We found that agonist dissociation from desensitized GABA receptors was dominated by a single exponential component with a time constant of 30 ± 2 sec, whereas GABA receptors recovered from functional desensitization with a time constant of 124 ± 26 sec. These data indicate that a basic model of GABA receptor desensitization must be cyclical and include the ability of agonist to dissociate directly from the desensitized receptor (AD → D in Scheme I). In fact, this must be the dominant pathway for recovery, otherwise the rate of recovery and the rate of dissociation would be the same. For example, receptors recovering from AD to AR (Scheme I) will move along a pathway from which they can be reactivated. Some reactivation may occur, and this is evident in Figure3A in which the tail of the GABA-activated current, because of the increased level of desensitization, is slowed with increasing GABA concentration.
Jones and Westbrook (1995) observed that GABA receptor deactivation was slowed by desensitization and modeled their data using a noncyclical model. Because they did not examine binding and the relationship between dissociation and functional recovery, we do not know whether a cyclical model would be more appropriate. The desensitization and recovery in the Jones and Westbrook study were dominated by rates faster than we could resolve in our binding and electrophysiological analyses. The question remains as to how their findings relate to the present study. It should be kept in mind that they probed a heterogeneous population of native GABA receptors in mammalian neurons with brief GABA applications, whereas we examined a homogeneous population of recombinant receptors expressed in oocytes desensitized for extended time periods. Furthermore, we are studying GABA receptors in an exogenous expression system that may lack components (cytoskeletal elements, linker proteins, and kinases) that contribute to the properties of receptor activation and desensitization. The relationship between their mechanism and the mechanism we presented here and the degree to which this difference can be attributed to the expression system, receptor subtype, and/or temporal resolution must await future studies. More recently, a slow component of desensitization (τrecovery ∼15 sec) was investigated in hippocampal neurons and shown to influence synaptic inhibition by decreasing GABA receptor availability. Therefore, slow components of desensitization may serve a physiological role (Overstreet et al., 2000).
Does desensitization increase the affinity of the receptor for agonist?
Indeed, evidence has supported the hypothesis that desensitized receptors have a higher agonist affinity than the resting closed state (Rang and Ritter, 1970; Weber et al., 1975; Weiland et al., 1975; Quast et al., 1978; Sine and Taylor, 1979; Neubig et al., 1982; Heidmann et al., 1983), although these measurements have been typically performed in membrane or vesicle preparations in which the functional integrity of the receptors was unknown. Using the single oocyte binding technique, we have been able to compare the apparent affinity of the same set of surface receptors, before and after receptor desensitization. In both cases, the relationship between the concentration of [3H]muscimol and the specific binding was described by the sum of two Hill equations: a component with an apparent KD of ∼8.0 μm and a higher-affinity component with a apparent KD of ∼0.20 μm. The fraction of the specific binding in the high-affinity component increased with receptor desensitization. The simplest interpretation of these data are that desensitization shifted receptors from a low- to high-affinity state. Although a minimum of two apparent affinities seems inescapable from the data in Figure5B, caution must be exercised in interpreting the parameters of these components. First, by the nature of the binding assay, the relative amplitudes of the two components reflect a partial recovery of the receptors from desensitization. Second, some fraction of the receptors were desensitized by the [3H]muscimol incubation used to assess the level of desensitization. Third, in the case of the specific binding before receptor desensitization in which the high-affinity component was relatively small, it was necessary to constrain the apparent KD for the fitting algorithm to converge. Finally, caution must be exercised in interpreting the apparent KD values because they do not represent true binding affinities, and their values must be considered in light of a specific kinetic scheme for receptor desensitization (Colquhoun, 1998). It should be mentioned that, at steady state, only a single component would be observed in the dose dependence of agonist binding. However, these measurements were not taken at steady state, thus permitting the appearance of the two apparent affinities.
Working hypothesis for desensitization of GABAA receptors
Although the consideration of a complete model with transitions between the resting, open, and desensitized states is beyond the scope of this study, we can consider select transitions to and from the desensitized states, begin to define the most likely pathways, and put some limitations on select rates. Scheme

2 is similar to Scheme 1 but has been modified to account for the two agonist binding steps. In this allosteric scheme,KR represents the affinity of the resting state (the open states are not depicted in this scheme),KD represents the affinity for binding to the desensitized states, M is [R]/[D], and a isKD/KR.
In a previous study, taking advantage of a mutation that promotes spontaneous opening of the α1β2γ2 GABA receptor, we were able to estimate KR (Chang and Weiss, 1999b). The desensitized states were not included in the derivation of these parameters. If entry into desensitized states are significant along the activation pathway (R → AR → A2R → A2O), then desensitization would affect the derivation of these values. Nevertheless, we estimatedKR to be ∼78.5 μm.
In the present study, we examined receptors under conditions that favor receptor desensitization. We approximated theKD for muscimol (Fig. 5B), but, because a majority of the experiments we presented in this study were with GABA, we set out to determine the apparent affinity of the desensitized receptor for [3H]GABA (Fig.7A). In this case, the binding was performed with a 30 min incubation at each concentration of [3H]GABA to ensure that equilibrium was obtained. The relationship between specific binding and concentration was well described by a single Hill equation with a apparentKD of 0.04 μmand a Hill coefficient of 1.22. In contrast to muscimol (Fig.5B), a second component was not seen because the resting state affinity (∼78 μm) would be too low to resolve in this assay. Early studies examining [3H]GABA binding in synaptic membrane fractions revealed a KD on the order of 100 nm (Zukin et al., 1974). Subsequently, a heterogeneity of binding sites was observed withKD values in the 10–20 and 100–200 nm range (Olsen et al., 1981). Whether these two affinities represent an interconvertible pool or distinct receptor populations was unclear. The interpretation ofKD values determined from membrane preparations is further complicated by a study in which the apparent binding affinity of nicotine to nACh receptors expressed in oocytes was compared in both intact oocytes and membranes prepared from these oocytes. In this case, the membrane preparation demonstrated an ∼25-fold higher affinity for nicotine, suggesting that membrane isolation may alter the apparent receptor affinity or uncover new high-affinity binding sites (Fenster et al., 1999).
Fig. 7.
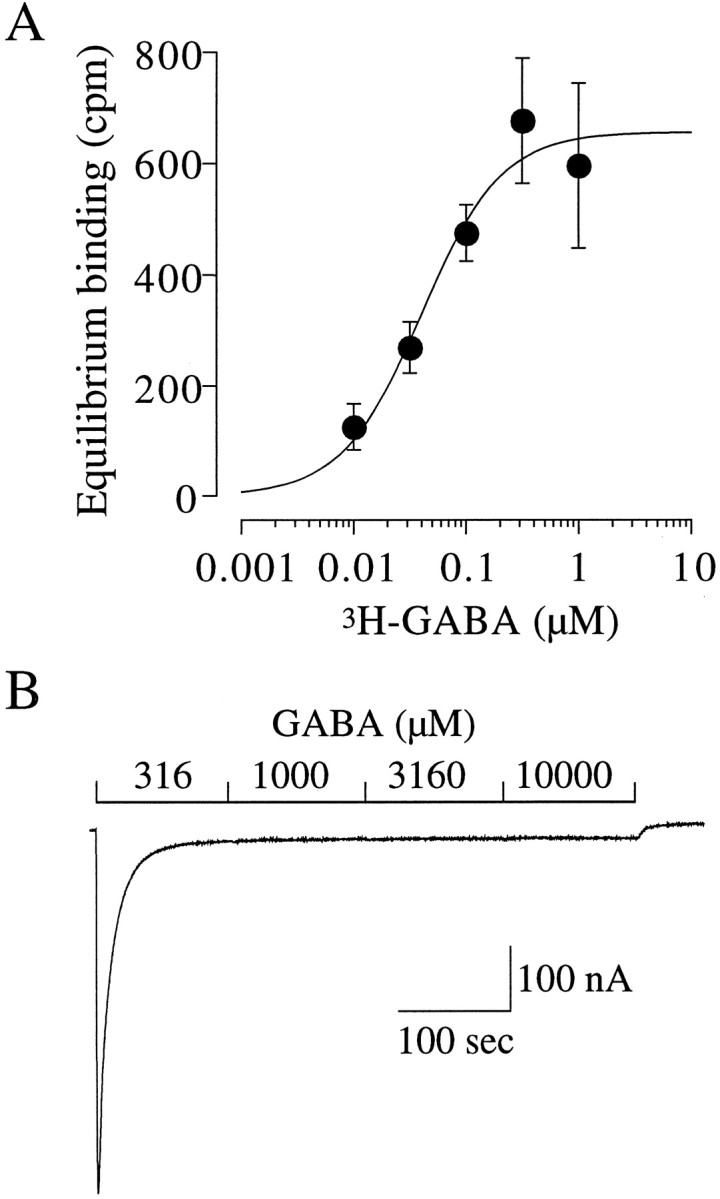
Equilibrium binding and steady-state desensitization. A, Equilibrium binding of [3H]GABA. Oocytes were incubated for 30 min with the indicated concentrations of [3H]GABA. Thefilled symbols represent the average of three oocytes. The continuous line is a fit of the Hill equation of these data yielding a apparent KD of 0.040 μm and a Hill coefficient of 1.22. B, Determination of the steady-state level of desensitization. An oocyte was voltage clamped at −70 mV, and the indicated concentrations of GABA were applied successively. All four concentrations of GABA were saturating in terms of desensitization. The amplitude of the baseline current at the end of the application was used to calculate the maximum fraction of receptors in the desensitized state, which was 0.049 of the peak GABA-activated current.
The KD is the ratio of the microscopic off and on rates. Because we determined the dissociation rate for GABA (∼0.03 sec−1) (Fig. 6), thekon for the desensitized state would be ∼0.75 μm−1 sec−1. In Scheme 2, a =KD/KRor 0.00051. At saturating concentrations of GABA, the receptors would be at equilibrium between A2D and A2R. We can estimate the population of state A2D from the experiment shown in Figure7B in which receptors were exposed to 316, 1000, 3160, and 10000 μm GABA. These are all saturating concentrations of GABA in terms of desensitization because the steady-state current level did not change. At the end of the series of applications, 0.049 of the peak current remained. This value is an upper limit because we likely underestimated the peak attributable to the necessarily slow application rate of GABA to oocytes. Nevertheless, after correction for the maximum open probability of 0.84 (Chang and Weiss, 1999b) based on Scheme 2, Ma2= [A2R]/[A2D] = 0.043 and therefore M = 165320. This model suggests that, in the absence of GABA, 6.0 × 10−6 of the receptors would be in the desensitized state. Again, these parameters are only approximations, and the equilibrium constants would depend on the specific kinetic scheme. For example, it is conceivable that the desensitized states communicate mainly with the open states. Nevertheless, our demonstration of a cyclical model for desensitization would still hold, as would our estimation of the apparent affinity of the desensitized state. From our previous analysis of receptor activation and the present consideration of desensitization, we provided estimates of the apparent KDvalues of the resting, open, and desensitized states to be ∼78.5 μm, 120 nm, and 40 nm, respectively. It is interesting that a biochemical study of [3H]GABA binding in native membranes from bovine cortex suggested three sites in terms of affinity with KD values of 1 μm, 100 nm, and 10 nm. These affinities were hypothesized to represent the resting, open, and desensitized states, respectively (Olsen et al., 1984).
Can Scheme 2 account for our experimental data? Using Scheme 2, the derived equilibrium constants, and, in Equation 3, we predicted an EC50 for functional desensitization of 19.4 μm (see Materials and Methods) that is close to the experimentally observed value of 9.8 μm. Clearly this is a simplified model because it does not include the open states. Inclusion of transitions from the open states to desensitized states could shift the predicted estimate toward the observed value. A complete description of the experimental results must await a more comprehensive model of receptor activation and desensitization.
Footnotes
This work was supported in part by National Institutes of Health Grants NS35291, NS36195 (D.S.W.), and DK07545 (Y.C.).
Correspondence should be addressed to Dr. David S. Weiss, Department of Neurobiology, University of Alabama at Birmingham, 1719 Sixth Avenue South, CIRC410, Birmingham, AL 35294-0021. E-mail:dweiss@nrc.uab.edu.
REFERENCES
- 1.Amin J, Weiss DS. GABAA receptor needs two homologous domains of the β-subunit for activation by GABA but not by pentobarbital. Nature. 1993;366:565–569. doi: 10.1038/366565a0. [DOI] [PubMed] [Google Scholar]
- 2.Amin J, Weiss DS. Homomeric ρ1 GABA channels: activation properties and domains. Receptors Channels. 1994;2:227–236. [PubMed] [Google Scholar]
- 3.Birnir B, Tieney ML, Dalziel JE, Cox GB, Gage PW. A structural determinant of desensitization and allosteric regulation by pentobarbitone of the GABAA receptor. J Membr Biol. 1997;155:157–166. doi: 10.1007/s002329900167. [DOI] [PubMed] [Google Scholar]
- 4.Boyd ND, Cohen JB. Kinetics of binding of [3H]acetylcholine to Torpedo postsynaptic membranes: association and dissociation rate constants by rapid mixing and ultrafiltration. Biochemistry. 1980;19:5353–5358. doi: 10.1021/bi00564a032. [DOI] [PubMed] [Google Scholar]
- 5.Chang Y, Weiss DS. Channel opening locks agonist onto the GABAC receptor. Nat Neurosci. 1999a;2:219–225. doi: 10.1038/6313. [DOI] [PubMed] [Google Scholar]
- 6.Chang Y, Weiss DS. Allosteric activation mechanism of the α1β2γ2 GABAA receptor revealed by mutation of the conserved M2 leucine. Biophys J. 1999b;77:2542–2551. doi: 10.1016/s0006-3495(99)77089-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Changeux J-P, Edelstein SJ. Allosteric receptors after 30 years. Neuron. 1998;21:959–980. doi: 10.1016/s0896-6273(00)80616-9. [DOI] [PubMed] [Google Scholar]
- 8.Colquhoun D. Binding, gating, affinity, and efficacy: the interpretation of structure-activity relationships for agonists and of the effects of mutating receptors. Br J Pharmacol. 1998;125:923–948. doi: 10.1038/sj.bjp.0702164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Edelstein SJ, Changeux J-P. Allosteric proteins after thirty years: the binding and state functions of the neuronal α7 nicotinic acetylcholine receptors. Experientia. 1996;52:1083–1090. doi: 10.1007/BF01952106. [DOI] [PubMed] [Google Scholar]
- 10.Fenster CP, Whitworth TL, Sheffield EB, Quick MW, Lester RAJ. Upregulation of surface α4β2 nicotinic receptors is initiated by receptor desensitization after chronic exposure to nicotine. J Neurosci. 1999;19:4804–4814. doi: 10.1523/JNEUROSCI.19-12-04804.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heidmann T, Bernhardt J, Neumann E, Changeux JP. Rapid kinetics of agonist binding and permeability response analyzed in parallel on acetylcholine rich membranes from Torpedo marmorata. Biochemistry. 1983;22:5452–5459. doi: 10.1021/bi00292a029. [DOI] [PubMed] [Google Scholar]
- 12.Jones MV, Westbrook GL. Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron. 1995;15:181–191. doi: 10.1016/0896-6273(95)90075-6. [DOI] [PubMed] [Google Scholar]
- 13.Katz B, Thesleff S. A study of the desensitization produced by acetylcholine at the motor end-plate. J Physiol (Lond) 1957;138:63–80. doi: 10.1113/jphysiol.1957.sp005838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liman ER, Tytgat J, Hess P. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron. 1992;9:861–871. doi: 10.1016/0896-6273(92)90239-a. [DOI] [PubMed] [Google Scholar]
- 15.Neubig RR, Boyd ND, Cohen JB. Conformations of Torpedo acetylcholine receptor associated with ion transport and desensitization. Biochemistry. 1982;21:3460–3467. doi: 10.1021/bi00257a032. [DOI] [PubMed] [Google Scholar]
- 16.Olsen RW, Bergman MO, Van Ness PC, Lummis SC, Watkins AE, Napias C, Greenlee DV. γ-aminobutyric acid receptor binding in mammalian brain. heterogeneity of binding sites. Mol Pharmacol. 1981;19:217–227. [PubMed] [Google Scholar]
- 17.Olsen RW, Wong EHF, Stauber GB, King RG. Biochemical pharmacology of the γ-aminobutyric acid receptor/ionophore protein. Fed Proc. 1984;43:2773–2778. [PubMed] [Google Scholar]
- 18.Overstreet LS, Jones MV, Westbrook GL. Slow desensitization regulates the availability of synaptic GABAA receptors. J Neurosci. 2000;20:7914–7921. doi: 10.1523/JNEUROSCI.20-21-07914.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quast U, Schimerlik M, Witzemann V, Blanchard S, Raftery MA. Ligand-induced conformation changes in Torpedo californica membrane-bound acetylcholine receptor. Biochemistry. 1978;17:2405–2414. doi: 10.1021/bi00605a024. [DOI] [PubMed] [Google Scholar]
- 20.Rang HP, Ritter JM. On the mechanism of desensitization at cholinergic receptors. Mol Pharmacol. 1970;6:357–382. [PubMed] [Google Scholar]
- 21.Sine SM, Taylor P. Functional consequences of agonist-mediated state transitions in the cholinergic receptor. Studies in cultured muscle cells. J Biol Chem. 1979;254:3315–3325. [PubMed] [Google Scholar]
- 22.Tia S, Wang JF, Kotchabhakdi N, Vicini S. Distinct deactivation and desensitization kinetics of recombinant GABAA receptors. Neuropharmacology. 1996;35:1375–1382. doi: 10.1016/s0028-3908(96)00018-4. [DOI] [PubMed] [Google Scholar]
- 23.Weber M, David-Pfeuty T, Changeux J. Regulation of binding properties of the nicotinic receptor protein by cholinergic ligands in membrane fragments from Torpedo marmorata. Proc Natl Acad Sci USA. 1975;72:3443–3447. doi: 10.1073/pnas.72.9.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weiland G, Georgia B, Lappi S, Chignell C, Taylor P. Kinetics of agonist-mediated transitions in state of the cholinergic receptor. J Biol Chem. 1975;252:7648–7656. [PubMed] [Google Scholar]
- 25.Zhu WJ, Vicini S. Neurosteroid prolongs GABAA channel deactivation by altering kinetics of desensitized states. J Neurosci. 1997;17:4022–4031. doi: 10.1523/JNEUROSCI.17-11-04022.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zukin SR, Young AB, Snyder SH. Gamma-aminobutyric acid binding to receptor sites in the rat central nervous system. Proc Natl Acad Sci USA. 1974;71:4802–4807. doi: 10.1073/pnas.71.12.4802. [DOI] [PMC free article] [PubMed] [Google Scholar]