

Abstract
The role of small GTPases of the Rho family in synaptic functions has been addressed by analyzing the effects of lethal toxin (LT) fromClostridium sordellii strain IP82 (LT82) on neurotransmitter release at evoked identified synapses in the buccal ganglion of Aplysia. LT82 is a large monoglucosyltranferase that uses UDP-glucose as cofactor and glucosylates Rac (a small GTPase related to Rho), and Ras, Ral, and Rap (three GTPases of the Ras family). Intraneuronal application of LT (50 nm) rapidly inhibits evoked acetylcholine (ACh) release as monitored electrophysiologically. Injection of the catalytic domain of the toxin similarly blocked ACh release, but not when key amino acids needed for glucosylation were mutated. Intraneuronal application of competitive nucleotide sugars that differentially prevent glucosylation of Rac- and Ras-related GTPases, and the use of a toxin variant that affects a different spectrum of small GTPases, established that glucosylation of Rac is responsible for the reduction in ACh release. To determine the quantal release parameters affected by Rac glucosylation, we developed a nonstationary analysis of the fluctuations in postsynaptic response amplitudes that was performed before and after the toxin had acted or during toxin action. The results indicate that neither the quantal size nor the average probability for release were affected by lethal toxin action. ACh release blockage by LT82 was only caused by a reduction in the number of functional release sites. This reveals that after docking of synaptic vesicles, vesicular Rac stimulates a membrane effector (or effectors) essential for the fusion competence of the exocytotic sites.
Keywords: Aplysia, synapse, exocytosis, quantal parameters, synaptic vesicle fusion, Rho-GTPases, clostridial toxins, fluctuation analysis
Rho proteins (Rho, Rac, Cdc42) are widely expressed monomeric GTPases. They cycle between a soluble, GDP-bound inactive state and a membrane-associated GTP-bound state that stimulates downstream effectors. The effectors include protein kinase N, Rho-kinase, the myosin-binding subunit of myosin phosphatase, several phosphatidylinositol protein kinases (PI3-kinase, PI4-kinase, PI5-kinase), and phospholipase C and phospholipase D (PLD). Despite the ubiquitous distribution of cytosolic Rho-GTPases, their translocation to specific membrane domains allows them to intervene in distinct biological functions such as regulation of actin cytoskeletal dynamics, cell cycle progression, gene transcription, and membrane transport (Hall, 1998; Bishop and Hall, 2000; Ridley, 2001). Recently, Rho, Rac, and Cdc42 have been implicated in the regulation of neuronal morphogenesis (axonal guidance, maintenance of dendritic spines and branching) (Luo, 2000; Nakayama et al., 2000; Dickson, 2001; Liu and Strittmatter, 2001; Redmond and Ghosh, 2001), control of neurotransmitter-receptor density (Meyer et al., 2000), and induction of neuronal apoptosis (Linseman et al., 2001; Mota et al., 2001). Our previous findings that RhoA, RhoB, Rac1, and Cdc42 are present in nerve terminals and that Rac1 is associated with synaptic vesicles (SVs) (Doussau et al., 2000) have suggested that these GTPases may also play a role in neurotransmitter release.
The cellular function of endogenous Rho and Ras proteins can be acutely manipulated by using bacterial protein toxins that modify and inactivate them in a highly specific manner (Busch and Aktories, 2000;Just and Boquet, 2000). Clostridium sordellii lethal toxin (LT) glucosylates a conserved threonine in the effector domain of Rac (which is related to Rho), and Ras, Rap, and Ral, (which are members of the Ras family). LT variants have been reported to also affect Cdc42. When glucosylated, the Rho-GTPases can associate with target membranes but cannot activate their effectors, thereby silencing the downstream pathways (Just et al., 1996; Popoff et al., 1996; Busch and Aktories, 2000; Just and Boquet, 2000).
We have found previously that intraneuronal injection of LT or toxins that glycosylate or ADP-ribosylate Rho-related GTPases blocks acetylcholine (ACh) release at Aplysia synapses (Doussau et al., 2000). This indicates that members of the Rho family play a role in neurotransmitter exocytosis and extends to neurons the idea that Rho-related proteins control secretion mechanisms (Prepens et al., 1996; Kowluru et al., 1997; Brown et al., 1998; Gasman et al., 1998,1999; Djouder et al., 2000; Hong-Geller and Cerione, 2000; Guo et al., 2001).
To identify the key GTPase(s) whose inactivation is responsible for the LT-blocking action on ACh release, we have investigated the causal relationship between the glycosylating activity of two LT variants that each affect a distinct set of GTPases and inhibition of neurotransmitter release at identified Aplysia synapses. We found that ACh release blockage is caused by glucosylation of Rac GTPase. To determine the release step(s) altered after LT application, we analyzed the fluctuations in postsynaptic response amplitudes before and during LT-induced blocking action. Our results show a decrease in the number of active release sites, but quantal size and release probability are unaltered. This reveals that the Rac-mediated pathway is implicated in the activation of release sites.
MATERIALS AND METHODS
Preparation of toxins and determination of their glucosylating activity
The toxins were purified from C. sordellii IP82 (LT82) and VPI9048 (LT9048) strains as described previously (Popoff, 1987). The DNA coding for the 546 N-terminal amino acids, which contain the enzymatic site (Hofmann et al., 1998), was amplified by PCR from C. sordellii IP82 strain. Primers were taken from the LT gene sequence (Green et al., 1995), and xSalI and XhoI sites were added at the 5′ and 3′ ends of the coding sequence, respectively. The resulting PCR DNA fragment was cloned into the corresponding sites of pET28b (Novagen). The plasmid was transformed into the Escherichia coli BL21 strain, and production of the 6 His-tag fusion protein was induced with isopropyl-1-thio-β-d-galactopyranoside. Purification was performed with a cobalt column (Novagen) according to the recommendations of the manufacturer. An inactive mutant of the LT82 N-terminal part (Ala-286-Asp, Ala-288-Asp) was prepared as described previously (Busch et al., 1998), and the protein was produced and purified as mentioned above.
Recombinant small GTPases were produced in E. coli, purified with glutathione-Sepharose 4B (Amersham Biosciences), and prepared by thrombin digestion according to the recommendations of the manufacturer. Rho, Rac1, and Cdc42 clones were a generous gift from Dr. Alan Hall (London, UK). H-Ras, Rap1a, and RalC clones were kindly donated by Dr. Robert Cool (Max Plank Institute, Dortmund, Germany).
In vitro glucosylation of small GTPases was performed in 50 mm triethanolamine, pH 7.5, containing 2 μl of UDP-14C-glucose (DuPont NEN; 286.2 mCi/mmol; final concentration 7 μm), 2 mm MgCl2, 1 mm DTT, 0.3 mm GDP, 1 μg of recombinant GTPase, and 5 μg/ml LT82, LT82 recombinant fragment, or LT9048. The reaction was performed for 1 hr at 37°C and stopped by adding 10 μl of sample buffer (three times) followed by boiling for 3 min. Then samples were electrophoresed on a 15% SDS-PAGE and autoradiographed. When necessary, UDP-mannose, ADP-glucose, and TDP-glucose (Sigma) were used to prevent glucosylation.
Detection and glucosylation of small GTPases in Aplysia
Aplysia californica (70–120 gm body weight; Marinus Inc., Long Beach, CA) were anesthetized by injection of 50–75 ml of solution containing 400 mmMgCl2, and the nervous system was removed. Freshly dissected nerve ganglia (buccal, cerebral, pleural, pedal, and abominal) were homogenized in Tris buffer containing 10 mm Tris, pH 7.5, 2 mmMgCl2, 1 mm DTT, 10 μg/ml leupeptin, 1 μm pepstatin, and 0.1 mm PMSF. Complete lysis was achieved by three cycles of freeze thawing.
Detection of Aplysia GTPases. Total proteins were extracted in Tris buffer containing 1% Triton X-100 for 20 min at room temperature. For Western blotting, samples containing 20 μg Aplysia proteins were subjected to SDS-PAGE on 15% gels, transferred to nitrocellulose, and incubated with a 1:50 dilution of antibodies against small GTPases in PBS-milk at room temperature for 16 hr. Polyclonal rabbit antibodies against Rho, Ras, Rap, and Ral were from Santa Cruz Biotechnology; anti-Rac1 was from Upstate Biotechnology. Polyclonal rabbit antibodies directed against Cdc42 were kindly provided by Dr. Philippe Chavrier (Institut Curie, Paris, France). Immunoblots were processed with peroxidase conjugate and chemiluminescence kit (ECL, Amersham) and analyzed by autoradiography.
Glucosylation of Aplysia GTPases. Lysates were centrifuged (15,000 rpm for 15 min) to separate a pellet containing membrane fractions [denoted as insoluble (I) fraction] and a fraction containing cytosolic and crude vesicle fractions [denoted as soluble (S) fraction]. The pellet was washed four times with distilled water and extracted with the buffer described above, but containing 1% Triton X-100, for 20 min at room temperature. Total proteins were assayed using a manufactured protein assay (Bio-Rad Life Science, Marne la Coquette, France). Glucosylation was performed on samples (100 μg protein) of the soluble and insoluble fractions as described above.
Acetylcholine release and electrical recordings atAplysia synapses
Electrophysiological experiments were performed at identified, chloride-dependent, inhibitory cholinergic synapses in dissected buccal ganglia of Aplysia as described previously (Poulain et al., 1986, 1988; Schiavo et al., 1992; Doussau et al., 1998, 2000; Humeau et al., 2001a,b). Two presynaptic cholinergic interneurons termed B4 and B5 (Gardner, 1971) and one postsynaptic neuron (either B3 or B6) were impaled with two glass microelectrodes (3 m KCl, Ag/AgCl2, 2–10 MΩ). To initiate action potentials and evoke ACh release, the presynaptic neurons were depolarized by a square pulse of 50 msec duration and appropriate intensity. To avoid overlap of the postsynaptic responses originating from the two B4 and B5 presynaptic neurons, the stimulus protocols were alternated, and each presynaptic neuron was stimulated every 40 sec (0.025 Hz).
Evoked ACh release was monitored by measuring the amplitude of the evoked IPSCs using the conventional two-electrodes voltage-clamp technique (AxoClamp2B, Axon Instruments). The postsynaptic currents are purely chloride dependent (Gardner and Stevens, 1980; Simonneau et al., 1980; Kehoe and McIntosh, 1998). Moreover, to measure accurately the amplitude of the postsynaptic response, the holding potentialVh was maintained at 30 mV above the reversal potential of postsynaptic responses,Vrev.Vrev was determined every 5 min. The recordings were digitized at 40 kHz and filtered at a cutoff frequency of 250 Hz using an eight-pole low-pass Bessel type filter (902LPS, Frequency Device).
To express the amplitude of the IPSC as a value proportional to the amount of released ACh but independent of the driving force for Cl−, the IPSC amplitude was converted as an apparent membrane conductance (G) by taking into account the reversal potential Vrev(at these synapses, Vrev =ECl−) of the evoked response according to the equation G =I/(Vh −Vrev). However, although the postsynaptic response amplitude has the dimension of a membrane conductance, we refer to it in this paper as the IPSC amplitude,I (nanosiemens).
Extracellular media
Dissected buccal ganglia were maintained at 22°C using a Peltier-plate system and superfused continuously (10 ml/hr) with a physiological medium containing (in mm): NaCl 460, KCl 10, CaCl2 43.8, MgCl2 76.2, MgSO4 28, Tris Buffer 10, pH 7.5. This high concentration in divalent cations was used to minimize spontaneous neuron firing activity as described (Humeau et al., 2001a). This medium corresponds to an extracellular [Ca2+/Mg2+] ratio of 0.42. To change [Ca2+]e, the extracellular [Ca2+/Mg2+] ratio was modified by changing the concentrations of CaCl2 and MgCl2 but not that of MgSO4 (to keep the sulfates unmodified), as described previously (Doussau et al., 1998; Humeau et al., 2001a).
Intraneuronal injection procedure
Injection electrodes were pulled from glass tubing without a capillary and contained a silver wire to allow the electrophysiological monitoring of the impalement. Samples to be injected were mixed with a vital dye (fast green FCF, 10% v/v; Sigma) as detailed elsewhere (Poulain et al., 1986; Doussau et al., 1998; Humeau et al., 2001a). The samples were air pressure-injected using a picopump PV820 (World Precision Instruments) under visual and electrophysiological monitoring. The injected volume was in the range of 1–2% of the cell body volume. Therefore, assuming a homogenous distribution of the injected material, the final intraneuronal concentration was ∼1% of that in the injection micropipette. After injection, the micropipette was removed carefully, and only presynaptic neurons with no alterations in action potentials and with membrane potentials between −60 and −45 mV were analyzed. The time of injection is denoted in most experiments as time zero. In several sets of experiments, we verified that the various buffers used for dissolving toxin samples had no effect on neurotransmitter release.
Determination of IPSC amplitude and rise and decay times
IPSC amplitude (I) was determined as the peak current of recorded IPSC. The time to rise from 20 to 80% of the maximal IPSC amplitude was determined and denoted as “IPSC rise time.” IPSC rise times were determined from at least 10 IPSCs recorded under the same experimental condition and averaged. The IPSC decay time constant was determined by fitting the IPSC with multi-exponential regression I(t) =Ipeak*[w1*exp(−t/τ1)+w2*exp(−t/τ2)], in which τ1 and τ2 are the time constants and w1 andw2 are the respective weights of the two components. In nearly all experiments analyzed,w1 → 1 andw2 → 0, indicating single exponential decay for IPSCs.
Estimation of the quantal parameters: theoretical aspects
In brief, consider the synaptic inputs attributable to the multiple contacts between a presynaptic neuron and its postsynaptic target and consider the three parameters: q, the amplitude of elementary postsynaptic response, n, the mean number of independent release sites, and p, the mean release probability at these sites. First, we assume that at each release sitep is the product of the output probabilitypo (i.e., the probability for a release-ready SV to fuse in response to presynaptic stimulus) and the probability pA that the site is eligible for release (i.e., that a primed SV is available for release) (Brown et al., 1976; Zucker, 1989; Quastel, 1997; Scheuss and Neher, 2001). Second, we assume that release probability and quantal size are uniform, that quanta sum up linearly, and that quantal release follows a simple binomial distribution. Therefore, following these assumptions, the average amplitude of IPSC is given by following equation:
| Equation 1 |
and the fluctuations of the responses around the mean have a variance:
| Equation 2 |
Similar to the ion channel analysis developed by Sigworth (1980), the analysis of the Var =f(Imean) relationship when the release probability is experimentally modified permits an evaluation of the quantal parameters n and q(Quastel, 1997; Silver et al., 1998; Reid and Clements, 1999;Oleskevich et al., 2000; Clements and Silver, 2000; Meyer et al., 2001;Scheuss et al., 2002). Although this does not allow separate evaluations of pA andpo (Scheuss and Neher, 2001), modifications of the productpo*pA= p can be detected. The simple binomial model of synaptic transmission predicts a parabolic relationship between Var andf(Imean). Indeed, this relationship can be reexpressed as a function ofImean:
| Equation 3 |
which can be fitted by a simple parabola of equation:
| Equation 4 |
This allows determination of two parameters: A, the initial slope of the parabola, and B, the extent factor of the parabola. Parameter A refers to q. However quantal variability (denoted as CVq,CV being the variation coefficient) also contributes to Var. Therefore, A provides an overestimate of q(Silver et al., 1998; Reid and Clements, 1999; Oleskevich et al., 2000,Scheuss and Neher, 2001):
| Equation 5 |
Parameter B refers to 1/n.However, probability parameterspo and pA are heterogeneous between the release sites at vertebrate synapses (Rosenmund et al., 1993; Murthy et al., 1997), and this is also likely to be the case at Aplysia synapses. Therefore, 1/B underestimates n according to equation:
| Equation 6 |
in which, CVp is the average variation ofpo*pA and βCVqinter is the fraction of quantal variance attributable to intersite variability (Brown et al., 1976; Silver et al., 1998; Meyer et al., 2001; Scheuss and Neher, 2001).
Determination of Imean and Var: practical aspects
Stationary conditions. When under a given experimental condition the amplitude of the IPSCs was stable with time (i.e., during control recordings or when ACh release was stable after a change in extracellular [Ca2+/Mg2+] or stimulation frequency), Imean and Var were calculated directly from the IPSC amplitude values determined in recording epochs of at least 25 IPSCs. Because background noise can contribute to Var, its variance, Varnoise, was calculated from the baseline fluctuations in IPSC recordings, and subtracted from Var. Then the Var =f(Imean) plots were constructed.
Nonstationary conditions. When IPSC amplitude does not stabilize, determination of Imean and Var can be performed by nonstationary analysis, as reported previously for analysis of channel fluctuations during the course of evoked postsynaptic responses (Robinson et al., 1991; Traynelis et al., 1993). In essence, the aim of the nonstationary analysis is to determineImean at each time ofI = f(t) plot. The result of subtracting the set of Imean toI = f(t) plot generates the set of the fluctuations of I data aroundImean, the variance of which is then determined. The accurate determination ofImean is critical for Var determination. In the earlier studies of Robinson et al. (1991) andTraynelis et al. (1993), Imean was approximated by the mean of fitting procedures or by averaging the recorded postsynaptic responses. We could not use this latter approach because each experiment had a unique time course, which could not be adjusted by simple mathematical equations. As an alternative, we estimated Imean at each timet of the I = f(t) plots as follows: I = f(t) plots were submitted to local linear fitting using a modified built-in procedure of SigmaPlot5 software (SPSS Inc., Chicago, IL) based on least squared estimation of parameters. The local linear fitting was performed on a window of w data (Fig.1A,B,straight line into brackets) and repeated for the whole data range by moving the window by a step of one IPSC data. The mid-value of each local fit was considered as the estimate ofImean at the corresponding time. The set of Imean fits theI = f(t) plot (Fig.1B, solid line with small black dots). The result of subtracting the set ofImean to I =f(t) plot (I −Imean) shows fluctuations around the zero level. I − Imeanvalues are denoted by ΔI in Figure 1C. The variance, Var, of these fluctuations was then calculated in a window of W data that was moving, with a step of one, along the whole data range to be analyzed. Var was corrected for background noise variance (Varnoise) (see above). Then, the Var =f(Imean) plots were constructed (Fig. 1D).
Fig. 1.

Determination of quantal parameters by nonstationary analysis as tested on a simulated experiment.A, IPSC amplitude =f(t) values (○) were calculated as the product binomial (n,pt)*q in whichn = 600 sites and q = 2 nS. A sigmoid decrease of release probability p = po*pA with time, t, was simulated using equationpt = 0.8/[1 + exp(−(t−20)/−3.5)0.5]. The solid line denotes the set of the Imeanvalues determined at each t (for details see Materials and Methods). B, Magnification of boxshown in A. The window width used for local linear fitting (w) is indicated withinbrackets (here w = 9). Linear fit is indicated by solid straight line. The mid-values of the local linear fits are the estimates of Imeanand are indicated by small dots on a solid line. C, Fluctuations in IPSC amplitude are approximated at each time t of the I= f(t) plot, by subtractingImean from corresponding Ivalue (○). The Imean values determined by nonstationary analysis deviate from the trueImean. This difference was determined by subtracting the determined Imean values from the predicted Imean and is represented by asolid thick line. The brackets denote the window width W used for Var determination (hereW = 16). D, The corresponding Var = f(Imean) plot. Solid line denotes the adjustment of the data by simple parabola (Eq. 4). The predicted parabola (dashed line) was calculated using Equations 1 and 2 and the above-mentioned quantal parameters. The difference between both parabolas generates errors in the determinations of qand n. The extent of this error can be minimized by optimizing the window width (w) used for local linear fitting of IPSC =f(t). Error in estimation ofq is shown in E[Δq(%)] as a function of w, for two “experimental” conditions: when simulated IPSC amplitude decreases rapidly (t1/2 = 25 min) or slowly (t1/2 = 75 min). These situations are denoted by ● and ▪, respectively. For these two conditions, optimalw values are indicated by arrows.
For each experiment analyzed, the width of the windows used for linear local fitting (w) and the variance calculation (W) needed to be adapted to the kinetics of the IPSC amplitude changes. Indeed, except in the case of a linear change in IPSC amplitude, when the window (w) used for local linear fitting is too wide, the local fit deviates from the actualImean, and this results in an overestimation of Var (i.e., the problem of fitting a curve with a straight line); in contrast, when w is too narrow, the local fit adapts to local fluctuations but leads to an underestimation of Var. The window width used for variance calculation, W, depended also on the kinetics of IPSC amplitude changes. Here, when the window is too wide, Var is calculated from the fluctuations in amplitude of IPSCs corresponding to very different mean amplitude, and the Var = f(Imean) plot is significantly distorted. To determine theW and w to be used, experiments in which either product ofpo*pA,n or q was changed were simulated. They were submitted to the nonstationary analysis described above. The most appropriate w and W were determined as those minimizing the deviation of estimated quantal parameters from the parameters used for simulation. For the faster changes in IPSC amplitude (such as those caused by a rapid fall in extracellular [Ca2+/Mg2+]), a good compromise was to use w = 9 or 11 data andW = 15–17 (Fig. 1E). For slower IPSC changes, w and W were increased. Even in these conditions, there was a remaining deviation of overall estimate ofImean (as determined by local moving linear fitting) with the true Imean(as calculated by the equation that we used to simulate IPSC amplitude change). This is illustrated by the thick dotted orhatched line in Figure 1C. A consequence of this deviation was that the Var =f(Imean) plots were fitted by parabolas (Fig. 1D, solid line) that were not exactly superimposable onto the theoretical calculated parabolas (Fig. 1D, dashed line). The uncertainty in the determination of parabola parameters Aand B in the faster experiments was ∼10%, whereas in slower experiments this error remained marginal.
Parabola adjustment and determination of quantal parameters at Aplysia synapses
Var = f(Imean) plots were fitted by Equation 4 constrained to pass the origin using a built-in procedure of SigmaPlot5 software. The coefficient of determination r2 is reported in the Figures or corresponding text to indicate how good the fit is.CVq was not determined in this study because of the difficulty in distinguishing miniature events issuing from B4 or B5 presynatic neurons from the inhibitory inputs caused by the many other presynaptic neurons, andCVp was not estimated. Therefore, to provide insights on possible changes in q, we determined the parabola fitting parameter A (see Eq. 5), and Bprovides an estimate of 1/n (see Eq. 6). According to Equation 1,po*pA =Imean/(n*q), and we calculated its approximation, P, according to the equation:
| Equation 7 |
P differs from actual values of average release probabilitypo*pA by the contribution of quantal variability and probability variability (Eq. 5, 6).
If not stated otherwise, results are presented as means ± SEM. When appropriate, the significance was tested by the paired or unpaired Student's t test.
RESULTS
Effect of the intraneuronal injection of lethal toxin fromC. sordellii on evoked ACh release
To restrict the action of the toxin to a single presynaptic element, LT82, which in vitro glucosylates recombinant Ras, Rac, Ral, and Rap but not Cdc42 and Rho, was pressure injected into either B4 or B5 cholinergic neurons to give a final intraneuronal concentration of ∼50 nm. This induced a rapid decrease in IPSC amplitude, indicating inhibition of ACh release (Fig.2A–C). No inhibition of ACh release was detected with the noninjected control neurons (Fig. 2A,C). In all experiments, the inhibitory action of LT manifested after an average delay of 15.2 ± 3.9 (SD, n = 53) min after the injection. This may represent the time needed for LT molecules [molecular weight (MW) ∼250 kDa] to diffuse up to the nerve terminals that are distant by 300–500 μm from the presynaptic soma. This delay is comparable with that (5–10 min) observed for tetanus and botulinum neurotoxins (MW ∼150 kDa) at the same synapses (Poulain et al., 1988; Schiavo et al., 1992).
Fig. 2.

Inhibition of evoked ACh release by LT toxins that inactivate small GTPases. A, A representative experiment is illustrated. ACh release was evoked at identified synapses in the buccal ganglion of Aplysia californica. Seeinset for a schematic drawing of the neuronal connections. The amplitude of the IPSCs (percentage of average IPSC in control period) is plotted against time, before and after pressure injection of lethal toxin from C. sordellii(LT) into one of the two presynaptic cholinergic neurons (●). The final concentration of LT in the cell body was ∼50 nm. The second presynaptic neuron (○) was not injected.B, Recordings of presynaptic action potentials and IPSC at the time indicated before and after injection of LT. The mean IPSC amplitude (C) and mean percentage changes in the IPSC rise time and decay time (D, E) were determined 180 min after the time of toxin injection for noninjected (open bars) and LT82-injected (filled bars) neurons and normalized with respect to the mean values observed before the time of toxin injection. The numberof experiments is indicated. C–E, ** denotes significant difference (p < 0.001) as compared with the controls; other comparisons are nonsignificant (p > 0.5).
Typical recordings, illustrated in Figure 2B, exhibit no detectable alteration in the action potentials recorded in the injected neurons at a time when IPSC amplitude was strongly depressed. This suggests that the inhibitory action of LT on ACh release does not result from an alteration in presynaptic membrane excitability. We also found no change in either the IPSC rise or decay time after LT had exerted its blocking action (Fig.2D,E). This latter finding indicated that release kinetics and synchronization of released quanta remained unchanged after LT action.
Blocking of ACh release needs an LT intact catalytic site
The impressive blocking action of LT (Fig.2A,C) was comparable, on a molar basis, to that produced by intraneuronal application of tetanus or botulinum neurotoxins (Poulain et al., 1988; Schiavo et al., 1992). This raised the question of whether the blocking action of LT was causally linked to its well characterized glucosylating enzymatic activity. Glucosylation of small GTPases by LT involves an enzymatic domain localized in the N-terminal region of the toxin (for review, seeBusch and Aktories, 2000). A recombinant polypeptide (amino acids 1–546, denoted as LT82 NH2) was produced in E. coli. Similar to its parent holotoxin, this peptide retained the ability to glycosylate mammalian recombinant Rac, Ras, Ral, and Rap, but neither Rho nor Cdc42, in vitro (Fig.3A, compare the top two autoradiograms). Intraneuronal application of LT82 NH2 (50 nm, final intrasomatic concentration) also blocked ACh release in a manner comparable to the holotoxin (Figs.2C, 3B) (no significant difference:p = 0.33). In contrast, when a double mutation (aspartate to alanine exchange at positions 286 and 288) was introduced to abolish Mn2+ coordination and UDP-glucose binding (Busch et al., 1998), the recombinant peptide (Fig.3, LT82 NH2 mutant) had no glucosyltransferase activity on recombinant GTPases, in vitro (Fig. 3A,bottom panel), and no blocking action on neurotransmitter release when applied intraneuronally (50 nm, final intrasomatic) (Fig. 3B,white bar).
Fig. 3.

Mutations in catalytic domain of LT82 abolish both GTPase-glucosylating and ACh release-blocking activities.A, Protein substrate specificity of glucosylation by LT82 (top panel), recombinant LT82 fragment (amino acids 1–546) containing an intact catalytic domain (middle panel; LT82 NH2) or a D to A exchange at positions 286 and 288 (bottom panel;LT82 NH2 mutant). Recombinant GTPases (1 μg of each) were incubated with LT or fragments (5 μg/ml) in the presence of UDP-14C-glucose for 60 min at 37°C. Labeled proteins were analyzed by SDS-PAGE and autoradiography. Position of molecular weight markers is indicated. B, The effect of recombinant fragments on ACh release. Same presentation of the data as in Figure2C. ** denotes significant difference (p < 0.001).
LT glucosylates small GTPases in Aplysia
Figure 4A shows a Western blot analysis of the small GTPases present inAplysia nerve tissue. These immunoreactivities were detected using specific antibodies raised against members of the Rho and Ras families and correspond to proteins migrating with an apparent molecular weight of 20–30 kDa (data not shown). They cannot be strictly assigned as being Rho, Rac, Cdc42, Ras, Ral, or Rap. Indeed, because of possible divergent evolution of Rho and Ras GTPases, cross-reactivities of the mammalian antibodies with several GTPases cannot be excluded.
Fig. 4.

LT induces glucosylation of GTPases inAplysia. A, Immunochemical detection of small GTPases present in Aplysia nerve tissue. Total protein homogenates (20 μg) were subjected to gel electrophoresis and transferred to nitrocellulose sheets, and the GTPases were detected using anti-Rho, anti-Rac1, anti-Cdc42, anti-Ras, anti-Ral, and anti-Rap antibodies. B, Samples of Aplysia nerve tissue fractions (S, soluble; I, insoluble) containing 100 μg of neuronal proteins were incubated with LT82 or LT9048 (5 μg/ml each) and UDP-14C-glucose alone (denoted as −) or in the presence of 10 mm UDP-mannose (denoted as +), submitted to SDS-PAGE, and autoradiographed. The position of molecular weight markers is indicated.
The intraneuronal targets affected by LT were examined by determining incorporation of 14C-glucose intoAplysia proteins. Because UDP-glucose, the LT cosubstrate, cannot cross the plasma membrane, these experiments were performed onAplysia neuronal tissue fractions. Fractions containing either soluble (i.e., mostly cytosolic) or insoluble (i.e., membrane associated) protein material were treated with LT82 or LT9048 (5 μg/ml for 1 hr) in the presence of 7 μmUDP-14C-glucose, and then proteins were separated by SDS-PAGE. Figure 4B, (first,second, fifth, and sixth lanes fromleft) shows that 14C-glucose incorporation was detectable only in proteins of 18–30 kDa MW and may correspond to the glucosylation of the Aplysia monomeric GTPases revealed by Western blotting (Fig. 4A).
Blocking activity of LT on ACh release is causally linked to glucosylation of Rac
Substitution of glucose in the nucleotide-sugar by other sugar moieties inhibits the glucosylation reaction (Hofmann et al., 1998;Busch et al., 2000). Moreover, the nucleotide moiety is also important: for example, UDP-mannose but not GDP-mannose competitively inhibits Ras glucosylation, in vitro (Busch et al., 2000). We found that incubation of 10 mm UDP-mannose completely prevented LT-induced incorporation of14C-UDP-glucose into protein extracts ofAplysia (Fig. 4B, third,fourth, seventh, and eighth lanes fromleft) or recombinant small GTPases (Fig.5A, lane 2). In good agreement with this observation, we observed that intraneuronal coapplication of UDP-mannose and LT82 (1 mm and 50 nm final, respectively) did not block ACh release (Fig. 5B, second bar fromleft). This provided further support for a causal link between glucosylation of GTPases and inhibition of exocytosis. Moreover, when UDP-mannose (1 mm) was injected into neurons preinjected with LT82, UDP-mannose immediately froze the blocking action of LT82 (a representative experiment of three in shown in Fig. 5C), but no recovery of ACh release was observed (i.e., for at least 6 hr after injection of UDP-mannose).
Fig. 5.

Glucosylation of Rac and inhibition of ACh release by LT. A, The substrate specificity of LT82 and LT9048 was determined as described in Figure 3A except thatin vitro glucosylation was performed in the absence or presence of glucosylation competitors: 1 mm unlabeled UDP-mannose (UDP-man.) or 10 mm unlabeled TDP-glucose (TDP-gluc.). The position of molecular weight markers is indicated. B, The ACh release inhibition determined 90 min after injection of LT82 alone (50 nm, final intrasomatic) or together with UDP-mannose (1 mm final) or TDP-glucose (10 mm final), or LT9048 (50 nm final). Data are expressed as a percentage of the average IPSC amplitude determined before injection. Thehorizontal dashed line means no inhibition. Thenumber of experiments in each conditions is indicated. ** denotes significant difference (p < 0.001) as compared with the other conditions. All other comparisons are nonsignificant. C, Same kind of experiment as in Figure2A, except that both presynaptic neurons were injected with LT82 (50 nm, final, white arrow), but one of them (●) was later injected with UDP-mannose (1 mm, final) at the time denoted by theblack arrow (similar results were obtained in 5 experiments).
Interestingly, we found that the glucosylation reaction could be assigned more selectively to Rac by using ADP- or TDP-glucose as competitive inhibitors for UDP-glucose. Indeed, when the glucosylation reaction was performed, in vitro, in the presence of 10 mm ADP-glucose (data not shown) or TDP-glucose (Fig. 5A), 14C-glucose incorporation into Ras-related proteins (Ras, Rap, and, at a lesser extent, Ral) was markedly reduced, whereas glucosylation of Rac was mostly unaffected. Therefore, intraneuronal application of TDP-glucose (10 mm, final) with LT82 (50 nm, final) was used to minimize intraneuronal glucosylation of Ras, Rap, and Ral. In this experimental condition, we observed that the release of ACh was blocked to a similar extent as that produced by LT82 alone (Fig. 5B; comparefirst and fourth bar fromleft).
To determine whether the residual glucosylation of Ral participates in the LT82-induced blockage of ACh release, we used LT9048, which has hardly any effect on Ral but efficiently glucosylates Rac, Ras, and Rap as LT82, but also Cdc42 (Fig. 5A, fourth lanefrom left). The blocking action of LT9048 (50 nm, final) was comparable to that induced by injection of LT82 (Fig. 5B, fourth bar fromleft). This indicated that glucosylation of Ral by LT82 does not contribute to the LT-induced blockage of ACh release. To summarize, these combinations of treatments and toxins indicate that the glucosylation of Rac is sufficient to explain the inhibitory action of LT on ACh release.
The second part of the study was aimed at determining the release process altered by LT (i.e., implicating Rac-mediated pathway). Previously, we showed that ACh release can recover from near total inhibition to initial values in ∼1 sec when sustained high-frequency stimulations (> 10 Hz) are applied to poisoned nerve terminals (Doussau et al., 2000). This time duration is shorter than the average SV cycle in active synaptic buttons (∼11 sec) and average residency time of docked SVs at active zones (∼2–5 sec) (Klingauf et al., 1998; Murthy and Stevens, 1999). Therefore, the SVs participating in ACh release recovery are likely to be recruited in a pool that is already tethered or docked at the active zone but cannot undergo fusion. Therefore, LT action may disrupt a membrane step of the exocytotic process. To investigate this possibility, we analyzed the possible modifications of the quantal release parameters by LT.
Determination of quantal size and number of active release sites at the identified cholinergic synapses in the buccal ganglion ofAplysia
To estimate the quantal size, q, and number of functional release sites, n, we generated IPSC variance versus mean plots (see Materials and Methods) by submitting buccal ganglion synapses to protocols aimed at changing average release probability, (i.e., the productpo*pA) and thus ACh release. Average productpo*pA was altered by two different manipulations. The first one involved a change in the extracellular [Ca2+/Mg2+] ratio and was aimed at changing the effectiveness of an action potential in activating a release site (i.e., to alter mainly but not exclusively po). The second manipulation was based on changes in stimulation frequency inducing a variable degree of depression (Silver et al., 1998) and was expected to affect mainly but not exclusively pA. Indeed, the probability for a site to be filled with a SV ready for release depends on the balance between the release rate and the replenishing kinetics of the release site and also from the priming mechanisms that allow a docked SV to become fully releasable.
In the first series of seven experiments, the release probability was changed by manipulating the extracellular [Ca2+/Mg2+] ratio. In the first part of the experiments, we analyzed (stationary analysis) the fluctuations in amplitude of IPSCs during stable epochs of ∼30 min after the ganglia had been superfused with medium containing different [Ca2+/Mg2+] ratios (five distinct levels between 2.1 and 0.14) (Fig.6A). In the second part of these experiments, nonstationary analysis was performed on the fluctuations in IPSC amplitude during the reduction in ACh release caused by a fast transition between high (2.1) and low (0.14) extracellular [Ca2+/Mg2+] ratios (Fig. 6B). As shown in Figure 6, Cand D, the A and B parameters of the parabolas (Eq. 4) adjusting the Var =f(Imean) plots were the same for both protocols (compare black-filled and open bars). The duration of the second protocol (45–60 min) was short enough to be performed twice on the same neuron, before and after injection of LT (see below).
Fig. 6.

Stationary and nonstationary analysis of the fluctuations in IPSC amplitude when release probability is modified. The experiment illustrated in A and B is representative of a series of seven performed under the same experimental conditions. The Aplysia ganglion was superfused with physiological medium containing modified extracellular [Ca2+/Mg2+] (as indicated inA1 andB1) to change release probability.A1, The I =f(t) plot reporting IPSC amplitude (I) measured after stabilization or evoked ACh release at the indicated extracellular [Ca2+/Mg2+].B1, The I =f(t) plot during transition between high and low extracellular [Ca2+/Mg2+]. A2, B2, The result of subtracting Imean from the corresponding I as determined by stationary (A) or nonstationary analysis using a window for local linear fitting w = 9, and variance window width w = 16 (B). For details, see Materials and Methods and Figure 1.A3,B3, The corresponding Var =f(Imean) plots. Thesolid line denotes the approximation of the data by a simple parabola using Equation 4. The regression coefficientsr2 are indicated to indicate how good the fits are. A and B are taken from the same experiment in which the corresponding values of Athat refer to q are 2.91 nS (A3) and 2.83 nS (B3) and 1/B that refer to n are 426 sites (A3) and 389 sites (B3). C,D, Averaged parabola parameters A and 1/B. Black-filled bars, open bars, and gray-filled bars refer toA and 1/B determined by stationary and nonstationary analysis when extracellular [Ca2+/Mg2+] was changed as described in A and B and when stimulation rate was modified as described in E, respectively. These two experimental conditions are denoted as[Ca2+/Mg2+]and Stim. rate. The number of experiments is indicated, and ns denotes nonsignificant difference.E, To change release probability, presynaptic neurons were submitted to various stimulation frequencies. A representative experiment of a series of six is illustrated. IPSC amplitudes from epochs of 50 recordings at the indicated stimulation frequency were reported against the total number of IPSCs analyzed.E2, Using stationary analysis, the fluctuation of IPSC around Imean was determined. E3, The corresponding Var = f(Imean) plot. The solid line denotes the parabola adjusted to the plot using Equation 4.
In a second series of experiments, we attempted to modify productpo*pA by changing the rate of stimulation. Increasing the stimulation rate resulted in synaptic depression (Fig.6E1), which is thought to reflect synaptic vesicle depletion at release sites. After stabilization of ACh release in each stimulation condition, the amplitude of fluctuation in IPSC around Imean was determined using stationary analysis. The Var =f(Imean) plots (Fig.6E3) were satisfactorily fitted (r2 > 0.90) by a simple parabola (Eq. 4). This graphically confirmed our assumption (see above) that when the rate of stimulation is increased, release probability is diminished. Figure 6, C and D(gray-filled bars), shows that the A andB parameters determined by this protocol were not significantly different from those determined by manipulating extracellular [Ca2+/Mg2+] ratios to change productpo*pA (Fig.6C,D, compare the gray-filled barswith open and black-filled bars).
The average A values (Fig. 6C) as determined by these three procedures indicate a quantal size, q, of ∼2.5–2.8 nS at the studied synapse. This value overestimates trueq because quantal variability (CVq) contributes to IPSC fluctuations (Eq. 5). Nevertheless, the A value is comparable to the uncorrected q values determined previously (1.5–2 nS) at the same Aplysia synapse using a different approach (Simonneau et al., 1980; Poulain et al., 1986). The parameterB (expressed as 1/B in Fig. 6Dto refer to n) suggests that the number of independent release sites at which quantal release can take place is between 400 and 500 at the studied Aplysia synapse. Because variability in release probability (CVp) and intersite quantal variability (CVq inter) were not determined, 1/B is likely to underestimate n (Eq.6).
Quantal parameters determined by manipulating extracellular [Ca2+/Mg2+] after LT action
To determine which of the quantal parameters were modified and accounted for the decrease in IPSC amplitude that characterizes LT action, the fast change in extracellular [Ca2+/Mg2+] protocol was applied before and after LT82 had significantly inhibited ACh release. Figure 7A shows a typical experiment from a series of five. To minimize the difficulty in analyzing the combined blocking action of LT82 and the reduction in synaptic efficacy caused by [Ca2+/Mg2+] change, the protocol was applied at a time when the LT-induced block was at least 80% (on average by 84%) (Fig.8A). During the high to low [Ca2+/Mg2+] transition protocol (40–60 min), we determined that the blockage caused by LT82 was 11.7 ± 2% (SEM) as compared with the 91.8 ± 1% decrease in IPSC amplitude observed when [Ca2+/Mg2+] was lowered from 2.1 to 0.14. The corresponding nonstationary analysis of fluctuations in IPSC amplitude and Var =f(Imean) plots is shown in Figure 7, B and C. After LT had blocked ACh release, the Var =f(Imean) relationship still appeared parabolic (Figs. 7D, 8B). The initial slope of the parabola, A, remained unmodified (Figs.7D,E, 8C), suggesting that quantal size, q, was not changed. However, if the studied synapse is saturating, it is conceivable that small changes in the presynaptic component of q (SV content) may not have been detected.
Fig. 7.
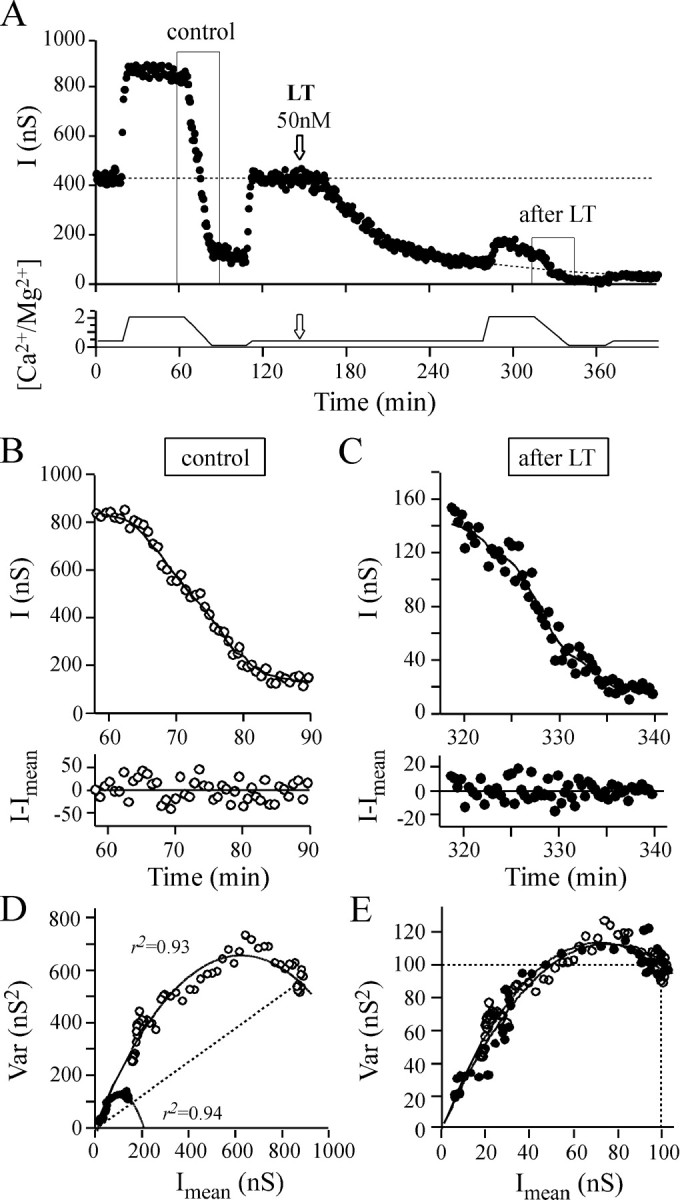
The effect of changing extracellular [Ca2+/Mg2+] before and during LT action. The experiment illustrated is representative of a series of five performed under the same experimental conditions.A, To determine the quantal parameters before and after LT82 application, extracellular [Ca2+/Mg2+] was modified between 2.1 and 0.14 as indicated in the bottom panel.Arrow denotes the time of LT injection. Solid lines denote the set of Imeandetermined by nonstationary analysis (with w = 9). The horizontal, long-dashed line denotes average I in the control period with [Ca2+/Mg2+] = 0.42.Dashed line (after LT) denotes the extrapolated I values (by sigmoid fitting) that would have been obtained without changing extracellular [Ca2+/Mg2+]. B,C, Top panels, Magnification of theI = f(t) plot portions boxed in A;bottom panels, the result of subtracting I values from the Imean estimated using nonstationary analysis. D, E, The corresponding Var = f(Imean) plots before (○) and after (●) LT action. The dashed straight line indicates that the initial release probability observed with [Ca2+/Mg2+] = 2.1 was similar before and after LT. In D, the plots have been scaled to the Imean observed before changing [Ca2+/Mg2+] and to the corresponding averaged Var. The identical initial slope of the parabolas indicates that quantal size, q, is not modified by LT.
Fig. 8.
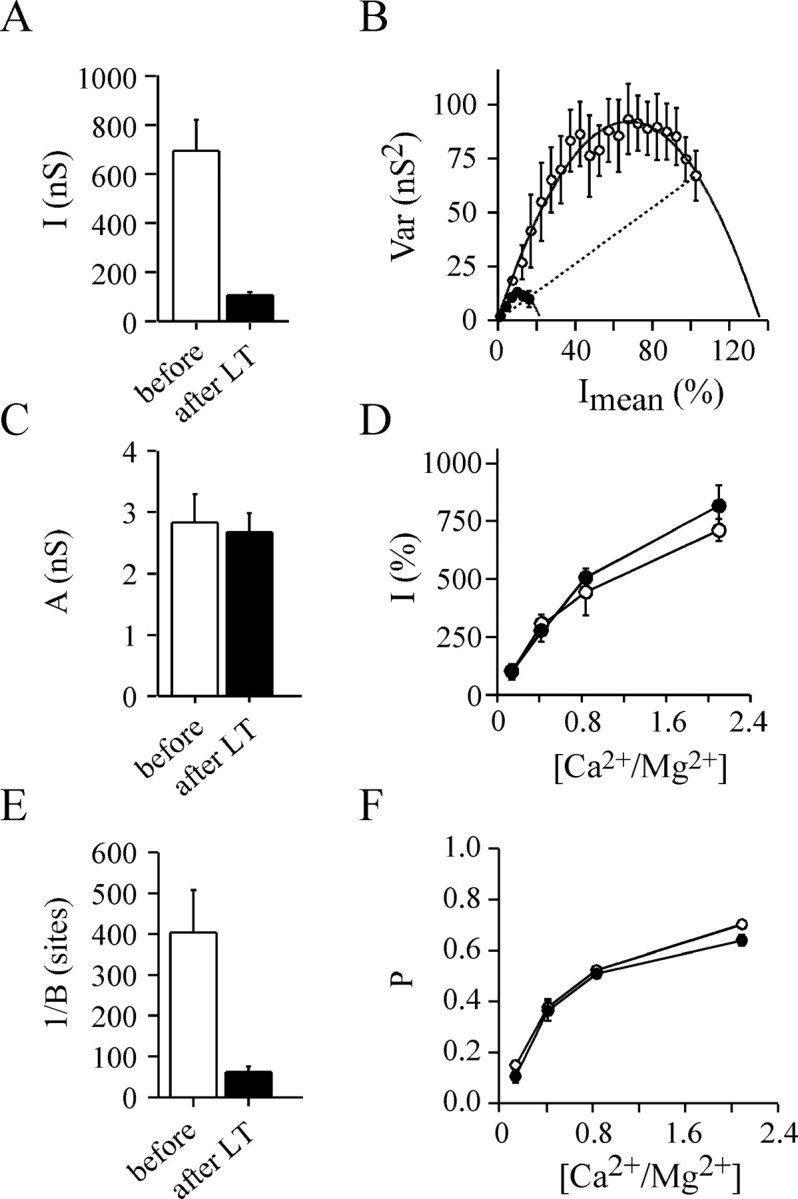
Determination of the parabola parameters affected by LT action using the fast change in extracellular [Ca2+/Mg2+] protocol. Five experiments similar to that illustrated in Figure6B were analyzed before (open barsor open symbols) and 190 ± 20 min (mean ± SD) after LT injection (closed symbols or filled bars). A, The mean IPSC amplitude.B, Mean Var =f(Imean) plot was obtained by averaging the Var values forImean over a 5% amplitude interval.C, The average parameter A of the adjusted parabolas, which refers to quantal size, q.D, The relationships between IPSC amplitude and extracellular [Ca2+/Mg2+]. The data were normalized to values observed at [Ca2+/Mg2+] = 0.14 to highlight possible differences. E, The average 1/Bratio, which refers to n. F, Relationships between P parameter (see Eq. 8), which refers topo*pA, and extracellular [Ca2+/Mg2+]. InA, B, and E, a significant difference (p < 0.001) was found compared with controls;C, D, and F show a nonsignificant difference (p > 0.5).
The relationship between ACh release and extracellular Ca2+ was examined by plotting theI =f([Ca2+/Mg2+]). To highlight possible changes in this relationship, the IPSC amplitude values were normalized to the amplitudes observed at the lowest extracellular [Ca2+/Mg2+] used. However, no apparent modification could be found after LT treatment compared with the control period (Fig. 8D). In all experiments, we observed that the portion of the parabola depicted in the Var =f(Imean) plots during decreasing extracellular [Ca2+/Mg2+] started at the same initial average probability as that observed before applying LT (Figs. 7E, 8B, straight lines), thereby indicating that average productpo*pA was unchanged. Moreover, the relationship between P (which refers to productpo*pA, Eq. 7) and extracellular [Ca2+/Mg2+] did not significantly change before and after LT (Fig.8F). This suggests that the coupling between Ca2+ influx and exocytosis is unlikely to be altered by LT action.
After LT, the only difference that we detected was the decrease in parabola parameter B. Indeed, 1/B (Fig.8E) was reduced to an extent similar to the reduction in mean IPSC amplitude induced by LT (Fig.8A). This suggests that the blocking action of LT action is caused by a diminution in n, the number of active release sites.
Intracellular application of the Ca2+-buffer EGTA has been shown to interfere with LT action (Doussau et al., 2000). Moreover, in rat basophilic leukemia (RBL) mast cells, LT application as well as expression of dominant-negative Rac results in the disruption of Ca2+ mobilization from internal stores (Djouder et al., 2000; Hong-Geller and Cerione, 2000). Hence, we cannot exclude the possibility that the change in extracellular [Ca2+/Mg2+] protocol used above for determining the quantal parameters did not interfere with LT inhibitory action leading to false deduction that only n was modified. To test this possibility, we next analyzed the variance-mean plots generated by changing the stimulation frequency to alter average productpo*pA, as described in Figure 6E.
Quantal parameters determined by changing stimulation frequency after LT action
The “change in stimulation rate” protocol was applied before and after LT82 had inhibited ACh release (Fig.9A). In preliminary experiments, we noticed that a significant inhibitory effect of LT developed during the long duration needed for this protocol (120–180 min to collect enough data for variance determination). To avoid this problem, the effects of LT were stopped by intraneuronal injection of UDP-mannose (1–10 mm, final) after that ACh release was inhibited by ∼80% (Figs. 9A,10A). Because of the technical difficulty in injecting the same neuron twice without trauma, only four experiments could be analyzed. The “stimulation rate protocol” was performed ∼60 min after the UDP-mannose injection to verify that LT action was indeed stopped. Fluctuations in IPSC amplitude were analyzed before (control) and after LT injection by the stationary procedure (Fig.9B,C).
Fig. 9.
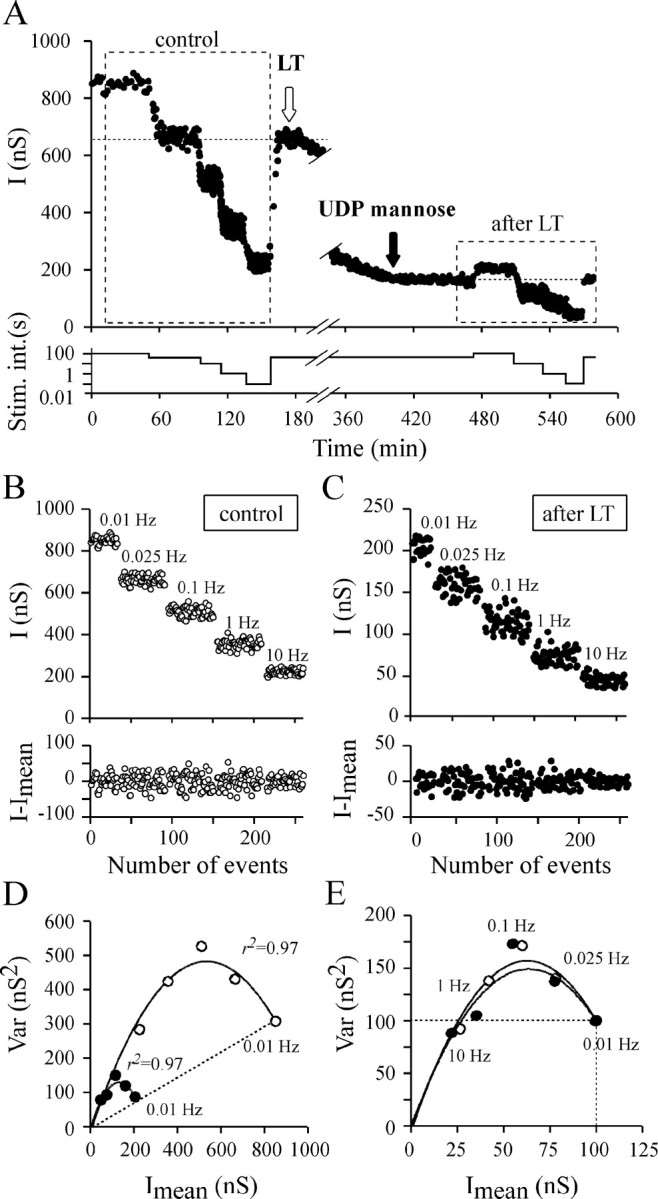
The effects of changing stimulation rate before and during LT action. A representative experiment of a series of four is illustrated. A, Top panel, To determine the quantal parameters before and after LT82 injection (white arrow), ACh release was modified by increasing the stimulation rate between 0.01 and 10 Hz to change release probability. The inhibitory action of LT was arrested by injection (black arrow) of UDP-mannose (10 mm, final).Bottom panel indicates the different stimulation intervals used during the experiment. B,C, Top panels, The IPSC amplitudes are observed at the indicated stimulation rate and correspond to the databoxed in A. Bottom panels, The result of subtracting I values fromImean (stationary analysis).D, The corresponding Var =f(Imean) plots before (○) and after (●) LT action. The dashed straight line indicates that release probability observed at a stimulation rate of 0.01 Hz is similar before and after LT.E, The plots reported in D have been scaled to the Imean observed at 0.01 Hz and to the corresponding averaged Var. The identical initial slope of the parabolas indicates that quantal size is not modified by LT.
Fig. 10.
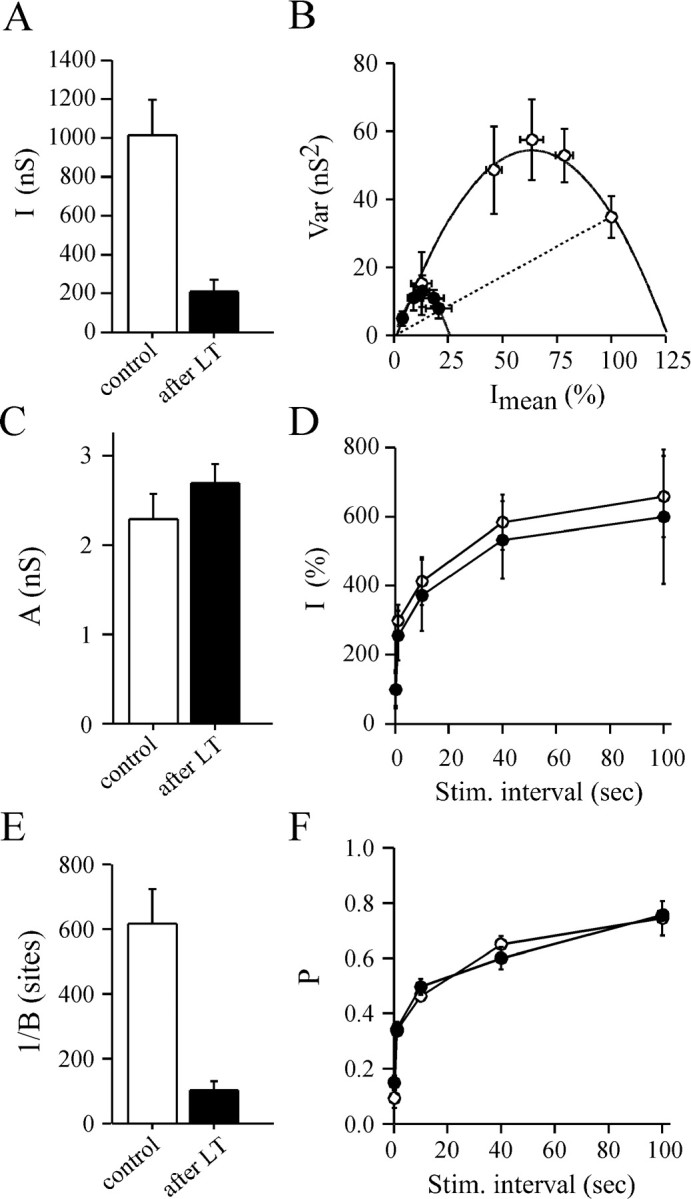
Determination of the parabola parameters before and after LT action using the change in stimulation rate protocol. Same presentation as in Figure 8, except that changes in the stimulation rate were used to modify release probability. InB, Imean and Var values were averaged for similar stimulation rate conditions. Averages were made from four successful experiments. In A, B, andE, a significant difference (p < 0.001) was found compared with controls; C, D, andF show a nonsignificant difference (p > 0.5).
The Var =f(Imean) plots were well fitted (r2 > 0.90) by a simple parabola (Figs. 9D,E,10B). As expected, the parabola parameterA was not significantly modified by LT treatment, confirming that quantal size is unchanged (Fig. 10C). Here also, the Var = f(Imean) plots depicted the same portion of parabola (Figs. 9D,10B) before and after blockage of ACh release by LT. Neither the shape of the Imean =f (stimulation interval) plots (Fig. 10D) nor the relationships between P and the stimulation interval were modified after LT injection, as compared with controls (Fig.10F). We could not explore the responsiveness of the synapse to very high frequency because at a stimulation rate over 10 Hz, LT-induced block is relieved (Doussau et al., 2000). These findings confirm that our deduction made above that the productpo*pA is not changed by LT action. Here also, the only parameter modified after LT was B (Fig. 10E), indicating thatn is reduced by LT action.
Variance to mean relationship during LT-induced blockage of ACh release confirms a reduction in the number of release sites
If LT82-induced blockage of ACh exocytosis is caused by a decrease in n, this should also manifest as a linear relationship between Imean and Var in the Var = f(Imean) plots made by nonstationary analysis of IPSC amplitude fluctuations during the ACh release blockage produced by LT injection. Indeed, when IPSC amplitude changes as the result of a reduction of n, Var should decrease linearly with Imean according to equation:
| Equation 8 |
A fall in IPSC amplitude caused by a change in the productpo*pA should be described by Equation 3, which is a parabola (see Eq. 4), similarly as this has been illustrated above whenpo*pA was experimentally modified by distinct procedures. After progressive q changes, the Var =f(Imean) relationship should be described by a quadratic function of positive curvature of equation:
| Equation 9 |
Data were submitted to a nonlinear regression analysis using the quadratic function Var =a*Imean +b*I2mean. Indeed, this procedure avoids any a priori on the linearity or nonlinearity of the Var =f(Imean) relationship. A linear slope should manifest with b = 0 anda > 0 (Var =a*Imean). Ifa = 0 and b >0, Var =b*I2mean, and the corresponding Var =f(Imean) plot would display a positive curvature pinpointing a change in q size (Eq. 9). If a > 0 and b < 0, Var =f(Imean) plot would exhibit a negative curvature indicating a change in productpo*pA (Eq.3).
In Figure11A3, the parameters for nonlinear regression analysis of the Var =f(Imean) plot werea = 0.68 and b = 10−4(r2 = 0.88) (dashed line). The mean adjustment parameter for the 11 LT82 and 5 LT9048 experiments analyzed (Fig. 11B,C) were a = 0.83, b = 1.4 10−3,r2 = 0.78, anda = 1.12, b = −0.8 10−3,r2 = 0.76, respectively. In all cases, b approaches to 0, indicating that Var =f(Imean) is of the form Var = a*Imean. Accordingly, good adjustments of the Var =f(Imean) plots were also obtained using linear regressions: Figure11A3: a = 0.73,r2 = 0.83; Figure11B: a = 0.97,r2 = 0.77; Figure11C: a = 1.03,r2 = 0.75. Therefore, the relationships between Var and Imeanwhen LT82 or LT9048 blocks ACh release are likely to be linear, and this graphically confirms that inactivation of Rac by LT causes inhibition of ACh release by reducing the number of release sites.
Fig. 11.

Graphical determination of n as the quantal parameter modified by LT action. A, IPSC fluctuations during the course of LT82-induced blockage of ACh release were analyzed by nonstationary analysis using a window width for local linear fitting w = 33, and variance windowW = 21. A1,I = f(t) plot. The set of Imean is represented by asolid white line. A2, The result of subtracting I values fromImean. A3, The corresponding Var =f(Imean) plot.B, C, The Var =f(Imean) plots from experiments in which either LT82 (n = 11) or LT9048 (n = 5) was injected. Individual plots are indicated by different symbols and are scaled to theImean and Var values determined before LT injection. For clarity, only 1 point of 10 is plotted.A3–C, The adjustment of the data by linear (solid straight line) or nonlinear regression analysis (dashed curves) is presented. Forr2 values see Results. The linear Var =f(Imean) relationships indicate that Imean diminution is caused by a decrease in n, the number of active release sites.
As an outcome of this study, one may anticipate that the graphical procedure described above [see also Humeau et al. (2001b)] may help to identify the quantal parameter(s) modified when synaptic efficacy is changed in response to a treatment.
DISCUSSION
Rac is the LT target by which glucosylation inhibits neurotransmitter release
LT potently inhibits exocytosis in chromaffin cells (Gasman et al., 1999), RBL mast cells (Djouder et al., 2000), and neurons (Doussau et al., 2000; this study). Our results demonstrate that the blocking action of LT on neuroexocytosis is causally linked to glucosylation of small GTPases but not to other toxin activities. LT82 (which has effects on Rac, Ras, Ral, and Rap), LT9048 (which acts on Rac, Ras, Cdc42, and Rap), and Toxin B from Clostridium difficile(which glucosylates Rho, Rac, and Cdc42) share common characteristics: all three block ACh release (Doussau et al., 2000; this study), and this inhibitory action can be transiently relieved by stimulating neurons at a high frequency [Toxin B and LT82 (Doussau et al., 2000); LT9048 (data not shown)]. Because Rac is the only GTPase modified by these toxins, inactivation of Rac is likely to be responsible for the inhibition of ACh release.
Glucosylation of the other GTPases present in nerve tissue does not seem to contribute to the blocking action of LT. First, Ras and Rap are absent or minor components of nerve terminals, and Ras is preferentially localized at postsynaptic sites (Kim et al., 1990; Huber at al., 1994). Second, although Ral is abundant in nerve terminals and is associated with vesicles (Bielinski et al., 1993; Huber et al., 1994; Luo et al., 1998), LT9048, which has no activity against Ral (El Hadj et al., 1999; this paper), and LT82, which glucosylates Ral, exert similar block on ACh release. Moreover, TDP-glucose, which prevents glucosylation of Ras, Ral, and Rap but not Rac, does not antagonize LT inhibitory action, whereas UDP-mannose, which inhibits glucosylation of Ras, Ral, Rap, and Rac, completely prevents blockage of ACh exocytosis by LT. These data provide further support for the deduction that glucosylation of only Rac causes inhibition of exocytosis.
Functional consequences of the glucosylation of Rac
The effect of glucosylation on the GTPase cycle has been studied extensively (Herrmann et al., 1998; Sehr et al., 1998; Busch and Aktories, 2000; Just and Boquet, 2000; Vetter et al., 2000). To summarize, only GDP-bound GTPase is susceptible to glucosylation by LT. Glucosylated GTPase can bind to its target membrane and undergo GDP–GTP exchange stimulated by guanylate exchange factors. However, the GTP-bound form of glucosylated GTPase cannot stimulate protein effectors. The silencing of the downstream pathways is likely to be persistent because glucosylated GTPase does not cycle normally: its interaction with guanylate- activating protein is mostly reduced, and glucosylation affects a residue (Thr 35 in Rac and Ras) essential for GTP hydrolysis. The GTPase remains GTP bound but inactive and cannot be removed from target membranes by guanylate dissociation inhibitor. Therefore, GTP-bound glucosylated Rac should behave as a dominant negative mutant of Rac. This is fully consistent with our findings. Although UDP-mannose immediately arrests LT action, we observed no recovery of ACh release over the next 6 hr.
How can Rac glucosylation cause reduction in the number of active release sites?
To identify the release step(s) altered by LT action (i.e., the release step controlled by Rac), we determined the quantal release parameters modified after LT action. We found no change in either quantal size (q) or average release probability (po*pA). Only the number of active release sites, n, appears to be strongly reduced. As discussed above, incorporation of glucosylated Rac in the release machinery is likely to cause a prolonged inactivation of the release site (i.e., that appears permanent or irreversible over the duration of the experiment). Therefore, does the reduction inn mean that release sites have been physically eliminated (true reduction in n) or that they have been inactivated in a long-lasting manner? Indeed, setting in an all-or-none manner the product po*pAat a given release site to 0 should result in an apparent reduction inn. These possibilities are discussed below.
Mutations that affect interaction of Rho proteins (Rho, Rac, Cdc42) with effectors or GTP hydrolysis are known to induce remodeling of neuron morphology and diminution in neurite branching (Luo, 2000;Nakayama et al., 2000; Liu and Strittmatter, 2001). However, the reduction in number of active release sites is unlikely to result from retraction of synaptic buttons. Indeed, ACh release can recover initial levels in <1 sec by applying high-frequency stimulations (Doussau et al., 2000). This indicates that release sites are physically present but inactivated.
Rac is a well known regulator of the actin-based cytoskeleton (Hall, 1998; Bishop and Hall 2000), and inactivation of Ca2+ influx has been reported after actin-based cytoskeleton has been severed (Johnson and Byerly, 1993,1994). Therefore, the possibility that inactivation of release site is caused by an altered Ca2+ influx should be considered. Consistent with our previous observation that paired-pulse facilitation is unaffected by LT (Doussau et al., 2000), we found that average po*pAremains unchanged after LT (Figs. 8, 9). This shows that the coupling between Ca2+ influx and exocytosis is intact at the sites that are still able to release after LT treatment. What about the inactivated release sites? At certain synapses, only one channel needs to open to trigger transmitter release (for review, seeStanley, 1997; Augustine, 2001). In this case, the permanent disabling of this channel after LT action should set localpo*pA to 0, leading to the apparent elimination of the release site (i.e., expressed as a reduction in n). However, at other synapses, several channels are required for transmitter release (Borst and Sakmann, 1996; Schneggenburger and Neher, 2000). This is likely to be the case at Aplysia synapses. Indeed, we observed that the slow Ca2+-buffer EGTA blocks transmitter release (Doussau et al., 1998). This suggests that (1) the open Ca2+ channels and fusion particle are distant and (2) several channels contribute to the calcium microdomain that triggers release. Therefore, the mechanism by which LT inhibits ACh release cannot be caused by the inactivation of a fraction of the Ca2+ channels. Indeed, a decrease in localpo*pA should have been detected at the macroscopic level as a decrease in averagepo*pA but not as a change in n.
Because LT-poisoned nerve terminals can respond normally to changes in stimulation frequency, SV traffic and priming are normal (i.e., also the unchangedpo*pA) at the sites that remain active after LT treatment. Nevertheless, this does not preclude the possibility that an alteration in the supply of “ready to fuse” SV to release sites is the cause of the functional elimination of release sites. Indeed, if the supply of certain release sites is permanently blocked (i.e., localpo*pA → 0), this should express as an apparent n modification. Several possibilities should be considered. Actin filaments are present in the highly structured presynaptic density (Phillips et al., 2001). Consistent with the implication of Rac in actin cytoskeleton organization, it is conceivable that glucosylation of Rac may have disrupted the SV traffic along actin filaments. However, it is not known whether a release site is connected with a single actin filament. Recently, Rac and also Rho, Cdc42, Ral, and PLD1 have been shown to participate in a multiprotein complex that comprises Sec6/Sec 8 proteins, termed “exocyst” (Brymora et al., 2001; Guo et al., 2001;Moskalenko et al., 2002; Short and Barr, 2002), which is present in nerve terminals and growth cones (Hazuka et al., 1999; Brymora et al., 2001; Vega and Hsu, 2001). The exocyst is believed to regulate polarized secretion and docking of vesicles to regions of the plasma membrane specialized in exocytosis, possibly in a one-to-one stoichiometry with fusion sites (for review, see Short and Barr, 2002). Therefore, inactivation of Rac in the exocyst might be responsible for disrupting the supply of SV to release sites in an all-or-none manner. However, the data available on exocyst function suggest that it plays a major role in tethering vesicles to plasma membrane. This is not compatible with our previous observation that full recovery of ACh release can be obtained in ∼1 sec by applying tetanic stimulation to LT-poisoned terminals (Doussau et al., 2000). One second is shorter than the mean residency time (2–5 sec) for the SV docking stage before fusion at release sites (Klingauf et al., 1998;Murthy and Stevens, 1999). Thus, the SVs recruited for recovery are likely to be tethered or docked at an active zone. Therefore, glucosylated Rac is likely to act downstream of the step regulated by exocyst.
Rac: a regulator of the fusion competence of plasma membrane?
Two Rac-mediated pathways compatible with a post-docking role for Rac need to be considered. First, Rac stimulates phosphatidylinositol kinases (Hartwig et al., 1995; Bokoch et al., 1996), and phosphoinositides are key regulators for exo–endocytosis because they can specifically target proteins to fusion sites (Martin, 2001; Osborne et al., 2001). Second, PLD1 is inhibited at LT-poisoned cells (El Hadj et al., 1999), and Rac regulates PLD1 activity (Hammond et al., 1997). PLD1 is associated with release sites and has recently been implicated in exocytosis in both chromaffin cells (Vitale et al., 2001) andAplysia neurons. In the latter case, PLD activity was shown to be crucial for determining the number of active release sites (Humeau et al., 2001b). Therefore, during tethering or docking of SVs, SV-associated Rac may stimulate hydrolysis of membrane phosphatidylcholine by PLD1. This generates phosphatidic acid, which favors formation of lipid bilayer intermediates during membrane fusion (Chernomordik et al., 1995; Monck and Fernandez, 1996). To conclude, the present analysis of LT action on presynaptic mechanisms reveals an essential role for Rac in the exocytotic mechanism, possibly by regulating lipid composition at the fusion site.
Footnotes
This work was supported by grants from the Association Française contre les Myopathies, DSP contract 99-34-038 to B.P., and Programme de Recherche Fondamentale en Microbiologie et sur les Maladies Infectieuses et Parasitaires 2000 (to B.P. and M.R.P.). We thank Drs. Fabio Benfenati (Università di Genova, Italy) and Nancy Grant (Unité Propre de Recherche 2356 du Centre National de la Recherche Scientifique, Strasbourg, France) for critical reading of this manuscript, and Tamou Thahouly for excellent technical assistance.
Correspondence should be addressed to Bernard Poulain, Neurotransmission et Sécrétion Neuroendocrine, UnitéPropre de Recherche 2356 du Centre National de la Recherche Scientifique, 5, rue Blaise Pascal, F-67084 Strasbourg Cedex, France. E-mail: poulain@neurochem.u-strasbg.fr.
Y. Humeau's present address: PharmaZentrum, Klingelbergstrasse 50, CH-4056 Basel, Swizerland.
H. Kojima's present address: Department of Molecular Biology, Princeton University, Princeton, NJ 08544.
REFERENCES
- 1.Augustine GJ. How does calcium trigger neurotransmitter release? Curr Opin Neurobiol. 2001;11:320–326. doi: 10.1016/s0959-4388(00)00214-2. [DOI] [PubMed] [Google Scholar]
- 2.Bielinski DF, Pyun HY, Linko-Stentz K, Macara IG, Fine RE. Protein Ral and Rab3a are major GTP-binding proteins of axonal rapid transport and synaptic vesicles and do not redistribute following depolarization stimulated synaptosomal exocytosis. Biochim Biophys Acta. 1993;1151:246–256. doi: 10.1016/0005-2736(93)90109-d. [DOI] [PubMed] [Google Scholar]
- 3.Bishop AL, Hall A. Rho GTPases and their effector proteins. Biochem J. 2000;348:241–255. [PMC free article] [PubMed] [Google Scholar]
- 4.Bokoch GM, Vlahos CJ, Wang Y, Knaus UG, Traynor-Kaplan AE. Rac GTPase interacts specifically with phosphatidylinositol 3-kinase. Biochem J. 1996;315:775–779. doi: 10.1042/bj3150775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borst JG, Sakmann B. Calcium influx and transmitter release in a fast CNS synapse. Nature. 1996;383:431–434. doi: 10.1038/383431a0. [DOI] [PubMed] [Google Scholar]
- 6.Brown AM, O'Sullivan AJ, Gomperts BD. Induction of exocytosis from permeabilized mast cells by the guanosine triphosphatases Rac and Cdc42. Mol Biol Cell. 1998;9:1053–1063. doi: 10.1091/mbc.9.5.1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown TH, Perkel DH, Feldman MW. Evoked neurotransmitter release: statistical effects of nonuniformity and nonstationarity. Proc Natl Acad Sci USA. 1976;73:2913–2917. doi: 10.1073/pnas.73.8.2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brymora A, Valova VA, Larsen MR, Roufogalis BD, Robinson PJ. The brain exocyst complex interacts with RalA in a GTP-dependent manner: identification of a novel mammalian Sec3 gene and a second Sec15 gene. J Biol Chem. 2001;276:29792–29797. doi: 10.1074/jbc.C100320200. [DOI] [PubMed] [Google Scholar]
- 9.Busch C, Aktories K. Microbial toxins and the glucosylation of rho family GTPases. Curr Opin Struct Biol. 2000;10:528–535. doi: 10.1016/s0959-440x(00)00126-3. [DOI] [PubMed] [Google Scholar]
- 10.Busch C, Hofmann F, Selzer J, Munro S, Jeckel D, Aktories K. A common motif of eukaryotic glycosyltransferases is essential for the enzyme activity of large clostridial cytotoxins. J Biol Chem. 1998;273:19566–19572. doi: 10.1074/jbc.273.31.19566. [DOI] [PubMed] [Google Scholar]
- 11.Busch C, Hofmann F, Gerhard R, Aktories K. Involvement of a conserved tryptophan residue in the UDP-glucose binding of large clostridial cytotoxin glycosyltransferases. J Biol Chem. 2000;275:13228–13234. doi: 10.1074/jbc.275.18.13228. [DOI] [PubMed] [Google Scholar]
- 12.Chernomordik L, Chanturiya A, Green J, Zimmerberg J. The hemifusion intermediate and its conversion to complete fusion: regulation by membrane composition. Biophys J. 1995;69:922–929. doi: 10.1016/S0006-3495(95)79966-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clements JD, Silver RA. Unveiling synaptic plasticity: a new graphical and analytical approach. Trends Neurosci. 2000;23:105–113. doi: 10.1016/s0166-2236(99)01520-9. [DOI] [PubMed] [Google Scholar]
- 14.Dickson BJ. Rho GTPases in growth cone guidance. Curr Opin Neurobiol. 2001;11:103–110. doi: 10.1016/s0959-4388(00)00180-x. [DOI] [PubMed] [Google Scholar]
- 15.Djouder N, Prepens U, Aktories K, Cavalie A. Inhibition of calcium release-activated calcium current by Rac/Cdc42-inactivating clostridial cytotoxins in RBL cells. J Biol Chem. 2000;275:18732–18738. doi: 10.1074/jbc.M001425200. [DOI] [PubMed] [Google Scholar]
- 16.Doussau F, Clabecq A, Henry J-P, Darchen F, Poulain B. Calcium-dependent regulation of Rab3 in short-term plasticity. J Neurosci. 1998;18:3147–3157. doi: 10.1523/JNEUROSCI.18-09-03147.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Doussau F, Gasman S, Humeau Y, Vitiello F, Popoff M, Boquet P, Bader MF, Poulain B. A Rho-related GTPase is involved in Ca2+-dependent neurotransmitter exocytosis. J Biol Chem. 2000;275:7764–7770. doi: 10.1074/jbc.275.11.7764. [DOI] [PubMed] [Google Scholar]
- 18.El Hadj NB, Popoff MR, Marvaud JC, Payrastre B, Boquet P, Geny B. G-protein-stimulated phospholipase D activity is inhibited by lethal toxin from Clostridium sordellii in HL-60 cells. J Biol Chem. 1999;274:14021–14031. doi: 10.1074/jbc.274.20.14021. [DOI] [PubMed] [Google Scholar]
- 19.Gardner D. Bilateral symmetry and interneuronal organization in the buccal ganglia of Aplysia. Science. 1971;173:550–553. doi: 10.1126/science.173.3996.550. [DOI] [PubMed] [Google Scholar]
- 20.Gardner D, Stevens CF. Rate-limiting step of inhibitory post-synaptic current decay in Aplysia buccal ganglia. J Physiol (Lond) 1980;304:145–164. doi: 10.1113/jphysiol.1980.sp013316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gasman S, Chasserot-Golaz S, Hubert P, Aunis D, Bader MF. Identification of a potential effector pathway for the trimeric Go protein associated with secretory granules. Go stimulates a granule-bound phosphatidylinositol 4-kinase by activating RhoA in chromaffin cells. J Biol Chem. 1998;273:16913–16920. doi: 10.1074/jbc.273.27.16913. [DOI] [PubMed] [Google Scholar]
- 22.Gasman S, Chasserot-Golaz S, Popoff MR, Aunis D, Bader MF. Involvement of Rho GTPases in calcium-regulated exocytosis from adrenal chromaffin cells. J Cell Sci. 1999;112:4763–4771. doi: 10.1242/jcs.112.24.4763. [DOI] [PubMed] [Google Scholar]
- 23.Green GA, Schue V, Monteil H. Cloning and characterization of the cytotoxin L-encoding gene of Clostridium sordellii: homology with Clostridium difficile cytotoxin B. Gene. 1995;161:57–61. doi: 10.1016/0378-1119(95)00263-6. [DOI] [PubMed] [Google Scholar]
- 24.Guo W, Tamanoi F, Novick P. Spatial regulation of the exocyst complex by Rho1 GTPase. Nat Cell Biol. 2001;3:353–360. doi: 10.1038/35070029. [DOI] [PubMed] [Google Scholar]
- 25.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 26.Hammond SM, Jenco JM, Nakashima S, Cadwallader K, Gu Q, Cook S, Nozawa Y, Prestwich GD, Frohman MA, Morris AJ. Characterization of two alternately spliced forms of phospholipase D1. Activation of the purified enzymes by phosphatidylinositol 4, 5-bisphosphate, ADP-ribosylation factor, and Rho family monomeric GTP-binding proteins and protein kinase C-alpha. J Biol Chem. 1997;272:3860–3868. doi: 10.1074/jbc.272.6.3860. [DOI] [PubMed] [Google Scholar]
- 27.Hartwig JH, Bokoch GM, Carpenter CL, Janmey PA, Taylor LA, Toker A, Stossel TP. Thrombin receptor ligation and activated Rac uncap actin filament barbed ends through phosphoinositide synthesis in permeabilized human platelets. Cell. 1995;82:643–653. doi: 10.1016/0092-8674(95)90036-5. [DOI] [PubMed] [Google Scholar]
- 28.Hazuka CD, Foletti DL, Hsu SC, Kee Y, Hopf FW, Scheller RH. The sec6/8 complex is located at neurite outgrowth and axonal synapse-assembly domains. J Neurosci. 1999;19:1324–1334. doi: 10.1523/JNEUROSCI.19-04-01324.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Herrmann C, Ahmadian MR, Hofmann F, Just I. Functional consequences of monoglucosylation of Ha-Ras at effector domain amino acid threonine 35. J Biol Chem. 1998;273:16134–16139. doi: 10.1074/jbc.273.26.16134. [DOI] [PubMed] [Google Scholar]
- 30.Hofmann F, Busch C, Aktories K. Chimeric clostridial cytotoxins: identification of the N-terminal region involved in protein substrate recognition. Infect Immunol. 1998;66:1076–1081. doi: 10.1128/iai.66.3.1076-1081.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hong-Geller E, Cerione RA. Cdc42 and Rac stimulate exocytosis of secretory granules by activating the IP(3)/calcium pathway in RBL-2H3 mast cells. J Cell Biol. 2000;148:481–494. doi: 10.1083/jcb.148.3.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huber LA, Ullrich O, Takai Y, Lutcke A, Dupree P, Olkkonen V, Virta H, de Hoop MJ, Alexandrov K, Peter M, Zerial M, Simons K. Mapping of Ras-related GTP-binding proteins by GTP overlay following two-dimensional gel electrophoresis. Proc Natl Acad Sci USA. 1994;91:7874–7878. doi: 10.1073/pnas.91.17.7874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Humeau Y, Doussau F, Vitiello F, Greengard P, Benfenati F, Poulain B. Synapsin controls both reserve and releasable synaptic vesicle pools during neuronal activity and short-term plasticity in Aplysia. J Neurosci. 2001a;21:4195–4206. doi: 10.1523/JNEUROSCI.21-12-04195.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Humeau Y, Vitale N, Chasserot-Golaz S, Dupont JL, Du G, Frohman MA, Bader MF, Poulain B. A role for phospholipase D1 in neurotransmitter release. Proc Natl Acad Sci USA. 2001b;98:15300–15305. doi: 10.1073/pnas.261358698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson BD, Byerly L. A cytoskeletal mechanism for Ca2+ channel metabolic dependence and inactivation by intracellular Ca2+. Neuron. 1993;10:797–804. doi: 10.1016/0896-6273(93)90196-x. [DOI] [PubMed] [Google Scholar]
- 36.Johnson BD, Byerly L. Ca2+ channel Ca(2+)-dependent inactivation in a mammalian central neuron involves the cytoskeleton. Pflügers Arch. 1994;429:14–21. doi: 10.1007/BF02584025. [DOI] [PubMed] [Google Scholar]
- 37.Just I, Boquet P. Large clostridial cytotoxins as tools in cell biology. Curr Top Microbiol Immunol. 2000;250:97–107. doi: 10.1007/978-3-662-06272-2_5. [DOI] [PubMed] [Google Scholar]
- 38.Just I, Selzer J, Hofmann F, Green GA, Aktories K. Inactivation of Ras by Clostridium sordellii lethal toxin-catalyzed glucosylation. J Biol Chem. 1996;271:10149–10153. doi: 10.1074/jbc.271.17.10149. [DOI] [PubMed] [Google Scholar]
- 39.Kehoe J, McIntosh JM. Two distinct nicotinic receptors, one pharmacologically similar to the vertebrate α7-containing receptor, mediate Cl currents in Aplysia neurons. J Neurosci. 1998;18:8198–8213. doi: 10.1523/JNEUROSCI.18-20-08198.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim S, Mizoguchi A, Kikuchi A, Takai Y. Tissue and subcellular distributions of the smg-21/rap1/Krev-1 proteins which are partly distinct from those of c-ras p21s. Mol Cell Biol. 1990;10:2645–2652. doi: 10.1128/mcb.10.6.2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klingauf J, Kavalali ET, Tsien RW. Kinetics and regulation of fast endocytosis at hippocampal synapses. Nature. 1998;394:581–585. doi: 10.1038/29079. [DOI] [PubMed] [Google Scholar]
- 42.Kowluru A, Li G, Rabaglia ME, Segu VB, Hofmann F, Aktories K, Metz SA. Evidence for differential roles of the Rho subfamily of GTP-binding proteins in glucose- and calcium-induced insulin secretion from pancreatic beta cells. Biochem Pharmacol. 1997;54:1097–1108. doi: 10.1016/s0006-2952(97)00314-6. [DOI] [PubMed] [Google Scholar]
- 43.Linseman DA, Laessig T, Meintzer MK, McClure M, Barth H, Aktories K, Heidenreich KA. An essential role for Rac/Cdc42 GTPases in cerebellar granule neuron survival. J Biol Chem. 2001;276:39123–39131. doi: 10.1074/jbc.M103959200. [DOI] [PubMed] [Google Scholar]
- 44.Liu BP, Strittmatter SM. Semaphorin-mediated axonal guidance via Rho-related G proteins. Curr Opin Cell Biol. 2001;13:619–626. doi: 10.1016/s0955-0674(00)00260-x. [DOI] [PubMed] [Google Scholar]
- 45.Luo JQ, Liu X, Frankel P, Rotunda T, Ramos M, Flom J, Jiang H, Feig LA, Morris AJ, Kahn RA, Foster DA. Functional association between Arf and RalA in active phospholipase D complex. Proc Natl Acad Sci USA. 1998;95:3632–3637. doi: 10.1073/pnas.95.7.3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luo L. Rho GTPases in neuronal morphogenesis. Nat Rev Neurosci. 2000;1:173–180. doi: 10.1038/35044547. [DOI] [PubMed] [Google Scholar]
- 47.Martin TF. PI(4, 5)P(2) regulation of surface membrane traffic. Curr Opin Cell Biol. 2001;13:493–499. doi: 10.1016/s0955-0674(00)00241-6. [DOI] [PubMed] [Google Scholar]
- 48.Meyer AC, Neher E, Schneggenburger R. Estimation of quantal size and number of functional active zones at the calyx of Held synapse by nonstationary EPSC variance analysis. J Neurosci. 2001;21:7889–7900. doi: 10.1523/JNEUROSCI.21-20-07889.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meyer DK, Olenik C, Hofmann F, Barth H, Leemhuis J, Brunig I, Aktories K, Norenberg W. Regulation of somatodendritic GABAA receptor channels in rat hippocampal neurons: evidence for a role of the small GTPase Rac1. J Neurosci. 2000;20:6743–6751. doi: 10.1523/JNEUROSCI.20-18-06743.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Monck JR, Fernandez JM. The fusion pore and mechanisms of biological membrane fusion. Curr Opin Cell Biol. 1996;8:524–533. doi: 10.1016/s0955-0674(96)80031-7. [DOI] [PubMed] [Google Scholar]
- 51.Moskalenko S, Henry DO, Rosse C, Mirey G, Camonis JH, White MA. The exocyst is a Ral effector complex. Nat Cell Biol. 2002;4:66–72. doi: 10.1038/ncb728. [DOI] [PubMed] [Google Scholar]
- 52.Mota M, Reeder M, Chernoff J, Bazenet CE. Evidence for a role of mixed lineage kinases in neuronal apoptosis. J Neurosci. 2001;21:4949–4957. doi: 10.1523/JNEUROSCI.21-14-04949.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murthy VN, Stevens CF. Reversal of synaptic vesicle docking at central synapses. Nat Neurosci. 1999;2:503–507. doi: 10.1038/9149. [DOI] [PubMed] [Google Scholar]
- 54.Murthy VN, Sejnowski TJ, Stevens CF. Heterogeneous release properties of visualized individual hippocampal synapses. Neuron. 1997;18:599–612. doi: 10.1016/s0896-6273(00)80301-3. [DOI] [PubMed] [Google Scholar]
- 55.Nakayama AY, Harms MB, Luo L. Small GTPases Rac and Rho in the maintenance of dendritic spines and branches in hippocampal pyramidal neurons. J Neurosci. 2000;20:5329–5338. doi: 10.1523/JNEUROSCI.20-14-05329.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oleskevich S, Clements J, Walmsley B. Release probability modulates short-term plasticity at a rat giant terminal. J Physiol (Lond) 2000;524:513–523. doi: 10.1111/j.1469-7793.2000.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Osborne SL, Meunier FA, Schiavo G. Phosphoinositides as key regulators of synaptic function. Neuron. 2001;32:9–12. doi: 10.1016/s0896-6273(01)00455-x. [DOI] [PubMed] [Google Scholar]
- 58.Phillips GR, Huang JK, Wang Y, Tanaka H, Shapiro L, Zhang W, Shan WS, Arndt K, Frank M, Gordon RE, Gawinowicz MA, Zhao Y, Colman DR. The presynaptic particle web: ultrastructure, composition, dissolution, and reconstitution. Neuron. 2001;32:63–77. doi: 10.1016/s0896-6273(01)00450-0. [DOI] [PubMed] [Google Scholar]
- 59.Popoff MR. Purification and characterization of Clostridium sordellii lethal toxin and cross-reactivity with Clostridium difficile cytotoxin. Infect Immunol. 1987;55:35–43. doi: 10.1128/iai.55.1.35-43.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Popoff MR, Chaves-Olarte E, Lemichez E, von Eichel-Streiber C, Thelestam M, Chardin P, Cussac D, Antonny B, Chavrier P, Flatau G, Giry M, de Gunzburg J, Boquet P. Ras, Rap, and Rac small GTP-binding proteins are targets for Clostridium sordellii lethal toxin glucosylation. J Biol Chem. 1996;271:10217–10224. doi: 10.1074/jbc.271.17.10217. [DOI] [PubMed] [Google Scholar]
- 61.Poulain B, Baux G, Tauc L. Presynaptic transmitter content controls the number of quanta released at a neuro-neuronal cholinergic synapse. Proc Natl Acad Sci USA. 1986;83:170–173. doi: 10.1073/pnas.83.1.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Poulain B, Tauc L, Maisey EA, Wadsworth JD, Mohan PM, Dolly JO. Neurotransmitter release is blocked intracellularly by botulinum neurotoxin, and this requires uptake of both toxin polypeptides by a process mediated by the larger chain. Proc Natl Acad Sci USA. 1988;85:4090–4094. doi: 10.1073/pnas.85.11.4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Prepens U, Just I, von Eichel-Streiber C, Aktories K. Inhibition of Fc epsilon-RI-mediated activation of rat basophilic leukemia cells by Clostridium difficile toxin B (monoglucosyltransferase). J Biol Chem. 1996;271:7324–7329. doi: 10.1074/jbc.271.13.7324. [DOI] [PubMed] [Google Scholar]
- 64.Quastel DM. The binomial model in fluctuation analysis of quantal neurotransmitter release. Biophys J. 1997;72:728–753. doi: 10.1016/s0006-3495(97)78709-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Redmond L, Ghosh A. The role of Notch and Rho GTPase signaling in the control of dendritic development. Curr Opin Neurobiol. 2001;11:111–117. doi: 10.1016/s0959-4388(00)00181-1. [DOI] [PubMed] [Google Scholar]
- 66.Reid CA, Clements JD. Postsynaptic expression of long-term potentiation in the rat dentate gyrus demonstrated by variance-mean analysis. J Physiol (Lond) 1999;518:121–130. doi: 10.1111/j.1469-7793.1999.0121r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ridley AJ. Rho family proteins: coordinating cell responses. Trends Cell Biol. 2001;11:471–477. doi: 10.1016/s0962-8924(01)02153-5. [DOI] [PubMed] [Google Scholar]
- 68.Robinson HP, Sahara Y, Kawai N. Nonstationary fluctuation analysis and direct resolution of single channel currents at postsynaptic sites. Biophys J. 1991;59:295–304. doi: 10.1016/S0006-3495(91)82223-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rosenmund C, Clements JD, Westbrook GL. Nonuniform probability of glutamate release at a hippocampal synapse. Science. 1993;262:754–757. doi: 10.1126/science.7901909. [DOI] [PubMed] [Google Scholar]
- 70.Scheuss V, Neher E. Estimating synaptic parameters from mean, variance, and covariance in trains of synaptic responses. Biophys J. 2001;81:1970–1989. doi: 10.1016/S0006-3495(01)75848-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Scheuss V, Schneggenburger R, Neher E. Separation of presynaptic and postsynaptic contributions to depression by covariance analysis of successive EPSCs at the calyx of Held synapse. J Neurosci. 2002;22:728–739. doi: 10.1523/JNEUROSCI.22-03-00728.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schiavo G, Benfenati F, Poulain B, Rossetto O, Polverino de Laureto P, DasGupta BR, Montecucco C. Protein tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature. 1992;359:832–835. doi: 10.1038/359832a0. [DOI] [PubMed] [Google Scholar]
- 73.Schneggenburger R, Neher E. Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature. 2000;406:889–893. doi: 10.1038/35022702. [DOI] [PubMed] [Google Scholar]
- 74.Sehr P, Joseph G, Genth H, Just I, Pick E, Aktories K. Glucosylation and ADP ribosylation of rho proteins: effects on nucleotide binding, GTPase activity, and effector coupling. Biochemistry. 1998;37:5296–5304. doi: 10.1021/bi972592c. [DOI] [PubMed] [Google Scholar]
- 75.Short B, Barr FA. Membrane traffic: exocyst III–makes a family. Curr Biol. 2002;12:R18–20. doi: 10.1016/s0960-9822(01)00641-8. [DOI] [PubMed] [Google Scholar]
- 76.Sigworth FJ. The variance of sodium current fluctuations at the node of Ranvier. J Physiol (Lond) 1980;307:97–129. doi: 10.1113/jphysiol.1980.sp013426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Silver RA, Momiyama A, Cull-Candy SG. Locus of frequency-dependent depression identified with multiple-probability fluctuation analysis at rat climbing fibre-Purkinje cell synapses. J Physiol (Lond) 1998;510:881–902. doi: 10.1111/j.1469-7793.1998.881bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Simonneau M, Tauc L, Baux G. Quantal release of acetylcholine examined by current fluctuation analysis at an identified neuro-neuronal synapse of Aplysia. Proc Natl Acad Sci USA. 1980;77:1661–1665. doi: 10.1073/pnas.77.3.1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Stanley EF. The calcium channel and the organization of the presynaptic transmitter release face. Trends Neurosci. 1997;20:404–409. doi: 10.1016/s0166-2236(97)01091-6. [DOI] [PubMed] [Google Scholar]
- 80.Traynelis SF, Silver RA, Cull-Candy SG. Estimated conductance of glutamate receptor channels activated during EPSCs at the cerebellar mossy fiber-granule cell synapse. Neuron. 1993;11:279–289. doi: 10.1016/0896-6273(93)90184-s. [DOI] [PubMed] [Google Scholar]
- 81.Vega IE, Hsu SC. The exocyst complex associates with microtubules to mediate vesicle targeting and neurite outgrowth. J Neurosci. 2001;21:3839–3848. doi: 10.1523/JNEUROSCI.21-11-03839.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vetter IR, Hofmann F, Wohlgemuth S, Herrmann C, Just I. Structural consequences of mono-glucosylation of Ha-Ras by Clostridium sordellii lethal toxin. J Mol Biol. 2000;301:1091–1095. doi: 10.1006/jmbi.2000.4045. [DOI] [PubMed] [Google Scholar]
- 83.Vitale N, Caumont AS, Chasserot-Golaz S, Du G, Wu S, Sciorra VA, Morris AJ, Frohman MA, Bader MF. Phospholipase D1: a key factor for the exocytotic machinery in neuroendocrine cells. EMBO J. 2001;20:2424–2434. doi: 10.1093/emboj/20.10.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]