

Abstract
Ceruloplasmin is a ferroxidase that oxidizes toxic ferrous iron to its nontoxic ferric form. We have previously reported that a glycosylphosphatidylinositol-anchored form of ceruloplasmin is expressed in the mammalian CNS. To better understand the role of ceruloplasmin in iron homeostasis in the CNS, we generated a ceruloplasmin gene-deficient (Cp−/−) mouse. AdultCp−/− mice showed increased iron deposition in several regions of the CNS such as the cerebellum and brainstem. Increased lipid peroxidation was also seen in some CNS regions. Cerebellar cells from neonatalCp−/− mice were also more susceptible to oxidative stress in vitro.Cp−/− mice showed deficits in motor coordination that were associated with a loss of brainstem dopaminergic neurons. These results indicate that ceruloplasmin plays an important role in maintaining iron homeostasis in the CNS and in protecting the CNS from iron-mediated free radical injury. Therefore, the antioxidant effects of ceruloplasmin could have important implications for various neurodegenerative diseases such as Parkinson's disease and Alzheimer's disease in which iron deposition is known to occur.
Keywords: iron, neurodegeneration, free radicals, lipid peroxidation, ferroxidase, aceruloplasminemia, Parkinson's disease, oxidative stress
Iron plays an important role in many biological processes. It is an essential cofactor for various enzymes, including ribonucleotide reductase and aconitase, and its presence in heme imparts hemoglobin the ability to transport oxygen and enables cytochrome oxidase to reduce oxygen to water. The oxidation-reduction reactions of iron are fundamental to its role as a cofactor. However, this also makes free iron highly toxic because of its ability to generate free radicals. In particular, ferrous [Fe (II)] iron can generate the highly toxic hydroxyl and superoxide free radicals in the presence of hydrogen peroxide or molecular oxygen. Consequently, iron metabolism is tightly regulated by various proteins that transport, sequester, and mobilize iron (De Silva et al., 1996).
Transferrin, the major iron-transporting protein in plasma, transports iron from sites of storage, such as the liver, to tissues using iron. The ferroxidase ceruloplasmin (Cp), which is produced by the liver and secreted into the plasma, also plays an important role in the movement of iron. By oxidizing the ferrous [Fe (II)] form of iron to the ferric [Fe (III)] form, Cp promotes iron loading onto transferrin, which only binds the ferric form of the metal (Osaki et al., 1966). In addition, Cp is an effective antioxidant, because of its ability to oxidize highly toxic ferrous iron to the relatively nontoxic ferric form and thus help prevent oxidative damage to proteins, lipids, and DNA (Gutteridge, 1992).
Ceruloplasmin is expressed by astrocytes in the brain, cerebellum, and retina and by the epithelial cells of the choroid plexus (Klomp et al., 1996; Patel and David, 1997; Patel et al., 2000). Although hepatocytes in the liver produce a secreted form of Cp found in serum that does not cross the blood–brain barrier, astrocytes express an alternatively spliced, glycosylphosphatidylinositol (GPI)-anchored form of Cp (Patel and David, 1997; Patel et al., 2000). This GPI-anchored form of Cp is the predominant form expressed in the brain (Patel et al., 2000) and is likely to play an important role in iron homeostasis and antioxidant defense in the CNS. The full-length cDNA for human GPI–Cp has been reported recently (Hellman et al., 2002).
The importance of Cp in vivo is most clearly illustrated by studies of patients with aceruloplasminemia, a hereditary deficiency of Cp caused by mutations in the Cp gene (Miyajima et al., 1987; Harris et al., 1995; Morita et al., 1995; Yoshida et al., 1995;Gitlin, 1998). Patients with this disorder have massive iron deposition in a number of organs, including the brain and liver. The iron deposition in the brain leads to neurodegeneration and neurological symptoms, such as motor incoordination and other motor deficits, between the ages of 45 and 55 years. The severe iron accumulation in these patients suggests that Cp prevents iron accumulation in vivo. Indeed, a recent study ofCp−/− mice showed that the iron that accumulates in the liver can be released when these mice are treated with ceruloplasmin (Harris et al., 1999).
We now report that increased iron accumulation and free radical injury occur in the CNS of Cp−/−mice. In addition, we present in vitro data that provide direct evidence that CNS tissue fromCp−/− mice is more susceptible to iron-mediated free radical injury. TheCp−/− mouse offers a good model for the study of the human disease aceruloplasminemia and can provide insights into the pathogenesis and secondary damage that may occur in other neurodegenerative diseases, such as Parkinson's disease, Alzheimer's disease, and amyotrophic lateral sclerosis, in which iron deposition occurs.
MATERIALS AND METHODS
Generation of Cp−/−mice. To construct a gene-targeting vector, a mouse 129/Sv genomic library in Lambda Dash II (Stratagene) was screened with a probe to the first exon of the rat Cp gene (Fleming and Gitlin, 1990). The two longest positive clones, containing inserts of 11 and 12 kb, were mapped with restriction enzymes. ABglII–NheI fragment containing the first exon was replaced with a neo cassette (Promega) in the orientation opposite to that of the Cp gene to produce the targeting vector (see Fig. 1A). The linearized targeting construct was electroporated into R1 embryonic stem (ES) cells as described (Joyner, 1993).
Fig. 1.
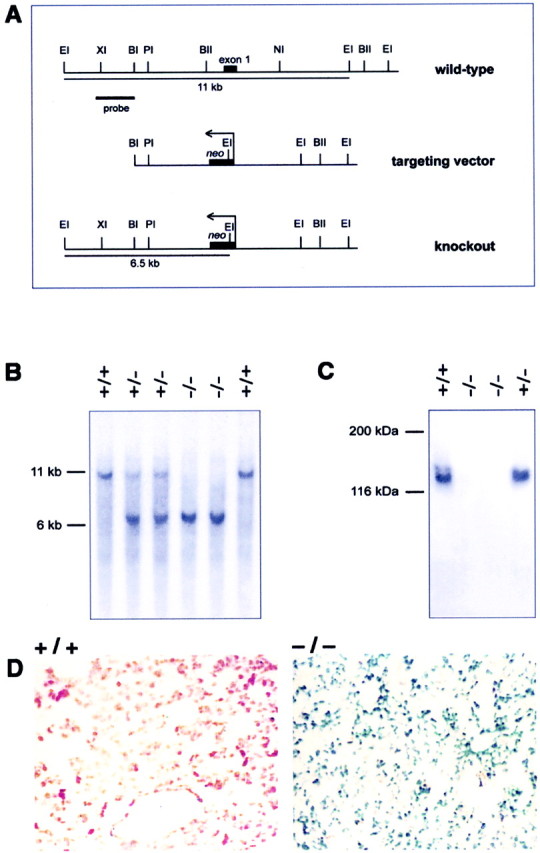
Generation ofCp−/− mice. A, The targeting vector was generated by replacing the 3 kbBglII–NheI fragment containing the first exon of the Cp gene with a neo cassette. Homologous recombination of the targeting vector in ES cells leads to the generation of a 6.5 kb EcoRI fragment compared with a 11 kb wild-type fragment detected using a 1.5 kb probe upstream of the targeting vector (probe). BI, BglI;BII, BglII; EI,EcoRI; NI, NheI;PI, PstI; XI,XhoI. B, Southern blot ofEcoRI-digested genomic DNA from F2 animals using the probe shown in A. C, Western blot of serum using a rabbit anti-Cp polyclonal antibody. BothCp+/+ andCp+/− mice contain large amounts of Cp in the serum, whereasCp−/− mice lack Cp.D, Histological sections of the livers of 16-month-oldCp+/+ andCp−/− mice stained for iron using Perl's stain. The sections were counterstained with neutral red. The liver from a Cp+/+ mouse (+/+) does not show staining for iron. Only theneutral red counterstaining is visible. In contrast, the liver from a Cp−/−(−/−) mouse shows strong labeling for iron, which appears as blue staining.
ES cell clones resistant to G418 (Life Technologies) were selected. Genomic DNA was isolated from these clones and digested withEcoRI. Southern blot analysis was performed using a probe to the mouse genomic sequence upstream of the sequence used in the targeting construct to identify clones with a homologous recombination event. Two positive ES cell clones were microinjected into C57BL/6J blastocysts that were then implanted into pseudopregnant BALB/c mice. The resulting male chimeras were then mated with C57BL/6J females, and the resulting F1 animals were genotyped by Southern blot analysis to identify heterozygotes, which were back-crossed to C57BL/6J animals. After one or more rounds of back-crossing, the resulting heterozygous mice were mated, and the resulting mice were genotyped by Southern blot analysis or PCR and subsequently used for analysis. All protocols for mice were in accordance with the Canadian Council on Animal Care guidelines and approved by the McGill University Animal Care Committee.
Western blot analysis. Western blots were performed using serum from Cp+/+ andCp−/− mice to determine whether Cp protein expression was absent inCp−/− mice. Serum (0.1 μl) diluted in Tris-buffered saline was subjected to SDS-PAGE, and Western blot analysis was performed using a rabbit anti-human Cp antibody (Dako) as described previously (Patel and David, 1997).
Iron histochemistry. Mice were perfused with 0.1m phosphate buffer, pH 7.2, followed by 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.2. Iron was detected in cryostat sections of the liver using a modified Perl's stain for iron (Smith et al., 1997). For this, tissue sections were rehydrated in deionized water and incubated with a 7% solution of potassium ferrocyanide in 3% HCl for 60 min. Sections were then rinsed and counterstained with 1% neutral red and coverslipped. Iron histochemistry on the cerebellum and brainstem was performed using an enhanced Perl's stain that is more sensitive than the one described above. For this, tissue sections were washed in PBS and then incubated in 4% potassium ferrocyanide in 4% HCl for 1 hr. After rinsing in PBS, tissue sections were incubated with unactivated diaminobenzidine (DAB) containing 1% nickel chloride for 15 min. This was followed by a 15 min incubation with activated DAB and counterstaining with neutral red. Tissue sections of wild-type and knock-out mice were picked up on the same slide to permit monitoring and detection of nonspecific staining.
Immunohistochemistry. Tyrosine hydroxylase (TH) immunohistochemistry was done to detect dopaminergic neurons in the midbrain. Cryostat sections (15–20 μm) of 4% paraformaldehyde-fixed brain from Cp+/+ andCp−/− mice were processed for immunohistochemistry as described previously (Ousman and David, 2000). Tissue sections were incubated overnight with a polyclonal rabbit anti-TH antibody (1:200; Chemicon, Temecula, CA). Binding of the primary antibody was detected using a biotinylated secondary antibody followed by avidin–biotin complex conjugated to peroxidase and visualized as described previously (Ousman and David, 2000).
Total non-heme iron determination. Total non-heme iron (TNHI) was measured as described previously by Torrance and Bothwell (1980), with slight modifications. Briefly, tissues were removed from mice after perfusion with PBS. Tissues were dried for 48 hr at 45°C, weighed, and digested for 48 hr in 10% trichloroacetic acid/10% HCl at 65°C. Two hundred microliters of the extract were then added to 1 ml of chromogen solution (0.01% bathophenanthroline-disulfonic acid, 0.1% thioglycolic acid, 7 m sodium acetate) and incubated for 10 min, and the absorbance was measured at 535 nm. A certified iron standard from Sigma (kit 565A) was used to determine iron levels. A standard curve performed for iron concentrations between 10 and 500 μg/ml revealed a linearity of response with a slope of ∼1. Samples were diluted appropriately to fall within the linear range, and a 100 μm FeCl3solution was used as an additional internal control. TNHI values were expressed as micrograms of iron per gram of dry weight.
Lipid peroxidation assay. Lipid peroxidation was assessed using the thiobarbituric acid reactive substances (TBARS) assay that detects malondialdehyde (MDA), an end product of the peroxidation of polyunsaturated fatty acids and related esters (Bowie et al., 1997). Briefly, tissues were homogenized with 0.5% SDS in PBS, and 100 μl of the sample was incubated with 900 μl TBARS reagent (0.4% 2-thiobarbituric acid, 0.5% SDS, 10% acetic acid, pH 3.5), incubated for 60 min at 95°C, and centrifuged, and the absorbance of the supernatant was read at 532 nm. The amount of TBARS was quantified using a standard curve of MDA [malonaldehyde bis (dimethyl acetal)] and expressed as nanomoles of MDA per gram of protein.
Serum iron measurement and hematological analysis. Serum iron levels and total iron-binding capacity were determined using a kit from Sigma. Hematological parameters, such as hematocrit, hemoglobin levels, red blood cell counts, white blood cell counts, and platelet counts, were performed using an automated counter using fresh heparinized blood.
Behavioral assays. To assess motor skills, mice were put on a 3-cm-diameter rod that was covered with a thin rubber mat for grip and rotated at 20 rpm (rotary rod assay) (Steele et al., 1998). The mice were given a training period and subsequently put on the rod for a maximum duration of 3 min. The mice were allowed to rest for 2 min, and the test was repeated five more times.
To assess whether the Cp−/−mice had impaired activity levels, mice were placed in cages containing a hamster wheel that was connected to a meter (Liu et al., 1997). The total distance for the 12 hr night cycle was recorded for each mouse, and the test was performed for two additional nights. The average distance per 12 hr night cycle for each mouse was used for the data analysis.
To determine whether theCp−/− mice had reduced muscle strength, the mice were placed on a thin metal grid attached to weights of increasing mass. After the mice had grasped the grid, they were gently raised by the base of their tails (Frey et al., 2000), and the maximum weight held by the mice for a period of 5 sec was recorded.
In vitro H2O2toxicity assay. Dissociated cultures of the cerebellum were prepared from postnatal day 9 mice using previously described protocols (Mittal and David, 1994). Dissociations were performed using 0.25% trypsin in HBSS for 10 min at room temperature. Cells were plated into poly-l-lysine-coated 24-well tissue culture plates (Linbro) at a density of 8 × 105 cells per well and grown in DMEM containing 10% fetal bovine serum, vitamins, and penicillin/streptomycin. These cultures contain a mixture of mainly neurons and astrocytes, with the latter expressing the GPI-anchored form of Cp (Patel and David, 1997).
After 4 d in culture, the cerebellar cell cultures were washed twice with DMEM, and fresh serum-free Neurobasal Medium containing G-5 supplement, l-glutamine, vitamins, and penicillin/streptomycin (Life Technologies) was added. After 2 hr, fresh serum-free medium containing H2O2 at either 50 or 250 μm was added to the cultures in quadruplicate wells. In some experiments, the iron chelator desferrioxamine mesylate (DFO) was included at 125 μm and added immediately before the addition of H2O2. After 24 hr, cultures were washed with DMEM and incubated with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide [(MTT); 0.5 mg/ml in DMEM] for 90 min (Hansen et al., 1989). The medium was then removed, and the cells were solubilized with 1 ml isopropanol/0.1N HCl. The blue color reaction in the solution was quantified with a spectrophotometer at 570 nm, and the results were normalized to control cultures not treated with H2O2.
For immunostaining, the cerebellar cells were plated on poly-l-lysine-coated 15-mm-round glass coverslips at 5 × 105 cells per coverslip. Cells were cultured and treated with 50 μmH2O2 as described above. At the termination of the experiment, cultures were fixed and permeabilized and double labeled with a monoclonal anti-neurofilament antibody, RT97, to label neurons and a rabbit polyclonal anti-glial fibrillary acidic protein antibody to label astrocytes. Primary antibodies were visualized using appropriate rhodamine- and fluorescein-conjugated secondary antibodies, and the nuclei were labeled with nuclear yellow (Hoechst S769121).
RESULTS
To generate Cp-deficient mice, a targeting vector that contained aneo gene cassette flanked on either side by a 3 kb ceruloplasmin genomic sequence was used to replace a 3 kb endogenous fragment containing the first exon of the ceruloplasmin gene (Fig.1A). This strategy eliminates both the transcription start site as well as the first exon that codes for the signal peptide, thus eliminating the possibility that a truncated protein might be produced. Southern blotting demonstrated the successful generation ofCp−/− mice and was used to genotype mice for analysis (Fig. 1B). Western blotting of the serum confirmed the absence of Cp inCp−/− mice (Fig.1C).
Iron homeostasis is disrupted inCp−/− mice
Because severe hepatic iron accumulation is a hallmark of Cp deficiency in humans, and because the clinical symptoms are generally detected later in life between the ages of 45 and 55 years, we examined the liver of 16-month-oldCp−/− mice for iron content. Abundant iron accumulation was observed within hepatocytes in the liver of Cp−/− mice using the Perl's stain (Fig. 1D). In contrast, very little staining was seen in the liver ofCp+/+ mice (Fig.1D).
Cp−/− mice were examined for changes in various hematological parameters associated with a lack of Cp. Compared with Cp+/+ mice,Cp−/− mice had a severe reduction in serum iron levels (Table 1). This ∼6.5-fold reduction in serum iron levels is also reflected in the lower transferrin saturation (the percentage of total iron binding sites in transferrin loaded with iron) in these mice (4 vs 27%). No significant changes in serum hemoglobin levels (Table 1) or in several other hematological parameters, such as hematocrit or white blood cell count, were observed (data not shown). Using an assay that measures TNHI, iron levels were found to be highly elevated in the liver ofCp−/− mice compared withCp+/+ controls.Cp−/− mice had a 10.6-fold increase in hepatic TNHI at 16 months (Table 1). Iron levels in the spleen were not statistically significantly different between the two groups of mice.
Table 1.
Serum and tissue iron content in wild-type and ceruloplasmin knock-out mice
| Mice | Serum iron (μg/dl) | TIBC (μg/dl) | Transferrin saturation (%) | Hb (gm/dl) | Liver TNHI (μg/gm) | Spleen TNHI (μg/gm) | Kidney TNHI (μg/gm) |
|---|---|---|---|---|---|---|---|
| Cp+/+ | 106 ± 24 | 399 ± 77 | 26.6 ± 6.6 | 13.7 ± 0.8 | 203 ± 24 | 4385 ± 1456 | 213 ± 18 |
| Cp−/− | 16 ± 5.3* | 399 ± 89 | 3.9 ± 1.9* | 13.5 ± 1.8 | 2156 ± 482* | 1389 ± 616 | 248 ± 31 |
Values are expressed as the mean ± SE; n = 4 for each group at 16 months of age. TIBC, Total iron binding capacity; Hb, hemoglobin; TNHI, total non-heme iron; Significant differences are observed between Cp+/+ andCp−/− mice for serum iron, transferrin saturation, and liver TNHI (
p ≤ 0.05, Student's t test).
Iron accumulates in the CNS ofCp−/− mice
Because iron accumulation in the CNS is a characteristic feature of patients with aceruloplasminemia, we examined various regions of the nervous system, including different parts of the brain and spinal cord and the retina, for iron accumulation. The total non-heme iron content of the brainstem and cerebellum was significantly elevated in theCp−/− mice at 16 months, with the brainstem showing a 103% increase and the cerebellum a 35% increase over Cp+/+ mice (Fig.2A). The spinal cord was also affected in Cp−/−mice, with the cervical spinal cord demonstrating a 116% increase and the thoracic spinal cord an 84% increase overCp+/+ mice (Fig.2A). The retina showed a 93% elevation in iron content in the Cp−/− mice. The levels of iron in other parts of the nervous system, such as the cortex, caudate/putamen, olfactory bulb, and lumbar spinal cord, were not significantly different betweenCp−/− andCp+/+ mice.
Fig. 2.

Iron content and lipid peroxidation in CNS.A, The total non-heme iron content of different brain regions from Cp+/+ and Cp−/− mice. Values are shown as the mean ± SE. n = 4 for both groups of mice (*p ≤ 0.05; Student's t test).B, Levels of lipid peroxidation as assessed by measuring the levels of 2-thiobarbituric acid reactive substances in tissue homogenates of different brain regions from wild-type and knock-out mice are displayed. Values are shown as the mean ± SE.n = 4 for both groups of mice (*p ≤ 0.05; Student's ttest).
Iron deposition in the cerebellum and midbrain was also assessed histochemically using a modified Perl's technique. Marked iron deposition was detected in both regions in 24-month-oldCp−/− mice. The deposits in the cerebellum were prominent in the granule cell layer (Fig.3A,B) and the deep cerebellar nuclei. Iron staining was also seen in the region of the substantia nigra (data not shown). Iron levels are known to be high in the normal substantia nigra although this iron is bound to protein and therefore not toxic. On the other hand, the iron deposits in Cp−/− mice are likely to be in the redox active state because the viability of these neurons is severely reduced (see below).
Fig. 3.
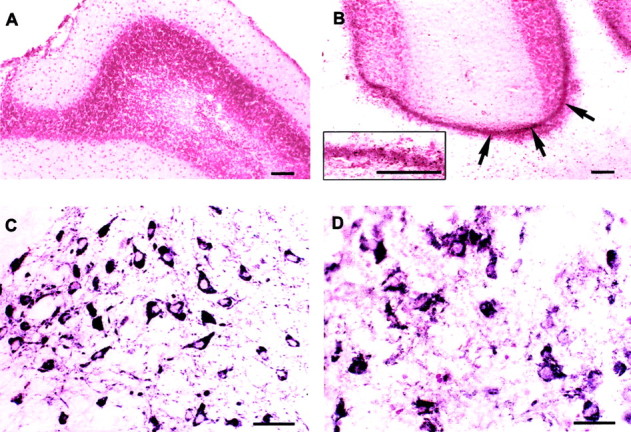
Iron histochemistry and Neurodegeneration.A, B, Iron histochemistry of the cerebellum of Cp+/+(A) and Cp−/−mice (B). Iron deposits, which appearbrown in color (arrows), are seen in the granule cell layer in Cp−/− mice (B). The inset in Bshows these iron deposits at higher magnification. Scale bar, 100 μm.C, D, TH+ dopaminergic neurons in the substantia nigra ofCp+/+ (C) andCp−/− mice (D). There are fewer TH+neurons in the null mice (D), and those present appear to be undergoing degeneration. Scale bars, 100 μm.
Increased free radical-mediated damage occurs in the CNS ofCp−/− mice
Because the lack of Cp leads to increased iron deposition in some regions of the CNS and because Cp is an antioxidant, we assessed lipid peroxidation in different parts of the CNS as evidence of oxidative damage. Significant elevations in lipid peroxidation were observed in the olfactory bulb (84% increase) and the cervical spinal cord (58% increase) (Fig. 2B) ofCp−/− mice at 16 months. Despite having elevated levels of iron inCp−/− mice, the cerebellum, brainstem, thoracic spinal cord, and retina did not show significant increases in lipid peroxidation by the assay that was used. It is possible that the gradual accumulation of iron over a period of months may result in a low level of cell damage occurring over a prolonged period of time, which may not be detected by the assay used. This is supported by the findings that despite the lack of an increase in lipid peroxidation products in the brainstem and retina, there was loss of tyrosine hydroxylase-positive dopaminergic neurons in the brainstem (Fig. 3C,D) and histological evidence of neurodegeneration in the inner nuclear layer of the retina (Fig.4A–D). Ceruloplasmin mRNA has been shown to be expressed mainly in this layer of the retina (Klomp and Gitlin, 1996).
Fig. 4.

Retinal degeneration. A,B, Retina of 18-month-oldCp+/+ (A) andCp−/− mice (B) in Epon-embedded sections stained with toluidine blue. Neurodegenerative changes are seen in the inner nuclear layer (INL) of Cp−/−mice (B). ONL, Outer nuclear layer; GCL, ganglion cell layer. C,D, Higher magnification of the inner nuclear layer ofCp+/+ andCp−/− mice, respectively. Neurons in this layer in wild-type mice are large rounded cells with a prominent nucleolus (C). In contrast, inCp−/− mice many of the cells in this layer show condensed chromatin and dark cytoplasmin (D, arrows). Cells with small, irregularly shaped nuclei that are likely to be macrophages are also present in Cp−/− mice (D). Scale bars (shown in B forA, B): 50 μm; (shown inD for C, D): 20 μm.
On the other hand it is also possible that in some CNS regions other antioxidant mechanisms may protect cells from iron-mediated oxidative damage. This possibility is supported by the fact that although massive iron accumulation occurs in the liver ofCp−/− mice, it does not show a significant increase in lipid peroxidation. In addition, although the olfactory bulb did not show a significant increase in iron content inCp−/− mice, it showed a marked elevation in lipid peroxidation, suggesting that in some CNS regions there may be a change in the redox active state of iron.
Neural cells from Cp−/− mice are more susceptible to free radical injury
Because Cp is an antioxidant and GPI-anchored Cp is expressed by astrocytes, we examined whether neural cells fromCp−/− mice have an increased susceptibility to oxidative stress. Dissociated neonatal cerebellar cell cultures from Cp−/− andCp+/+ mice were treated with H2O2, and their viability was assessed 24 hr later. Cerebellar cultures fromCp+/+ mice were not affected by 50 μmH2O2. In contrast, cultures from Cp−/− mice showed a 45% reduction in viability compared with untreated cultures using the MTT assay (Fig. 5A). Cell counts showed a 90% loss in viability ofCp−/− cultures (588 ± 17/mm2 untreated; 60 ± 5/mm2 treated) but no loss in cultures of cells from Cp+/+ mice (530 ± 22/mm2 untreated; 533 ± 30/mm2 treated). The more severe effect detected with the cell counts is likely caused by the loss of very weakly attached cells that are lost because of the extra washing steps required to stain the cells for counting. Double-immunofluorescence staining for astrocytes and neurons showed that although the viability of both cell types was reduced inCp−/− by such treatment, the neuronal loss was more severe (Fig. 6). At the higher (250 μm) concentration of H2O2, cultures from bothCp−/−and Cp+/+ mice displayed a drastic reduction in viability (Fig. 5A). This result suggests that at this high concentration of H2O2, the protection mediated by Cp is insufficient to prevent cellular injury. Alternatively, Cp itself might be damaged by the high H2O2 and would therefore no longer be protective. The latter possibility is supported by the observation that H2O2 at high concentrations leads to the release of redox-active copper atoms within Cp and the fragmentation of the protein (Choi et al., 2000).
Fig. 5.
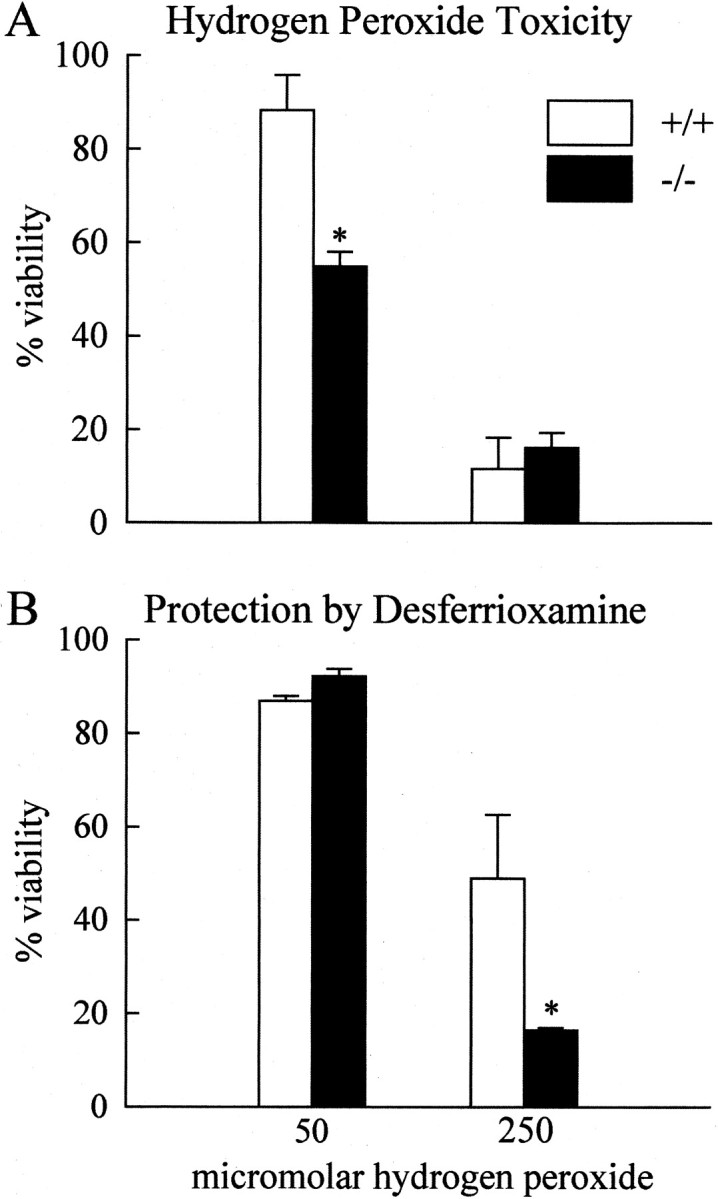
Susceptibility of cerebellar cultures to H2O2 toxicity. A, Cultures of dissociated cerebellum were treated with H2O2, and their viability was assessed using MTT assay. Viability is expressed relative to untreated (0 μm H2O2) control cultures and is shown as the mean ± SE. Results are from three separate experiments. There is a significant decrease in cell viability in cultures of cerebellar cells fromCp−/− mice as compared with those from Cp+/+ mice treated with 50 μm H2O2 (*p≤ 0.05; Student's tTest).B, Cultures of dissociated cerebellum were treated with H2O2 in the presence of 125 μmDFO mesylate. Viability was assessed as above and is the result of three separate experiments. DFO treatment eliminated the decrease in viability of the knockout cultures mediated by the 50 μmH2O2 concentration. DFO partially prevented the decrease in viability caused by the 250 μmH2O2 concentration, and it rescued the cultures from Cp+/+ mice to a significantly greater extent (*p ≤ 0.05; Student'st test).
Fig. 6.

Counts of astrocyte and neuronal populations in cerebellar cultures from Cp+/+ andCp−/− mice treated with 50 μm H2O2 indicate that although the viability of both cell types is reduced in cultures ofCp−/− mice, the loss of neurons is more drastic than that of astrocytes. Results are presented as percentage viability compared with untreated cultures.
The reduction in viability of the cerebellar cultures fromCp−/− mice caused by H2O2in vitro is likely to be caused by the production of toxic hydroxyl radicals. H2O2 can react with a number of different redox-active metals to generate this free radical (Winterbourn, 1995). To determine whether the toxicity of H2O2 was mediated by iron, cerebellar cultures were treated with H2O2 in the presence of the iron chelator DFO, and cell viability was assessed 24 hr later. DFO completely prevented the decrease in viability of theCp−/− cultures treated with 50 μmH2O2 (Fig. 5B). The iron chelator only partially prevented the decrease in viability of both the Cp−/− andCp+/+ cultures treated with 250 μmH2O2. However, the level of rescue was higher in cultures fromCp+/+ control mice compared with those from Cp−/− mice. High concentrations of H2O2can promote the release of iron from the iron–DFO complex (Borg and Schaich, 1986) and may underlie the partial rescue at the higher H2O2 concentration.
Cp−/−mice have impaired motor coordination
To assess whether the increased accumulation of iron and increased oxidative injury in the CNS ofCp−/− mice affect motor behavior, we performed the rotary rod assay on mice at 16 months.Cp−/− mice had an impaired ability to remain on the rotary rod compared with theCp+/+ mice. Although theCp+/+ mice remained on the rotary rod for >3 min, theCp−/− mice remained on the rod for <25 sec, indicating a severe loss of motor coordination in these mice (Fig. 7).Cp+/+ andCp−/− mice did not demonstrate statistically significant differences in strength (121 ± 13 vs 103 ± 9 gm) or locomotory endurance (3823 ± 733 vs 6062 ± 2647 m), the latter assessed using the hamster wheel assay. These results suggest that the motor deficit inCp−/− mice observed using the rotary rod assay was not caused by increased fatigability or decreased strength.
Fig. 7.

Tests of motor coordination. Motor coordination ofCp+/+ andCp−/− mice was assessed using the rotary rod assay. Mice were placed on a rotating rod, and the time, in seconds, that the mice remained on the rod without falling off, up to a maximum of 180 sec, was recorded. Mice of 16 months of age were used. The results are the shown as the mean ± SE; n= 4 for each group. A significant impairment in the ability ofCp−/− mice to remain on the rotary rod compared with Cp+/+ mice is observed (p ≤ 0.05; Student'st test).
DISCUSSION
Increased iron accumulation in the CNS ofCp−/− mice
Iron accumulates in different parts of the CNS in Cp-deficient mice to varying extents. The accumulation is high in the brainstem, retina, and cervical and thoracic spinal cord, with approximately a doubling of the iron content. A significant increase in iron is also found in the cerebellum. The iron deposition seen in these mice is similar in some respects to that observed in patients with aceruloplasminemia, because these patients also display iron accumulation in the brainstem, cerebellum, and retina (Miyajima et al., 1987; Harris et al., 1995; Morita et al., 1995; Yoshida et al., 1995). However, other regions such as the caudate and putamen, which show iron accumulation in aceruloplasminemia patients, appeared to have normal iron levels in Cp null mice. Overall, the extent of iron accumulation in the CNS ofCp−/− mice appears to be less than that observed in patients with aceruloplasminemia, because in the latter the iron accumulation is observable at the gross anatomical level in autopsy samples of the brain (Morita et al., 1995). In humans, neurological symptoms are manifested only between the age of 45 and 55 years (Miyajima et al., 1987; Morita et al., 1995; Yoshida et al., 1995), indicating that severe iron accumulation in the CNS takes several decades. The shorter life span of mice may account for the lower levels of iron accumulation inCp−/− mice.
The iron accumulation in some regions of the CNS of Cp−/−mutant mice, such as the cerebellum and brainstem, may contribute to the deficit in motor coordination observed using the rotary rod assay. This motor deficiency is somewhat similar to the behavioral deficits observed in patients with aceruloplasminemia (Yoshida et al., 1995; Okamoto et al., 1996). Although an increase in lipid peroxidation was not observed in the cerebellum of Cp−/− mice, thein vitro experiments indicate that cerebellar cells are susceptible to iron-mediated oxidative stress. Iron does not accumulate in tissues such as muscle or kidney ofCp−/− mice (data not shown), indicating that iron accumulation is not a generalized nonspecific phenomenon. In addition, iron accumulation in the CNS ofCp−/− mice is not caused by high serum load, because serum iron is markedly decreased.
Cp prevents oxidative damage in the CNS
Because the increase in iron in the CNS might lead to increased oxidative stress caused by the reaction of ferrous iron with oxygen and endogenously produced peroxides, we looked for signs of increased oxidative injury inCp−/− mice. Surprisingly, significantly elevated levels of lipid peroxidation were seen only in the olfactory bulb and the cervical spinal cord. Although the increased lipid peroxidation in the cervical spinal cord was associated with an accumulation of iron, the olfactory bulb did not show an increase in iron. Because Cp is a ferroxidase that oxidizes highly toxic ferrous [Fe (II)] iron to the ferric [Fe (III)] form, it is a potent antioxidant. Therefore, the increased lipid peroxidation observed in the olfactory bulb may be attributable to an increase in the ratio of Fe (II)/Fe (III), caused by the absence of the ferroxidase activity of Cp, rather than to an increase in the quantity of iron.
Despite showing iron accumulation, the cerebellum, brainstem, thoracic spinal cord, and retina did not display a significant elevation in lipid peroxidation. This may be attributable to regional differences in the capacity to manage iron and oxidative stress. For example, different regions of the CNS vary in their susceptibility to iron-dependent lipid peroxidation and in their antioxidant potential (Hussain et al., 1995; Surai et al., 1999). In addition, different regions of the CNS also vary in the expression of ferritin and thus may differ in their capacity to bind iron and render it nontoxic (Han et al., 2000). On the other hand, the gradual accumulation of iron over months may result in a low level of cell and tissue damage over a prolonged period that may be difficult to detect by measuring lipid peroxidation products. This is supported by the evidence of neurodegeneration of dopaminergic neurons in the substantia nigra in the brainstem and of neurons in the inner nuclear layer of the retina. Interestingly, in the retina, ceruloplasmin mRNA expression is detected mainly in the inner nuclear layer (Klomp and Gitlin, 1996).
That Cp is an effective antioxidant in vivo is supported by the in vitro experiments using neonatal cerebellar cultures. These cultures contain astrocytes that express Cpin vivo (Klomp and Gitlin, 1996; Klomp et al., 1996) andin vitro (Zahs et al., 1993; Patel and David, 1997). We have shown previously that GPI-anchored Cp, which is expressed by astrocytes, is the predominant form of this protein expressed in the CNS (Patel et al., 2000). Cerebellar cultures fromCp−/− mice were susceptible to 50 μmH2O2, demonstrating a significant decrease in viability. In contrast,Cp+/+ cultures were unaffected, indicating that Cp protects these cells from free radical injury. The decrease in viability of cerebellar cultures fromCp−/− mice mediated by 50 μmH2O2 could be completely prevented by DFO, a cell-permeable iron chelator. These data indicate that the H2O2 toxicity is mediated by iron, most likely involving the production of highly toxic hydroxyl radicals via the Fenton reaction (Borg and Schaich, 1986).
Role of Cp in vivo
Our data on Cp−/− mice support the earlier findings by Harris et al. (1999) indicating that Cp is essential for iron homeostasis in non-neural tissues. In addition, we show that Cp plays a similar role in the CNS. Cp likely plays an important role in loading iron onto transferrin, and this helps mobilize iron out of hepatocytes. This is supported by the observation that Cp enhances the efflux of iron out of hepatocyte cell lines in combination with transferrin in vitro (Young et al., 1997;Richardson, 1999). Furthermore, Harris et al. (1999) reported recently that Cp administered to Cp knock-out mice can mobilize iron out of the liver and temporarily restore normal iron homeostasis. As expected, we observed a marked reduction in serum iron in ourCp−/− mice that is similar to that seen in patients with aceruloplasminemia. However, Harris et al. (1999) did not observe any differences in serum iron in theirCp−/− and Cp+/+ mice. Differences in the mouse strains (C57BL/6J and black Swiss-Webster) may underlie the observed differences in serum iron levels, but the mechanism whereby the mice ofHarris et al. (1999) are able to maintain apparently normal serum iron levels is not known. Although there are differences in the gene targeting strategies that were used in the two studies, ceruloplasmin could not be detected by Western blotting in the serum of either type of Cp−/− mice.
How Cp prevents iron accumulation in the CNS is not known. Iron uptake by the brain appears to be mediated primarily by transferrin receptors located on the brain capillary endothelial cells (Malecki et al., 1999) as well as on certain neural cells (Roskams and Connor, 1992). Other non-transferrin-mediated iron uptake mechanisms may also exist, such as via the iron transporter DMT1 (Williams et al., 2000). GPI–Cp could play a role in limiting the amount of ferrous iron available for transport via DMT1, which has specificity for ferrous iron (Andrews, 1999). The efflux of iron out of CNS cells may occur via transporters such as Ireg1/ferroportin1, a membrane protein that has been shown to transport ferrous iron out of intestinal epithelial cells (Donovan et al., 2000; McKie et al., 2000). GPI–ceruloplasmin in the brain could also potentially play a role in controlling the efflux of ferrous iron via Ireg1 by rapidly oxidizing it to the ferric form as it exits at the cell surface. Such a mechanism may prevent iron accumulation in the normal CNS and lead to accumulation in the Cp-null mice and in humans with aceruloplasminemia. Hephaestin, an integral membrane protein with ferroxidase activity that is found in intestinal epithelial cells, has homology to Cp and appears to mediate iron efflux from gut epithelial cells (Vulpe et al., 1999; Frazer et al., 2001). Mice lacking hephaestin accumulate iron in the gut caused by impaired egress of iron from intestinal enterocytes (Vulpe et al., 1999). In the CNS, GPI-anchored Cp, which is expressed on the surface of astrocytes, could play a similar role in the efflux of iron out of the CNS.
CONCLUSIONS
Our data indicate that Cp is an effective antioxidant in the CNSin vivo and protects neural cells from oxidative stress. The antioxidant function of Cp is likely to be particularly crucial during CNS injury, such as ischemia or mechanical trauma, when levels of free iron and reactive oxygen species, including H2O2, increase (Hyslop et al., 1995; Palmer et al., 1999). During such injuries, the levels of Cp are also likely to increase because Cp is an acute-phase protein. Indeed, levels of Cp increase in the retina after optic nerve crush (Levin and Geszvain, 1998).
The pathological changes observed in patients with aceruloplasminemia and in Cp−/− mice share similarities with other neurodegenerative diseases. Iron accumulation occurs in the substantia nigra in Parkinson's disease, in the cortex and amyloid plaques in Alzheimer's disease, and in the spinal cord in amyotrophic lateral sclerosis (Gerlach et al., 1994). In addition, levels of free radicals and markers of oxidative injury are also elevated in these disorders (Olanow, 1993; Boll et al., 1999). Whether a reduction in Cp levels contributes to the pathology of these neurodegenerative diseases is not yet known. However, levels of Cp are reduced in the cortex in Alzheimer's disease (Connor et al., 1993), and the ferroxidase activity of Cp is reduced in the cerebrospinal fluid in Parkinson's disease (Boll et al., 1999). It is therefore possible that a reduction in Cp may contribute to the neurodegenerative process in these disorders by leading to an increase in the levels of ferrous iron, which can promote the generation of toxic free radicals. Thus, in addition to providing insights into the human disease aceruloplasminemia, Cp−/−mice could also serve as a useful model to study the role of Cp in the more common neurodegenerative diseases. Furthermore, these mice could potentially be used to test novel therapeutic strategies to prevent iron-mediated free radical injury in the CNS.
Footnotes
This work was supported by a grant from the Canadian Institutes of Health Research and the Parkinson's Disease Foundation (S.D.). B.N.P. was supported by studentships from the National Centers of Excellence NeuroScience Network and the David and Dorothy Lam Award from the NeuroScience Canada Foundation.
Correspondence should be addressed to Dr. Samuel David, Centre for Research in Neuroscience, Montreal General Hospital Research Institute, 1650 Cedar Avenue, Montreal, Quebec, Canada H3G 1A4. E-mail:sdavid11@po-box.mcgill.ca.
REFERENCES
- 1.Andrews NC. The iron transporter DMT1. Int J Biochem Cell Biol. 1999;31:991–994. doi: 10.1016/s1357-2725(99)00065-5. [DOI] [PubMed] [Google Scholar]
- 2.Boll M-C, Sotelo J, Otero E, Alcaraz-Zubeldia M, Rios C. Reduced ferroxidase activity in the cerebrospinal fluid from patients with Parkinson's disease. Neurosci Lett. 1999;265:155–158. doi: 10.1016/s0304-3940(99)00221-9. [DOI] [PubMed] [Google Scholar]
- 3.Borg DC, Schaich KM. Prooxidant action of desferrioxamine: Fenton-like production of hydroxyl radicals by reduced ferrioxamine. J Free Radic Biol Med. 1986;2:237–243. doi: 10.1016/s0748-5514(86)80004-6. [DOI] [PubMed] [Google Scholar]
- 4.Bowie AG, Moynagh PN, O'Neill LAJ. Lipid peroxidation is involved in the activation of NF-kappaB by tumor necrosis factor but not interleukin-1 in the human endothelial cell line ECV304. Lack of involvement of H2O2 in NF-kappaB activation by either cytokine in both primary and transformed endothelial cells. J Biol Chem. 1997;272:25941–25950. doi: 10.1074/jbc.272.41.25941. [DOI] [PubMed] [Google Scholar]
- 5.Choi SY, Kwon HY, Kwon OB, Eum WS, Kang JH. Fragmentation of human ceruloplasmin induced by hydrogen peroxide. Biochimie. 2000;82:175–180. doi: 10.1016/s0300-9084(00)00380-1. [DOI] [PubMed] [Google Scholar]
- 6.Connor JR, Tucker P, Johnson M, Snyder B. Ceruloplasmin levels in the human superior temporal gyrus in aging and Alzheimer's disease. Neurosci Lett. 1993;159:88–90. doi: 10.1016/0304-3940(93)90805-u. [DOI] [PubMed] [Google Scholar]
- 7.De Silva DM, Askwith CC, Kaplan J. Molecular mechanisms of iron uptake in eukaryotes. Physiol Rev. 1996;76:31–47. doi: 10.1152/physrev.1996.76.1.31. [DOI] [PubMed] [Google Scholar]
- 8.Donovan A, Brownlie A, Zhou Y, Shepard J, Pratt SJ, Moynihan J, Paw BH, Drejer A, Barut B, Zapata A, Law TC, Brugnara C, Lux SE, Pinkus GS, Pinkus JL, Kingsley PD, Palis J, Fleming MD, Andrews NC, Zon LI. Positional cloning of zebrafish ferroportin1 identifies a conserved vertebrate iron exporter. Nature. 2000;403:776–781. doi: 10.1038/35001596. [DOI] [PubMed] [Google Scholar]
- 9.Fleming RE, Gitlin JD. Primary structure of rat ceruloplasmin and analysis of tissue-specific gene expression during development. J Biol Chem. 1990;265:7701–7707. [PubMed] [Google Scholar]
- 10.Frazer DM, Vulpe CD, McKie AT, Wilkins SJ, Trinder D, Cleghorn GJ, Anderson GJ. Cloning and gastrointestinal expression of rat hephaestin: relationship to other iron transport proteins. Am J Physiol Gastrointest Liver Physiol. 2001;281:G931–G939. doi: 10.1152/ajpgi.2001.281.4.G931. [DOI] [PubMed] [Google Scholar]
- 11.Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci. 2000;20:2534–2542. doi: 10.1523/JNEUROSCI.20-07-02534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gerlach M, Ben-Shachar D, Riederer P, Youdim MBH. Altered brain metabolism of iron as a cause of neurodegenerative diseases? J Neurochem. 1994;63:793–807. doi: 10.1046/j.1471-4159.1994.63030793.x. [DOI] [PubMed] [Google Scholar]
- 13.Gitlin JD. Aceruloplasminemia. Pediatr Res. 1998;44:271–276. doi: 10.1203/00006450-199809000-00001. [DOI] [PubMed] [Google Scholar]
- 14.Gutteridge JM. Iron and oxygen radicals in brain. Ann Neurol. 1992;32:S16–S21. doi: 10.1002/ana.410320705. [DOI] [PubMed] [Google Scholar]
- 15.Han J, Day JR, Thomson K, Connor JR, Beard JL. Iron deficiency alters H- and L-ferritin expression in rat brain. Cell Mol Biol. 2000;46:517–528. [PubMed] [Google Scholar]
- 16.Hansen MB, Nielsen SE, Berg K. Reexamination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J Immunol Methods. 1989;119:203–210. doi: 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
- 17.Harris ZL, Takahashi Y, Miyajima H, Serizawa M, MacGillivary RT, Gitlin JD. Aceruloplasminemia: molecular characterization of this disorder of iron metabolism. Proc Natl Acad Sci USA. 1995;92:2539–2543. doi: 10.1073/pnas.92.7.2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harris ZL, Durley AP, Man TK, Gitlin JD. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc Natl Acad Sci USA. 1999;96:10812–10817. doi: 10.1073/pnas.96.19.10812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hellman NE, Kono S, Miyajima H, Gitlin JD. Biochemical analysis of a missense mutation in aceruloplasminemia. J Biol Chem. 2002;277:1375–1380. doi: 10.1074/jbc.M109123200. [DOI] [PubMed] [Google Scholar]
- 20.Hussain S, Slikker W, Jr, Ali SF. Age-related changes in antioxidant enzymes, superoxide dismutase, catalase, glutathione peroxidase and glutathione in different regions of mouse brain. Int J Dev Neurosci. 1995;13:811–817. doi: 10.1016/0736-5748(95)00071-2. [DOI] [PubMed] [Google Scholar]
- 21.Hyslop PA, Zhang Z, Pearson DV, Phebus LA. Measurement of striatal H2O2 by microdialysis following global forebrain ischemia and reperfusion in the rat: correlation with the cytotoxic potential of H2O2 in vitro. Brain Res. 1995;671:181–186. doi: 10.1016/0006-8993(94)01291-o. [DOI] [PubMed] [Google Scholar]
- 22.Joyner AL. Gene targeting: a practical approach. Oxford UP; Oxford: 1993. [Google Scholar]
- 23.Klomp LW, Gitlin JD. Expression of the ceruloplasmin gene in the human retina and brain: implications for a pathogenic model in aceruloplasminemia. Hum Mol Genet. 1996;5:1989–1996. doi: 10.1093/hmg/5.12.1989. [DOI] [PubMed] [Google Scholar]
- 24.Klomp LW, Farhangrazi ZD, Dugan LL, Gitlin JD. Ceruloplasmin gene expression in the murine central nervous system. J Clin Invest. 1996;98:207–215. doi: 10.1172/JCI118768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Levin LA, Geszvain KM. Expression of ceruloplasmin in the retina: induction after optic nerve crush. Invest Ophthalmol Vis Sci. 1998;39:157–163. [PubMed] [Google Scholar]
- 26.Liu C, Weaver DR, Strogatz SH, Reppert SM. Cellular construction of a circadian clock: period determination in the suprachiasmatic nuclei. Cell. 1997;91:855–860. doi: 10.1016/s0092-8674(00)80473-0. [DOI] [PubMed] [Google Scholar]
- 27.Malecki EA, Devenyi AG, Beard JL, Connor JR. Existing and emerging mechanisms for transport of iron and manganese to the brain. Neurosci Res. 1999;56:113–122. doi: 10.1002/(SICI)1097-4547(19990415)56:2<113::AID-JNR1>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 28.McKie AT, Marciani P, Rolfs A, Brennan K, Wehr K, Barrow D, Miret S, Bomford A, Peters TJ, Farzaneh F, Hediger MA, Hentze MW, Simpson RJ. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol Cell. 2000;5:299–309. doi: 10.1016/s1097-2765(00)80425-6. [DOI] [PubMed] [Google Scholar]
- 29.Mittal B, David S. A monoclonal antibody that recognizes an adhesion molecule expressed by certain cells of neuroectodermal and mesenchymal origin. Mol Cell Neurosci. 1994;5:63–77. doi: 10.1006/mcne.1994.1006. [DOI] [PubMed] [Google Scholar]
- 30.Miyajima H, Nishimura Y, Mizoguchi K, Sakamoto M, Shimizu T, Honda N. Familial apoceruloplasmin deficiency associated with blepharospasm and retinal degeneration. Neurology. 1987;37:761–767. doi: 10.1212/wnl.37.5.761. [DOI] [PubMed] [Google Scholar]
- 31.Morita H, Ikeda S, Yamamoto K, Morita S, Yoshida K, Nomoto S, Kato M, Yanagisawa N. Hereditary ceruloplasmin deficiency with hemosiderosis: a clinicopathological study of a Japanese family. Ann Neurol. 1995;37:646–656. doi: 10.1002/ana.410370515. [DOI] [PubMed] [Google Scholar]
- 32.Okamoto N, Wada S, Oga T, Kawabata Y, Baba Y, Habu D, Takeda Z, Wada Y. Hereditary ceruloplasmin deficiency with hemosiderosis. Hum Genet. 1996;97:755–758. doi: 10.1007/BF02346185. [DOI] [PubMed] [Google Scholar]
- 33.Olanow CW. A radical hypothesis for neurodegeneration. Trends Neurosci. 1993;16:439–444. doi: 10.1016/0166-2236(93)90070-3. [DOI] [PubMed] [Google Scholar]
- 34.Osaki S, Johnson DA, Frieden E. The possible significance of the ferrous oxidase activity of ceruloplasmin in normal human serum. J Biol Chem. 1966;241:2746–2751. [PubMed] [Google Scholar]
- 35.Ousman S, David S. Lysophosphatidylcholine induces rapid recruitment and activation of macrophages in the adult mouse spinal cord. Glia. 2000;30:92–104. [PubMed] [Google Scholar]
- 36.Palmer C, Menzies SL, Roberts RL, Pavlick G, Connor JR. Changes in iron histochemistry after hypoxic-ischemic brain injury in the neonatal rat. J Neurosci Res. 1999;56:60–71. doi: 10.1002/(SICI)1097-4547(19990401)56:1<60::AID-JNR8>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 37.Patel BN, David S. A novel glycosylphosphatidylinositol-anchored form of ceruloplasmin is expressed by mammalian astrocytes. J Biol Chem. 1997;272:20185–20190. doi: 10.1074/jbc.272.32.20185. [DOI] [PubMed] [Google Scholar]
- 38.Patel BN, Dunn RJ, David S. Alternative RNA splicing generates a glycosylphosphatidylinositol-anchored form of ceruloplasmin in mammalian brain. J Biol Chem. 2000;275:4305–4310. doi: 10.1074/jbc.275.6.4305. [DOI] [PubMed] [Google Scholar]
- 39.Richardson DR. Role of ceruloplasmin and ascorbate in cellular iron release. J Lab Clin Med. 1999;134:454–465. doi: 10.1016/s0022-2143(99)90166-x. [DOI] [PubMed] [Google Scholar]
- 40.Roskams AJ, Connor JR. Transferrin receptor expression in myelin deficient (md) rats. J Neurosci Res. 1992;31:421–427. doi: 10.1002/jnr.490310304. [DOI] [PubMed] [Google Scholar]
- 41.Smith MA, Harris PL, Sayre LM, Perry G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci USA. 1997;94:9866–9868. doi: 10.1073/pnas.94.18.9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Steele PM, Medina JF, Nores WL, Mauk MD. Using genetic mutations to study the neural basis of behavior. Cell. 1998;95:879–882. doi: 10.1016/s0092-8674(00)81712-2. [DOI] [PubMed] [Google Scholar]
- 43.Surai PF, Speake BK, Noble RC, Sparks NH. Tissue-specific antioxidant profiles and susceptibility to lipid peroxidation of the newly hatched chick. Biol Trace Elem Res. 1999;68:63–78. doi: 10.1007/BF02784397. [DOI] [PubMed] [Google Scholar]
- 44.Torrance JD, Bothwell TH. Tissue iron stores. In: Cook JD, editor. Methods in hematology: iron. Churchill Livingstone; New York: 1980. pp. 90–115. [Google Scholar]
- 45.Vulpe CD, Kuo YM, Murphy TL, Cowley L, Askwith C, Libina N, Gitschier, Anderson GJ. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat Genet. 1999;21:195–199. doi: 10.1038/5979. [DOI] [PubMed] [Google Scholar]
- 46.Williams K, Wilson MA, Bressler J. Regulation and developmental expression of the divalent metal-iron transporter in the rat brain. Cell Mol Biol. 2000;46:563–571. [PubMed] [Google Scholar]
- 47.Winterbourn CC. Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol Lett. 1995;82–83:969–974. doi: 10.1016/0378-4274(95)03532-x. [DOI] [PubMed] [Google Scholar]
- 48.Yoshida K, Furihata K, Takeda S, Nakamura A, Yamamoto K, Morita H, Hiyamuta S, Ikeda S, Shimizu N, Yanagisawa N. A mutation in the ceruloplasmin gene is associated with systemic hemosiderosis in humans. Nat Genet. 1995;9:267–272. doi: 10.1038/ng0395-267. [DOI] [PubMed] [Google Scholar]
- 49.Young SP, Fahmy M, Golding S. Ceruloplasmin, transferrin and apotransferrin facilitate iron release from human liver cells. FEBS Lett. 1997;411:93–96. doi: 10.1016/s0014-5793(97)00478-x. [DOI] [PubMed] [Google Scholar]
- 50.Zahs KR, Bigornia V, Deschepper CF. Characterization of “plasma proteins” secreted by cultured rat macroglial cells. Glia. 1993;7:121–133. doi: 10.1002/glia.440070202. [DOI] [PubMed] [Google Scholar]