

Abstract
Serotonin uptake, mediated by the serotonin transporter (SERT), is blocked acutely by antidepressants such as the selective serotonin reuptake inhibitors (SSRIs), but such blockade does not correlate temporally with the onset of therapeutic improvement. Treatment with SSRIs for 21 d induced downregulation of the SERT (Benmansour et al., 1999). The time course of SERT downregulation as well as the time course for its recovery after cessation of treatment with the SSRI sertraline were investigated using tritiated cyanoimipramine to measure SERT binding sites. To determine if there was a temporal correlation between the time when sertraline induced downregulation of the SERT and when marked alteration in SERT function occurred, clearance of locally applied 5-HT into the CA3 region of hippocampus was achieved using in vivo electrochemistry. After 4 or 10 d treatment with sertraline, SERT binding sites decreased very little (15–30%), and the chronoamperometric signals for serotonin in sertraline-treated rats were comparable with ones obtained in control animals. By contrast, after 15 d of treatment, when SERT binding sites were markedly reduced by 80%, there was robust decrease in the clearance of 5-HT. Moreover, the functional consequences of SERT downregulation as measured by chronoamperometry were significantly greater than those seen after acute blockade of the SERT by SSRIs. SERT binding sites decreases are not a consequence of reduced SERT gene expression, as revealed by in situhybridization measurements. SSRI-induced downregulation of the SERT may be a key component for the clinical response to SSRIs.
Keywords: serotonin transporter, antidepressant, sertraline, chronoamperometry, downregulation, mRNA
It is widely believed that the onset of beneficial drug effect in depression is delayed for 2–3 weeks (Gelenberg and Chesen, 2000), although some behavioral dimensions of the illness may respond more quickly (Katz et al., 1996, 1997). Early research on antidepressants (ADs) focused on their acute effects on noradrenergic and/or serotonergic systems. The idea that there is a delay in therapeutic effect led to studies of their longer-term pharmacologic effects, with much of this work focused on receptor regulation (Mongeau et al., 1997; Piñeyro and Blier, 1999). More recently, this work has been extended to studies of protein kinases, transcription factors, and gene regulation (Duman et al.,1997, 1999;Popoli et al., 2000). What seems clear is that the acute pharmacologic enhancement of serotonergic and/or noradrenergic transmission is what initiates the cascade of events that eventually produces clinical improvement (Delgado et al., 1999), although acute enhancement seems insufficient. Initially, enhanced transmission was thought to occur acutely as a direct consequence of AD-induced blockade of either the serotonin transporter (SERT) and/or the norepinephrine transporter. However, a variety of rapid compensatory mechanisms also occur that diminish the ability of transporter blockade alone to significantly enhance synaptic transmission. Subsequent to such rapid compensatory changes, regulatory responses then occur, primarily thought to involve receptor desensitization, that permit synaptic transmission to be enhanced (Artigas et al., 2001). The time required for such secondary compensatory effects to occur and to enhance synaptic transmission has been speculated to account for the delay in therapeutic benefit (Artigas et al., 2001).
It is now recognized, however, that biogenic amine transporters are the key cellular elements regulating the concentration of these transmitters in the extracellular fluid (Giros et al., 1996) and, furthermore, that these transporters can be regulated in vitro. It has been shown that activation of protein kinase C (PKC) induces SERT phosphorylation and sequestration and that this is associated with a decrease in the SERT activity. This effect was shown to be modulated in vitro by 5-HT and selective serotonin reuptake inhibitors (SSRIs) (Ramamoorthy and Blakely, 1999). Also recently it has been shown that SERT forms a complex with the catalytic subunit of protein phosphatase 2A. These heteromeric assemblies are subject to regulation by PKC activation or protein phosphatase inhibition, which leads to phosphorylation and downregulation of the SERT (Bauman et al., 2000).
We found recently (Benmansour et al., 1999) that 21 d of treatment of rats with SSRIs, at clinically relevant and stable serum concentrations (achieved by the use of osmotic minipumps), caused robust downregulation of the SERT. In this study, the time course for such downregulation and its recovery is measured as well as potential mechanisms producing these effects. Importantly, the consequences of SERT downregulation on the functioning of the SERTin vivo was measured directly using chronoamperometry. The use of this technique revealed for the first time that such downregulation produces a much more marked inhibition of 5-hydroxytryptamine (5-HT; serotonin) clearance in vivo than that seen after acute blockade of the SERT with SSRIs.
MATERIALS AND METHODS
Animals. Male Sprague Dawley rats (Harlan, Indianapolis, IN) weighing 175–200 gm at the time of initiation of drug treatment were housed individually on a 12 hr light/dark cycle with lights on at 7:00 A.M. and with food and water provided ad libitum. All animal procedures were in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All efforts were made to minimize both the number of animals used and stress or discomfort to the animal during the experimental procedure.
Chronic drug treatments. Schedule of treatment for onset effects: rats were treated with sertraline (Pfizer, Groton, CT), 7.5 mg · kg−1 · d−1for 4, 10, 15, or 21 d, subcutaneously by means of osmotic minipumps (2ML4; Alza, Palo Alto, CA) to produce stable serum concentrations, and experiments were performed after 2 d of drug washout to reduce serum concentration to <2 ng/ml (Benmansour et al., 1999). Control groups received vehicle (50% ethanol:water).
Schedule of treatment for the time course of recovery: rats were treated with sertraline for 21 d followed by either 2, 6, 8, 10, or 16 d of washout.
Autoradiographic procedures. After completing the treatment regimen and, for some rats, in vivo chronoamperometry recordings, rats were decapitated, and their brains were frozen quickly on dry ice and stored at −80°C until sectioning. Serotonin uptake sites were measured using [3H]-cyanoimipramine (CN-IMI) as a ligand (Kovachich et al., 1988; Benmansour et al., 1999). Brain sections (20 μm) were incubated with 1 nm[3H]-CN-IMI (80–85 Ci/mmol; American Radiolabeled Chemicals, St. Louis, MO) in a buffer consisting of 50 mm Tris, pH 7.4, and 120 mmNaCl at 4°C for 24 hr. Nonspecific binding was defined using 5 μm sertraline and was ∼5% of total binding. Dried slide-mounted sections were placed into spring-loaded cassettes and apposed to tritium-sensitive film ([3H] Ultrofilm; Amersham Pharmacia Biotech, CEA AB, Sweden) at room temperature for 12–14 d. Autoradiograms were analyzed using NIH Image (National Institutes of Health (NIH), Bethesda, MD) and using the Scion software package. Quantitation was achieved using plastic-embedded tritium standards (American Radiolabeled Chemical) calibrated using brain mash sections. Measurements were taken at the level of plate 33 of the atlas ofPaxinos and Watson (1986). The concentration of [3H]-CN-IMI used is approximately eight time its KD value (Kovachich et al., 1988), so the values obtained approximateBmax values.
In vivo chronoamperometry. This procedure was performed as described previously (Daws et al., 1998; Benmansour et al., 1999). Carbon fiber electrodes (30 μm tip diameter; 95–175 μm length in the active region) were coated with Nafion (a perfluorinated ion exchange resin), tested for sensitivity to 5-hydroxyindole acetic acid (250 μm; Sigma, St. Louis, MO), and calibratedin vitro to 5-HT. After completing the appropriate treatment and washout time, rats were anesthetized with chloralose (70 mg/kg)/urethane (700 mg/kg) and the electrode, attached to a multibarrel pipette, was positioned in the CA3 region of the dorsal hippocampus. The internal diameter of each micropipette tip was 8–12 μm, and the distance between electrode and pipette tips was 275–325 μm. Multibarrel micropipettes were filled with either 5-HT (200 μm; Sigma), or fluvoxamine (400 μm; Pharmacia–Upjohn, Kalamazoo, MI). The pH of all solutions was 7.4. 5-HT was delivered by pressure ejection in a volume of 26, 52, or 78 nl. High-speed chronoamperometric recordings were made using the Fast-12 system (Censet, Lexington, KY). Oxidation potentials consisted of 100 msec pulses of +0.55 V versus Ag–AgCl that were delivered one per second; the electrode was held at a resting potential of 0.0 V between measurement. The reference electrode was positioned in the superficial cortex. Oxidation and reduction currents were digitally integrated during the last 80 msec of each 100 msec voltage pulse.
Several parameters are obtained from the electrochemical signal produced by exogenous applications of 5.2, 10.4, or 15.6 pmol of 5-HT. Parameters analyzed were signal amplitude; T80, the time it takes for the peak amplitude to be reduced by 80%; and the total time course (t-course), the total time for the signal to return to baseline from the time of application of 5-HT.
In situ hybridization. Animals treated chronically with sertraline (7.5 mg · kg−1 · d−1) or vehicle (used for the in vivo chronoamperometry experiments) were killed by rapid decapitation. Brains were frozen in isopentane on dry ice and stored at −70°C. Alternate series of 20 μm sections were cut through the dorsal raphe, thaw mounted onto silane-coated glass microscope slides, fixed for 15 min in 4% paraformaldehyde, dehydrated, and stored at −70°C.
Methods for in situ hybridization were as described previously (Domyancic and Morilak, 1997; Benmansour et al., 1999). The 660 nucleotide SERT riboprobe (obtained from Dr. Stanley Watson University of Michigan) was transcribed with the addition of α-35S-UTP (New England Nuclear, Boston, MA), to a specific activity of 2 × 109 cpm/μg. Brain sections were hydrated, acetylated, and rinsed in 2× SSC (1× SSC is 150 mm sodium chloride, 15 mmsodium citrate, pH 7.2), then dehydrated, delipidated, and air-dried. Sections were incubated in hybridization buffer containing 4 × 107 cpm/ml radiolabeled riboprobe (∼20 ng/ml), overnight in a sealed humidified chamber at 60°C.
All posthybridization solutions contained 1 mmdithiothreitol. After rinsing in four washes of 2× SSC and digesting with RNase A (20 μg/ml, 30 min at 37°C), sections were taken through a series of increasingly stringent washes: 10 min each in 1×, 0.5×, and 0.2× SSC at 24°C, followed by 3 × 1 hr in 0.1× SSC at 60°C. They were then rinsed, dehydrated, and apposed, along with 14C-radioactive standards, to Kodak Biomax MR x-ray film for 24 hr before developing. Integrated signal density overlying the dorsal raphe, calibrated from the standards on each film, and expressed in standard units of nanocuries per milligram, was measured in three to six sections per brain corresponding approximately to plate 49 in the atlas of Paxinos and Watson (1986), using the NIH Image software package (version 1.55; Wayne Rasband, NIH).
Serum levels of sertraline. Serum was collected after 10 or 15 d treatment with sertraline to measure steady-state levels. Serum concentrations of sertraline were determined by HPLC as described previously (Benmansour et al., 1999).
Statistical analysis. Data were analyzed by ttest or by one-way ANOVA followed by Newman–Keuls post hocmultiple comparisons, with significance determined atp < 0.05 or by Mann–Whitney U Test for percentage comparison.
RESULTS
Downregulation of the SERT
Chronic treatment with sertraline induced a time-dependent downregulation of SERT binding sites. Figure1 shows data obtained from the CA3 region of hippocampus, parietal cortex, and basolateral amygdala nucleus; similar data were observed throughout the rest of brain. Sertraline caused only a modest reduction in SERT binding site density during the first 10 d of treatment (not more than a 25% decrease in [3H]-CN-IMI binding). This reduction became statistically significant at 10 d of treatment. After 15 d, a marked loss (>70%) of SERT binding sites occurred, and this persisted with further treatment up to 21 d. Shown also in Figure 1 is the recovery of binding sites for the SERT after cessation of sertraline treatment. Recovery was slow, taking at least 10 d after cessation of treatment to approach values obtained in vehicle-treated rats (Fig. 1).
Fig. 1.
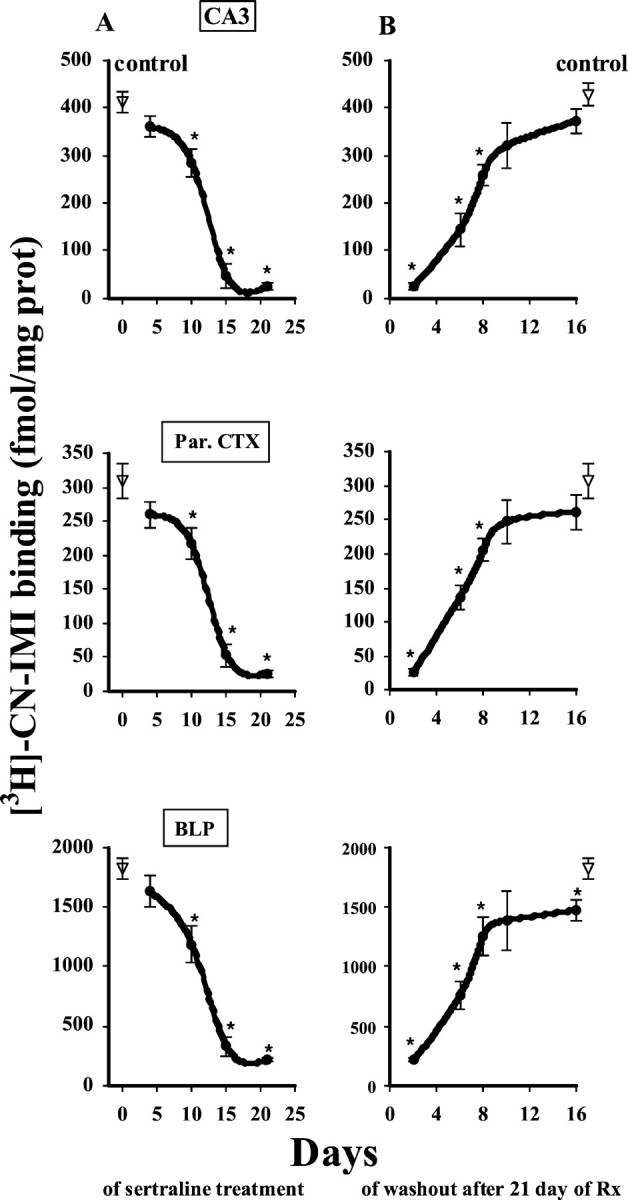
Time course of the reduction (A) and recovery (B) of the binding of [3H]-CN-IMI (1 nm) after sertraline treatment. To study the onset of effects (A), rats were treated by osmotic minipump with sertraline (7.5 mg · kg−1 · d−1, s.c.) or vehicle (for control rats) for 4, 10, 15, or 21 d, always followed by a 2 d washout. To study the time course of recovery (B), rats were treated for 21 d with sertraline or vehicle followed by 2, 6, 8, 10, or 16 d of washout. Serotonin uptake sites were measured using quantitative autoradiography for [3H]-cyanoimipramine binding, as described in Materials and Methods. Results obtained were similar throughout the brain. Shown for illustration are results obtained in the CA3 region of the hippocampus, the parietal cortex (Par. CTX), and the basolateral amygdaloid nucleus posterior (BLP). The number of animals in each drug-treated group was four; the control group included eight animals. *p < 0.05 comparison of each time point for the treatment group with the corresponding time point of the control group. ANOVA, followed by Newman–Keulspost hoc comparison.
Clearance of 5-HT
Given that the time course for marked loss of SERT binding sites (10–15 d) corresponds to the time when drug-induced behavioral improvement becomes evident in patients, it may be that the loss of SERT binding sites is important for achieving long-lasting and marked enhancement of serotonergic transmission with subsequent behavioral improvement. Consequently, the functional consequences of SSRI-induced loss of SERT binding sites was studied in detail. Measurement of SERT function was performed by in vivo chronoamperometry in rats treated with vehicle or sertraline for 4, 10, or 15 d. The primary measure in this study was the clearance of 5-HT after pressure-ejection of exogenous 5-HT (5.2, 10.4, 15.6 pmol) into the CA3 region of the hippocampus of vehicle- or sertraline-treated rats. The CA3 region was selected for the study because previous work has shown that the active clearance of 5-HT from the extracellular fluid of this brain area is caused primarily by activity of the SERT (Daws et al., 1998). Shown in Figure 2 is the chronoamperometric signal caused by 5-HT in a control rat or in a rat treated with sertraline for 15 d, when robust downregulation of the SERT has occurred. After 15 d of sertraline treatment and the 2 d washout period, the 5-HT signal was augmented markedly for each amount of serotonin applied into the CA3 region (Fig. 2). In the sertraline-treated rat, each amount of 5-HT produced much larger peak signal amplitude and a slower decline of the chronoamperometric signal than that measured in the control rat.
Fig. 2.

Representative 5-HT electrochemical signals from the CA3 region of the dorsal hippocampus in a control rat (dashed lines) and in one treated with sertraline for 15 d (solid lines). In vivochronoamperomeric measurements were performed, as described in Materials and Methods, in rats treated with sertraline by osmotic minipump (7.5 mg · kg−1 · d−1, s.c.) for 15 d followed by a 2 d washout. Once reproducible electrochemical signals from 5-HT were obtained, the response to three different amounts of 5-HT (5.2, 10.4, 15.6 pmol) was tested in each drug-treated or control rat. For clarity, only oxidation current curves are shown. Each amount of serotonin produced a greater response in the sertraline-treated rat than in the control one.
In contrast, these effects were not seen in rats treated for 4 or 10 d with sertraline. Analysis of three representative serotonin signal parameters (amplitude, T80, and time course) showed no statistically significant increases either in the peak amplitudes of the signals produced by 5-HT or in the time course of clearance of 5-HT in rats treated with sertraline for 4 or 10 d (Fig.3). However, marked, significant increases in all these parameters were measured in rats treated with sertraline for 15 d. The greater effect on 5-HT clearance in rats treated with sertraline for 15 d as compared with those treated for 10 d is not caused by differences in the serum concentration of sertraline obtained in these animals. In the 10 d-treated rats, the serum concentration of sertraline was 37.6 ± 5.5 ng/ml (n = 6), whereas it was 26 ± 3.4 ng/ml (n = 6) in those treated for 15 d (p > 0.05).
Fig. 3.
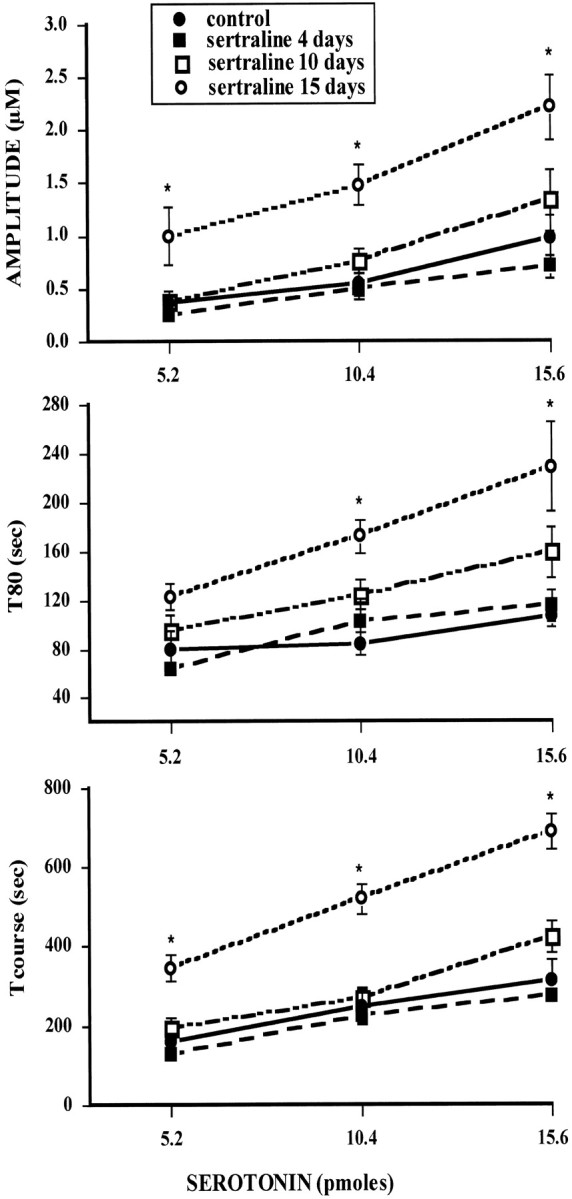
Effect of increasing the amount of 5-HT on the serotonin signal of control and sertraline-treated rats. Rats were treated with vehicle or sertraline for 4, 10, or 15 d followed by 2 d of washout, as described in Materials and Methods. Several parameters are obtained from the electrochemical signal in response to local application of equal amounts of 5-HT (5.2, 10.4, or 15.6 pmol) into the CA3 region of the hippocampus of these rats. Parameters analyzed here were signal amplitude, T80, the time it takes for the peak amplitude to be reduced by 80%; total time course (t-course), the total time for the signal to return to baseline from the time of application of 5-HT. Each point represents the means ± SEM of an n of five to eight rats. *p < 0.05 comparison of each time point in the treatment groups with the corresponding time in the control group; ANOVA, followed by Newman–Keuls post hoccomparison.
The effect that sertraline-induced downregulation of the SERT has on the chronoamperometric signal caused by 5-HT was compared with that caused by acute SSRI-induced pharmacologic blockade of the SERT. To do this, rats treated with vehicle at the same time others rats were treated for 15 d with sertraline, were given local application of both 5-HT and fluvoxamine into the CA3 region of the hippocampus. As shown previously (Benmansour et al., 1999; Daws et al., 2000), local administration of an SSRI such as fluvoxamine into the CA3 region caused a modest, statistically significant effect on the clearance of 5-HT, as reflected by the increase in the T80 value, but had no significant effect on peak amplitude (Table1). Similarly, acute systemic administration of the SSRI paroxetine (10 mg/kg, i.p.) caused an increase in the 5-HT clearance parameter, T80, comparable with that seen with local application of fluvoxamine directly into the CA3 region, while having no significant effect on signal amplitude (Table1). However, chronic treatment of rats with sertraline caused a significantly greater effect on the T80 value than that seen with either local or acute systemic administration of an SSRI, and furthermore, such chronic treatment caused a significant increase in signal amplitude as well (Table 1).
Table 1.
Effects of SSRIs on serotonin signal parameters
| Treatments | Amplitude (μm) | T80 (sec) | T80 (% of pre or control value) |
|---|---|---|---|
| Local fluvoxamine (n = 6) | Pre 0.55 ± 0.09a | 96 ± 7.50 | |
| Post 0.50 ± 0.11 | 135 ± 15.46* | (142 ± 15.26%)b | |
| Systemic paroxetine (n = 6) | Pre 0.73 ± 0.15 | 72 ± 7.70 | |
| Post 0.67 ± 0.09 | 105 ± 8.22* | (148 ± 7.40%) | |
| Chronic vehicle (n = 8) | 0.55 ± 0.12 | 84 ± 9.62 | |
| Chronic sertraline (n = 6) | 1.48 ± 0.19** | 172 ± 13.74** | (205 ± 16.42%)*** |
Comparative analysis of the effect of acute (local or systemic) or chronic treatment with SSRIs. Fluvoxamine (72 pmol) was applied locally into the CA3 region of hippocampus of rats that received vehicle chronically, as described in Materials and Methods. It was pressure-ejected 90 sec before application of 5-HT (10.4 pmol). Data from these experiments (local fluvoxamine) were performed at the same time as the experiments on rats treated for 15 d with sertraline shown in the last two rows of the table. Paroxetine (10 mg/kg, i.p.) was given to naive rats and induced a decrease in clearance of 5-HT (18.2 ± 4.6 pmol) that reached a maximum at 40–60 min after injection, which is when the values shown in this table were measured. The data shown were obtained using amounts of 5-HT to produce comparable peak signal amplitudes before SSRI administration to that measured in the rats treated chronically with vehicle.
Mean ± SEM.
*p<0.05, paired t test comparing postdrug treatment with the corresponding pretreatment value.
**p<0.05, Student's t test comparing values in animals chronically treated with sertraline with those in animals chronically treated with vehicle.
Values in parentheses are the percentage of increase produced by the SSRI in comparison with the pre or control value.
***p < 0.05 in comparison with the other percentage increases, Mann–Whitney U test.
mRNA for the SERT
Previously, we have shown that 21 d of treatment with the SSRI paroxetine had no effect on SERT gene expression (Benmansour et al., 1999). To determine if changes occurred at earlier stages during treatment, mRNA for the SERT was measured in the dorsal raphe nucleus (DRN) by in situ hybridization in the same rats used for the [3H]-CN-IMI binding experiments. Message levels for the SERT increased slowly, reaching a statistically significant increase, by a maximum of 29% after 10 d of treatment, then decreased back to baseline after 21 d of treatment (Fig. 4). Message levels again rose significantly 6 d after 21 d of treatment was terminated, and then returned back to control levels rapidly (Fig. 4). Shown also for comparative purposes in Figure 4 are SERT binding sites in the DRN. Transient increases in mRNA early in the course of treatment may have opposed the SSRI-induced downregulation of SERT binding sites. Only when message levels had declined after the initial increase did SERT binding sites show a marked reduction. However, the short-lived increase in message after the cessation of treatment is accompanied by a more sustained increase in binding sites.
Fig. 4.

Time course of the effect of sertraline treatment and cessation of treatment on the density and mRNA levels of the SERT in the dorsal raphe nucleus. SERT density (solid line) was measured by quantitative autoradiography of [3H]-CN-IMI (1 nm) binding, and mRNA levels for the SERT (dashed line) were measured byin situ hybridization in the same group of animals treated with sertraline, as described in Materials and Methods. Each point is expressed as a percentage of respective control values ± SEM of an N of 4–11 rats. SEM for percentage of control binding is shown by the dashed horizontal lines, and the one for percentage of control mRNA is shown by the solid horizontal lines. Control values for [3H]-CN-IMI in the DRN were 3097 ± 70 fmol/mg of protein, and the mean integrated density for mRNA levels in control rats was 2.27 ± 0.3 nCi/mg. The x-axis of the graph represents the number of days of treatment plus the washout time. Up to 23 d, this represents a variable number of treatment days plus a fixed 2 d washout time. After day 23, the numbers represent a fixed 21 d treatment plus a variable number of washout days. *p < 0.05 comparison of each time point of treatment with the appropriate time point of the control group; ANOVA followed by Newman–Keuls post hoccomparisons.
DISCUSSION
These results demonstrate that sertraline-induced decreases in SERT binding site density are not caused by decreased SERT gene expression, but recovery of binding sites after cessation of treatment may be attributable to increased synthesis of the SERT after an increase in gene expression. The time course of recovery of binding sites is consistent with the turnover rate of the SERT (Vicentic et al., 1999). Most importantly, these data show that the time-dependent loss of SERT binding sites has a much greater effect on the 5-HT electrochemical signal than that seen after acute blockade of the transporter with SSRIs, either given locally or systemically.
Although a small increase of serotonin peak signal amplitude (∼30%) after local application of fluvoxamine has been observed earlier in our laboratory (Daws et al., 1998), this small effect on amplitude is no longer observed consistently as we have refined the technique (Benmansour et al., 1999; Daws et al., 2000) and was not observed in the present study. This lack of effect of fluvoxamine on serotonin signal amplitude is not caused by the use of low dose of fluvoxamine (Daws et al., 1998, 2000). Furthermore, differences in 5-HT clearance observed in rats treated for 10 versus 15 d with sertraline is not caused by these rats achieving different serum concentrations of this SSRI and therefore, is unlikely to be caused by differences in occupancy of the SERT at this times.
Consistent with the inability of acute local application of an SSRI to raise the peak amplitude of the 5-HT electrochemical signal (Benmansour et al., 1999; Daws et al., 2000), there was also little to no change in maximal amplitude of the 5-HT electrochemical signal evoked in a brain slice preparation by either a single electrochemical pulse or a train of pulses (Bunin and Wightman, 1998; Bunin et al., 1998). Thus, the inability of acute blockade of the SERT to increase peak signal amplitude is seen both with exogenously administered 5-HT and with the evoked release of endogenous 5-HT.
The loss of SERT binding sites has a much greater effect on the 5-HT electrochemical signal than that seen with acute blockade. Peak signal amplitude was markedly increased (approximately twofold), and clearance of 5-HT was delayed even more after SERT downregulation than that seen with acute blockade (Table 1). These effects are similar to those seen after lesions of serotonin neurons with the neurotoxin 5,7-dihydroxytryptamine (Daws et al., 1998). It seems likely that this marked effect on 5-HT clearance is caused by the “absence” of SERT.
The parameters used to analyze quantitatively the serotonin electrochemical signal (e.g., T80 values, total time course) were selected because they clearly and reproducibly detect effects of uptake inhibitors and are also known to reflect primarily the uptake process rather than metabolism or diffusion (Cass et al., 1993). However, the detection of neurotransmitter injected into brain by a sensor located some distance from the site of injection is a complex phenomenon. Factors that influence the shape of the signals detected include not only uptake of the transmitter but also its diffusion in obstructed extracellular space (Nicholson and Phillips, 1981), as well, perhaps, as metabolism. In addition, experimental factors such as administration of material by, for example, microiontophoresis versus pressure ejection (Gerhardt and Palmer, 1987), the internal diameter of the ejection pipette, and the distance between the ejection site and the detector (Nicholson, 1995) can strongly influence the results obtained. Finally, with respect to the uptake process itself, which is characterized with Michaelis–Menten kinetics, theVmax orKm of the transporter in the detection areas can also influence the data obtained (Wightman and Zimmerman, 1990; Nicholson, 1995). Perhaps the best attempt to model such data is the elegant paper of Nicholson (1995), who derived numerical solutions to this three-dimensional diffusion problem with nonlinear uptake using the integral equation approach of Tosaka and Miyake (1982). Of the theoretical curves generated for dopamine using this approach, none are exactly like the electrochemical signals measured in vivofor serotonin (Fig. 2), but this may be attributable to their analysis being based on microiontophoretic application versus the pressure-ejection technique used in this study. Nevertheless, comparison of our signals with the theoretical curves obtained byNicholson (1995) shows quite clearly that we are working at a distance between the micropipette ejection tip and the carbon fiber electrode where uptake does contribute to the clearance and where measurable signals are detected. Under our experimental conditions of pressure-ejection of 5-HT, uptake is strongly related to the magnitude of Vmax. Consequently, reduction ofVmax to 0, i.e., the “no uptake” condition in Nicholson's model (which could approximate in our case situations of SERT loss after either chronic SSRI treatment or 5,7-DHT lesions), increases signal amplitude and slows clearance very substantially, much more so than increases inKm as a result of local application of a competitive inhibitor. Our results then, are in good agreement with Nicholson's model.
Our data are consistent with that obtained using in vivomicrodialysis in which chronic treatment of rats with SSRIs caused much greater increases in extracellular 5-HT than seen with single acute systemic administration of an SSRI (Bel and Artigas, 1993; Rutter et al., 1994; Kreiss and Lucki, 1995; Tanda et al., 1996; Hervás et al., 2001). The interpretation of these microdialysis results has, to date, focused on the hypothesis that chronic SSRI treatment produces desensitization of somatodendritic 5-HT1A autoreceptors that normally mediate feed-back inhibition of 5-HT release (Rutter et al., 1994;Invernizzi et al., 1994; Kreiss and Lucki, 1995). Thus, somatodendritic autoreceptor desensitization has been speculated to play a key role in the ability of SSRIs to enhance serotonergic transmission over time to initiate behavioral improvement. The data in this report, though, describe for the first time an alternative or, at least, an additional mechanism for the elevated levels of extracellular 5-HT seen with chronic drug administration, i.e., SERT downregulation facilitating the entry of 5-HT into the extracellular fluid to a greater extent than can be achieved by acute uptake inhibition.
Studies of the effect of chronic antidepressants on the SERT have resulted in inconsistent reports (Owens and Nemeroff, 1998). Among the factors that may contribute to such inconsistency is the route of drug administration. In most studies of chronic administration of ADs to rats, the drugs are given either intraperitoneally or subcutaneously, either once or twice daily. As rats metabolize these drugs more rapidly than humans, such dosage schedules can result in appreciable fluctuations in the serum concentration of drug throughout the day. For certain pharmacologic effects, sustained drug action may be needed. A good example of this is a recent study (Cremers et al., 2000) of citalopram, which has a half-life in the rat of 3–5 hr (Fredricson Overo, 1982; Melzacka et al., 1984). It was shown that chronic treatment with citalopram induced a marked subsensitivity of somatodendritic 5-HT1A receptors only when given by osmotic minipump as opposed to daily injection. It seems likely also that sustained high occupancy of the SERT by SSRIs is needed to demonstrate regulatory effects (Piñeyro et al., 1994; Blier and Bouchard, 1994; El Mansari et al., 1995; Benmansour et al., 1999). Importantly, because the half-lives of many SSRIs in humans are >20 hr (Hiemke and Hartter, 2000), it is likely that these drugs are producing sustained pharmacologic effects throughout the day in patients.
To the extent that effects measured using chronoamperometry and exogenous administration of transmitter reflect changes in synaptic serotonergic transmission, it would seem that SSRIs cause only a modest increase of transmission early in treatment. Only after sustained treatment, when these drugs induce a loss of SERT binding sites, would there be a marked enhancement of transmission. Importantly, the time required for this to occur approximates the time required for clinical improvement in behavior to become manifest (Quitkin et al., 1987). Thus, the loss of SERT binding sites may be an important mechanism to further increase the initial modest elevation in serotonergic transmission that occurs after acute pharmacologic blockade of the transporter. Such mechanisms could involve phosphorylation and sequestration of transporter, as shown in vitro (Ramamoorthy and Blakely, 1999; Bauman et al., 2000), but this has yet to be demonstrated in vivo. Understanding the mechanisms leading to SERT downregulation may permit the development of drugs that can induce this effect more rapidly, allowing perhaps a more rapid course of clinical improvement.
Footnotes
This work was supported by Research Funds from Department of Veterans Affairs and United State Public Health Service Grants MH57001 (A.F.) and MH53851 (D.A.M.). We thank Drs. Georgianna G. Gould and Martin A. Javors for providing help in measuring serum concentrations of sertraline.
Correspondence should be addressed to Dr. Saloua Benmansour, Department of Pharmacology, University of Texas Health Science Center at San Antonio, 7703 Floyd Curl Drive, San Antonio, TX 78229-3900. E-mail:benmansour@uthscsa.edu.
REFERENCES
- 1.Artigas F, Celada P, Laruelle M, Adell A. How does pindolol improve antidepressant action? Trends Pharmacol Sci. 2001;22:224–228. doi: 10.1016/s0165-6147(00)01682-5. [DOI] [PubMed] [Google Scholar]
- 2.Bauman AL, Apparsundaram S, Ramamoorthy S, Wadzinski BE, Vaughan RA, Blakely RD. Cocaine and antidepressant-sensitive biogenic amine transporters exist in regulated complexes with protein phosphatase 2A. J Neurosci. 2000;20:7571–7578. doi: 10.1523/JNEUROSCI.20-20-07571.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bel N, Artigas F. Chronic treatment with fluvoxamine increases extracellular serotonin in frontal cortex but not in raphe nuclei. Synapse. 1993;15:243–245. doi: 10.1002/syn.890150310. [DOI] [PubMed] [Google Scholar]
- 4.Benmansour S, Cecchi M, Morilak DA, Gerhardt GA, Javors MA, Gould GG, Frazer A. Effects of chronic antidepressant treatments on serotonin transporter function, density, and mRNA level. J Neurosci. 1999;19:10494–14501. doi: 10.1523/JNEUROSCI.19-23-10494.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blier P, Bouchard C. Modulation of 5-HT release in the guinea-pig brain following long-term administration of antidepressant drugs. Br J Pharmacol. 1994;113:485–495. doi: 10.1111/j.1476-5381.1994.tb17015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bunin MA, Wightman RM. Quantitative evaluation of 5-hydroxy-tryptamine (serotonin) neuronal release and uptake: an investigation of extrasynaptic transmission. J Neurosci. 1998;18:4854–4860. doi: 10.1523/JNEUROSCI.18-13-04854.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bunin MA, Prioleau C, Mailman RB, Wightman RM. Release and uptake rates of 5-hydroxytryptamine in the dorsal raphe and substantia nigra reticulata of the rat brain. J Neurochem. 1998;70:1077–1087. doi: 10.1046/j.1471-4159.1998.70031077.x. [DOI] [PubMed] [Google Scholar]
- 8.Cass WA, Zahniser NR, Flach KA, Gerhardt GA. Clearance of exogenous dopamine in rat dorsal striatum and nucleus accumbens: role of metabolism and effects of locally applied uptake inhibitors. J Neurochem. 1993;61:2269–2278. doi: 10.1111/j.1471-4159.1993.tb07469.x. [DOI] [PubMed] [Google Scholar]
- 9.Cremers TI, Spoelstra EN, de Boer P, Bosker FJ, Mork A, den Boer JA, Westerink BH, Wikstrom HV. Desensitisation of 5-HT autoreceptors upon pharmacokinetically monitored chronic treatment with citalopram. Eur J Pharmacol. 2000;397:351–357. doi: 10.1016/s0014-2999(00)00308-3. [DOI] [PubMed] [Google Scholar]
- 10.Daws LC, Toney GM, Gerhardt GA, Frazer A. In vivo chronoamperometric measures of extracellular serotonin clearance in rat dorsal hippocampus: contribution of serotonin and norepinephrine transporters. J Pharmacol Exp Ther. 1998;286:967–976. [PubMed] [Google Scholar]
- 11.Daws LC, Gould GG, Teicher SD, Gerhardt GA, Frazer A. 5-HT(1B) receptor-mediated regulation of serotonin clearance in rat hippocampus in vivo. J Neurochem. 2000;75:2113–2122. doi: 10.1046/j.1471-4159.2000.0752113.x. [DOI] [PubMed] [Google Scholar]
- 12.Delgado PL, Miller HL, Salomon RM, Licinio J, Krystal JH, Moreno FA, Heninger GR, Charney DS. Tryptophan-depletion challenge in depressed patients treated with desipramine or fluoxetine: implications for the role of serotonin in the mechanism of antidepressant action. Biol Psychiatry. 1999;46:212–220. doi: 10.1016/s0006-3223(99)00014-1. [DOI] [PubMed] [Google Scholar]
- 13.Domyancic AV, Morilak DA. Distribution of alpha1A adrenergic receptor mRNA in the rat brain visualized by in situ hybridization. J Comp Neurol. 1997;386:358–378. doi: 10.1002/(sici)1096-9861(19970929)386:3<358::aid-cne3>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 14.Duman RS, Heninger GR, Nestler EJ. A molecular and cellular theory of depression. Arch Gen Psychiatry. 1997;54:597–606. doi: 10.1001/archpsyc.1997.01830190015002. [DOI] [PubMed] [Google Scholar]
- 15.Duman RS, Malberg J, Thome J. Neural plasticity to stress and antidepressant treatment. Biol Psychiatry. 1999;46:1181–1191. doi: 10.1016/s0006-3223(99)00177-8. [DOI] [PubMed] [Google Scholar]
- 16.El Mansari M, Bouchard C, Blier P. Alteration of serotonin release in the guinea pig orbito-frontal cortex by selective serotonin reuptake inhibitors. Relevance to treatment of obsessive-compulsive disorder. Neuropsychopharmacology. 1995;13:117–127. doi: 10.1016/0893-133X(95)00045-F. [DOI] [PubMed] [Google Scholar]
- 17.Gelenberg AJ, Chesen CL. How fast are antidepressants? J Clin Psychiatry. 2000;61:712–721. doi: 10.4088/jcp.v61n1002. [DOI] [PubMed] [Google Scholar]
- 18.Gerhardt GA, Palmer MR. Characterization of the techniques of pressure-ejection and microiontophoresis using in vivo electrochemistry. J Neurosci Methods. 1987;22:147–159. doi: 10.1016/0165-0270(87)90009-4. [DOI] [PubMed] [Google Scholar]
- 19.Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- 20.Fredricson Overo K. Kinetics of citalopram in test animals; drug exposure in safety studies. Prog Neuropsychopharmacol Biol Psychiatry. 1982;6:297–309. doi: 10.1016/s0278-5846(82)80180-2. [DOI] [PubMed] [Google Scholar]
- 21.Hervás I, Vilaro MT, Romero L, Scorza MC, Mengod G, Artigas F. Desensitization of 5-HT(1A) autoreceptors by a low chronic fluoxetine dose effect of the concurrent administration of WAY-100635. Neuropsychopharmacology. 2001;24:11–20. doi: 10.1016/S0893-133X(00)00175-5. [DOI] [PubMed] [Google Scholar]
- 22.Hiemke C, Hartter S. Pharmacokinetics of selective serotonin reuptake inhibitors. Pharmacol Ther. 2000;85:11–28. doi: 10.1016/s0163-7258(99)00048-0. [DOI] [PubMed] [Google Scholar]
- 23.Invernizzi R, Bramante M, Samanin R. Chronic treatment with citalopram facilitates the effect of a challenge dose on cortical serotonin output: role of presynaptic 5-HT1A receptors. Eur J Pharmacol. 1994;260:243–246. doi: 10.1016/0014-2999(94)90344-1. [DOI] [PubMed] [Google Scholar]
- 24.Katz MM, Koslow SH, Frazer A. Onset of antidepressant activity: reexamining the structure of depression and multiple actions of drugs. Depress Anxiety. 1996/1997;4:257–267. doi: 10.1002/(SICI)1520-6394(1996)4:6<257::AID-DA1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- 25.Kovachich GB, Aronson CE, Brunswick DJ, Frazer A. Quantitative autoradiography of serotonin uptake sites in rat brain using [3H]cyanoimipramine. Brain Res. 1988;454:78–88. doi: 10.1016/0006-8993(88)90805-0. [DOI] [PubMed] [Google Scholar]
- 26.Kreiss DS, Lucki I. Effects of acute and repeated administration of antidepressant drugs on extracellular levels of 5-hydroxytryptamine measured in vivo. J Pharmacol Exp Ther. 1995;274:866–876. [PubMed] [Google Scholar]
- 27.Melzacka M, Rurak A, Adamus A, Daniel W. Distribution of citalopram in the blood serum and in the central nervous system of rats after single and multiple dosage. Pol J Pharmacol Pharm. 1984;36:675–682. [PubMed] [Google Scholar]
- 28.Mongeau R, Blier P, de Montigny C. The serotonergic and noradrenergic systems of the hippocampus: their interactions and the effects of antidepressant treatments. Brain Res Brain Res Rev. 1997;23:145–195. doi: 10.1016/s0165-0173(96)00017-3. [DOI] [PubMed] [Google Scholar]
- 29.Nicholson C. Interaction between diffusion and Michaelis-Menten uptake of dopamine following iontophoresis in striatum. Biophys J. 1995;68:1699–1715. doi: 10.1016/S0006-3495(95)80348-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nicholson C, Phillips JM. Ions diffusion modified by tortuosity and volume fraction in the extracellular microenvironment of the rat cerebellum. J Physiol (Lond) 1981;321:225–257. doi: 10.1113/jphysiol.1981.sp013981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Owens MJ, Nemeroff CB. The serotonin transporter and depression. Depress Anxiety. 1998;8:5–12. [PubMed] [Google Scholar]
- 32.Paxinos G, Watson C. The rat brain in stereotaxic coordinates. Academic; New York: 1986. [DOI] [PubMed] [Google Scholar]
- 33.Piñeyro G, Blier P. Autoregulation of serotonin neurons: role in antidepressant drug action. Pharmacol Rev. 1999;51:533–591. [PubMed] [Google Scholar]
- 34.Piñeyro G, Blier P, Dennis T, de Montigny C. Desensitization of the neuronal 5-HT carrier following its long-term blockade. J Neurosci. 1994;14:3036–3047. doi: 10.1523/JNEUROSCI.14-05-03036.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Popoli M, Brunello N, Perez J, Racagni G. Second messenger-regulated protein kinases in the brain: their functional role and the action of antidepressant drugs. J Neurochem. 2000;74:21–33. doi: 10.1046/j.1471-4159.2000.0740021.x. [DOI] [PubMed] [Google Scholar]
- 36.Quitkin FM, Rabkin JD, Markowitz JM, Stewart JW, McGrath PJ, Harrison W. Use of pattern analysis to identify true drug response. A replication. Arch Gen Psychiatry. 1987;44:259–264. doi: 10.1001/archpsyc.1987.01800150071009. [DOI] [PubMed] [Google Scholar]
- 37.Ramamoorthy S, Blakely RD. Phosphorylation and sequestration of serotonin transporters differentially modulated by psychostimulants. Science. 1999;285:763–766. doi: 10.1126/science.285.5428.763. [DOI] [PubMed] [Google Scholar]
- 38.Rutter JJ, Gundlah C, Auerbach SB. Increase in extracellular serotonin produced by uptake inhibitors is enhanced after chronic treatment with fluoxetine. Neurosci Lett. 1994;171:183–186. doi: 10.1016/0304-3940(94)90635-1. [DOI] [PubMed] [Google Scholar]
- 39.Tanda G, Frau R, Di Chiara G. Chronic desipramine and fluoxetine differentially affect extracellular dopamine in the rat prefrontal cortex. Psychopharmacology (Berl) 1996;127:83–87. doi: 10.1007/BF02805978. [DOI] [PubMed] [Google Scholar]
- 40.Tosaka N, Miyake S. Analysis of nonlinear diffusion problem with Michaelis-Menten kinetics by an integral equation method. Bull Math Biol. 1982;44:841–849. doi: 10.1007/BF02465184. [DOI] [PubMed] [Google Scholar]
- 41.Vicentic A, Battaglia G, Carroll FI, Kuhar MJ. Serotonin transporter production and degradation rates: studies with RTI-76. Brain Res. 1999;841:1–10. doi: 10.1016/s0006-8993(99)01761-8. [DOI] [PubMed] [Google Scholar]
- 42.Wightman RM, Zimmerman JB. Control of dopamine extracellular concentration in rat striatum by impulse flow and uptake. Brain Res Rev. 1990;15:135–144. doi: 10.1016/0165-0173(90)90015-g. [DOI] [PubMed] [Google Scholar]