

Abstract
To clarify whether inhibitory transmission in the superficial dorsal horn of the spinal cord is reduced after peripheral nerve injury, we have studied synaptic transmission in lamina II neurons of an isolated adult rat spinal cord slice preparation after complete sciatic nerve transection (SNT), chronic constriction injury (CCI), or spared nerve injury (SNI). Fast excitatory transmission remains intact after all three types of nerve injury. In contrast, primary afferent-evoked IPSCs are substantially reduced in incidence, magnitude, and duration after the two partial nerve injuries, CCI and SNI, but not SNT. Pharmacologically isolated GABAAreceptor-mediated IPSCs are decreased in the two partial nerve injury models compared with naive animals. An analysis of unitary IPSCs suggests that presynaptic GABA release is reduced after CCI and SNI. Partial nerve injury also decreases dorsal horn levels of the GABA synthesizing enzyme glutamic acid decarboxylase (GAD) 65 kDa ipsilateral to the injury and induces neuronal apoptosis, detected by terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling staining in identified neurons. Both of these mechanisms could reduce presynaptic GABA levels and promote a functional loss of GABAergic transmission in the superficial dorsal horn.
Keywords: neuropathic pain, GAD, CCI, SNI, cell death, disinhibition
By modulating both synaptic inflow and dorsal horn neuronal excitability, presynaptic and postsynaptic inhibition regulates the transfer of information from the periphery to the CNS. Reduction or elimination of spinal cord inhibition could be expected, therefore, to augment sensory and motor responses to certain signals from the periphery and could play a role in generating pathological excitability. One example of pathological excitation in the somatosensory system is the spontaneous pain and tactile hypersensitivity that can follow peripheral nerve injury. Multiple mechanisms appear to be responsible for this injury-induced pain, including ectopic activity in primary afferents, induction of central sensitization in the dorsal horn in response to such inputs, and sprouting of the central terminals of large myelinated primary afferents (Devor and Seltzer, 1999; Woolf and Mannion, 1999). Another mechanism that has been proposed to contribute to neuropathic pain is peripheral nerve injury-induced loss of inhibition in the spinal cord (Bennett et al., 1989; Dubner, 1991). Indirect evidence suggests that peripheral nerve injury promotes a loss of inhibitory transmission in the spinal cord (Moore et al., 2001). Dorsal root potentials and primary afferent depolarization, indicators of presynaptic inhibition at the central terminals of low-threshold myelinated fibers, are diminished after complete sciatic nerve axotomy (Wall and Devor, 1981). Similarly, large A-fiber-mediated inhibition of C-fiber-evoked responses in dorsal horn neurons is also diminished (Woolf and Wall, 1982). Furthermore, reduced levels of GABA (Castro-Lopes et al., 1993;Ibuki et al., 1997; Eaton et al., 1998) and its receptors (Bhisitkul et al., 1990; Castro-Lopes et al., 1995) have been reported after axotomy and chronic constriction injury (CCI) of the sciatic nerve, as have indications of degenerative changes in the superficial dorsal horn (Sugimoto et al., 1990). What has not been established, however, is whether there is any nerve injury-induced loss of inhibition in the superficial dorsal horn, the site of termination of small caliber primary afferents, and whether such a loss results from diminished presynaptic or postsynaptic inhibition.
To investigate this, primary afferent-evoked EPSCs and IPSCs, respectively, have been analyzed in lamina II neurons of an isolated adult rat spinal cord preparation with an attached dorsal root. The primary afferent-evoked currents in lamina II neurons were recorded in slices from naive rats and after three types of peripheral nerve injury, complete sciatic nerve transection (SNT), CCI (Bennett and Xie, 1988), and spared nerve injury (SNI) (Decosterd and Woolf, 2000). Although excitatory transmission remained intact in all three models, GABAergic inhibition was markedly reduced in the two partial peripheral nerve injury models, CCI and SNI.
MATERIALS AND METHODS
Peripheral nerve injury models. Adult male Sprague Dawley rats were anesthetized with halothane (2.5%), and the left sciatic nerve was exposed. For complete SNT, the sciatic nerve was ligated and severed in the popliteal fossa. For CCI, four 4-0 chromic gut sutures spaced ∼1 mm apart were tied loosely around the sciatic nerve proximal to the trifurcation (Bennett and Xie, 1988). For SNI, the tibial and common peroneal nerves were tightly ligated with 5-0 silk and sectioned distal to the ligation (Decosterd and Woolf, 2000). Rats were allowed to recover in their normal environment for ∼2 weeks after injury.
Spinal cord slice preparation and electrophysiological recording. Under urethane anesthesia (1.5 gm/kg, i.p.), the lumbar spinal cord was removed from nerve-injured rats (2 weeks after injury) or age-matched naive adults and placed in preoxygenated ice-cold Krebs' solution (composition in mm: NaCl 117, KCl 3.6, CaCl2 2.5, MgCl21.2, NaH2PO4 1.2, NaHCO3 25, and glucose 11). The pia arachnoid membrane as well as all ventral and dorsal roots, except the L4 dorsal root ipsilateral to the injury, were removed before cutting a 650-μm-thick transverse slice with the L4 dorsal root (10–20 mm) intact (Yoshimura and Nishi, 1993; Kohno et al., 1999). The slice was placed on a nylon mesh in the recording chamber and perfused with Krebs' solution for at least 30 min before recording.
For whole-cell recording, the resistance of patch pipettes was 5–10 MΩ when filled with internal solution (composition in mm: Cs2SO4 110, CaCl2 0.5, MgCl2 2, EGTA 5, HEPES 5, TEA 5, ATP-Mg salt 5). Electrodes were positioned in lamina II, which is identifiable as a distinct translucent band across the superficial dorsal horn under a dissecting microscope with transmitted illumination. Placing the electrode in such a manner targets a heterogeneous group of intrinsic stalk and islet neurons in inner and outer lamina II (Baba et al., 1999). Graded intensity dorsal root stimulation sufficient to recruit Aβ, Aδ, and C-fibers was applied with a suction electrode linked to a constant current stimulator, and evoked EPSCs and IPSCs were recorded in lamina II neurons voltage clamped to −70 or 0 mV, respectively. EPSCs were classified as monosynaptic if response latency remained constant with high-frequency (20 Hz) stimulation. Synaptic currents were amplified using an Axopatch 200A (Axon Instruments). Signals were filtered at 2 kHz and digitized at 5 kHz. Data were collected using pCLAMP (Axon Instruments) and analyzed using pCLAMP and Mini-analysis software (Synaptosoft).
Western blots. The ipsilateral L4 dorsal horn from naive, CCI, and SNI rats was homogenized in lysis buffer to yield a concentration of 40 μg/μl. Proteins were separated on 4–15% SDS gradient polyacrylamide gels and then transferred to nitrocellulose membranes (Immobilon-P, Millipore). Membranes were incubated with mouse anti-glutamic acid decarboxylase (GAD) 65 kDa (1:2000; Chemicon) (Chang and Gottlieb, 1988) or rabbit anti-GAD67 (1:4000; Chemicon) (Kaufman et al., 1991), followed by horseradish peroxidase (HRP)-conjugated goat anti-mouse (1:15,000; Jackson Laboratories) or donkey anti-rabbit (1:2000; Amersham). Proteins were detected with the enhanced chemiluminescence system (NEN), and then membranes were stripped and reprobed with rabbit anti-p42 MAP kinase (ERK42) as a loading control. Molecular weight markers were used to confirm that the size of GAD65, GAD67, and ERK42 bands were consistent with their predicted sizes. Band intensity was measured densitometrically using NIH Image, normalized to ERK42 levels, and expressed as a percentage of naive. Because ERK42 expression is unaffected by partial peripheral nerve injury (R. R. Ji and C. J. Woolf, personal communication), it is a suitable loading control for comparing GAD expression in naive and injured rats.
Immunohistochemistry. After perfusion with Zamboni's fixative (2% paraformaldehyde and 15% picric acid in 0.1m phosphate buffer, pH 7.4), spinal cord samples were collected from naive and nerve-injured rats. Samples were postfixed for 2 hr, cryoprotected overnight (20% sucrose in PBS), embedded in OCT compound, and frozen. Cryostat sections (20 μm) were thaw mounted onto cold Superfrost microscope slides (Fisher). Sections were incubated overnight at 4°C with mouse anti-GAD65 (1:1000), rabbit anti-GAD67 (1:1000), or mouse anti-GABAA receptor β2/β3 subunit [1:20; Boehringer Mannheim; this antibody should detect most dorsal horn GABAA receptors (Alvarez et al., 1996)], followed by secondary antibody, anti-rabbit FITC, or anti-mouse FITC (1:200; Vector) for 1 hr at room temperature. Fluorescent images were captured using a SPOT camera (Diagnostic Instruments Inc.), and staining intensity was measured using IPLab Image Analysis software (Scanalytics Inc.). Data were normalized to naive.
Detection of apoptosis. Triple fluorescent staining using terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL), bisbenzimide (Hoechst 33342), and neuronal nuclei (NeuN) immunohistochemistry was performed as described (Lewis et al., 1999) on 10 randomly selected 10 μm transverse sections of the L4 segment of the dorsal horn from six animals 7 d after SNI.
Data analysis. Data are expressed as mean ± SEM. Student's paired and unpaired t test and the ztest were used where appropriate. p < 0.05 was considered significant.
RESULTS
Peripheral nerve injury (CCI, SNI, or SNT) does not reduce primary afferent-evoked EPSCs in lamina II neurons
In the L4 segment of spinal cord slices from naive adult rats, all lamina II neurons studied had either Aδ- or C-fiber-evoked fast EPSCs (n = 83). Such primary afferent-evoked excitatory responses remained present in all neurons studied after partial (CCI,n = 57; SNI, n = 46) or complete (SNT,n = 34) sciatic nerve lesions (Fig.1A). Although the activation threshold for A-fiber responses was shifted after nerve injury (naive = 32 ± 2 μA, n = 81; SNT = 22 ± 2 μA, n = 32, p< 0.0001; CCI = 23 ± 2 μA, n = 54,p = 0.0003; SNI = 49 ± 4 μA,n = 37, p < 0.0001), when Aβ and Aδ fibers were fully activated (100 μA, 0.05 msec), EPSC amplitudes were similar in all four populations (Table1). Furthermore, the resting membrane potential was not affected by any of the sciatic nerve injuries (Table1).
Fig. 1.

Partial peripheral nerve injury (CCI or SNI) reduces primary afferent-evoked IPSCs in lamina II neurons.A, Representative neuron from a CCI animal with a monosynaptic Aδ-fiber-evoked EPSC but no evoked IPSC. After CCI or SNI, excitatory transmission remains intact, but the proportion of neurons with no evoked IPSC increases (Naive = 1/65; SNT = 3/32, p = 0.20;CCI = 7/41, p = 0.006;SNI = 9/32, p < 0.0001). *p < 0.05 compared with naive. B, Representative A-fiber-evoked IPSCs recorded in spinal cord slices from naive, CCI, and SNI rats. Note the reduction in amplitude and duration in CCI and SNI traces. C, Amplitude distributions for IPSCs recorded from naive rats and rats subjected to SNT, CCI, or SNI. In both partial nerve injury models, IPSC amplitudes were significantly decreased compared with the naive.Arrows indicate means. D, Frequency distributions of IPSC decay time constants (τ) from naive and nerve-injured rats, as well as pharmacologically isolated glycine (insensitive to 5–10 μm bicuculline;n = 18) and GABAA (bicuculline sensitive; n = 16) IPSCs.
Table 1.
EPSC amplitudes and resting membrane potentials (RMP) in lamina II neurons from naive and nerve-injured rats
| Naive | SNT | CCI | SNI | |
|---|---|---|---|---|
| EPSC amplitude (pA) | 196 ± 15 (61) | 160 ± 16 (30) | 186 ± 20 (31) | 202 ± 32 (36) |
| RMP (mV) | −62 ± 1 mV (60) | −64 ± 1 mV (32) | −65 ± 1 mV (25) | −64 ± 2 mV (25) |
EPSC amplitudes and RMPs were unaffected by complete (SNT) or partial (CCI, SNI) sciatic nerve injury. Numbers of neurons are indicated in parentheses.
Partial peripheral nerve injury (CCI or SNI) reduces primary afferent-evoked IPSCs in lamina II neurons
In contrast to excitatory transmission that remains intact after nerve injury, the proportion of superficial dorsal horn neurons with primary afferent-evoked IPSCs was significantly decreased by 17% after CCI and by 28% after SNI (Fig. 1A). Moreover, the IPSCs remaining in CCI and SNI animals were significantly reduced in both amplitude and duration (Fig. 1B–D, Table 2). Complete sciatic nerve section had no detectable effect on inhibitory synaptic transmission in lamina II of the dorsal horn. The proportion of SNT neurons with IPSCs (Fig.1A) did not differ from naive, nor did IPSC amplitudes and kinetics (Fig. 1C,D, Table 2). In the CCI group, those cells with no primary afferent-evoked IPSCs were further analyzed to determine whether they had spontaneous IPSCs (sIPSCs). In this subgroup of neurons, sIPSCs were present and of normal amplitude, but their frequency was reduced 2.4 ± 0.4 Hz (n = 5) compared with that found in neurons with evoked IPSCs (4.7 ± 1.3 Hz; n = 5).
Table 2.
IPSC amplitudes, rise times, and decay τ values in lamina II neurons from naive and nerve-injured rats
| IPSC | Naive | SNT | CCI | SNI |
|---|---|---|---|---|
| Amplitude (pA) | 159 ± 13 (64) | 157 ± 15 (31) | 113 ± 12 (34)* | 47 ± 7 (23)* |
| Rise time (msec) | 5.1 ± 0.2 (62) | 5.0 ± 0.4 (24) | 4.2 ± 0.2 (34)* | 4.2 ± 0.4 (21)* |
| Decay τ (msec) | 30.4 ± 0.3 (67) | 26.2 ± 2.8 (25) | 17.5 ± 0.2 (32)* | 11.0 ± 0.3 (17)* |
The IPSCs remaining in CCI and SNI animals were significantly reduced in amplitude, rise time, and duration. Numbers of neurons are indicated in parentheses.
p < 0.05 compared with naive.
Selective loss of GABAA receptor-mediated IPSCs in CCI and SNI models
Multiple inhibitory inputs converge on dorsal horn neurons, and some inhibitory interneurons in lamina II coexpress GABA and glycine (Todd and Sullivan, 1990). Therefore, coapplication of both bicuculline (BCC) (5–10 μm; a GABAA receptor antagonist) and strychnine (0.5 μm; a glycine receptor antagonist) was required to completely block primary afferent-evoked IPSCs in lamina II neurons from naive rats (Fig.2A). The kinetics of the GABAA and glycine receptor-mediated IPSCs differed considerably, however. Glycine IPSCs, isolated by BCC to block GABAA receptors, were short lasting, peaking at 4.8 ± 0.6 msec (n = 9) and decaying with a time constant (τ) of 9.2 ± 0.8 msec (n = 18) (Fig.1D). GABAA IPSCs, isolated by strychnine to block glycine receptors, had a slower time to peak (6.9 ± 0.8 msec; n = 13) and decayed with a longer τ (30.8 ± 3.2 msec; n = 16) (Fig. 1D). Although ∼80% of IPSCs in naive slices had both GABAA- and glycine-mediated components, the predominant influence was GABAA receptor-mediated, as shown by both the kinetics (Fig. 1D) and the high sensitivity of these composite IPSCs to BCC (Fig. 2A). After partial, but not complete, sciatic nerve injury, the IPSC kinetics shifted closer to those resembling pure glycinergic currents (Fig. 1D), and the BCC-sensitive (GABAA) component was reduced significantly (Fig. 2A,B).
Fig. 2.
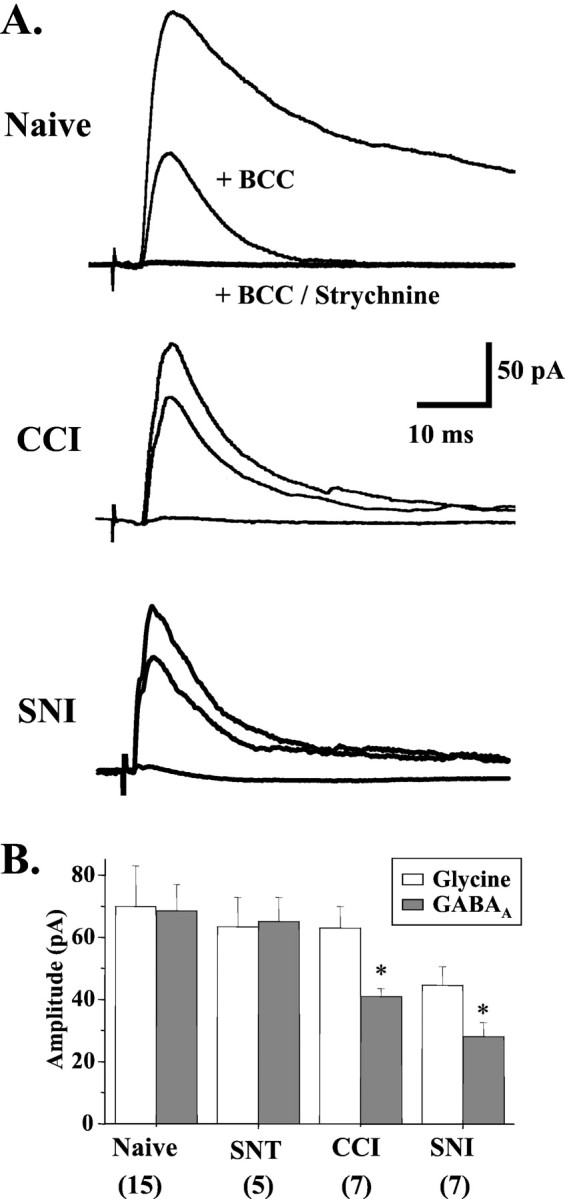
Selective loss of GABAA IPSCs in CCI and SNI models. A, In naive neurons, the GABAA receptor antagonist bicuculline (5–10 μm) blocked a large fraction of the primary afferent-evoked IPSC (n = 14). However, after CCI (n = 13) or SNI (n = 7), bicuculline had little effect. Coapplication of bicuculline and strychnine (0.5 μm) reduced IPSCs to 4.3 ± 1.1 pA (n = 19). B, The amplitude of the GABAA component, but not the glycine component, of evoked IPSCs was significantly reduced after CCI (p= 0.014) or SNI (p = 0.002), but not SNT (p = 0.745). Numbers of neurons are indicated in parentheses. p< 0.05 compared with naive.
Reduced GABA release accounts for the loss of inhibition
To determine whether presynaptic or postsynaptic factors are responsible for the attenuation of the GABAAreceptor-mediated inhibition in lamina II neurons, the frequency and amplitude of sIPSCs and miniature IPSCs, respectively, were investigated. The overall frequency of sIPSCs was not significantly affected by CCI or SNI, but the frequency of the strychnine-insensitive, GABAA receptor-mediated sIPSCs was significantly reduced by both partial nerve injuries (Table3), consistent with a presynaptic locus for the loss of inhibition. In contrast, the frequency of BCC-insensitive, glycine receptor-mediated sIPSCs was unaffected by CCI or SNI, as were the amplitude distributions of both GABAAand glycine sIPSCs (Table 3). A reduction in GABAAreceptor-mediated sIPSC frequency, in the absence of an effect on sIPSC amplitude, reflects either a loss of action potential-driven synaptic currents or diminished activity-independent (miniature) synaptic release. Analysis of miniature GABAA receptor-mediated IPSCs revealed a decrease in frequency, but not amplitude, after CCI or SNI (Fig. 3), consistent with diminished presynaptic GABA release.
Table 3.
The frequency of GABAA sIPSCs is reduced after CCI and SNI
| Spontaneous IPSCs | Naive | CCI | SNI |
|---|---|---|---|
| Amplitude (pA) | |||
| All sIPSCs | 17.3 ± 2.4 (10) | 14.6 ± 2.2 (5) | 19.8 ± 3.0 (9) |
| GABAA | 12.1 ± 1.5 (7) | 13.0 ± 2.0 (6) | 16.3 ± 2.7 (8) |
| Glycine | 14.2 ± 2.9 (6) | 13.1 ± 1.1 (5) | 12.3 ± 2.3 (5) |
| Frequency (Hz) | |||
| All sIPSCs | 6.3 ± 1.2 (10) | 4.7 ± 1.3 (5) | 4.4 ± 1.0 (9) |
| GABAA | 5.8 ± 0.6 (7) | 3.9 ± 0.4 (7)3-150 | 1.8 ± 0.3 (8)3-150 |
| Glycine | 2.8 ± 1.2 (6) | 2.4 ± 0.6 (5) | 2.5 ± 1.1 (5) |
GABAA and glycine receptor-mediated sIPSCs were isolated pharmacologically with strychnine (0.5 μm) or bicuculline (5–10 μm), respectively. The frequency of GABAA sIPSCs decreased significantly after CCI (p = 0.02) and SNI (p < 0.0001). Numbers of neurons are indicated in parentheses.
F3-150: p < 0.05 compared with naive.
Fig. 3.

GABA transmission is reduced by a presynaptic mechanism after partial nerve injury. The frequency of miniature GABAA IPSCs (recorded in the presence of 0.5 μm TTX and 0.5 μm strychnine) was significantly decreased from 4.0 ± 0.3 Hz (n= 5) in naive slices to 2.2 ± 0.5 Hz (n = 8;p = 0.024) in CCI slices and 2.0 ± 0.6 Hz (n = 6; p = 0.020) in SNI slices. The amplitude of miniature GABAA IPSCs was unaffected by partial nerve injury (Naive = 11.7 ± 1.8 pA; CCI = 10.5 ± 0.6 pA;SNI = 13.8 ± 2.3 pA). p < 0.05 compared with naive.
Partial nerve injury reduces GAD65 levels
Western blot analysis of the two isoforms of the GABA synthesizing enzyme, GAD65 and GAD67, revealed a small (20–40%), but significant, decrease in GAD65 protein levels after both CCI and SNI. In contrast, GAD67 was generally unaffected (Fig. 4). Immunohistochemical analysis revealed that GAD65 was downregulated throughout the ipsilateral dorsal horn (laminas I–IV) (Fig.4B); however, no reduction was observed in the contralateral dorsal horn (Fig. 4B), implying that reduced GABA release may be a general feature in all laminas of the dorsal horn ipsilateral, but not contralateral, to the lesion. Furthermore, Western blotting demonstrates that GAD65 downregulation was time dependent. In CCI rats, GAD65 levels drop within 6 d of injury and slowly return to baseline within 4 weeks of the injury (Fig.4A). In SNI rats, GAD65 also falls within 6 d of injury, but in contrast to CCI rats, it remains depressed at 4 weeks (Fig. 4A).
Fig. 4.
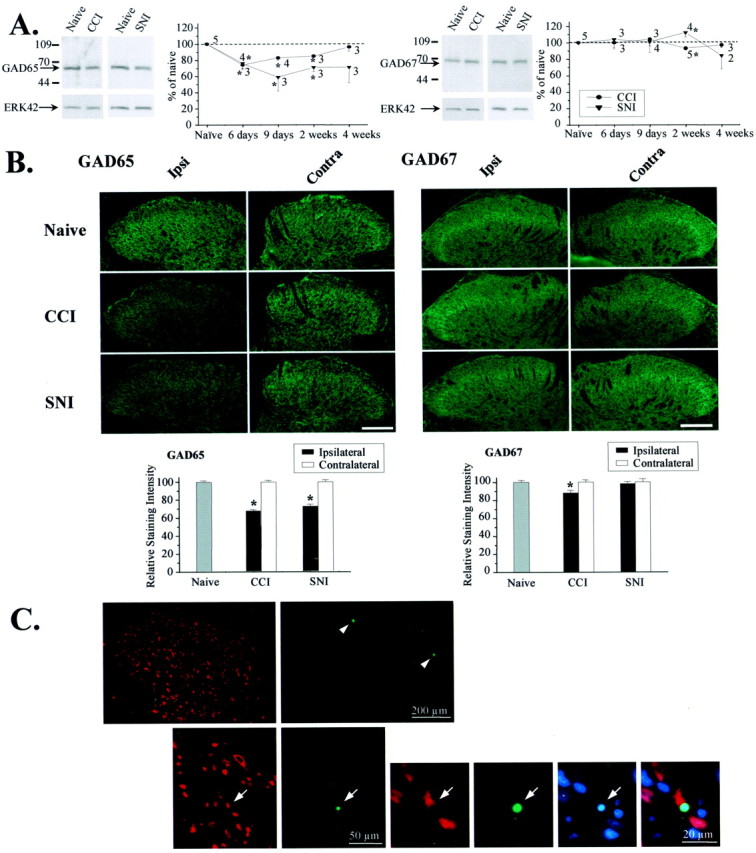
Partial nerve injury reduces GAD65 levels and induces neuronal apoptosis. A, The 65 kDa, but not the 67 kDa, isoform of GAD was reduced in the lumbar (L4) dorsal horn ipsilateral to CCI or SNI when measured by Western blotting. Thegraph shows the time course of regulation. Data are normalized to ERK42 and expressed as a percentage of naive. *p < 0.05 compared with naive. B, Immunohistochemical analysis revealed diminished GAD65 levels in the ipsilateral dorsal horn (laminas I–IV) 2 weeks after CCI (n = 46 sections from 4 rats; p< 0.0001) and SNI (n = 31 sections from 6 rats;p < 0.0001). A small decrease in the levels of GAD67 was detectable after CCI (n = 52 sections from 4 rats; p = 0.002). Scale bar, 100 μm. *p < 0.05 compared with naive. C, TUNEL (green) and chromatin staining with bisbenzimide (blue), combined with immunostaining for the neuronal marker NeuN (red), revealed apoptotic cell death in the superficial dorsal horn of spinal cord segment L4 1 week after SNI. The top row shows a section through the ipsilateral dorsal horn with TUNEL-positive cells in laminas I and II (arrowheads). An example of neuronal apoptosis in lamina II is given below. Arrows point to a TUNEL-positive neuron with condensed chromatin structure, indicating nuclear pyknosis.
Neuronal death occurs in the dorsal horn after partial peripheral nerve injury
A reduction in the releasable GABA pool, reflected by the diminution of both miniature GABAA IPSC frequency (Fig. 3) and evoked IPSC amplitude (Figs. 1, 2), might reflect decreased GABA production within the axon terminals of GABAergic interneurons attributable to GAD65 downregulation (Fig.4A). Alternatively, or additionally, it could also reflect selective degeneration of this population of inhibitory interneurons resulting from excitotoxic cell death induced by excessive nerve discharge at the time of the injury (Sugimoto et al., 1990). Although no significant loss of neurons is observed in lamina II after a complete sciatic nerve section (Coggeshall et al., 2001), TUNEL-positive profiles indicative of apoptosis peak within 1 week after CCI (Whiteside and Munglani, 2001). The extent to which these profiles represent neurons or glia is unclear. We have now found TUNEL-positive profiles with pyknotic nuclei, detected by chromatin staining, in the L4 segment of the dorsal horn 7 d after an SNI lesion (1.48 ± 0.08 profiles per 10 μm section). Many of the TUNEL-positive profiles were located in the superficial dorsal horn and were not stained by NeuN, a specific neuronal marker. Nevertheless, apoptotic neuronal (TUNEL-positive/NeuN-positive) profiles were found in laminas I and II after SNI (0.16 ± 0.03 profiles per 10 μm section) (Fig. 4C). The non-NeuN-stained population may represent dying non-neuronal cells or neurons that have lost NeuN immunoreactivity.
GABAA receptors remain intact after CCI and SNI
In addition to changes in GABA release, dynamic regulation of GABAA receptor density might contribute to spinal disinhibition. The analysis of miniature GABAA IPSCs showing a reduction in frequency, but not amplitude (Fig. 3), suggests that synaptic GABAA receptors remain present and functional after both partial nerve injuries. Immunohistochemistry confirmed that GABAA receptor expression was not decreased after either partial nerve injury (Fig. 5).
Fig. 5.
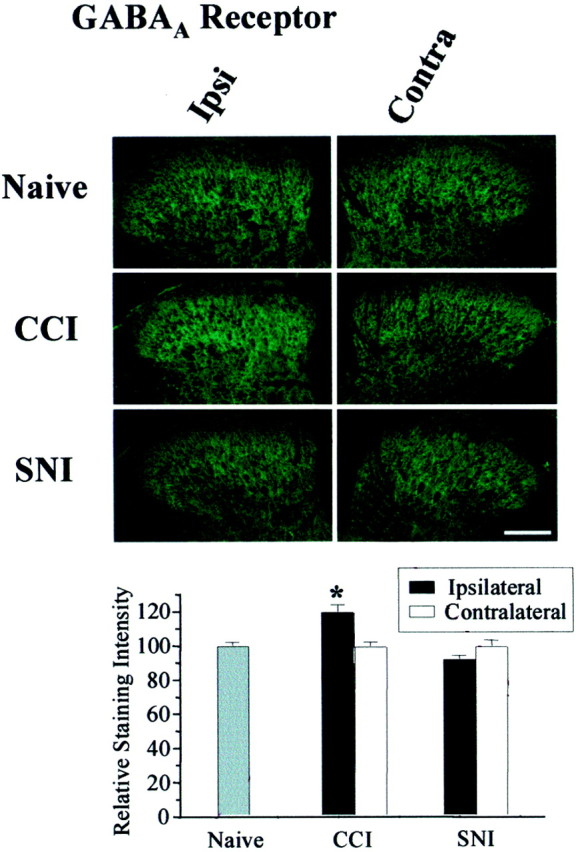
GABAA receptors remain after partial nerve injury. Immunohistochemical analysis for GABAAreceptor in the lumbar dorsal horn revealed a small upregulation after CCI (n = 27 sections from 3 rats;p = 0.031) but not SNI (n = 55 sections from 6 rats). Scale bar, 100 μm. Data are expressed as a percentage of naive. p < 0.05 compared with naive.
DISCUSSION
We have found a loss of postsynaptic GABAAreceptor-mediated inhibition in the superficial dorsal horn in two different partial peripheral nerve injury models. Because similar changes did not occur after a complete section of the sciatic nerve, it appears that some intact input is required for the loss of inhibition to occur. After either CCI or SNI nerve lesion, GABAergic inhibitory transmission in lamina II neurons was diminished, whereas glycinergic transmission remained mostly intact. Most IPSCs observed in slices from naive rats had both GABAA- and glycine receptor-mediated components. However, the duration of the GABAA IPSCs was up to three times longer than that of the glycine IPSCs, suggesting that the GABAA component may normally play a more critical role in limiting neuronal firing frequency and controlling long latency or polysynaptic inputs to the dorsal horn. Therefore, a loss of GABAergic postsynaptic inhibition might be expected to amplify excitatory responses of lamina II neurons to afferent inputs. The overall functional significance for sensory processing in the spinal cord will depend on whether similar changes also occur in other dorsal horn laminas, as well as the connectivity and transmitter content of the affected lamina II neurons, a heterogeneous population that may include excitatory and inhibitory interneurons.
After either CCI or SNI, GABAA receptors remained present, but unitary GABA release was significantly decreased, suggesting that presynaptic, rather than postsynaptic, changes account for the loss of GABAergic inhibition in lamina II. GABA synthesis relies on two GAD isoforms, GAD65 and GAD67, each of which is encoded by a distinct, differentially regulated gene. GAD isoforms are commonly coexpressed in GABAergic interneurons; however, GAD67 is primarily cytosolic, localized to neuronal cell bodies and dendrites, whereas GAD65 is preferentially targeted to membranes and nerve endings where it may preferentially synthesize GABA for vesicular release (Soghomonian and Martin, 1998). Our immunohistochemical analysis illustrates a differential distribution pattern for the two isoforms. Further studies will be required to determine whether these isoforms are coexpressed in the same neurons in the dorsal horn.
In both partial nerve injury models, GAD65 protein levels declined significantly in all laminas of the ipsilateral dorsal horn [see alsoEaton et al. (1998)]. This decrease may reflect reduced transcription/translation of GAD65 and a loss of GABAergic interneurons caused by cell death. Relative to GAD65, GAD67 is generally maintained after partial peripheral nerve injury, although some loss may occur after CCI. Several possible explanations could account for the selective diminution of GAD65. GABAergic interneurons may undergo apoptosis, leading to loss of both GAD65 and GAD67 followed by compensatory upregulation of GAD67, but not GAD65, in those interneurons that survive (Dumoulin et al., 1996). Alternatively, if the two isozymes are not expressed in the same neurons, death of those interneurons that express GAD65 with survival of those that express GAD67 could account for the selective attenuation of GAD65. Finally, GAD65 may be downregulated in response to partial peripheral nerve injury independent of cell death. Reduction of GAD65 may be one of several transient mechanisms that contribute to the early/intermediate phases of hyperalgesia that follow nerve injury. Early changes in the dorsal horn (e.g., reduced inhibition caused by reduced GABA synthesis) and in primary afferent physiology (e.g., ectopic discharge in injured and neighboring noninjured sensory fibers) might help to set the stage for mechanisms that underlie the persistent phases of nerve injury-induced pain hypersensitivity, such as death of inhibitory interneurons.
After SNT, where we failed to detect a loss of inhibition, there is also an absence of neuronal loss in lamina II, as demonstrated recently by unbiased stereological counts in electron microscopic sections (Coggeshall et al., 2001). In contrast, after CCI, TUNEL-positive cells indicating apoptosis have been identified in the ipsilateral dorsal horn, although the cell type (i.e., whether they represent neurons or not) was not specified (Whiteside and Munglani, 2001). We have now found that apoptosis also occurs in the superficial dorsal horn after SNI, and although most of the profiles do not express the neuronal marker NeuN, positively identified apoptotic neurons were observed. It is uncertain, however, whether this includes GABAergic interneurons, because immunoreactivity may be lost as cells die. The number and nature of neurons that are lost after partial nerve injury require further investigation.
Diminished GABA release will reduce both presynaptic (influencing synaptic inflow) and postsynaptic (modulating dorsal horn neuron excitability) GABAA receptor-mediated inhibition, as well as GABAB receptor-mediated inhibition. Here, we have demonstrated a decrease in GABAA receptor-mediated postsynaptic inhibition. Earlier studies have reported that primary afferent depolarization and dorsal root potentials, indirect indicators of presynaptic inhibition, are reduced in large A-fibers after complete sciatic nerve axotomy (Wall and Devor, 1981). It is highly probable from our findings that GABA-mediated presynaptic inhibition of Aδ- and C-fiber nociceptors that terminate in the superficial dorsal horn will also be reduced after partial peripheral nerve injury as a result of the reduction in the releasable GABA pool.
GABAergic transmission to lamina II neurons was reduced after two distinct partial peripheral nerve lesions, CCI and SNI, both of which generate marked pain hypersensitivity. Pharmacological antagonism of GABAergic and glycinergic inhibitory transmission in the spinal cord in normal animals also generates a neuropathic pain-like tactile hypersensitivity (Yaksh, 1989; Sivilotti and Woolf, 1994;Khandwala et al., 1997). These two observations raise the key question: does diminished inhibitory transmission in the dorsal horn contribute to heightened pain sensitivity after partial nerve injury? Although it is not yet possible to answer this definitively, therapeutic implications for patients could be substantial. To increase efficacy, neuropathic pain management may need to include treatment directed at preventing a loss of endogenous inhibitory control systems or augmenting those that remain.
K.A.M. and T.K. contributed equally to this work.
This work was supported by National Institutes of Health (NIH) Grant R01, National Institute of Neurological Disorders and Stroke Grant NS38253-01 (C.J.W.), NIH National Research Service Award NS11076–01 (K.A.M.), and Alexander von Humboldt Foundation Grant V-3FLF-DEU/106 9701 (J.S.). We thank Drs. Katia Befort and Keiko Mizuno for technical advice.
Correspondence should be addressed to Dr. Tatsuro Kohno, Neural Plasticity Research Group, Department of Anesthesia and Critical Care, Massachusetts General Hospital and Harvard Medical School, 149 13th Street, Charlestown, MA 02129. E-mail:Kohno@helix.mgh.harvard.edu.
K. A. Moore's present address: Department of Cellular and Molecular Pharmacology, University of California San Francisco, San Francisco, CA 94143-0450.
H. Baba's present address: Department of Anesthesiology, Niigata University School of Medicine, Niigata, 951-8510, Japan.
REFERENCES
- 1.Alvarez FJ, Taylor-Blake B, Fyffe RE, De Blas AL, Light AR. Distribution of immunoreactivity for the β2 and β3 subunits of the GABAA receptor in the mammalian spinal cord. J Comp Neurol. 1996;365:392–412. doi: 10.1002/(SICI)1096-9861(19960212)365:3<392::AID-CNE5>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 2.Baba H, Doubell TP, Woolf CJ. Peripheral inflammation facilitates Aβ fiber-mediated synaptic input to the substantia gelatinosa of the adult rat spinal cord. J Neurosci. 1999;19:859–867. doi: 10.1523/JNEUROSCI.19-02-00859.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- 4.Bennett GJ, Kajander KC, Saraha Y, Iadarola MJ, Sugimoto T. Neurochemical and anatomical changes in the dorsal horn of rats with an experimental peripheral neuropathy. In: Cervero F, Bennett GJ, Headley PM, editors. Processing of sensory information in the superficial dorsal horn of the spinal cord. Plenum; New York: 1989. pp. 463–471. [Google Scholar]
- 5.Bhisitkul RB, Kocsis JD, Gordon TR, Waxman SG. Trophic influence of the distal nerve segment on GABAA receptor expression in axotomized adult sensory neurons. Exp Neurol. 1990;109:273–278. doi: 10.1016/s0014-4886(05)80017-2. [DOI] [PubMed] [Google Scholar]
- 6.Castro-Lopes JM, Tavares I, Coimbra A. GABA decreases in the spinal cord dorsal horn after peripheral neurectomy. Brain Res. 1993;620:287–291. doi: 10.1016/0006-8993(93)90167-l. [DOI] [PubMed] [Google Scholar]
- 7.Castro-Lopes JM, Malcangio M, Pan BH, Bowery NG. Complex changes of GABAA and GABAB receptor binding in the spinal cord dorsal horn following peripheral inflammation or neurectomy. Brain Res. 1995;679:289–297. doi: 10.1016/0006-8993(95)00262-o. [DOI] [PubMed] [Google Scholar]
- 8.Chang YC, Gottlieb DI. Characterization of the proteins purified with monoclonal antibodies to glutamic acid decarboxylase. J Neurosci. 1988;8:2123–2130. doi: 10.1523/JNEUROSCI.08-06-02123.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Coggeshall RE, Lekan HA, White FA, Woolf CJ. A-fiber sensory input induces neuronal cell death in the dorsal horn of the adult rat spinal cord. J Comp Neurol. 2001;435:276–282. doi: 10.1002/cne.1029. [DOI] [PubMed] [Google Scholar]
- 10.Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87:149–158. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- 11.Devor M, Seltzer Z. Pathophysiology of damaged nerves in relation to chronic pain. In: Wall PD, Melzack R, editors. Textbook of pain. Churchill Livingstone; Edinburgh: 1999. pp. 129–164. [Google Scholar]
- 12.Dubner R. Neuronal plasticity in the spinal cord medullary dorsal horn: a possible role in central pain mechanisms. In: Casey KL, editor. Pain and central nervous system disease. Raven; New York: 1991. pp. 143–155. [Google Scholar]
- 13.Dumoulin A, Alonso G, Privat A, Feldblum S. Biphasic response of spinal GABAergic neurons after a lumbar rhizotomy in the adult rat. Eur J Neurosci. 1996;8:2553–2563. doi: 10.1111/j.1460-9568.1996.tb01549.x. [DOI] [PubMed] [Google Scholar]
- 14.Eaton MJ, Plunkett JA, Karmally S, Martinez MA, Montanez K. Changes in GAD- and GABA- immunoreactivity in the spinal dorsal horn after peripheral nerve injury and promotion of recovery by lumbar transplant of immortalized serotonergic precursors. J Chem Neuroanat. 1998;16:57–72. doi: 10.1016/s0891-0618(98)00062-3. [DOI] [PubMed] [Google Scholar]
- 15.Ibuki T, Hama AT, Wang XT, Pappas GD, Sagen J. Loss of GABA-immunoreactivity in the spinal dorsal horn of rats with peripheral nerve injury and promotion of recovery by adrenal medullary grafts. Neuroscience. 1997;76:845–858. doi: 10.1016/s0306-4522(96)00341-7. [DOI] [PubMed] [Google Scholar]
- 16.Kaufman DL, Houser CR, Tobin AJ. Two forms of the γ-aminobutyric acid synthetic enzyme glutamate decarboxylase have distinct intraneuronal distributions and cofactor interactions. J Neurochem. 1991;56:720–723. doi: 10.1111/j.1471-4159.1991.tb08211.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khandwala H, Hodge E, Loomis CW. Comparable dose-dependent inhibition of AP-7 sensitive strychnine-induced allodynia and paw pinch-induced nociception by mexiletine in the rat. Pain. 1997;72:299–308. doi: 10.1016/s0304-3959(97)00021-3. [DOI] [PubMed] [Google Scholar]
- 18.Kohno T, Kumamoto E, Higashi H, Shimoji K, Yoshimura M. Actions of opioids on excitatory and inhibitory transmission in substantia gelatinosa of adult rat spinal cord. J Physiol (Lond) 1999;518:803–813. doi: 10.1111/j.1469-7793.1999.0803p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewis SE, Mannion RJ, White FA, Coggeshall RE, Beggs S, Costigan M, Martin JL, Dillmann WH, Woolf CJ. A role for HSP27 in sensory neuron survival. J Neurosci. 1999;19:8945–8953. doi: 10.1523/JNEUROSCI.19-20-08945.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moore KA, Kohno T, Scholz J, Karchewski LA, Woolf CJ. Spinal disinhibition: a role in neuropathic pain? Neurosci News. 2001;4:5–10. [Google Scholar]
- 21.Sivilotti L, Woolf CJ. The contribution of GABAA and glycine receptors to central sensitization: disinhibition and touch-evoked allodynia. J Neurophysiol. 1994;72:169–179. doi: 10.1152/jn.1994.72.1.169. [DOI] [PubMed] [Google Scholar]
- 22.Soghomonian JJ, Martin DL. Two isoforms of glutamate decarboxylase: why? Trends Pharmacol Sci. 1998;19:500–505. doi: 10.1016/s0165-6147(98)01270-x. [DOI] [PubMed] [Google Scholar]
- 23.Sugimoto T, Bennett GJ, Kajander KC. Transsynaptic degeneration in the superficial dorsal horn after sciatic nerve injury: effects of a chronic constriction injury, transection, and strychnine. Pain. 1990;42:205–213. doi: 10.1016/0304-3959(90)91164-E. [DOI] [PubMed] [Google Scholar]
- 24.Todd AJ, Sullivan AC. Light microscope study of the coexistence of GABA-like and glycine-like immunoreactivities in the spinal cord of the rat. J Comp Neurol. 1990;296:496–505. doi: 10.1002/cne.902960312. [DOI] [PubMed] [Google Scholar]
- 25.Wall PD, Devor M. The effect of peripheral nerve injury on dorsal root potentials and on transmission of afferent signals into the spinal cord. Brain Res. 1981;209:95–111. doi: 10.1016/0006-8993(81)91174-4. [DOI] [PubMed] [Google Scholar]
- 26.Whiteside GT, Munglani R. Cell death in the superficial dorsal horn in a model of neuropathic pain. J Neurosci Res. 2001;64:168–173. doi: 10.1002/jnr.1062. [DOI] [PubMed] [Google Scholar]
- 27.Woolf CJ, Mannion RJ. Neuropathic pain: aetiology, symptoms, mechanisms, and management. Lancet. 1999;353:1959–1964. doi: 10.1016/S0140-6736(99)01307-0. [DOI] [PubMed] [Google Scholar]
- 28.Woolf CJ, Wall PD. Chronic peripheral nerve section diminishes the primary afferent A-fibre mediated inhibition of rat dorsal horn neurones. Brain Res. 1982;242:77–85. doi: 10.1016/0006-8993(82)90497-8. [DOI] [PubMed] [Google Scholar]
- 29.Yaksh TL. Behavioral and autonomic correlates of the tactile evoked allodynia produced by spinal glycine inhibition: effects of modulatory receptor systems and excitatory amino acid antagonists. Pain. 1989;37:111–123. doi: 10.1016/0304-3959(89)90160-7. [DOI] [PubMed] [Google Scholar]
- 30.Yoshimura M, Nishi S. Blind patch-clamp recordings from substantia gelatinosa neurons in adult rat spinal cord slices: pharmacological properties of synaptic currents. Neuroscience. 1993;53:519–526. doi: 10.1016/0306-4522(93)90216-3. [DOI] [PubMed] [Google Scholar]