

Abstract
Ca2+ currents, especially those activated at low voltages (LVA), influence burst generation in thalamocortical circuitry and enhance the abnormal rhythmicity associated with absence epilepsy. Mutations in several genes for high-voltage-activated (HVA) Ca2+ channel subunits are linked to spike-wave seizure phenotypes in mice; however, none of these mutations are predicted to increase intrinsic membrane excitability or directly enhance LVA currents. We examined biophysical properties of both LVA and HVA Ca2+ currents in thalamic cells oftottering (tg; Cav2.1/α1A subunit), lethargic (lh; β4 subunit), and stargazer (stg; γ2 subunit) brain slices. We observed 46, 51, and 45% increases in peak current densities of LVA Ca2+ currents evoked at −50 mV from −110 mV in tg, lh, andstg mice, respectively, compared with wild type. The half-maximal voltages for steady-state inactivation of LVA currents were shifted in a depolarized direction by 7.5–13.5 mV in all three mutants, although no alterations in the time-constant for recovery from inactivation of LVA currents were found. HVA peak current densities intg and stg were increased by 22 and 45%, respectively, and a 5 mV depolarizing shift of the activation curve was observed in lh. Despite elevated LVA amplitudes, no alterations in mRNA expression of the genes mediating T-type subunits, Cav3.1/α1G, Cav3.2/α1H, or Cav3.3/α1I, were detected in the three mutants. Our data demonstrate that mutation of Cav2.1 or regulatory subunit genes increases intrinsic membrane excitability in thalamic neurons by potentiating LVA Ca2+ currents. These alterations increase the probability for abnormal thalamocortical synchronization and absence epilepsy in tg, lh, andstg mice.
Keywords: Ca2+ currents, thalamocortical relay cells, absence seizures, stargazer, tottering, lethargic, calcium ion channel mutation
Rhythmic burst firing characterizes cellular signaling behavior in the thalamus and depends on intrinsic membrane properties of thalamocortical relay (TC) neurons, as well as the synaptically linked neurons in the adjacent thalamic reticular nucleus (nRT) (McCormick and Bal, 1997; Steriade, 2000). In both cell types, membrane hyperpolarization produces a Ca2+-dependent low-threshold depolarization that serves as the generator potential for bursts of Na+ spikes (Llinás and Jahnsen, 1982). Voltage-clamp studies have identified the ion channel responsible for thalamic postinhibitory rebound burst firing as a T-type Ca2+ channel (Jahnsen and Llinás, 1984; Coulter et al., 1989a; Crunelli et al., 1989; Huguenard, 1996). Three genes, Cacna1g,Cacna1h, and Cacna1i, encoding T-type Cav3.1/α1G, Cav3.2/α1H, and Cav3.3/α1I α subunits have been localized in rat brain; Cav3.1 is predominantly expressed in thalamic relay nuclei and Cav3.3 in the nRT (Talley et al., 1999). The evidence directly linking this pathway to human absence epilepsy is based on thalamic involvement in spike-wave electrogenesis (Williams, 1953; Niedermeyer et al., 1969) and the ability of anti-absence drugs to reduce low-voltage-activated (LVA) currents (Coulter et al., 1989b; Gomora et al., 2001). Indirect evidence from experimental models provides additional support; LVA currents in nRT are increased in a rat model of absence epilepsy (Tsakiridou et al., 1995), and the threshold for spike-wave generation is significantly elevated in Cav3.1 −/− mice (Kim et al., 2001).
The identification of mutations in Ca2+ channel subunit genes intottering (tg; Cav2.1/α1A) (Fletcher et al., 1996),lethargic (lh; β4) (Burgess et al., 1997), and stargazer (stg; γ2) (Letts et al., 1998) mice and in humans (Escayg et al., 2000; Jouvenceau et al., 2001) that display an absence epilepsy phenotype provide an excellent opportunity to determine how defective Ca2+ signaling leads to thalamocortical epilepsy. In tg and its alleletgla, dramatic reductions in single-channel open probability and peak current density of high-voltage-activated (HVA) P/Q-type (Cav2.1/2.2) Ca2+currents in cerebellar Purkinje have been reported; however, it is difficult to explain how this defect could enhance intrinsic membrane excitability if present in the thalamocortical circuit (Dove et al., 1998; Lorenzon et al., 1998; Wakamori et al., 1998). In lhPurkinje cells, loss of functional β4 subunits did not alter Cav2.1/2.2 Ca2+ currents, probably attributable to compensatory interactions with alternative β1–3 subunits (Burgess et al., 1999; McEnery et al., 1998). Similarly, loss of the γ2subunit in stg, shown recently to directly interact with HVA channels (Kang et al., 2001), had no significant effect on Ca2+ currents in cerebellar granule cells, presumably attributable to rearrangements with alternative members of the γ subunit family (Chen et al., 2000; Burgess et al., 2001).
Because the behavior of Ca2+ currents observed in cerebellar cells cannot be generalized to thalamic neurons, which express their own pattern of interacting subunits and modulatory signals, the cellular mechanisms of epileptogenesis in these three mutants remain unclear. We therefore evaluated the functional properties of both LVA and HVA Ca2+currents in mutant thalamic neurons and examined in vivogene expression patterns of the three LVA channel genes. Our results demonstrate that elevated thalamic LVA currents are a common feature shared by three distinct absence epilepsy gene mutations.
MATERIALS AND METHODS
Preparation of brain slices
Coronal brain slices (350-μm-thick) were prepared from 14- to 19-d-old homozygous wild-type (C57BL/6J),tottering(C57BL/6J-Cacna1atg/tg),lethargic(B6EiC3H-a/A-Cacnβ4lh/lh), andstargazer(C57BL/6J-Cacnγ2stg/stg) mice. Genotypes of the three mutants were confirmed by PCR of tail DNA (Burgess et al., 1997). Slices were obtained at the level of the lateral dorsal nucleus (LDN) of the thalamus, selected because of its projection to frontal cortical regions in which cortical spike-wave discharges in these mutants predominate (J. L. Noebels, unpublished observations). Each slice was perfused with a solution containing (in mm) (Kapur et al., 1998): 125 choline-Cl, 3.0 KCl, 1.25 NaH2PO4, 25 NaHCO3, 1.0 Ca Cl2, 7.0 MgCl2, 10 dextrose, 1.3 ascorbate acid, and 3.0 pyruvate (bubbled with 95% O2–5% CO2). Slices were then incubated in an artificial CSF solution for 40 min at 37°C and then maintained at room temperature (22–25°C). The artificial CSF was gassed with 95% O2–5% CO2 and contained (in mm): 130 NaCl, 3.0 KCl, 2.0 MgCl2, 2.0 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose.
Electrophysiological recording
Macroscopic Ca2+ currents from thalamocortical cells in the LDN were recorded using the whole-cell configuration of the patch-clamp technique. A Zeiss (Oberkochen, Germany) Axioskop fitted with a 40× water-immersion objective and differential interference contrast optics was used to view the slices and to identify neurons for analysis. Voltage command pulses were generated by a computer using pClamp 8.02 software. Currents were recorded with an Axopatch-1D amplifier, filtered at 10 kHz (−3 dB), and compensated for series resistance (∼70%). Patch electrodes were drawn from borosilicate glass and coated with Sylgard. Ca2+ currents were corrected for leak and capacitive currents by subtracting a scaled current elicited by a 10 mV hyperpolarization from the standard holding potential of −70 mV. All recordings were performed at room temperature (22–25°C). The standard holding potential was −70 mV. To identify and stain the neurons in whole-cell recordings from the slice, 1% biocytin was included in the intracellular patch pipette solution. After recordings, the slices were cut into 50-μm-thick sections and then immediately fixed. A standard histochemical procedure was used to process the sections and stain the injected neurons (Huguenard and Prince, 1992).
Solutions. The recording bath solution consisted of (in mm): 115 NaCl, 3.0 KCl, 10 sucrose, 10 glucose, 26 NaHCO3, 2 MgCl2, 2.5 CaCl2, 0.5 4-aminopyridine, 5 CsCl, 10 tetraethylammonium-Cl (TEA-Cl), and 0.001 TTX, pH 7.4 (gassed with 95% O2–5% CO2). The intracellular pipette solution contained (in mm): 78 Cs-gluconate, 20 HEPES, 10 BAPTA-Cs4 (cell-impermeant), 0.5 CaCl2, 1.0 MgCl2, 4 Mg-ATP, 0.3 GTP-Tris, 6 phosphocreatine (Di-Tris salt), 4.0 NaCl, and 20 TEA-Cl, pH 7.3 (titrated with CsOH).
Voltage protocols. To generate Ca2+ channel current–voltage (I–V) curves, currents were elicited by applying voltage step commands (200 msec) to varying potentials from a 3 sec prepulse potential at −60 or −110 mV. The I–V protocol for HVA Ca2+ currents consisted of voltage steps from −80 to +60 mV in 5 mV increments triggered from a 3 sec prepulse potential at −60 mV. To define LVA Ca2+ currents, difference currents obtained by digital subtraction of the currents elicited during depolarizing voltage steps from −60 and −110 mV were used. Standard voltage protocols for steady-state activation (SSA) of HVA Ca2+ currents, as well as the steady-state inactivation (SSI) and recovery from inactivation of LVA currents, respectively, were applied, and are explained in further detail below (see figure legends). In our study, we did not find any significant time-dependent ICa2+ run down within 30–40 min after membrane breaking. Statistical data analysis was tested by one-way ANOVA with the post hoc test. Differences with p < 0.05 were scored as statistically significant. The data shown represent means ± SE
In situ hybridization
In situ hybridization of mRNAs encoding α subunits for three calcium channel subtypes mediating LVA T-type Ca2+ currents, Cav3.1/α1G, Cav3.2/α1H, and Cav3.3/α1I, was performed in 2- to 3-week-old homozygous tg, lh, andstg mutants and C57BL/6 +/+ mice using standard techniques described previously in detail (Burgess et al., 1999). Briefly, horizontal brain sections (12-μm-thick) from 14- to 19-d-old mice were fixed in 4% paraformaldehyde in PBS and dehydrated through an ascending ethanol series. Antisense oligonucleotide probes were end labeled using terminal deoxynucleotidyl transferase (Promega, Madison, WI) and [α-35S]dATP (1250 Ci/mmol; NEN, Boston, MA) to a specific activity of ∼109 dpm/μg. The hybridization solution contained 50% (v/v) formamide, 4× SSC, 25 mm sodium phosphate, 1 mmsodium pyrophosphate, 10% dextran sulfate (w/v), 5× Denhardt's solution, 200 μg/ml sonicated herring sperm DNA (Promega), 100 μg/ml polyadenylic acid [5′] (Sigma-Aldrich, Milwaukee, WI), and 5 × 102 dpm of [α-35S]dATP-labeled probe. Control sections were hybridized with an additional 100-fold excess of unlabeled oligonucleotide. The sequences of the 45-mer probes were as follows: Cacna1g, 5′-GATGCAGCTGGTGTCTGCTGGTTGGGAGTGAACAGACAAGATGG-3′; Cacna1h, 5′-CAAGAAGGTCAGGTTGTTGTTCCTGACGAAGGCGCTGTCCA-GGAA-3′; andCacna1i, 5′ GCGGATGGCTGACAGGTTGATGTTC-TGTAGGTCCAGAGAGTACTC-3′. The probes were hybridized to the sections overnight at 42°C, washed in 1× SSC (22°C, 20 min), 0.3× SSC (55°C, 40 min), and 2× SSC (22°C, 5 min), and then dehydrated and exposed to Kodak BioMax MR film (Eastman Kodak, Rochester, NY) for 1 week. Developed autoradiographs were digitized (Sprintscan 35; Polaroid, Cambridge, MA) and arranged using Photoshop 5.0 (Adobe Systems, San Jose, CA), and all images were processed simultaneously. Optical density reflecting relative abundance of mRNA was determined by Scion (Frederick, MD) Image–NIH Image software. The in situ results were collected from wild-type and mutant mice (three animals per group). Eight brain sections were obtained from each group. Multiple small square areas within several anatomically distinct brain areas were selected, including the lateral dorsal nucleus and the adjacent white matter as an internal standard. These values were used to determine the lateral dorsal/white matter density ratio between affected mice and the respective homozygous wild-type mice. Differences in the optical densities were analyzed for statistical significance using Student's t test.
RESULTS
Increased peak current density of LVA calcium channels in TC neurons of tottering, lethargic, andstargazer mutants
We investigated the effects of mutation of Cav2.1/α1A (tg), β4 (lh), and γ2 (stg) Ca2+ channel subunits on Ca2+ currents in mouse LDN thalamic neurons defined by intracellular staining of biocytin. In mouse, these cells show a round-shaped cell body with multiple dendrites, similar in morphology to TC neurons from rat ventrobasal nucleus (Destexhe et al., 1998) (Fig.1A). Figure1B shows representative traces of LVA Ca2+ currents in response to a test pulse to −50 mV from a 3 sec prepulse to −110 mV in TC neurons from wild-type, tg, lh, and stg mice. At a membrane potential of −50 mV, all LVA Ca2+ currents have recovered from inactivation and are thus available for opening in both wild-type and mutant neurons (Figs.2B, 3), whereas HVA Ca2+ currents in these cells have not yet started to activate (see Fig. 5A). The current traces of the LVA calcium channels show fast activation and inactivation, similar to those studied in vitro by expression of Cav3.1/α1G and Cav3.2/α1H T-type calcium channels (Lee et al., 1999; Delisle and Satin, 2000; Zhang et al., 2000), as well as native LVA currents from dissociated rat TC neurons (Destexhe et al., 1998). The peak current densities (i.e., normalized by cell capacitance) of LVA currents at a membrane potential of −50 mV increased by 46% in tg, 51% in lh, and 45% instg mutants compared with control (Fig. 1C). The mean peak current amplitude and peak current density were −926.3 ± 182.2 pA and 9.5 ± 1.3 pA/pF in control, −1571.9 ± 106.8* pA and 17.63 ± 1.6** pA/pF in tg, −1852.1 ± 118.9** pA and 19.6 ± 1.0** pA/pF in lh, and 1514.9 ± 142* pA and 17.4 ± 1.2** pA/pF in stgmice (*p < 0.05; **p < 0.01 vs control).
Fig. 1.

Increased LVA Ca2+ peak current in tottering, lethargic, andstargazer. A, Biocytin-filled thalamic neuron in the LDN of thalamus from wild-type mouse (C57BL/6J).B, Representative LVA current traces from TCs of the LDN in control, tottering, lethargic, andstargazer mice. The cell capacitance values of these four neurons were 101.25, 107.1, 95.5, and 95.34 pF, respectively. Holding potential, −70 mV. The membrane potential was prepulsed to −110 mV for 3 sec before stepping to −50 mV for 200 msec. Decay of the current was fitted by a single-exponential function (dotted line). No significant alterations in macroscopic current decay were found. The representative time constants τ for decay were 29.2, 29.7, 30.1, and 31.8 msec, respectively, in control, tg,lh, and stg mice. C, Elevated LVA Ca2+ current amplitude and peak current density from mutant TCs. LVA currents were evoked at the same membrane potential as described in B. The mean current amplitude and peak current densities were −926.3 ± 182.2 pA and 9.5 ± 1.3 pA/pF in control, 1514.9 ± 142 pA and 17.4 ± 1.2 pA/pF in stg, −1571.9 ± 106.8 pA and 17.63 ± 1.6 pA/pF in tg, and −1852.1 ± 118.9 pA and 19.6 ± 1.0 pA/pF in lh mice. *p < 0.05; **p < 0.01 versus control.
Fig. 2.
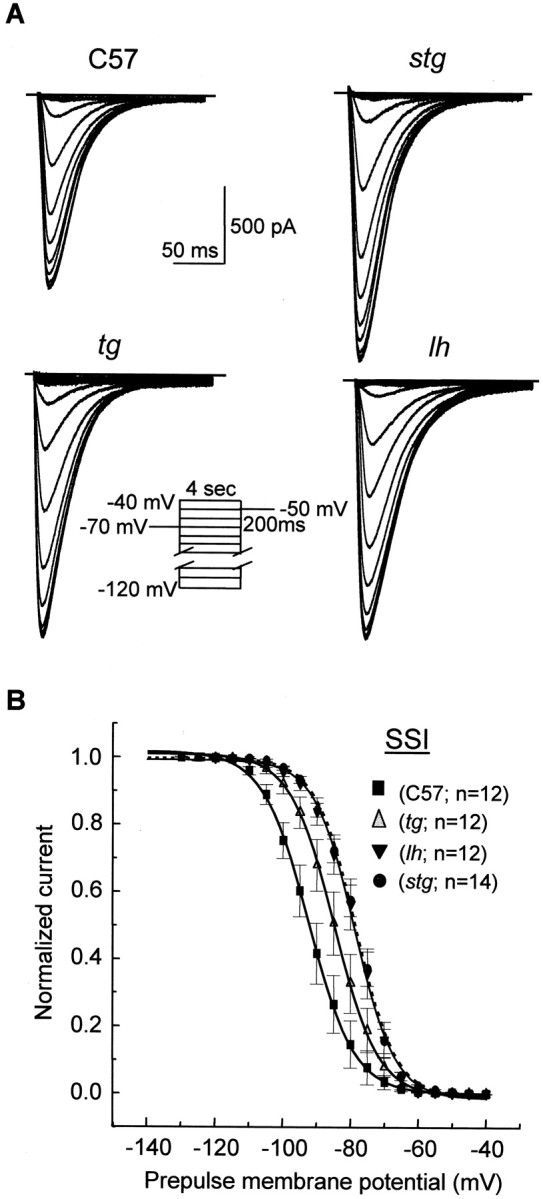
Depolarized shift of the voltage dependence of LVA calcium channel availability (steady-state inactivation) intottering, lethargic, andstargazer mutants. A, Representative current traces for SSI of LVA Ca2+ currents. A standard double-pulse protocol for steady-state inactivation was given from the holding potential of −70 mV. A 4 sec prepulse at potentials ranging from −120 to −40 mV preceded each depolarization, followed by a subsequent voltage step to −50 mV for 200 msec. The interpulse interval was 10 sec. B, Normalized current–voltage curves for SSI of LVA Ca2+ currents. Current amplitude from the inactivation protocol, normalized to maximum, was plotted as a function of prepulse membrane potentials and best fitted with a Boltzmann function: I/Imax = {1 + exp(V −V1/2)/k} − 1. The pooled half-maximal voltages (V1/2) and slopes (k) were −92.3 ± 0.16 and 6.8 ± 0.16 mV in control, −84.8 ± 0.17* and 6.51 ± 0.15 mV intg, −78.72 ± 0.3** and 6.0 ± 0.27 mV inlh, and −78.6 ± 0.3 ** and 6.0 ± 0.27 mV instg, respectively. *p < 0.05; **p < 0.01.
Fig. 3.
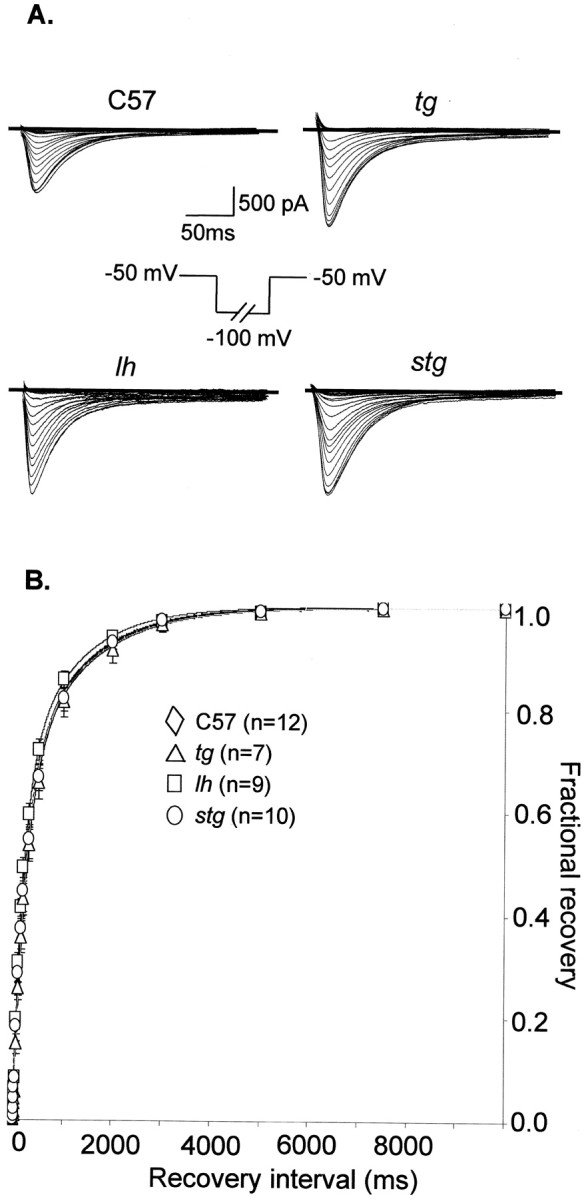
Recovery from inactivation of LVA Ca2+ currents. A, Representative current traces for recovery from inactivation of LVA currents in control, tg, lh, and stg. The holding potential was set to −50 mV, and 50 mV hyperpolarizations of incremental duration were applied. LVA peak amplitude was measured after returning to −50 mV. B, Recovery from inactivation curves. Recovery curves were established by plotting the normalized peak amplitude versus duration. The recovery curves followed a two-exponential time course, best fitted with fast time constant (τ1) of 230, 240, 196, and 225 msec for control,tg, lh, and stg, and slow time constant (τ2) of 1300, 1250, 1100, and 1270 msec for control, tg, lh, andstg, respectively.
Fig. 5.

Peak HVA Ca2+ currents are increased in tottering and stargazer but not lethargic mice. A, Current density–voltage curves for HVA Ca2+ currents, constructed by plotting the normalized current amplitude at various membrane potentials. The voltage protocol used was identical to that described in Figure 4. B, Peak current density and current amplitude from A. The mean peak current density was 10.12 ± 1.2, 13.03 ± 0.6, 9.42 ± 0.4, and 17.99 ± 2.1 pA/pF in control, tg,lh, and stg (**p < 0.01 vs control). The mean current amplitude was −885.51 ± 112.8, 1204.21 ± 86.4, −901.3 ± 48.7, and −1567.6 ± 188 pA in control, tg, lh, andstg mutants (**p < 0.01 vs control). C, SSA of HVA Ca2+ currents in control, tg, lh, andstg mice. The steady-state conductance (G) and voltage (V) data were transformed from I–V data shown inA. The solid and dotted curves are fits of the data to the Boltzmann equation of the following form: G/Gmax = 1/(1 + exp(V1/2 −V)/k), whereGmax is maximum conductance,V1/2 is half-maximal voltage, andk is the slope. The mean values ofV1/2 and slope for SSA of HVA currents are −22.0 ± 0.17 and 5.5 ± 0.15 mV in control, −21.7 ± 0.16 and 4.5 ± 0.14 mV in tg, −17.0 ± 0.11* and 4.7 ± 0.1 mV in lh mice, and −23.9 ± 0.18 and 4.6 ± 0.16 mV in stg, respectively (*p < 0.05 vs control).
The duration of macroscopic inactivation for LVA Ca2+ currents in both control and mutant mice is closer to the time scale observed in Cav3.1 but not Cav3.2 T-type calcium channels expressed in mammalian cells (Lee et al., 1999; Zhang et al., 2000). The decay of macroscopic LVA currents evoked at −50 mV was fitted by a single-exponential function (Fig. 1B). No significant alterations in macroscopic LVA current decay were found in any of the three mutants compared with the wild type. The time constants for decay were 29.2, 29.7, 30.1, and 31.8, respectively, in control, tg, lh, andstg mice.
Depolarizing shifts in voltage dependence of steady-state inactivation and kinetics for recovery from inactivation of LVA currents
We next examined the kinetics of LVA Ca2+ currents in both wild-type and mutant neurons. The current traces of SSI of LVA are shown in Figure2A. For the SSI protocol, we used a 4 sec prepulse to various membrane potentials before delivering a second test stimulus to −50 mV. The 4 sec prepulse was long enough to bring channels to a steady-state condition, because all LVA Ca2+ channels in TC neurons recover from inactivation within 3 sec (Fig. 3). As demonstrated in Figure2A, LVA currents elicited at −50 mV from different premembrane potentials in both control and mutants show fast inactivation and decay completely within 150 msec. We did not observe any sustained component for current decay in tg,lh, or stg mice. Nevertheless, we found a significant depolarizing shift of the steady-state inactivation curves of LVA currents in the mutants in contrast to wild-type neurons (Fig.2B). The mean half-maximal voltage (V1/2) and slope (k) for SSI curves were −92.3 ± 0.16 and 6.8 ± 0.16 in control, −84.8 ± 0.17* and 6.51 ± 0.15 mV in tg, −78.72 ± 0.3** and 6.0 ± 0.27 mV in lh, and −78.6 ± 0.3** and 6.0 ± 0.27 mV in stg, respectively (*p < 0.05; **p < 0.01). The 7.5–13.5 mV depolarizing shifts of the voltage dependence for SSI of LVA currents in TC neurons of the mutants suggest that, at physiological membrane potentials varying from −70 to −75 mV, a higher fraction of all LVA calcium channels are available for opening in the mutant relative to control mice. Recovery from inactivation of the LVA Ca2+ currents in all mice was complete within 3 sec (Fig. 3). The recovery from inactivation curve was best fitted with a two-exponential function, and the fast and slow time constant derived from curve fitting did not significantly differ in any of the mutants compared with control mice. Thus, the depolarizing shift of SSI curves for LVA currents provides an additional biophysical mechanism that may contribute to increased neuronal burst synchronization observed in tottering,lethargic, and stargazer mice.
To determine whether the dramatic depolarizing shifts of SSI curves in all three mutants involves phosphorylation of thalamic T-type channels, we examined the effects of a specific protein kinase A inhibitor (PKA-I), as well as a protein kinase C inhibitor (PKC-I) on the voltage dependence of SSI for LVA currents. PKA-I at 50 μm(fragment 6–22, amide) or 100 μm PKC-I (fragment 19–36) was loaded into the pipette solution as described previously (Zhang et al., 2000), and the SSI protocol for LVA channels was then initiated 15–20 min after membrane rupture. We observed that neither PKA-I nor PKC-I antagonized the shift of V1/2for steady-state inactivation of the LVA currents found in untreatedtg, lh, or stg mutants (data not shown). In addition, neither PKA-I nor PKC-I altered the baseline value of V1/2 for SSI in untreated wild-type neurons, suggesting that the resting level of PKA or PKC could be low in both control and mutant mice. These observations suggest that neither PKA nor PKC significantly modulate the voltage dependence of SSI for LVA channels in these mutants, although consensus sites for both PKA and PKC exist on Cav3.1/α1G and Cav3.2/α1H channels (Cribbs et al., 1998;Perez-Reyes et al., 1998).
High-voltage-activated Ca2+ peak currents are increased in tottering andstargazer but not lethargic mice
We then determined whether mutation of Cav2.1/α1A (tg), β4 (lh), or γ2 (stg) Ca2+ channel subunits affect HVA Ca2+ currents in LDN cells. HVA Ca2+ currents are mediated by pore-forming α1 subunits, with current amplitude and gating regulated by cytoplasmic β subunits and transmembrane α2δ and γ subunits (Ahlijanian et al., 1990; Chien et al., 1995; Witcher et al., 1995;Gurnett et al., 1996; Walker and De Waard, 1998; Yamaguchi et al., 1998; Meir et al., 2000; Kang et al., 2001).
The I–V relationships of Ca2+ currents for control and three mutants are shown in Figure 5A. Figure4 shows representative HVA Ca2+ current traces from wild-type,tg, lh, and stg mutant neurons. Both control and mutant HVA Ca2+ currents start to activate at approximately −45 mV. The currents reach a peak at −10 to −15 mV in control and mutant mice (Fig.5A). Pooled peak currents and peak current densities are shown in Figure 5B. The mean peak current density was 10.12 ± 1.2, 13.03 ± 0.6**, 9.42 ± 0.4, and 17.99 ± 2.1** pA/pF in control, tg,lh, and stg (**p < 0.01 vs control). The mean peak amplitude was −885.51 ± 112.8, 1204.21 ± 86.4**, −901.3 ± 48.7, and −1567.6 ± 188** pA in control, tg, lh, and stgmutants, respectively (**p < 0.01 vs control).
Fig. 4.

Representative superimposed HVA Ca2+ current traces from thalamocortical cells in control, tg, lh, and stgmice. The I–V protocol consisted of a 3 sec prepulse potential at −60 mV, followed by voltage steps (200 msec) ranging from −80 to +60 mV in 5 mV increments, with the holding potential maintained at −70 mV.
Voltage dependence of steady-state activation for HVA currents
The steady-state activation curves of HVA Ca2+currents in TC neurons are demonstrated in Fig 5C. TheV1/2 for steady-state activation of HVA currents in tottering and stargazer mutants did not change dramatically but shifted by 5 mV in a depolarized direction in lethargic mice relative to that in control. The mean values of V1/2 and slope for SSA of HVA channels were −22.0 ± 0.17 and 5.5 ± 0.15 mV in control, −21.7 ± 0.16 and 4.5 ± 0.14 mV intg, −17.0 ± 0.11* and 4.7 ± 0.1 mV inlh, and −23.9 ± 0.18 and 4.6 ± 0.16 mV instg, respectively (*p < 0.05 vs control). The depolarized shift of the SSA curve in lh neurons is consistent with the in vitro finding that heterologous coexpression of β4 subunits elicited a hyperpolarizing shift for the SSA of Cav2.1 HVA calcium channels (De Waard and Campbell, 1995).
Expression of T-type Ca2+ channel Cav3.1/α1G mRNA in tottering,lethargic, and stargazer thalamus
To ascertain whether alterations in T-type calcium channel gene expression could underlie the increased LVA currents observed in mutant TC neurons, we compared regional mRNA expression levels of Cav3.1/α1G, Cav3.2/α1H, and Cav3.3/α1I. Figure6 shows representative autoradiograms of coronal sections taken at the thalamic LDN level from mutant and wild-type brains. The three genes encoding neuronal T-type Ca2+ channels are expressed in distinct and primarily non-overlapping patterns that are consistent with localization patterns in adult rat brain (Talley et al., 1999). In wild-type mouse brain, Cav3.1 mRNA was expressed at high levels in thalamic relay nuclei, Cav3.3 mRNA was detected at high levels in nRT, and Cav3.2 mRNA was detected at only trace levels in either. No extrathalamic alterations were noted in any of the three mutants, and only the expression patterns of each gene in the thalamus are described.
Fig. 6.
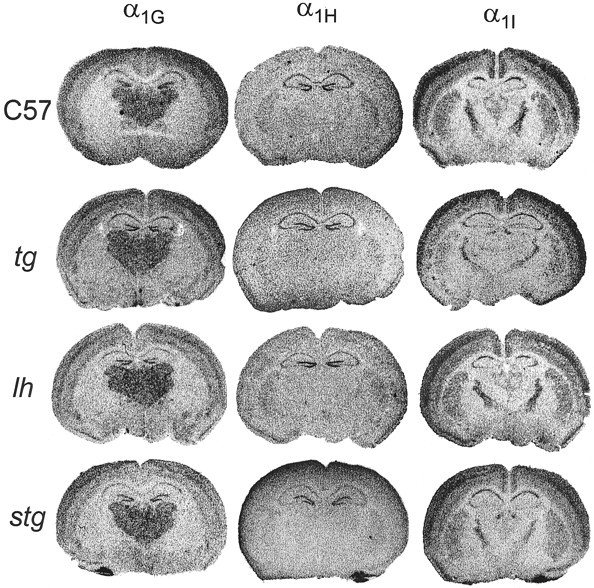
Expression of T-type calcium channel genes in tg, lh, and stgthalamus. The three genes, Cacna1g(Cav3.1/α1G),Cacna1h (Cav3.2/α1H), andCacna1i (Cav3.3/α1I) were expressed in distinct and primarily non-overlapping patterns in coronal sections of mouse brain. Cav3.1 mRNA was detected at highest levels in neocortex and thalamus and in hippocampal dentate granule cells, with lower levels in hippocampal CA1–CA2 regions. Cav3.2 expression appeared to be restricted to hippocampal dentate gyrus and CA1–CA3 regions in the sections shown above. Cav3.3 expression was the most broadly distributed of the three T-type genes in mouse brain, with high levels of mRNA observed in nRT, habenula, hippocampal CA1 region, neocortex, and striatum. These patterns were not appreciably altered in homozygous tg,lh, or stg mutant mice.
Cav3.1/α1G mRNA
Transcripts of this gene were detected at highest levels in thalamic relay nuclei, with barely detectable levels present in nRT. This pattern was not appreciably altered in homozygous tg,lh, or stg mutant mice.
Cav3.2/α1H mRNA
Only barely detectable levels of Cav3.2 appeared in the wild-type thalamic relay nuclei, and trace levels were found in nRT. This pattern was not visibly altered in the tg,lh, or stg mutant mice.
Cav3.3/α1I mRNA
Cav3.3 was the most broadly distributed of the three T-type genes in the mouse thalamus, with high levels of mRNA observed in nRT. Low and barely detectable levels of Cav3.3 mRNA were detected in principle thalamic relay nuclei. The pattern of this gene was not appreciably altered in homozygous tg,lh, or stg mutant mice. Optical densitometry confirmed that there were no significant differences in optical densities of thalamic expression of Cav3.1–3 mRNA between the three mutants and the wild-type mice.
Although the correlation between mRNA intensity and LVA current density is unknown, the lack of appreciable differences in the in situ hybridization results between mutant and control provides no evidence to support the hypothesis that the Ca2+ channel subunit mutations intg, lh, and stg mutants lead to functionally significant dysregulation of Cav3.1, Cav3.2, or Cav3.3 mRNA levels, and thus the ∼50% increases in thalamic T-type Ca2+ currents cannot be simply accounted for by a proportional upregulation of channel gene expression.
DISCUSSION
Our results demonstrate increased peak current densities and a depolarizing shift of the steady-state inactivation curves of LVA currents, as well as subunit-specific HVA current modifications intottering, lethargic, and stargazerthalamic neurons. The enhanced LVA currents could not be simply explained by major changes in thalamic Cav3.1, Cav3.2, or Cav3.3 gene expression in any of the three mutants. Both the increased LVA current density and shifted voltage dependence of inactivation may favor spike-wave burst firing, the excitability phenotype shared bytg, lh, and stg mice. An additional finding was that HVA peak current densities, which may also facilitate network oscillations, are increased in tg and stgbut not in lh thalamic neurons. This selectivity emphasizes the diversification of cellular compensatory mechanisms modulating Ca2+ currents after mutation of Cav2.1/α1A, β4, or γ2 Ca2+ channel subunits. Our study also provides a key mechanistic link between mutation of HVA subunits and the expression of thalamocortical hypersynchrony.
Functional properties of LVA Ca2+channels in mutant thalamic neurons
The similarity of the spontaneous six to seven per sec spike-wave phenotype in tg, lh, andstg mice (Noebels and Sidman, 1979; Noebels et al., 1990;Hosford et al., 1992) suggests that a common cellular mechanism, potentiation of LVA currents, may be responsible for enhanced neuronal synchronization attributable to the distinct Ca2+ channel subunit mutations. LVA currents in thalamic neurons play a major and perhaps essential role in the genesis of synchronized oscillations in this system by amplifying postinhibitory high-frequency rebound bursting that enables rhythmic firing patterns. The HVA Ca2+ channel Cav2.1 and β4 subunits have not been shown to physically interact with T-type channel proteins or influence T-type channel biophysical properties in central neurons, although there is emerging evidence that γ2 may interact with both HVA and LVA channel types. Nonetheless, we found dramatic increases (45–51%) in peak current densities of thalamic LVA currents in all three mutants. The increases favor augmented burst firing and membrane hyperexcitability, because T-type channels start to activate at relatively hyperpolarized membrane potentials (Huguenard and Prince, 1992; Delisle and Satin, 2000; Zhang et al., 2000), and thalamic neurons have hyperpolarized resting membrane potentials attributable to the rhythmic inputs from GABAergic nRT neurons (Steriade and Llinás, 1988). In our study, LVA currents started to activate at approximately −65 to −70 mV (data not shown). Interestingly, we also observed a 7.5–13.5 mV depolarizing shift in SSI of LVA channels in all three mutants compared with theV1/2 for SSI in wild-type mice. This very large depolarizing shift in SSI indicates a 7–30% elevation in LVA channel availability in the range of membrane potentials close to the resting membrane potential (−65 to −75 mV) (Fig. 2), and hence more channels will be open once the threshold for activation is reached.
Candidate mechanisms underlying LVA current alterations
We initially considered the possibility that peak LVA current increases might arise from additional de novo T-type channel synthesis, by either upregulation of native Cav3.1/α1G subunit expression or ectopic transcription of Cav3.2/α1H and Cav3.3/α1I subunits not normally expressed in these cells. Expression patterns of all three CaVT genes were unaltered, indicating that neither the mutant subunits nor the thalamocortical seizures they provoke are sufficient to induce CaVT gene transcription. The latter observation is consistent with the absence of c-Fos or c-Jun dysregulation in stg (Nahm and Noebels, 1998), confirming that the rhythmic oscillations during spike-wave synchronization are weak promoters of molecular plasticity compared with the prolonged, continuous depolarizations that stimulate expression of Ca2+ channels and other genes after convulsive seizures (Vigues et al., 1999). Although the increase in LVA peak current density detected in all three mutants is unlikely to result from altered CaVT gene expression, we cannot entirely exclude the possibility of persistent LVA channel elevation attributable to small increases of mRNA below the resolution of the method, increased mRNA translation efficiency, or decreased lability of the membrane protein. A mismatch between elevated LVA T-type Ca2+ currents and gene expression was observed previously in dissociated nRT neurons of the genetically undefined GAERS rat strain with absence epilepsy (Tsakiridou et al., 1995). In this model, one study detected no change in Cav3.1 expression (de Borman et al., 1999), whereas another reported small elevations of Cav3.1 mRNA in ventral posterior lateral thalamic relay nuclei and of Cav3.2 mRNA in nRT neurons (Talley et al., 2000).
Absence of thalamic CaVT gene upregulation suggests the existence of alternative nontranscriptional mechanisms for increased LVA current densities and shifted voltage dependence of SSI in tg, lh, and stg mutants. Because neither Cav2.1/α1A nor β4 proteins physically interact with Cav3.1/α1G subunits, the reduction of P/Q currents in tg and lh mutants must modify LVA current indirectly by altering a downstream Ca2+ signaling pathway. In the case ofstg mice, direct interactions between γ2 and α1G may occur in vivo, and lack of regulatory γ2 subunits could directly alter current density and SSI by allowing novel γ3–8 subunit interactions with LVA channel complexes (Dolphin et al., 1999;Green et al., 2001; Rousset et al., 2001). Other potential mechanisms for elevated LVA current density include the contribution of depolarizing shifts of SSI, which increase channel availability, or increased membrane insertion of channels or spatial redistribution of channels from dendritic to somatic compartments in which they could affect the voltage dependence of inactivation and maximally influence firing properties (Karst et al., 1993). Finally, although we found little evidence for abnormal modulation of T-type channels in mutant neurons by PKA or PKC, modulation by PKG or through other pathways, including pH (Delisle and Satin, 2000; Shan et al., 2001), G-protein (Matsushima et al., 1993; Park and Dunlap, 1998), or molecules such as the endogenous cannabinoid anandamide (Chemin et al., 2001), remain possible candidate mechanisms.
Differential effects of Cav2.1/α1A, β4, and γ2 channel subunit mutations on HVA currents
We found that thalamic Ca2+ currents in tg brain slices showed a 22% increase in HVA peak current density, and a similar result was demonstrated recently in dissociated TCs of Cav2.1-deficient mutants (Song et al., 2001). Because point mutation or deletion of the Cav2.1 gene reduces or eliminates P-type currents (Dove et al., 1998; Wakamori et al., 1998; Jun et al., 1999), the small increase in whole-cell HVA peak current density observed in our experiment likely represents upregulation of other HVA currents, such as L-, N-, or R-type. Indeed, decreased Ca2+ currents through tgP/Q-type channels significantly increases mRNA levels for L-type Ca2+ channels in cerebellar Purkinje cells (Campbell and Hess, 1999). In TC neurons, we found that L-type currents account for 55–70% of whole-cell HVA currents in both control and the three mutants (data not shown). Additional pharmacological studies with selective Ca2+ channel blockers will define which specific HVA channels account for the increases intg thalamic HVA currents.
The increased HVA Ca2+ peak current density in stg is consistent with recent functional analysis of γ2 subunit interactions with neuronal calcium channels, indicating that loss of the γ2 subunit may increase HVA currents (Letts et al., 1998; Klugbauer et al., 2000; Kang et al., 2001). These studies found that γ2/γ3 subunits cosedimented and coimmunoprecipitated with either Cav2.1/α1A or Cav2.2/α1B subunit from rabbit cerebellum and that γ2 coexpression in Xenopus oocytes significantly decreased the current amplitude of both Cav2.1 and Cav2.2 calcium channels. Of the neuronal Ca2+ channel γ subunits investigated so far, the γ2 and γ4 subunits shift the steady-state inactivation curve toward hyperpolarized potentials when coexpressed with Cav2.1 (Klugbauer et al., 2000). Thus, absence of γ2 in stg neurons should increase channel availability at membrane potentials (−40 to −30 mV) that start to activate HVA Ca2+ channels. Our data showed a 45% increase in HVA peak current density in TC neurons that is consistent with the non-neuronal expression studies.
In contrast to tg and stg, mutation of the β4 subunit did not significantly alter peak HVA current density inlh thalamic neurons. This result was unexpected, because β subunits regulate assembly and membrane incorporation of Cav2.1 subunits mediating HVA currents (Nishimura et al., 1993; Chien et al., 1995) and influence the amplitude of Ca2+ currents (De Waard and Campbell, 1995; Roche and Treistman, 1998) in vitro. β subunits have been also found to associate and functionally modify native P/Q-, L-, or N-type Ca2+ channels (Chien et al., 1995; Brice et al., 1997; Meir et al., 2000). Interestingly, the lack of an effect on HVA peak current in TCs oflh mice is consistent with the same negative effect on P/Q-type Ca2+ currents in dissociatedlh Purkinje cells (Burgess et al., 1999) and on P/Q channel function mediating transmitter release at lh hippocampal synapses (Qian and Noebels, 2000). Because alternative β subunits are widely localized in the brain, the negative impact of the β4 mutation on thalamic HVA current in lh may be explained by β subunit reshuffling (Burgess et al., 1999). Finally, the 5 mV depolarizing shift of SSA found in lethargic mice is consistent with in vitro data that β4 subunit interaction elicits a hyperpolarizing shift of inactivation curve for HVA currents (De Waard and Campbell, 1995). Because lh mice show both the LVA current increase and the spike-wave EEG phenotype, these findings suggest that the thalamic HVA alterations in tg andstg favor, but are not essential for, spike-wave generation.
In conclusion, our results are the first Ca2+ current defects to be described intg, lh, and stg thalamic neurons and provide supportive physiological evidence for the functional interaction of β and γ regulatory subunits in modulating LVA currents.
Footnotes
This work was supported by an American Epilepsy Society–Epilepsy Foundation Postdoctoral Fellowship (Y.Z.), National Institutes of Health Grant NS97209 (J.L.N.), and the Blue Bird Circle Foundation.
Correspondence should be addressed to Dr. Jeffrey L. Noebels, Department of Neurology, Baylor College of Medicine, One Baylor Plaza, Houston, TX 77030. E-mail: jnoebels@bcm.tmc.edu.
REFERENCES
- 1.Ahlijanian MK, Westenbroek RE, Catterall WA. Subunit structure and localization of dihydropyridine-sensitive calcium channels in mammalian brain, spinal cord, and retina. Neuron. 1990;6:819–832. doi: 10.1016/0896-6273(90)90135-3. [DOI] [PubMed] [Google Scholar]
- 2.Brice NL, Berrow NS, Campbell V, Page KM, Brickley K, Tedder I, Dolphin AC. Importance of the different beta subunits in the membrane expression of the alpha1A and alpha2 calcium channel subunits: studies using a depolarization-sensitive alpha1A antibody. Eur J Neurosci. 1997;9:749–759. doi: 10.1111/j.1460-9568.1997.tb01423.x. [DOI] [PubMed] [Google Scholar]
- 3.Burgess DL, Jones JM, Meisler MH, Noebels JL. Mutation of the Ca2+-channel β subunit gene Cchb4 is associated with ataxia and seizures in the lethargic (lh) mouse. Cell. 1997;88:385–392. doi: 10.1016/s0092-8674(00)81877-2. [DOI] [PubMed] [Google Scholar]
- 4.Burgess DL, Biddlecome GH, McDonough SI, Diaz ME, Zilinski CA, Bean BP, Campbell KP, Noebels JL. β subunit reshuffling modifies N- and P/Q type Ca2+ channel subunit compositions in lethargic mouse brain. Mol Cell Neurosci. 1999;13:293–311. doi: 10.1006/mcne.1999.0748. [DOI] [PubMed] [Google Scholar]
- 5.Burgess DL, Gefrides LA, Foreman PJ, Noebels JL. A cluster of three novel Ca2+ channel gamma subunit genes on chromosome 19q13.4: evolution and expression profile of the gamma subunit gene family. Genomics. 2001;71:339–350. doi: 10.1006/geno.2000.6440. [DOI] [PubMed] [Google Scholar]
- 6.Campbell DB, Hess EJ. L-type calcium channels contribute to the tottering mouse dystonic episodes. Mol Pharmacol. 1999;55:23–31. doi: 10.1124/mol.55.1.23. [DOI] [PubMed] [Google Scholar]
- 7.Chemin J, Monteil A, Perez-Reyes E, Nargeot J, Lory P. Direct inhibition of T-type calcium channels by the endogenous cannabinoid anadamide. EMBO J. 2001;20:7033–7040. doi: 10.1093/emboj/20.24.7033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bredt DS, Nicoll RA. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408:936–943. doi: 10.1038/35050030. [DOI] [PubMed] [Google Scholar]
- 9.Chien AL, Zhao X, Shirokov RE, Puri TS, Chang CF, Sun D, Rios E, Hosey MM. Role of a membrane-localized β subunit in the formation and targeting of functional L-type Ca2+ channels. J Biol Chem. 1995;270:30036–30044. doi: 10.1074/jbc.270.50.30036. [DOI] [PubMed] [Google Scholar]
- 10.Coulter DA, Huguenard JR, Prince DA. Calcium currents in rat thalamocortical relay neurons: kinetic properties of the transient, low-threshold current. J Physiol (Lond) 1989a;414:587–604. doi: 10.1113/jphysiol.1989.sp017705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coulter DA, Huguenard JR, Prince DA. Characterization of ethosuximide reduction of low-threshold calcium current in thalamic neurons. Ann Neurol. 1989b;25:582–593. doi: 10.1002/ana.410250610. [DOI] [PubMed] [Google Scholar]
- 12.Cribbs LL, Lee J-H, Yang J, Satin J, Zhang Y, Daud A, Barclay J, Williamson MP, Fox M, Rees M, Perez-Reyes E. Cloning and characterization of α1H from human heart, a member of the T-type calcium channel gene family. Circ Res. 1998;83:103–109. doi: 10.1161/01.res.83.1.103. [DOI] [PubMed] [Google Scholar]
- 13.Crunelli V, Lightowler S, Pollard CE. A T-type Ca2+ current underlies low-threshold Ca2+ potentials in cells of the cat and rat lateral geniculate nucleus. J Physiol (Lond) 1989;413:543–561. doi: 10.1113/jphysiol.1989.sp017668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Borman B, Lakaye B, Minet A, Zorzi W, Vergnes M, Marescaux C, Grisar T. Expression of mRNA encoding alpha1E and alpha1G subunit in the brain of a rat model of absence epilepsy. NeuroReport. 1999;10:569–574. doi: 10.1097/00001756-199902250-00023. [DOI] [PubMed] [Google Scholar]
- 15.Delisle BP, Satin J. pH Modulation of human T-type calcium channel gating. Biophys J. 2000;78:1895–1905. doi: 10.1016/S0006-3495(00)76738-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Destexhe A, Neubig M, Ulrich D, Huguenard J. Dendritic low-threshold currents in thalamic relay cells. J Neurosci. 1998;18:3574–3588. doi: 10.1523/JNEUROSCI.18-10-03574.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Waard M, Campbell KP. Subunit regulation of the neuronal α1A Ca2+ channel expressed in Xenopus oocytes. J Physiol (Lond) 1995;485:619–634. doi: 10.1113/jphysiol.1995.sp020757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dolphin AC, Wyatt CN, Richards J, Beattie RE, Craig P, Lee J-H, Cribbs LL, Volsen SG, Perez-Reyes E. The effect of α2-δ and other accessory subunits on expression and properties of the calcium channel α1G. J Physiol (Lond) 1999;519:35–45. doi: 10.1111/j.1469-7793.1999.0035o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dove LS, Abbott LC, Griffith WH. Whole-cell and single channel analysis of P-type calcium currents in cerebellar Purkinje cells of leaner mutant mice. J Neurosci. 1998;18:7687–7699. doi: 10.1523/JNEUROSCI.18-19-07687.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Escayg A, De Waard M, Lee DD, Bichet D, Wolf P, Mayer T, Johnston J, Baloh R, Sander T, Meisler MH. Coding and noncoding variation of the human calcium channel beta4-subunit gene CACANB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am J Hum Genet. 2000;66:1531–1539. doi: 10.1086/302909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fletcher CF, Lutz CM, O'Sullivan TN, Shaughnessy JDJ, Hawkes R, Frankel WN, Copeland NG, Jenkins NA. Absence epilepsy in tottering mutant mice is associated with calcium channel defects. Cell. 1996;87:607–617. doi: 10.1016/s0092-8674(00)81381-1. [DOI] [PubMed] [Google Scholar]
- 22.Gomora JC, Daud AN, Weiergraber M, Perez-Reyes E. Block of cloned human T-type calcium channels by succinimide antiepileptic drugs. Mol Pharmacol. 2001;60:1121–1132. [PubMed] [Google Scholar]
- 23.Green PJ, Warre R, Hayes PD, McNaughton NC, Medhurst AD, Pangalos M, Duckworth DM, Randall AD. Kinetic modification of the alpha(1I) subunit-mediated T-type Ca2+ channel by a human neuronal Ca2+ channel gamma subunit. J Physiol (Lond) 2001;533:467–478. doi: 10.1111/j.1469-7793.2001.0467a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gurnett CA, De Waard M, Campbell KP. Dual function of the voltage-dependent Ca2+ channel alpha 2 delta subunit in current stimulation and subunit interaction. Neuron. 1996;16:431–440. doi: 10.1016/s0896-6273(00)80061-6. [DOI] [PubMed] [Google Scholar]
- 25.Hosford DA, Clark S, Cao Z, Wilson WA, Jr, Lin FH, Morrisett RA, Huin A. The role of GABAB receptor activation in absence seizures of lethargic (lh/lh) mice. Science. 1992;257:398–401. doi: 10.1126/science.1321503. [DOI] [PubMed] [Google Scholar]
- 26.Huguenard JR. Low-threshold calcium currents in central nervous system neurons. Annu Rev Physiol. 1996;58:329–348. doi: 10.1146/annurev.ph.58.030196.001553. [DOI] [PubMed] [Google Scholar]
- 27.Huguenard JR, Prince DA. A novel T-type current underlies prolonged Ca2+-dependent bursts in GABAergic firing neurons of rat thalamic reticular nucleus. J Neurosci. 1992;12:3804–3817. doi: 10.1523/JNEUROSCI.12-10-03804.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jahnsen H, Llinás RR. Ionic basis for the electroresponsiveness and oscillatory properties of guinea-pig thalamic neurons in vitro. J Physiol (Lond) 1984;349:227–247. doi: 10.1113/jphysiol.1984.sp015154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jouvenceau A, Eunson LH, Spauschus A, Ramesh V, Zuberi SM, Kullmann DM, Hanna MG. Human epilepsy associated with dysfunction of the brain P/Q-type calcium channel. Lancet. 2001;358:801–807. doi: 10.1016/S0140-6736(01)05971-2. [DOI] [PubMed] [Google Scholar]
- 30.Jun K, Piedras-Renteria ES, Smith SM, Wheeler DB, Lee SB, Lee TG, Chin H, Adams ME, Scheller RH, Tsien RW, Shin HS. Ablation of P/Q-type Ca channel currents, altered synaptic transmission, and progressive ataxia in mice lacking the a1A-subunit. Proc Natl Acad Sci USA. 1999;96:15245–15250. doi: 10.1073/pnas.96.26.15245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kang MG, Chen CC, Felix R, Letts VA, Frankel WN, Mori Y, Campbell KP. Biochemical and biophysical evidence for gamma 2 subunit association with neuronal voltage-activated Ca2+ channels. J Biol Chem. 2001;276:32917–32924. doi: 10.1074/jbc.M100787200. [DOI] [PubMed] [Google Scholar]
- 32.Kapur A, Yeckel MF, Gray R, Johnston D. L-type calcium channels are required for one form of hippocampal mossy fiber LTP. J Neurophysiol. 1998;79:2181–2190. doi: 10.1152/jn.1998.79.4.2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Karst H, Joels M, Wadman WJ. Low-threshold calcium current in dendrites of the adult rat hippocampus. Neurosci Lett. 1993;164:154–158. doi: 10.1016/0304-3940(93)90880-t. [DOI] [PubMed] [Google Scholar]
- 34.Kim D, Song I, Keum S, Lee T, Jeong MJ, Kim SS, McEnery MW, Shin HS. Lack of the burst firing of thalamocortical relay neurons and resistance to absence seizures in mice lacking alpha(1G) T-type Ca2+ channels. Neuron. 2001;31:35–45. doi: 10.1016/s0896-6273(01)00343-9. [DOI] [PubMed] [Google Scholar]
- 35.Klugbauer N, Dai S, Specht V, Lacinova L, Marais E, Bohn G, Hofmann FJ. A family of gamma-like calcium channel subunits. FEBS Lett. 2000;470:189–197. doi: 10.1016/s0014-5793(00)01306-5. [DOI] [PubMed] [Google Scholar]
- 36.Lee J-H, Daud AN, Cribbs LL, Lacerda AE, Pereverzev A, Klockner U, Schneider T, Perez-Reyes E. Cloning and expression of a novel member of the low voltage-activated T-type calcium channel family. J Neurosci. 1999;19:1912–1921. doi: 10.1523/JNEUROSCI.19-06-01912.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Letts VA, Felix R, Biddlecome GH, Arikkath J, Mahaffey CL, Valenzuela A, Bartlett FS, Mori Y, Campbell KP, Frankel WN. The mouse stargazer gene encodes a neuronal Ca2+ channel gamma subunit. Nat Genet. 1998;19:340–347. doi: 10.1038/1228. [DOI] [PubMed] [Google Scholar]
- 38.Llinás R, Jahnsen H. Electrophysiology of mammalian thalamic neurons in vitro. Nature. 1982;297:406–408. doi: 10.1038/297406a0. [DOI] [PubMed] [Google Scholar]
- 39.Lorenzon NM, Lutz CM, Frankel WN, Beam KG. Altered calcium channel currents in Purkinje cells of the neurological mutant mouse leaner. J Neurosci. 1998;18:4482–4489. doi: 10.1523/JNEUROSCI.18-12-04482.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matsushima T, Tegner J, Hill RH, Grillner S. GABAB receptor activation causes a depression of low- and high-voltage-activated Ca2+ currents, postinhibitory rebound, and postspike afterhyperpolarization in lamprey neurons. J Neurophysiol. 1993;70:2606–2619. doi: 10.1152/jn.1993.70.6.2606. [DOI] [PubMed] [Google Scholar]
- 41.McCormick DA, Bal T. Sleep and arousal: thalamocortical mechanisms. Annu Rev Neurosci. 1997;20:185–215. doi: 10.1146/annurev.neuro.20.1.185. [DOI] [PubMed] [Google Scholar]
- 42.McEnery MW, Copeland TD, Courtney LV. Altered expression and assembly of N-type calcium channel α1β and β subunits in epileptic lethargic (lh/lh) mouse. J Biol Chem. 1998;273:21435–21438. doi: 10.1074/jbc.273.34.21435. [DOI] [PubMed] [Google Scholar]
- 43.Meir A, Dell DC, Stephens GL, Page KM, Dolphin AC. Calcium channel beta subunit promotes voltage-dependent modulation of alpha1B by G beta gamma. Biophys J. 2000;79:731–746. doi: 10.1016/S0006-3495(00)76331-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nahm WK, Noebels JL. Nonobligate role of early or sustained expression of immediate-early gene proteins c-Fos, c-Jun, and Zif/268 in hippocampal mossy fiber sprouting. J Neurosci. 1998;18:9245–9255. doi: 10.1523/JNEUROSCI.18-22-09245.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Niedermeyer E, Laws ER, Jr, Walker EA. Depth EEG findings in epileptics with generalized spike-wave complexes. Arch Neurol. 1969;21:51–58. doi: 10.1001/archneur.1969.00480130065007. [DOI] [PubMed] [Google Scholar]
- 46.Nishimura S, Takeshima H, Hofmann F, Flockerzi V, Imoto K. Requirement of the calcium channel β subunit for functional conformation. FEBS Lett. 1993;324:283–286. doi: 10.1016/0014-5793(93)80135-h. [DOI] [PubMed] [Google Scholar]
- 47.Noebels JL, Sidman RL. Inherited epilepsy: spike-wave and focal motor seizures in the mutant mouse yottering. Science. 1979;204:1334–1336. doi: 10.1126/science.572084. [DOI] [PubMed] [Google Scholar]
- 48.Noebels JL, Qiao X, Bronson RT, Spencer C, Davisson MT. Stargazer: a new neurological mutant on chromosome 15 in the mouse with prolonged cortical seizures. Epilepsy Res. 1990;7:129–135. doi: 10.1016/0920-1211(90)90098-g. [DOI] [PubMed] [Google Scholar]
- 49.Park D, Dunlap K. Dynamic regulation of calcium influx by G-proteins, action potential waveform, and neuronal firing frequency. J Neurosci. 1998;18:6757–6766. doi: 10.1523/JNEUROSCI.18-17-06757.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perez-Reyes E, Cribbs LL, Daud A, Lacerda AE, Barclay J, Williamson MP, Fox M, Rees M, Lee J-H. Molecular characterization of a neuronal low-voltage-activated T type calcium channel. Nature. 1998;391:896–900. doi: 10.1038/36110. [DOI] [PubMed] [Google Scholar]
- 51.Qian J, Noebels JL. Presynaptic Ca2+ influx at a mouse central synapse with Ca2+ channel subunit mutations. J Neurosci. 2000;20:163–170. doi: 10.1523/JNEUROSCI.20-01-00163.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roche JP, Treistman SN. The Ca2+ channel beta3 subunit differentially modulate G-protein sensitivity of alpha1A and alpha1B. J Neurosci. 1998;18:878–886. doi: 10.1523/JNEUROSCI.18-03-00878.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rousset M, Cens T, Restituito S, Barrere C, Black JL, III, McEnery MW, Charnet P. Functional roles of gamma2, gamma3 and gamma4, three new Ca2+ channel subunits, in P/Q-type Ca2+ channel expressed in Xenopus oocytes. J Physiol (Lond) 2001;532:583–593. doi: 10.1111/j.1469-7793.2001.0583e.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shan MJ, Meis S, Munsch T, Pape HC. Modulation by extracellular pH of low- and high-voltage-activated calcium currents of rat thalamic relay neurons. J Neurophysiol. 2001;85:1051–1058. doi: 10.1152/jn.2001.85.3.1051. [DOI] [PubMed] [Google Scholar]
- 55.Song I, Kim D, Jun K, Shin HS. Role of T-type calcium channels in the genesis of absence seizure in the mutant mice for α1A, the pore-forming subunit of the P/Q-type calcium channel. Soc Neurosci Abstr. 2001;27:151.21. [Google Scholar]
- 56.Steriade M. Corticothalamic resonance, states of vigilance and mentation. Neuroscience. 2000;101:243–276. doi: 10.1016/s0306-4522(00)00353-5. [DOI] [PubMed] [Google Scholar]
- 57.Steriade M, Llinás RR. The functional states of the thalamus and the associated neuronal interplay. Physiol Rev. 1988;68:649–742. doi: 10.1152/physrev.1988.68.3.649. [DOI] [PubMed] [Google Scholar]
- 58.Talley EM, Cribbs LL, Lee JH, Daud A, Perez-Reyes E, Bayliss DA. Differential distribution of three members of a gene family encoding low voltage-activated (T-type) calcium channels. J Neurosci. 1999;19:1895–1911. doi: 10.1523/JNEUROSCI.19-06-01895.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Talley EM, Solorzano G, Depaulis A, Perez-Reyes E, Bayliss DA. Low-voltage-activated calcium channel subunit expression in a genetic model of absence epilepsy in the rat. Mol Brain Res. 2000;75:159–165. doi: 10.1016/s0169-328x(99)00307-1. [DOI] [PubMed] [Google Scholar]
- 60.Tsakiridou E, Bertollini L, de Curtis M, Avanzini G, Page HC. Selective increase in T-type calcium conductance of reticular thalamic neurons in a rat model of absence epilepsy. J Neurosci. 1995;15:3110–3117. doi: 10.1523/JNEUROSCI.15-04-03110.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vigues S, Gastaldi M, Chabret C, Massacrier A, Cau P, Valmier J. Regulation of calcium channel alpha(1A) subunit splice variant mRNAs in kainate-induced temporal lobe epilepsy. Neurobiol Dis. 1999;6:288–301. doi: 10.1006/nbdi.1999.0248. [DOI] [PubMed] [Google Scholar]
- 62.Wakamori M, Yamazaki K, Matsunodaira H, Teramoto T, Tanaka I, Niidome T, Sawada K, Nishizawa Y, Sekiguchi N, Mori E, Mori Y, Imoto K. Single tottering mutations responsible for the neuropathic phenotype of the P-type calcium channel. J Biol Chem. 1998;273:34857–34867. doi: 10.1074/jbc.273.52.34857. [DOI] [PubMed] [Google Scholar]
- 63.Walker D, De Waard M. Subunit interaction sites in voltage-dependent Ca2+ channels: role in channel function. Trends Neurosci. 1998;21:148–154. doi: 10.1016/s0166-2236(97)01200-9. [DOI] [PubMed] [Google Scholar]
- 64.Williams DA. A study of thalamic and cortical rhythms in petit mal. Brain. 1953;76:50–69. doi: 10.1093/brain/76.1.50. [DOI] [PubMed] [Google Scholar]
- 65.Witcher DR, De Waard M, Liu H, Pragnell M, Campbell KP. Association of native β Ca2+ channel subunits with the α subunit interaction domain. J Biol Chem. 1995;270:18088–18093. doi: 10.1074/jbc.270.30.18088. [DOI] [PubMed] [Google Scholar]
- 66.Yamaguchi H, Hara M, Strobeck M, Fukasawa K, Schwartz A, Varadi G. Multiple modulation pathways of calcium channel activity by a β subunit participaiton in membrane trafficking of the subunit. J Biol Chem. 1998;273:19348–19356. doi: 10.1074/jbc.273.30.19348. [DOI] [PubMed] [Google Scholar]
- 67.Zhang Y, Cribbs LL, Satin J. Arachidonic acid modulation of α1H, a cloned human T-type calcium channel. Am J Physiol Heart Circ Physiol. 2000;278:H184–H193. doi: 10.1152/ajpheart.2000.278.1.H184. [DOI] [PubMed] [Google Scholar]