

Abstract
Bax is required for the apoptotic death of sympathetic neurons deprived of nerve growth factor (NGF). After NGF withdrawal, Bax translocates from the cytoplasm to the mitochondria of these cells and induces release of the proapoptotic protein cytochromec. Here, we report that withdrawing NGF from mouse sympathetic neurons caused an increase of mitochondria-derived reactive oxygen species (ROS). Suppressing these ROS inhibited apoptosis. Bax deletion blocked death and prevented the ROS burst. Inducing a pro-oxidant state similar to that in NGF-deprived, wild-type cells caused cytochrome c release even in neurons lacking Bax. A similar ROS burst in cerebellar granule neurons undergoing apoptosis was also blocked by Bax deletion. These findings indicate that Bax lies upstream from increased ROS in NGF-deprived neurons and suggest that the Bax-induced ROS burst is both necessary and sufficient for cytochrome c redistribution in these cells.
Keywords: apoptosis, bax, reactive oxygen, mitochondria, nerve growth factor, programmed cell death
From 20 to 80% of neurons born during vertebrate neurogenesis undergo programmed cell death (PCD), a process critical for sculpting the developing nervous system (Oppenheim, 1991). Availability of a required neurotrophic factor supplied by target or other tissues is the primary determinant of which neurons escape PCD. Those cells obtaining an adequate supply of neurotrophic factor live, whereas those that do not die by apoptosis (Martin et al., 1988; Deckwerth and Johnson, 1993; Deshmukh et al., 1996; Yuan and Yanker, 2001). Two culture models have been extensively used to examine the cellular and molecular events underlying neuronal PCD, sympathetic neurons deprived of nerve growth factor (NGF; Martin et al., 1988; Deckwerth and Johnson, 1993; Deshmukh et al., 1996; Neame et al., 1998; Martinou et al., 1999; Putcha et al., 2001) and cerebellar granule cells induced to undergo PCD by membrane potential repolarization and serum withdrawal (Miller and Johnson, 1996; Schulz et al., 1996; D'Mello et al., 1997). As with many kinds of neurons (White et al., 1998), the PCD of both of these cell types depends on the proapoptotic Bcl-2 family member Bax (Deckwerth et al., 1996;Miller et al., 1997). In both systems, Bax deletion prevents apoptosis. In healthy cells, most Bax resides in the cytoplasm and the outer mitochondrial membrane (OMM). During apoptotic death, Bax translocates from the cytoplasm to the OMM (Wolter et al., 1997; Putcha et al., 1999). The increased OMM Bax concentration then induces release of apoptogenic factors from the mitochondrial intermembrane space into the cytoplasm (Liu et al., 1996; Susin et al., 1999; Du et al., 2000; Li et al., 2001). The best characterized of these factors is cytochromec. Normally, the holo form of cytochrome c is found in cells only within the mitochondrial intermembrane space. Once released into the cytoplasm, holocytochrome c causes formation of the apoptosome and activation of caspase 9 (Li et al., 1997; Zou et al., 1997). The caspase 9 then cleaves and activates other caspases that attack many protein substrates, causing cell death (Li et al., 1997).
The mechanism by which Bax permeabilizes the OMM is unknown. Withdrawing NGF from rat sympathetic neurons in cell culture causes increased levels of mitochondrial-derived reactive oxygen species (ROS) in those cells (Greenlund et al., 1995; Dugan et al., 1997). We recently presented evidence that this ROS burst induces cytochromec release (Kirkland and Franklin, 2001). In this report, we demonstrate that withdrawing NGF from mouse sympathetic neurons in cell culture caused a similar increase of ROS in those cells. This ROS burst was absent in neurons from Bax-deficient mice, indicating that Bax lay upstream from the increased ROS. Bax deletion also abolished an ROS burst in wild-type cerebellar granule neurons undergoing PCD. Mimicking the ROS burst of NGF-deprived sympathetic neurons with H2O2 caused release of cytochrome c even in cells lacking Bax. These findings suggest that one mechanism by which Bax causes OMM permeabilization is by increasing ROS. These ROS then induce release of cytochromec and, perhaps, other intermembrane space proteins via a Bax-independent pathway.
MATERIALS AND METHODS
Reagents. Monochlorobimane, Mitotracker Red CMXRos, and 5-(and-6)-chloromethyl-2′, 7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) were from Molecular Probes (Eugene, OR). Nerve growth factor 2.5S was from Harlan Bioproducts (Indianapolis, IN). Unless otherwise stated, all other reagents were from Sigma (St. Louis, MO).
Breeding and genotyping of mice. Mice heterozygous for thebax allele (The Jackson Laboratory, Bar Harbor, ME; Knudson et al., 1995) were mated to generate knock-out (bax−/−), heterozygous (bax+/−), and wild-type (bax+/+) offspring. Genomic DNA was prepared from the tail of each pup using a Wizard Prep kit (Promega, Madison, WI). A single PCR amplified both the mutant and the wild-type allele. An exon 5 forward primer (0.64 μm,5′-TGATCAGAACCATCATG-3′) and an intron 5 reverse primer (0.64 μm, 5′-GTTGACCAGAGTGGCGTAGG-3′) amplified a 304 bp portion of the wild-type allele. A neo/pgk primer (0.16 μm, 5′-CCGCTTCCATTGCTCAGCGG-3′) and the same intron 5 reverse primer generated a 507 bp product from the mutant allele. The cycling parameters were 1 min at 94°C, 1 min at 57.5°C, and 1.5 min at 72°C for 30 cycles. This was followed by 7 min at 72°C to allow complete extension of amplification products. The primer ratio favored amplification of the wild-type allele to ensure correct genotyping of bax-deficient animals.
Cell culture. Neurons were dissociated from the superior cervical ganglia of newborn mice and plated on a collagen substrate as described previously (Franklin et al., 1995; Deckwerth et al., 1996). Separate cultures were established for the ganglia from each pup. Neurons for survival assays were plated in 24-well Costar tissue culture dishes (Corning, Corning, NY). Cells for microscopy experiments were plated on number 1 glass coverslips affixed with silicon medical adhesive (Dow Corning, Midland, MI) over holes cut in the bottoms of 35 mm Falcon culture dishes (Beckton Dickinson, Franklin Lakes, NJ). Cells from one-half to one ganglion were plated per culture for most experiments. Two ganglia were plated per culture for immunoblot experiments. Cultures were incubated in medium containing Eagle's minimum essential medium with Earle's salts withoutl-glutamine (Invitrogen, San Diego, CA) and supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 μg/ml streptomycin, 20 μm fluorodeoxyuridine, 20 μm uridine, 1.4 mml-glutamine, and 50 ng/ml 2.5S NGF. All cultures were maintained at 35°C in an incubator having a 95% air and 5% CO2atmosphere.
Nerve growth factor was withdrawn from cultures by incubating them in culture medium lacking NGF and containing an NGF-neutralizing antibody (Harlan Bioproducts; Franklin and Johnson, 1998). After survival experiments, treatment media were washed out and replaced with culture medium containing NGF. After 3–5 d, cultures were fixed for 30 min at 4°C with 4% paraformaldehyde in PBS, pH 7.2, and stained with 0.1% crystal violet. This procedure caused significant hypertrophy of living neurons, allowing easy determination of surviving cells (Kirkland and Franklin, 2001). Blinded counting determined the number of neurons that survived. Counts were normalized to the number of neurons in untreated sibling cultures maintained since the time of plating in NGF. All experiments were begun when cultures were 6–9 d old.
Cerebellar granule cell cultures were prepared as described by Miller and Johnson (1996). Briefly, cerebella were removed from 7- to 8-d-old mice. Suspensions of single cells were prepared from the cerebella and plated (2.5 × 105cells/cm2) on a poly-l-lysine (100 μg/ml) substrate coated onto the microscopy dishes described above. Cultures were maintained at 35°C in an incubator with a 5% CO2 and 95% air atmosphere. Culture medium consisted of basal Eagle's medium (Invitrogen) with 25 mmKCl, 10% dialyzed fetal bovine serum, 50 U/ml penicillin G sodium, and 50 μg/ml streptomycin sulfate (Invitrogen). Twenty-four hours after plating, the antimitotic agent aphidicolin (3.3 μg/ml) was added to the culture medium. Apoptotic death was induced by replacing the culture medium with similar medium lacking serum and containing 5 mm KCl. Cells were cultured for 6–9 d before experiments. All experiments with sympathetic and cerebellar granule neurons were done with cultures from three to seven separate platings unless otherwise stated.
Confocal and fluorescent microscopy. A Bio-Rad (Hercules, CA) MRC 1024 laser-scanning confocal microscope mounted on a Nikon Diaphot 200 inverted microscope and controlled by Laser Sharp software (version 3.0; Bio-Rad) was used for confocal microscopy (Kirkland and Franklin, 2001). Neurons, chosen at random, were scanned at 512 × 512 pixel resolution. Images were quantified by measuring the raw pixel intensity in a 60 μm2 area of stained neuronal somas with Sigmagel software (SPSS, Chicago, IL). The intensity of each neuron was normalized to that of NGF-maintainedbax+/+ neurons receiving the same concentration of dye for the same time as the experimental cells.
A Nikon TE300 inverted microscope was used for fluorescence microscopy. Light was provided by a xenon lamp, and images were captured by a CCD camera (MicroMAX; Princeton Instruments, Trenton, NJ) using Metamorphsoftware (Universal Imaging Corp., West Chester, PA). Filter cubes were changed manually. The 380 ± 15 nm excitation filter was controlled by a Lambda 10-2 optical filter changer (Sutter Instrument Co., Novato, CA). Fluorescence intensity was measured in a 33 μm2 area of the soma of each cell with the Metamorph elliptical region tool. All microscopy was done at room temperature.
Electron microscopy. Cultures, plated on collagen-coated number 1 coverslips, were fixed for 1–2 hr at room temperature in 0.1m phosphate buffer, pH 7.2, containing 2% glutaraldehyde and 2% formaldehyde. They were then postfixed for 1 hr with 0.1m phosphate buffer containing 2% osmium tetroxide and dehydrated in a graded series of alcohol solutions. After embedding in Durcupan (Fluka, Buchs, Switzerland), coverslips were removed by etching with hydrofluoric acid. Cut sections (70 nm) were poststained with uranyl acetate and lead citrate. A Philips CM120 transmission electron microscope was used for microscopy.
ROS measurement. We used the redox-sensitive dye CM-H2DCFDA to determine ROS levels. CM-H2DCFDA is readily membrane-permeant and is trapped in cells by binding of the chloromethyl group to cellular thiols. It becomes fluorescent when oxidized by hydrogen peroxide and downstream free radical products of hydrogen peroxide (Royall and Ischiropoulos, 1993). We previously characterized the use of this dye in rat sympathetic neurons (Kirkland and Franklin, 2001). Similar characterization for mouse sympathetic neurons is shown in Table1. Cultures were incubated for 20–25 min at 35°C in the appropriate experimental medium containing CM-H2DCFDA (10 μm) unless otherwise stated. They were then washed two or three times with Leibovitz's L-15 medium containing the experimental treatments and left in the last wash for confocal microscopy. To determine how well the dye was retained, cells loaded with CM-H2DCFDA were treated with 20 mm H2O2 to maximally oxidize the dye. After 10 min of this treatment, CM-H2DCFDA intensity was determined, and the H2O2 was washed out. There was no significant change in dye intensity at later times (p > 0.05; n = 23–31 neurons), indicating little dye leakage. The dye was excited with the 488 nm line of the confocal laser, and the FITC photomultiplier was used for image acquisition.
Table 1.
Characterization of CM-H2DCFDA
| Condition | Fold change in dye intensity after 30 min of H2O2 |
|---|---|
| 2 mmH2O2 | |
| bax+/+ + NGF | +2.82 ± 0.27 |
| bax+/−+ NGF | +2.31 ± 0.22 |
| bax−/− + NGF | +2.03 ± 0.23 |
| bax+/+ − NGF | +2.75 ± 0.26 |
| bax+/− − NGF | +2.37 ± 0.15 |
| bax−/− − NGF | +2.68 ± 0.38 |
| 10 mmH2O2 | |
| bax+/+ ROT − NGF | 62 ± 9.02 |
| bax+/+ ANT A − NGF | 59 ± 2.58 |
All cells were loaded for 20 min with 10 μmCM-H2DCFDA. They were then transferred to L-15 medium and exposed to H2O2. Intensity is shown as fold change from CM-H2DCFDA intensity measured in NGF-maintainedbax+/+ cells that were plated at the same time.n = 26-112 cells from two or three separate platings. There were no significant differences in dye intensities of cells treated with the same concentrations of H2O2(p > 0.05). These data demonstrate that the different Bax levels (±NGF) in the three genotypes did not interfere with the concentration of CM-H2DCFDA loaded into cells or with the ability of CM-H2DCFDA to be oxidized, and the compounds tested did not interfere with dye loading or ability of the dye to be oxidized. All NGF deprivation (−NGF) was for 18 hr.
Immunocytochemistry. Neurons were immunostained for Bax and cytochrome c by a modification of the techniques (Kirkland and Franklin, 2001) described by Easton et al. (1997), Deshmukh and Johnson (1998), and Putcha et al. (1999). Cultures were fixed for 30 min at 4°C with 4% paraformaldehyde in PBS, pH 7.2. They were next washed three times with Tris-buffered saline (TBS) containing 100 mm Tris-HCl, pH 7.6, and 0.9% NaCl. After washing, cultures were incubated for at least 30 min at room temperature in blocking buffer (TBS containing 0.3% Triton X-100 and 5% normal goat serum). They were then incubated overnight at 4°C in blocking buffer containing the anti-cytochrome c primary antibody (200 ng/ml; PharMingen, San Diego, CA) or the anti-Bax primary antibody (Santa Cruz Biotechnology, Santa Cruz, CA). Cultures were next washed three times with TBS and incubated for 2–4 hr at 4°C in blocking buffer containing an FITC-conjugated anti-mouse secondary antibody (1.7 μg/ml; PharMingen). Incubation with the secondary antibody was followed by washing two times with TBS.
Immunoblotting. Cultures were washed two times with TBS, pH 7.6. Cells were scraped from the substrate, transferred in 1.0 ml of ice-cold NaPO4 EDTA to 1.5 ml microfuge tubes, and pelleted at 5000 × g for 1 min. Supernatants were removed, and 50–100 μl of lysis buffer [25% glycerol, 60 mm Tris, pH 6.8, 100 mmdithiothreitol, 1 mm EDTA, 0.1% bromophenol blue, 0.2% SDS, and a 1:100 dilution of a protease inhibitor mixture (539134; Calbiochem, La Jolla, CA)] was added. Pellets were next homogenized for 15–20 sec with a disposable pellet pestle (Kontes, Vineland, NJ). The homogenized samples were boiled for 5 min and then returned to room temperature and spun for 1 min in an Eppendorf 5414 table-top centrifuge to remove debris. Lysates (20–30 μl), each containing one ganglion, were loaded into wells of 10% Tris-HCl precast gels (Bio-Rad). Gels were run for 40 min at 200 V. Proteins were transferred onto polyvinylidene difluoride membranes (Millipore, Bedford, MA) for 60 min at 4°C (100 V). Membranes were blocked in 100 mm TBS containing 0.1% Tween 20 (TBST) and 5% dry milk for 60 min. Membranes were next incubated in 400 ng/ml mouse Bax antibody (Santa Cruz Biotechnology) overnight at 4°C. They were then washed in TBST and incubated for 60 min at room temperature in the HRP-labeled secondary antibody (1:1000) provided with ECL kits (Amersham Biosciences, Piscataway, NJ). Membranes were then incubated in ECL substrate for 1 min followed by exposure to Eastman Kodak (Rochester, NY) X-OMAT film. Quantification of scanned blots was done with SigmaGel (SPSS).
Glutathione assay. The small amount of material available in sympathetic cultures from single p1 mice precluded use of enzymatic assays for glutathione concentration or the activity of enzymes in the glutathione pathway because of lack of assay sensitivity. Therefore, concentration of reduced glutathione (GSH) was estimated by fluorescence microscopic imaging of single cells stained with monochlorobimane (MCB), a dye that binds to GSH (Fernandez-Checa and Kaplowitz, 1990; Kirkland and Franklin, 2001). We have previously determined that changes in MCB staining intensity accurately reflect changes of GSH concentration measured in large cultures of rat sympathetic neurons by an enzymatic technique (Kirkland and Franklin, 2001). Cultures were stained with MCB (5 μm) in L-15 medium for 30 min at room temperature. They were next washed two times in L-15 medium lacking MCB and left in the second wash for imaging. MCB was excited at 380 ± 15 nm. The emission filter was 510 ± 20 nm. MCB intensity was quantified with Metamorph software. All intensities were normalized to average MCB intensities measured in NGF-maintained bax+/+ cells.
Statistics. Statistical analysis was done with SigmaStat 2.0 (SPSS). An appropriate statistical procedure was determined for each data set. In most cases, statistical comparisons were made with at test or Kruskal–Wallis one-way ANOVA on ranks with Dunn's multiple comparisons. Means in all figures are shown as ±SEM.
RESULTS
Increased ROS in NGF-deprived sympathetic neurons was Bax-dependent
We reported recently that withdrawing NGF from rat sympathetic neurons in culture increases the ROS concentration in those cells and presented evidence that this ROS burst promotes release of cytochromec from the mitochondrial intermembrane space into the cytoplasm (Kirkland and Franklin, 2001). Because Bax is also central to cytochrome c redistribution and apoptosis in these neurons (Deckwerth et al., 1996; Deshmukh and Johnson, 1998; Putcha et al., 1999), it was of interest to determine the relation of Bax to this ROS burst. To this end, we used cultures of sympathetic neurons taken frombax-deficient mice. Figure1A shows that neurons from NGF-maintained, bax+/+ mice displayed both faint homogeneous and intense punctate immunostaining for Bax, consistent with the known cytoplasmic and mitochondrial Bax locations (Putcha et al., 1999). Neurons frombax−/− mice showed little immunostaining. Western blot analysis found that the Bax concentration in neurons from bax+/− mice was approximately one-half that of neurons taken frombax+/+ animals and that Bax was absent in neurons taken frombax−/− mice (Fig.1B,C). Therefore, Bax protein concentration was determined by bax gene dosage.
Fig. 1.

Effect of gene dosage on expression of Bax protein in mouse sympathetic neurons in cell culture. A, Expression of Bax in neurons frombax+/+ andbax−/− mice. Top andbottom micrographs, Paired phase contrast and fluorescent micrographs of clusters of neurons maintained in the presence of NGF. B,bax+/+ gene dosage determined the level of expression of Bax protein. Western blot analysis (bottom) shows an absence of Bax in a culture taken from a bax−/− mouse and an intermediate level of Bax in a culture taken from abax+/− animal. Top, PCR banding pattern used to determine the bax genotype (see Materials and Methods). C, Quantification of Bax levels. The densities of Bax bands on Western blots were determined for each culture with SigmaGel software. Similar loading was verified by comparable tubulin densities (data not shown). n = 5–7 cultures.
To determine the effects of Bax on ROS production, we used the redox-sensitive dye CM-H2DCFDA. We previously characterized the use of this dye for semiquantitative measurement of ROS levels in rat sympathetic neurons in cell culture and found that it was superior to other commonly used redox-sensitive dyes (Kirkland and Franklin, 2001). To determine whether CM-H2DCFDA was equally useful for measuring ROS in mouse sympathetic neurons, we determined its characteristics in these cells (Table 1). This characterization showed that CM-H2DCFDA was equally effective for measurement of ROS in mouse sympathetic neurons and that none of the findings reported here can be explained by artifacts caused by dye leaking from cells, by differential dye loading in the different experimental conditions, or by alterations in the ability of the dye to respond to oxidation in the different conditions.
Figure 2, A and B, shows that withdrawal of NGF caused increased CM-H2DCFDA intensity in bothbax+/+ andbax+/− neurons. There was a small but significant (p < 0.001) increase of average CM-H2DCFDA intensity by 6 hr after NGF withdrawal in cells of both genotypes. By 12 hr after NGF deprivation, average CM-H2DCFDA intensity inbax+/+ neurons had increased approximately twofold (p < 0.001) compared with control cells maintained in NGF-containing medium. By 18 hr after NGF withdrawal, the time at which the first cells become committed to die (Deshmukh and Johnson, 1998), the average CM-H2DCFDA intensity in cells taken frombax+/+ animals was approximately threefold higher than control values (p < 0.001). This ROS level was maintained for the rest of the time course of apoptotic death. In contrast, the average CM-H2DCFDA intensity in neurons frombax−/− animals did not increase at any time after NGF withdrawal. The average CM-H2DCFDA intensities of NGF-maintained neurons from bax+/+ andbax−/− animals were similar (p > 0.05). The difference in CM-H2DCFDA intensities between NGF-deprived neurons from bax+/+ andbax−/− animals could not be explained by differences in the amount of dye loaded or differences in the ability of the dye to respond to oxidation with increased fluorescence in the two cell types. Neurons of both genotypes loaded equivalent amounts of dye and responded with similar increases of CM-H2DCFDA intensity on oxidation by the same concentrations of H2O2(Table 1, see Fig. 4A). The increase of CM-H2DCFDA intensity in NGF-deprived neurons frombax+/− mice was approximately one-half that in NGF-deprived cells frombax+/+ animals. This finding also could not be explained by differences in dye loading or dye properties in cells of the two genotypes, because there were none (Table 1, see Fig. 4A). These data indicate that ROS levels increased after NGF withdrawal and that this increase was dependent on the concentration of Bax in the cells.
Fig. 2.
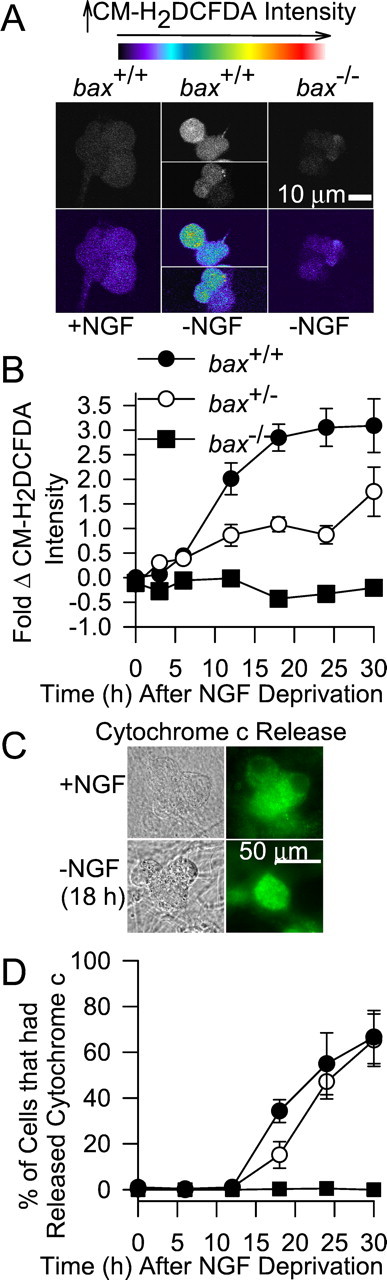
Bax lies upstream of increased ROS levels induced by NGF deprivation. A, Withdrawal of NGF caused increased ROS in sympathetic neurons taken frombax+/+ animals. Confocal micrographs show neuronal somas loaded with the redox-sensitive dye CM-H2DCFDA. Top micrographs, Raw, gray-scaled images of neurons taken frombax+/+ or bax−/− mice and maintained in culture medium containing NGF or deprived of NGF for 18 hr. Bottom micrographs, Same cells that have been pseudocolor-coded to indicate dye intensity. The barshows the relative intensities of the different colors. The more intense the dye, the higher the ROS levels. Note that only cells taken from bax+/+ animals exhibited increased CM-H2DCFDA intensity after NGF deprivation. The scale is the same for all micrographs. B, Quantification of changes in ROS (CM-H2DCFDA intensity) levels in NGF-deprived neurons. For each cell, CM-H2DCFDA intensity was normalized to the average CM-H2DCFDA intensity of NGF-maintained bax+/+ cultures that had been plated at the same time. ROS levels are shown as fold change from these values. This normalization method was used in all subsequent figures. n = 70–233 neurons. C, Micrographs illustrating release of cytochrome c after NGF deprivation. Left micrographs, Differential interference contrast (DIC) images. Right fluorescent micrographs, Same cells immunostained for cytochromec. Note the intense staining in cells that had been maintained in medium containing NGF and the faint, homogeneous staining in two of the three cells that were deprived of NGF for 18 hr. The remaining cell had not yet released cytochrome c into the cytoplasm. D, Time course of cytochromec redistribution in NGF-deprived neurons frombax+/+ andbax+/− animals. Cells frombax−/− mice did not release cytochrome c. Symbols indicate the same genotypes as in B. n = 3–6 cultures.
Fig. 4.

Increasing cellular GSH concentration inhibits death of NGF-deprived neurons frombax+/+ andbax+/− mice. A, Cells from bax+/− mice were saved from death by lower GSH ethyl ester concentrations than were cells frombax+/+ animals. Thebar shows survival ofbax−/− neurons deprived of NGF for the same period without GSH ethyl ester. n = 5–11 cultures for each time point. B, Effects of different concentrations of GSH ethyl ester on ROS levels (CM-H2DCFDA intensity measured 18 hr after NGF deprivation). Lower concentrations of GSH ethyl ester inhibited the ROS burst in neurons frombax+/− animals than in neurons frombax+/+ animals. Cells were exposed to the GSH ethyl ester from the time of NGF withdrawal. They were then washed and loaded with CM-H2DCFA for 20 min in medium lacking NGF and GSH ethyl ester. After washing, cultures were exposed to L-15 medium containing the appropriate concentrations of GSH ethyl ester (Kirkland and Franklin, 2001). n = 33–60 neurons for each time point. C, Time courses for rescue of NGF-deprived bax+/+ neurons by NGF and GSH ethyl ester (10 mm). Cells were deprived of NGF, and, at various times after withdrawal, medium containing either NGF or GSH ethyl ester (10 mm) was added. Forty-eight hours after withdrawal, the medium of all cultures was replaced with NGF-containing medium. Cultures were then allowed to recover 3–5 d, and survival was determined. The dotted line shows the GSH-mediated survival data scaled to 100% (at time 0) to allow easy comparison of the two rescue time courses. n = 9–12 cultures for the GSH time course. The time course for rescue by NGF was repeated only once (n = 7–12 cultures).
Redistribution of cytochrome c from the mitochondrial intermembrane space into the cytoplasm is central to the apoptotic death of NGF-deprived sympathetic neurons (Deshmukh and Johnson, 1998;Neame et al., 1998; Martinou et al., 1999). Loss of cytochromec from mitochondria can increase leakage of electrons from the mitochondrial electron transport chain to O2to form the free radical superoxide (O2·−;Turrens, 1997; Cai and Jones, 1998; Halliwell and Gutteridge, 1999;Kirkland and Franklin, 2001). Superoxide, in turn, is dismutated to form H2O2 that, along with its downstream free radical products, oxidizes CM-H2DCFDA (Royall and Ischiropoulos, 1993). Therefore, a possible mechanism underlying the Bax-mediated ROS burst after NGF withdrawal was increased formation of O2·− secondary to Bax-induced cytochromec release. To test this mechanism, we used immunocytochemistry to determine the time course of cytochromec redistribution after NGF withdrawal. Cytochromec appears to be rapidly released from most mitochondria in a cell during the apoptotic death of NGF-deprived sympathetic neurons (Deshmukh and Johnson, 1998; Neame et al., 1998; Kirkland and Franklin, 2001). Sympathetic neurons with mitochondria that are replete with cytochrome c exhibit intense, punctate immunocytochemical staining for cytochrome c. Cells with mitochondria depleted of cytochrome c show a faint, homogeneous staining pattern, apparently because cytochrome c is rapidly degraded once released into the cytoplasm. Figure 2C shows the criteria that were used to determine whether cytochrome c had been released from mitochondria into the cytoplasm after NGF withdrawal. When viewed by fluorescent microscopy, cells showing intense, punctate immunostaining for cytochrome c were scored as having retained cytochrome c in mitochondria, whereas those that showed faint, homogeneous staining were scored as having released cytochrome c. Using this technique, we found that no detectable cytochrome c redistribution occurred until ∼18 hr after NGF deprivation, and most cells had released cytochromec by 30 hr after withdrawal. The time course of release was similar to that reported previously for NGF-deprived sympathetic neurons (Deshmukh et al., 1996; Deshmukh and Johnson, 1998; Kirkland and Franklin, 2001). This time course was also nearly identical to the reported time courses for commitment to death and saving by caspase inhibitors in these cells (Deshmukh et al., 1996) further suggesting that little cytochrome c was released at earlier times after deprivation. Figure 2D shows the status of cytochromec in NGF-deprived bax+/+,bax+/−, andbax−/− neurons at different times after NGF withdrawal. As reported previously (Deshmukh and Johnson, 1998), cytochrome c was retained in the mitochondria ofbax−/− neurons after NGF withdrawal but was released from the mitochondria in neurons with the other two genotypes. The ROS burst began well before cytochromec redistribution in bothbax+/+ andbax+/− cells (Fig.2B,D). Indeed, release of cytochrome cappeared to have little effect on the ROS burst in these cells, because ROS levels reached a plateau by 18 hr after deprivation and changed little during the period of maximal cytochrome credistribution. Therefore, depletion of cytochrome c from the mitochondrial electron transport chain could not explain the increased ROS levels after NGF deprivation.
ROS derived from the mitochondrial electron transport chain
The most likely source for the increased ROS after NGF deprivation was the mitochondrial electron transport chain (Turrens, 1997; Tan et al., 1998). To examine the role of mitochondria in the ROS burst, we treated NGF-deprived bax+/+ neurons with compounds that block electron flow through mitochondrial respiratory complexes (Kirkland and Franklin, 2001). Acute application of rotenone (1 μm), a respiratory complex I inhibitor, potently suppressed the ROS burst occurring 18 hr after NGF withdrawal (Fig. 3A) (p < 0.001). Conversely, acute application of antimycin A (10 μm), a respiratory complex III inhibitor, did not significantly affect the ROS burst in NGF-deprived,bax+/+ cells (Fig. 3A) (p > 0.14). However, antimycin A did significantly enhance ROS levels inbax+/+ neurons maintained in NGF-containing medium (p < 0.001). Because the block of electron flow through complex I decreased ROS, whereas blocking complex III increased ROS in control cells and did not increase ROS in NGF-deprived cells (18 hr after deprivation), the data suggest that electrons entered the chain at complex I and leaked to O2 to form O2·− either within one of the complexes or between the two (Dugan et al., 1997;Halliwell and Gutteridge, 1999). The failure of antimycin A to enhance the ROS burst at 18 hr after NGF deprivation may indicate that ROS production was maximal at that time and could not be further increased.
Fig. 3.
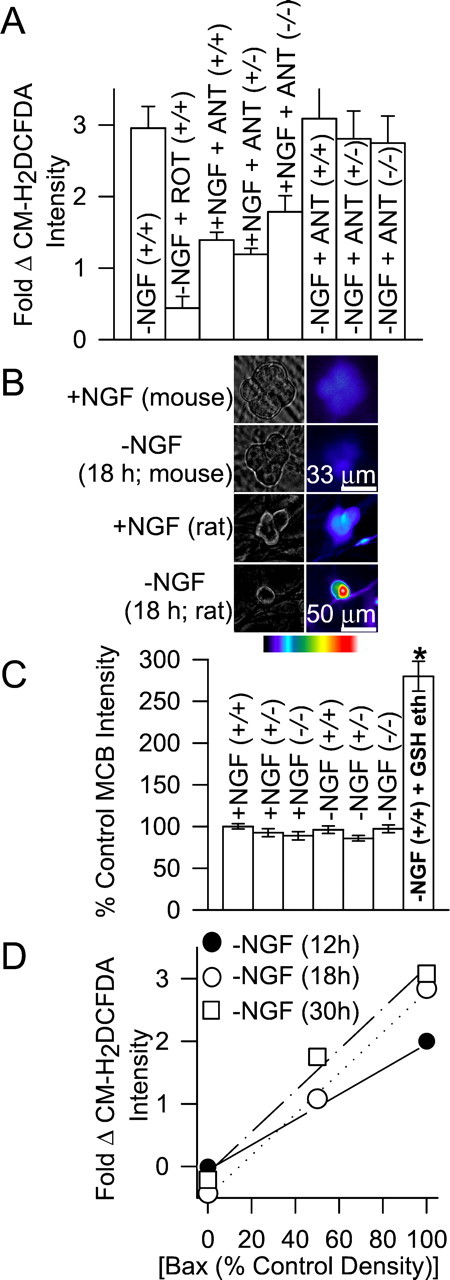
Increased ROS derives from the mitochondrial electron transport chain. A, Effects of mitochondrial agents on ROS levels. Cells were either maintained in NGF-containing medium before treatment (+NGF) with the compounds or were deprived of NGF for 18 hr before treatment (−NGF). ROT, Rotenone (10 μm); ANT, antimycin A (1 μm). There were no significant differences between the CM-H2DCFDA intensities in the ANT-exposed, NGF-maintained cells of the three genotypes (p > 0.27). However, the intensities of all three were significantly higher than in NGF-maintained neurons not exposed to ANT (p < 0.001). There were no significant differences between the CM-H2DCFDA intensities in NGF-deprived, ANT-exposed cells of the three genotypes and in NGF-deprived bax+/+ cells not exposed to ANT (p > 0.14). NGF-deprived cells exposed to ROT had much lower CM-H2DCFDA intensities than NGF-deprived neurons not exposed to ROT (p< 0.001). Compounds were added to the culture medium during the period of CM-H2DCFDA loading. After loading, the dye was washed out, and cultures were bathed in L-15 medium containing the same treatments for confocal microscopy. n = 58–233 neurons. B, Paired phase contrast (left) and fluorescent pseudocolored micrographs (right) of cells stained with the GSH-sensitive dye MCB. Unlike NGF-deprived rat sympathetic neurons (bottom), NGF-deprived mouse neurons (bax+/+) did not exhibit increased MCB staining after NGF withdrawal. The bar indicates relative pseudocolor values for MCB intensity (red,white are most intense and indicate higher GSH concentrations). The top and bottom scale bars are for mouse and rat neurons, respectively. C, GSH levels in the three mouse genotypes (as indicated by MCB staining) were similar and changed little after NGF withdrawal (18 hr). GSH ethyl ester (GSH eth; 10 mm for 18 hr) is a membrane-permeant form of GSH that was used as a positive control for MCB detection of GSH. n = 81–181 neurons.D, ROS levels were linearly related to Bax concentrations (Bax densities on Western blots). Bax data are taken from Figure 1C, and ROS data are from Figure2B. R = 0.99 for each deprivation time.
To determine whether mitochondria in cells of the three genotypes differed in their ability to generate ROS, we treatedbax+/− andbax−/− neurons with antimycin A (1 μm). There was no difference in the ability of antimycin A to increase ROS levels in NGF-maintained neurons with these genotypes from its ability to do so in NGF-maintained,bax+/+ cells (Fig. 3A) (p > 0.27). Therefore, there did not appear to be any difference in the ability of mitochondria in the three cell types to generate ROS. This assertion was further supported by experiments showing that treating NGF-deprived (18 hr)bax+/− andbax−/− neurons with antimycin A increased CM-H2DCFDA intensity to that measured in NGF-deprived bax+/+ cells (p > 0.14). The data in Table 1 demonstrate that there were no differences in the abilities of rotenone- and antimycin A-treated cells to load with CM-H2DCFDA or in the ability of the dye in these cells to respond to oxidation by similar concentrations of H2O2. These data indicate that the mitochondria in cells with the three genotypes were equally competent to produce ROS.
Although the dissimilarities in ROS levels in NGF-deprivedbax+/+,bax+/−, andbax−/− neurons apparently could not be explained by differences in the ability of mitochondria in the three cell types to generate ROS, it remained possible that dissimilar Bax concentrations in neurons of the different genotypes altered other cellular properties that affected ROS levels. The principal mechanism for detoxifying H2O2 in neurons, and most other cell types, involves oxidation of GSH (Halliwell and Gutteridge, 1999; Kirkland and Franklin, 2001). Increasing the GSH concentration can decrease H2O2 levels in sympathetic neurons (Kirkland and Franklin, 2001). Thus, it was possible that the disparate Bax concentrations in cells of the three genotypes altered the GSH concentration and, thereby, changed cellular H2O2 levels. To determine whether GSH concentrations were different inbax+/+,bax+/−, andbax−/− neurons, we stained them with the GSH-sensitive dye MCB (Fernandez-Checa and Kaplowitz, 1990;Ratan et al., 1994; Kirkland and Franklin, 2001). Figure 3,B and C, shows that MCB staining intensities were similar in NGF-maintained cells of each bax genotype, suggesting similar GSH concentrations. MCB staining intensity changed little after NGF deprivation. This contrasts with rat sympathetic neurons that have increased MCB staining intensities after NGF deprivation (Kirkland and Franklin, 2001). Therefore, the dissimilar CM-H2DCFDA intensities of neurons from the three genotypes probably cannot be explained by differences in GSH concentrations. Figure 3D shows that ROS levels after NGF deprivation were linearly related to Bax concentration. Together, the data suggest that, in NGF-deprived sympathetic neurons, Bax increases mitochondrial ROS production and that it is the cellular concentration of Bax that determines the amount of ROS produced.
A pro-oxidant state caused cytochrome c release and cell death independent of Bax
Increasing the cellular GSH concentration suppresses elevated ROS levels, cytochrome c release, and apoptotic death in NGF-deprived rat sympathetic neurons in cell culture (Kirkland and Franklin, 2001). To determine whether GSH had similar effects on NGF-deprived mouse sympathetic neurons, we artificially increased cellular GSH concentrations by treating NGF-deprived cultures frombax+/+ andbax+/− animals with GSH ethyl ester, a membrane-permeant form of GSH. Figure4A shows that GSH ethyl ester potently inhibited the apoptotic death of NGF-deprived neurons from bax+/+ andbax+/− mice. Interestingly, GSH ethyl ester supported survival of NGF-deprived,bax+/− neurons at lower concentrations than were required to support survival of NGF-deprivedbax+/+ cells. As reported previously (Deckwerth et al., 1996), all neurons frombax−/− mice survived without NGF in the culture medium.
It seemed likely to us that the reason GSH ethyl ester promoted survival of NGF-deprived bax+/−neurons at lower concentrations than those that were required to support survival of NGF-deprived,bax+/+ cells was related to the diminished ROS burst in the bax+/−neurons (Fig. 1B). Higher concentrations of GSH may have been required to suppress the larger ROS burst in thebax+/+ cells. To determine whether this was the case, we deprived cells of NGF in medium containing different concentrations of GSH ethyl ester and then measured CM-H2DCFDA intensities 18 hr after the initial treatments. Consistent with our hypothesis, higher concentrations of GSH ethyl ester were required to reduce CM-H2DCFDA intensities inbax+/+ than inbax+/− neurons (Fig.4B). To determine the time course over which GSH ethyl ester could prevent death after NGF deprivation, we deprivedbax+/+ neurons of NGF and then, at various times after the deprivation, exposed them to culture medium lacking NGF and containing GSH ethyl ester (10 mm). Figure 4C shows that the time course for rescue of NGF-deprived cells by GSH ethyl ester was similar to the time course for rescue by NGF. This rescue time course was similar to the time course of cytochrome c release (Fig.2D), suggesting that both NGF and GSH act close to the point of commitment to cell death, which in these cells is primarily determined by cytochrome c release (Deshmukh and Johnson, 1998). Moreover, both time courses are later than the reported time course for translocation of Bax to the mitochondria in these cells (Putcha et al., 1999), suggesting that both agents can block death downstream of Bax translocation.
The close correlation between the concentrations of GSH ethyl ester that blocked the ROS bursts inbax+/+ andbax+/− cells and that blocked death (Fig. 4A,B) suggested that increased ROS in NGF-deprived mouse sympathetic neurons was critical for cytochromec release and death. To more directly determine the effect of a pro-oxidant state on cytochrome c release and death, we treated cells with H2O2. Figure 5A shows the effects of different concentrations of H2O2 on CM-H2DCFDA intensities in NGF-maintainedbax+/+,bax+/−, andbax−/− neurons and on CM-H2DCFDA intensities in NGF-deprivedbax−/− cells. There were no differences in the effects of H2O2 on the CM-H2DCFDA intensities in any of these conditions (p > 0.1). These data further suggest that there were no Bax-induced changes in GSH concentration or in the activity of enzymes in the GSH pathway that are involved in detoxifying H2O2.
Fig. 5.
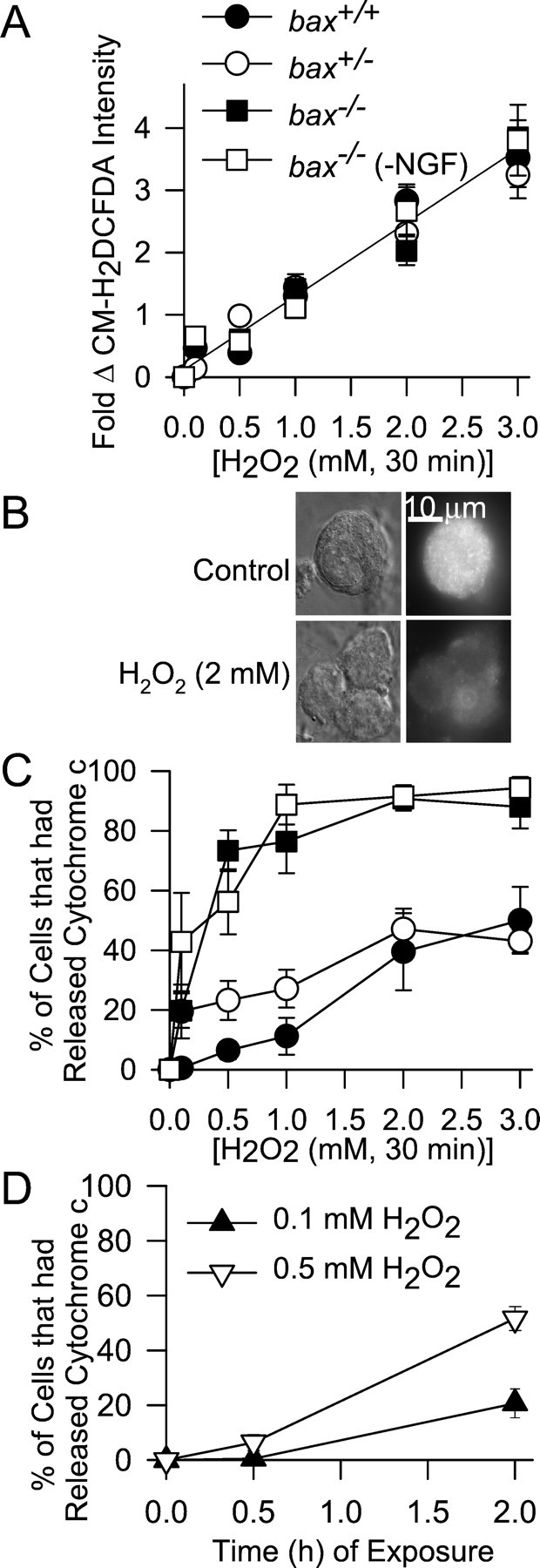
Increasing ROS with H2O2causes cytochrome c release independent of Bax.A, Effect of different concentrations of H2O2 on ROS levels (CM-H2DCFDA intensity) in NGF-maintained neurons from the three baxgenotypes and on ROS levels inbax−/− neurons deprived of NGF for 18 hr. After cells were loaded with CM-H2DCFDA, the dye was washed out, and cultures were exposed to H2O2 in the incubator for 20 min. They were then washed three times with L-15 medium and left in the second wash for confocal microscopy. The line shows a linear regression of all the data (R > 0.99).n = 45–128 neurons. B, Cytochromec was lost frombax−/− neurons after H2O2 exposure. Top panels, Paired DIC and fluorescent micrographs of a control (+NGF)bax−/− cell immunostained for cytochrome c. Bottom panels, Similar cells after treatment with H2O2 (2 mm). Cultures for were exposed to H2O2 in NGF-containing medium for 30 min. They were then washed twice with NGF-containing medium without H2O2 and returned to the incubator for 30 min before processing for immunocytochemistry. Western blots of total cytochrome c (mitochondrial and cytoplasmic) and tubulin in cultures exposed to H2O2 confirmed loss of cytochrome c without tubulin loss (data not shown).C, Effect of different concentrations of H2O2 on cytochrome c release in NGF-maintained neurons of the three bax genotypes and onbax−/− neurons deprived of NGF for 18 hr. Cells were exposed to the indicated H2O2concentrations for 30 min, washed two times with culture medium containing or lacking NGF, as appropriate, and returned to the incubator for another 30 min to assure degradation of released cytochrome c (Deshmukh et al., 1998; Neame et al., 1998; Kirkland and Franklin, 2001). Release was determined as in Figure 2. Symbols have the same meaning as inA. Data are from 3–11 cultures. D, Time-dependent effect of H2O2 on cytochromec release. Concentrations of H2O2 that had little effect on cytochromec redistribution after 30 min of exposure caused release when neurons (bax+/+) were treated with these concentrations for longer periods. H2O2 was replenished every 30 min. Data are from three to five cultures. The cytochrome c status of 50–100 neurons was determined for each culture in C andD.
We next treated cultures of the three genotypes with the same concentrations of H2O2 and determined cytochrome c status by immunocytochemistry. In all genotypes, 30 min of treatment with ≥0.5 mmH2O2 caused redistribution of cytochrome c from mitochondria into the cytoplasm (Fig.5B,C). Approximately 45% of both NGF-maintainedbax+/+ andbax+/− cells exposed to 3 mmH2O2 for 30 min had released cytochrome c from mitochondria into the cytoplasm. Surprisingly, all concentrations of H2O2 tested were more effective at causing cytochrome c release in NGF-maintained or NGF-deprived bax−/− cells than in the other genotypes. Concentrations of H2O2 ≥1 mm caused release of cytochrome c in 80–90% of NGF-maintained or -deprivedbax−/− cells. When cells were exposed to H2O2 for longer periods, cytochrome c was released at even lower concentrations (Fig. 5D). These data demonstrate that a pro-oxidant state induced cytochrome c release in these neurons. Moreover, the data show that the levels of ROS associated with the redistribution were similar to those occurring in NGF-deprived neurons (Fig. 2A), suggesting that ROS reached sufficient concentrations after NGF withdrawal to cause cytochromec release (Fig. 2A,B).
To determine whether H2O2induced redistribution of cytochrome c by causing nonspecific disruption of mitochondria, we exposed cells treated with H2O2 to Mitotracker Red CMXRos, a dye that concentrates in mitochondria having a membrane potential (Δψm; Neame et al., 1998). Figure6A shows that Mitotracker Red CMXRos intensely stained abax−/− neuron treated with H2O2 (2 mm for 30 min followed by 30 min recovery in H2O2-free medium). Moreover, this staining was punctate, consistent with mitochondrial localization of the dye. Ninety-nine percent ofbax−/− cells treated in this manner exhibited intense, punctate Mitotracker Red CMXRos staining (n = 144 cells). Similar results were observed in H2O2-treatedbax+/− andbax+/+ cells. Thus, mitochondria retained Δψm after H2O2 exposure, suggesting they were not significantly damaged. Further evidence that mitochondria were not damaged by H2O2exposure is shown in Figure 6B. Cells that had lost cytochrome c after transient exposure to H2O2 (2 mm for 30 min) became reconstituted with cytochrome c after 4 d of exposure to NGF-containing medium. One hundred percent ofbax+/+ cells (n = 130 neurons from 2 platings) and 99% ofbax−/− cells (n = 138 neurons from 2 platings) exhibited intense, punctate cytochromec staining after this treatment. Therefore, mitochondrial cytochrome c stores recovered from H2O2 exposure. Electron micrographs of mitochondria inbax−/− cells treated with H2O2 (2 mm for 30 min) appeared indistinguishable from mitochondria in control cells (Fig. 6C) (n = 20 mitochondria in 20 cells). Therefore, the concentrations of H2O2 that caused cytochromec release did not cause any obvious damage to mitochondria that could explain how it induced cytochrome credistribution. Deshmukh and Johnson (1998) reported that mouse sympathetic neurons must be deprived of NGF for several hours before cytoplasmic cytochrome c can induce apoptosis. Figure6D shows that although H2O2 caused rapid release of cytochrome c in NGF-maintained cells of the threebax genotypes, it did not kill them. However, ∼40% ofbax−/− neurons that had been deprived of NGF for 18 hr were killed by H2O2 treatment (Fig.6D). A similar number of cells were reported byDeshmukh and Johnson (1998) to be competent to die at this time.
Fig. 6.
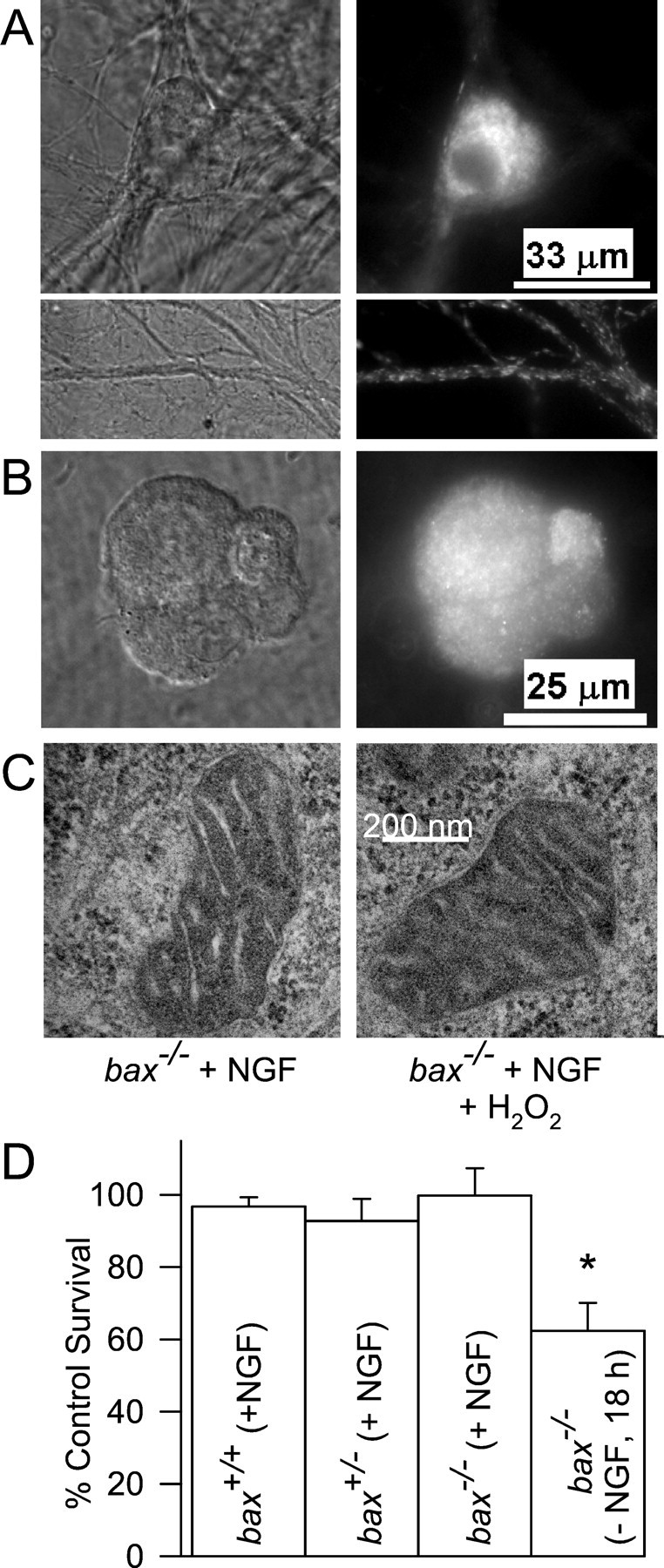
H2O2 treatment does not damage mitochondria. A, Treatment with H2O2 did not cause loss of Δψm. Shown are paired DIC and fluorescent micrographs of NGF-maintained cells (bax−/−) exposed to the Δψm-sensitive dye Mitotracker Red CMXRos (5 μm for 20 min in NGF-containing medium) after 30 min of H2O2 (2 mm) treatment followed by two washes. The intense punctate staining in the soma and neurites indicates retention of Δψm. B, Mitochondria in neurons depleted of cytochrome c by H2O2 treatment (2 mm for 30 min) became reconstituted with cytochrome c several days after NGF readdition. Shown are paired DIC and fluorescent micrographs of bax+/+ cells exposed for 30 min to H2O2 (2 mm), washed twice, and then maintained for 5 d in NGF-containing medium without H2O2. Immunocytochemical staining is for cytochrome c. bax−/−neurons treated in the same manner were visually indistinguishable from the bax+/+ cells shown.C, Electron micrographs of mitochondria in NGF-maintained neurons (bax−/−) and in NGF-maintained neurons (bax−/−) that had been treated for 30 min with H2O2 (2 mm). No morphological differences were noted.D, H2O2 induced death only inbax−/− neurons that had been deprived of NGF. NGF-maintained or -deprived neurons of the indicated genotypes were exposed to H2O2 (2 mm for 30 min). Cultures were then returned to NGF-containing medium without H2O2 for 3–5 d. Only the survival of NGF-deprived, bax−/− neurons was significantly lower than that of NGF-maintained cells (p< 0.001). Survival was normalized to that of untreated sibling cultures of NGF-maintained cells from the same mouse pup.n = 6–13 cultures.
Bax is important for the PCD of many types of neurons in the developing nervous system (White et al., 1998). To determine whether Bax affected cellular ROS levels during the PCD of neurons other than NGF-deprived sympathetic neurons, we used CM-H2DCFDA to measure ROS in cerebellar granule neurons induced to undergo apoptosis by membrane potential repolarization and serum withdrawal (Miller and Johnson, 1996). Cerebellar granule neurons frombax−/− mice do not die when treated in this manner, whereas those frombax+/+ animals undergo apoptosis (Miller et al., 1997). A ROS burst that occurs during the apoptotic death of bax+/+ cerebellar granule cells has been implicated in their death. As reported previously (Schulz et al., 1996), we found that cerebellar granule neurons frombax+/+ mice had increased ROS levels during apoptosis. The peak CM-H2DCFDA intensity occurred 9 hr after repolarization (replacing 25 mm KCl in the culture medium with 5 mm KCl) and serum withdrawal. There was no increase of CM-H2DCFDA intensity in cerebellar granule neurons from bax−/− mice (Fig. 7). Both depolarizedbax+/+ control cells maintained in high-potassium medium with serum andbax−/− neurons deprived of serum and repolarized for 12 hr by switching to low-potassium medium loaded equivalent amounts of CM-H2DCFDA (8.16 ± 2.6-fold and 7.85 ± 2.7-fold intensity increases, respectively, after 5 min exposure to 10 mmH2O2; p > 0.4; n = 77–100 neurons). Therefore, Bax also controls the ROS burst occurring during the apoptotic death of cerebellar granule cells, suggesting a general role for Bax in controlling the cellular redox state during neuronal apoptosis.
Fig. 7.

Bax deletion prevents increased ROS (CM-H2DCFDA intensity) during the PCD of cerebellar granule neurons. Apoptosis was induced by repolarization of membrane potential and serum withdrawal. Potential was repolarized by switching from a culture medium containing 25 mm KCl to one with 5 mm KCl. The method of CM-H2DCFDA loading and measurement was the same as for sympathetic neurons.n = 83–273 cells. Cells were from three platings, except for the bax−/− 6 and 12 hr time points, which were from two platings.
DISCUSSION
We conducted a study of the effect of Bax on the cellular redox state during the PCD of NGF-deprived mouse sympathetic neurons. Elevated ROS occurred in these cells within 6 hr of NGF withdrawal. ROS levels continued to increase, reaching a peak by 18 hr after deprivation and remaining at this level for the duration of PCD. The ROS burst was absent in neurons lacking Bax and was of intermediate amplitude in cells with one-half the normal Bax concentration. A similar ROS burst was absent in the PCD of cerebellar granule cells lacking Bax. Mimicking elevated ROS with H2O2 caused release of cytochrome c from mitochondria even in sympathetic neurons lacking Bax. Elevating the cellular GSH concentration inhibited both the elevated ROS and PCD. These findings suggest a central role for the Bax-mediated ROS burst in cytochrome crelease.
The role of Bax in ROS production
Depleting the mitochondrial electron transport chain of cytochromec increases leakage of electrons to O2to form O2·− (Turrens, 1997; Cai and Jones, 1998). Dismutation of the O2·− then forms H2O2, which can oxidize CM-H2DCFDA (Royall and Ischiropoulos, 1993). One possible explanation for how Bax caused elevated ROS after NGF deprivation was that O2·− production increased subsequent to Bax-induced cytochrome c redistribution. However, the ROS increase in NGF-deprived mouse sympathetic neurons occurred before cytochrome c release. Therefore, Bax-induced depletion of cytochrome c from the mitochondrial electron transport chain cannot explain how Bax increased ROS.
Rotenone, an inhibitor of mitochondrial respiratory complex I, greatly diminished the ROS burst after NGF withdrawal, suggesting that the ROS derived from the electron transport chain and that the electrons giving rise to the ROS entered the chain at complex I. Antimycin A, an inhibitor of mitochondrial respiratory complex III, caused a similar increase of ROS in NGF-maintained neurons of each baxgenotype, suggesting that there were no differences in the ability of their mitochondria to produce ROS. Antimycin A had no effect on ROS levels in NGF-deprived neurons frombax+/+ mice, possibly indicating that ROS production was maximal in these cells and could not be further increased. Antimycin A treatment elevated ROS in NGF-deprived neurons from bax+/− andbax−/− animals to concentrations similar to those measured in neurons from NGF-deprivedbax+/+ mice, also suggesting that there were no differences in the abilities of mitochondria in the three genotypes to produce ROS. Therefore, variation in the ROS levels of sympathetic neurons from bax+/+,bax+/−, andbax−/− mice after NGF withdrawal cannot be explained by differences in efficacy of mitochondrial ROS production. Glutathione levels were similar in cells of the three genotypes, suggesting there were no differences in their abilities to regulate ROS levels via the major cellular pathway for detoxifying H2O2. The CM-H2DCFDA intensities in cells from the three genotypes that were treated with the same concentrations of H2O2 increased to the same level, further suggesting that the cellular ability to detoxify H2O2 by the GSH and other pathways was not altered by Bax dosage.
The time course of ROS increase after NGF withdrawal was similar to that reported for Bax translocation to the mitochondria in these cells (Putcha et al., 1999) and the magnitude of the ROS burst was linearly related to Bax concentration. These data suggest that elevation of OMM Bax levels caused by ongoing Bax translocation was responsible for the time course of ROS increase after NGF withdrawal. Little is known about how Bax could cause mitochondria to increase production of ROS. Expression of Bax in yeast is toxic, although yeast lack molecular components of the apoptotic cascade (Gross et al., 2000; Harris et al., 2000). In some yeast strains, increased ROS appears critical for Bax-induced death (Gross et al., 2000). One suggestion for how Bax exerts its influence in both yeast and mammalian cells is that it restricts diffusion of compounds across the OMM that are required for respiration (Harris et al., 2000). Such an effect could, ostensibly, lead to increased ROS by limiting respiratory chain substrates. Another possibility is that Bax more directly regulates electron flow in mitochondria by interacting with respiratory complexes. However, there is currently no evidence for such an interaction. Finally, Bcl-2 acts as an antioxidant in some cells (Hockenbery et al., 1993; Kane et al., 1993) possibly by increasing cellular antioxidant capacity (Ellerby et al., 1996). Although it is possible that Bax blocks the antioxidant effects of antiapoptotic Bcl-2 family proteins and, secondarily, elevates ROS, the yeast data clearly demonstrate that Bax can increase ROS in cells lacking Bcl-2 family members.
Role of Bax and ROS in cytochrome c release
Early work demonstrating that Bcl-2 decreased cellular ROS or the effects of ROS during apoptosis led to the suggestion that a major pathway by which Bcl-2 family proteins regulate apoptotic death is by alteration of the cellular redox state (Hockenbery et al., 1993; Kane et al., 1993). This work was subsequently challenged by Shimizu et al. (1995) and Jacobson and Raff (1995), who showed that expression of Bcl-2 could slow death of several cell types even in the absence of molecular oxygen, a condition in which ROS should be markedly reduced. These authors concluded that ROS are not required for the survival-promoting effects of Bcl-2. However, later studies demonstrated that Bcl-2 has multiple sites of action and can block apoptosis both by inhibition of cytochrome c release and downstream from cytochrome c release (Clem et al., 1998;Rossé et al., 1998; Krebs et al., 1999). Therefore, the role of ROS regulation by Bcl-2 family members in apoptosis has not yet been fully elucidated. This report, as well as other recent work (Kirkland and Franklin, 2001; Madesh and Hajnóczky, 2001), suggests that alteration of cellular ROS levels by Bcl-2 family members regulates cytochrome c redistribution and is, therefore, a central component of the apoptotic cascade.
A major hypothesis for how proteins exit mitochondria during apoptosis involves opening of the permeability transition pore (PTP) of the inner mitochondrial membrane. This opening can cause mitochondria to swell and rupture the OMM, releasing intermembrane space proteins into the cytoplasm (Zamzami and Kroemer, 2001). The PTP can be regulated by ROS. However, in most cells, including NGF-deprived sympathetic neurons, mitochondrial swelling does not occur during apoptosis (Martinou et al., 1999; Von Ahsen et al., 2000). Therefore, it is unlikely that PTP is a major factor in most forms of apoptotic death. The proposed mechanism for release best supported by evidence is that an OMM pore forms during apoptosis and serves as the exit route for apoptogenic factors. Bax is a channel-forming protein (Schlesinger et al., 1997;Harris and Thompson, 2000; Antonsson et al., 2001). It can induce a high-conductance OMM channel that may be large enough to allow passage of cytochrome c and other proteins from the intermembrane space into the cytoplasm (Pavlov et al., 2001). However, the conductance of these channels is much higher than those produced by Bax in lipid bilayers, suggesting that Bax may regulate opening of pores in other OMM proteins. One OMM channel that has been implicated in cytochrome c release and that can be regulated by Bcl-2 family members is the voltage-dependent anion conductance (VDAC;Shimizu et al., 2000; Tsujimoto and Shimizu, 2000). Madesh and Hajnóczky (2001) reported that O2·− but not H2O2 induces VDAC-dependent permeabilization of the OMM in HepG2 cells independent of Bcl-2 family proteins. We found that H2O2 induced cytochromec release in sympathetic neurons, indicating that lack of a role for H2O2 in release cannot be generalized to all cell types. Both findings are consistent with the general idea that a pro-oxidant state can cause efflux of cytochrome c from mitochondria. Arguing against a role for VDAC in release, Pavlov et al. (2001) found that Bax induces a high conductance channel in yeast mitochondria lacking VDAC similar to the channel it induces in mitochondria containing VDAC. Therefore, at present, there is no clear answer as to what the actual OMM exit channel(s) is or how it could be regulated by ROS.
In conclusion, the findings reported here demonstrate that Bax caused a pro-oxidant state in mouse sympathetic and cerebellar granule neurons undergoing PCD. This pro-oxidant state was critical for Bax-induced cytochrome c release. We propose that a major mechanism by which Bax causes cytochrome c redistribution is by increasing production of ROS by mitochondria. These ROS then act as messenger molecules that regulate opening of a pathway(s) for passage of cytochrome c and, perhaps, other proteins across the OMM. These findings, along with earlier studies showing a role for superoxide in the apoptotic death of neurons (Greenlund et., 1995;Rabizadeh et al., 1995), suggest that oxidative stress is a central component of the molecular cascade leading to neuronal apoptosis.
Footnotes
This work was supported by National Institutes of Health Grant NS37110. We thank Dr. Peter Lipton for reviewing this manuscript.
Correspondence should be addressed to James L. Franklin, Department of Neurological Surgery, University of Wisconsin Medical School, 4640 Medical Sciences Center, 1300 University Avenue, Madison, WI 53706. E-mail: jlfrankl@facstaff.wisc.edu.
REFERENCES
- 1.Antonsson B, Montessuit S, Lauper S, Eskes R, Martinou JC. Bax oligomerization is required for channel-forming activity in liposomes and to trigger cytochrome c release from mitochondria. Biochem J. 2001;345:271–278. [PMC free article] [PubMed] [Google Scholar]
- 2.Cai J, Jones DP. Superoxide in apoptosis: mitochondrial generation triggered by cytochrome c loss. J Biol Chem. 1998;273:11401–11404. doi: 10.1074/jbc.273.19.11401. [DOI] [PubMed] [Google Scholar]
- 3.Clem RJ, Cheng EHY, Karp CL, Kirsch DG, Ueno K, Takahashi A, Kastan MB, Griffin DE, Earnshaw WC, Veliuona MA, Hardwick JM. Modulation of cell death by Bcl-xL through caspase interaction. Proc Natl Acad Sci USA. 1998;95:554–559. doi: 10.1073/pnas.95.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deckwerth TL, Johnson EM., Jr Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J Cell Biol. 1993;123:1207–1222. doi: 10.1083/jcb.123.5.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Deckwerth TL, Elliot JL, Knudson CM, Johnson EM, Jr, Snider WD, Korsmeyer SJ. Bax is required for neuronal death after trophic factor deprivation and during development. Neuron. 1996;17:401–411. doi: 10.1016/s0896-6273(00)80173-7. [DOI] [PubMed] [Google Scholar]
- 6.Deshmukh M, Johnson EM., Jr Evidence of a novel event during neuronal death: development of competence-to-die in response to cytoplasmic cytochrome c. Neuron. 1998;21:695–705. doi: 10.1016/s0896-6273(00)80587-5. [DOI] [PubMed] [Google Scholar]
- 7.Deshmukh M, Vasilakos J, Deckwerth TL, Lampe PA, Shivers BD, Johnson EM., Jr Genetic and metabolic status of NGF-deprived sympathetic neurons saved by an inhibitor of ICE-family proteases. J Cell Biol. 1996;135:1341–1354. doi: 10.1083/jcb.135.5.1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D'Mello SR, Borodezt K, Soltoff SP. Insulin-like growth factor and potassium depolarization maintain neuronal survival by distinct pathways: possible involvement of PI-3 kinase in IGF-1 signaling. J Neurosci. 1997;17:1548–1560. doi: 10.1523/JNEUROSCI.17-05-01548.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Du C, Fang M, Li Y, Li L, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 10.Dugan LL, Creedon DJ, Johnson EM, Jr, Holtzman DM. Rapid suppression of free radical formation by nerve growth factor involves the mitogen-activated protein kinase pathway. Proc Natl Acad Sci USA. 1997;94:4086–4091. doi: 10.1073/pnas.94.8.4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Easton RM, Deckwerth TL, Parsadanian AS, Johnson EM., Jr Analysis of the mechanism of loss of trophic factor dependence associated with neuronal maturation: a phenotype indistinguishable from Bax deletion. J Neurosci. 1997;17:9656–9666. doi: 10.1523/JNEUROSCI.17-24-09656.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ellerby LM, Ellerby HM, Park SM, Holleran AL, Murphy AN, Fiskum G, Kane DJ, Testa MP, Kayalar C, Bredesen DE. Shift of the cellular oxidation-reduction potential in neural cells expressing Bcl-2. J Neurochem. 1996;67:1259–1267. doi: 10.1046/j.1471-4159.1996.67031259.x. [DOI] [PubMed] [Google Scholar]
- 13.Fernandez-Checa JC, Kaplowitz N. The use of monochlorobimane to determine the hepatic GSH levels and synthesis. Anal Biochem. 1990;190:212–219. doi: 10.1016/0003-2697(90)90183-a. [DOI] [PubMed] [Google Scholar]
- 14.Franklin JL, Johnson EM., Jr Control of neuronal size homeostasis by trophic factor-mediated coupling of protein degradation to protein synthesis. J Cell Biol. 1998;142:1313–1324. doi: 10.1083/jcb.142.5.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Franklin JL, Sanz-Rodriguez C, Juhasz A, Deckwerth TL, Johnson EM., Jr Chronic depolarization prevents programmed death of sympathetic neurons in vitro but does not support growth: requirement for Ca2+ influx but not Trk activation. J Neurosci. 1995;15:643–664. doi: 10.1523/JNEUROSCI.15-01-00643.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Greenlund LJS, Deckwerth TL, Johnson EM., Jr Superoxide dismutase delays neuronal apoptosis: a role for reactive oxygen species in programmed neuronal death. Neuron. 1995;14:303–315. doi: 10.1016/0896-6273(95)90287-2. [DOI] [PubMed] [Google Scholar]
- 17.Gross A, Pilcher K, Blachly-Dyson E, Basso E, Jockel J, Bassik MC, Korsmeyer SJ, Forte M. Biochemical and genetic analysis of the mitochondrial response of yeast to Bax and Bcl-XL. Mol Cell Biol. 2000;20:3125–3136. doi: 10.1128/mcb.20.9.3125-3136.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Halliwell B, Gutteridge JMC. Free radicals in biology and medicine, Ed 3. Oxford UP; Oxford: 1999. [DOI] [PubMed] [Google Scholar]
- 19.Harris MH, Thompson CB. The role of the Bcl-2 family in the regulation of outer mitochondrial membrane permeability. Cell Death Differ. 2000;7:1182–1191. doi: 10.1038/sj.cdd.4400781. [DOI] [PubMed] [Google Scholar]
- 20.Harris MH, Vander Heiden MG, Kron SJ, Thompson CB. Role of oxidative phosphorylation in Bax toxicity. Mol Cell Biol. 2000;20:3590–3596. doi: 10.1128/mcb.20.10.3590-3596.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL, Korsmeyer SJ. Bcl-2 functions in an antioxidant pathway to prevent apoptosis. Cell. 1993;75:241–251. doi: 10.1016/0092-8674(93)80066-n. [DOI] [PubMed] [Google Scholar]
- 22.Jacobson MD, Raff MC. Programmed cell death and Bcl-2 protection in very low oxygen. Nature. 1995;374:814–816. doi: 10.1038/374814a0. [DOI] [PubMed] [Google Scholar]
- 23.Kane DJ, Sarafian TA, Anton R, Hahn H, Gralla EB, Valentine JS, Örd T, Bredesen DE. Bcl-2 inhibition of neural death: decreased generation of reactive oxygen species. Science. 1993;262:1274–1277. doi: 10.1126/science.8235659. [DOI] [PubMed] [Google Scholar]
- 24.Kirkland RA, Franklin JL. Evidence for redox regulation of cytochrome c release during programmed neuronal death: antioxidant effects of protein synthesis and caspase inhibition. J Neurosci. 2001;21:1949–1963. doi: 10.1523/JNEUROSCI.21-06-01949.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Knudson CM, Tung KSK, Tourtelotte WG, Brown GAJ, Korsmeyer SJ. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science. 1995;270:96–99. doi: 10.1126/science.270.5233.96. [DOI] [PubMed] [Google Scholar]
- 26.Krebs JF, Armstrong RC, Srinivasan A, Aja T, Wong AM, Aboy A, Sayers R, Pham B, Vu T, Hoang K, Karanewsky DS, Leist C, Schmitz A, Wu JC, Tomaselli KJ, Fritz LC. Activation of membrane-associated procaspase 3 is regulated by Bcl-2. J Cell Biol. 1999;144:915–926. doi: 10.1083/jcb.144.5.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412:95–99. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- 28.Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- 29.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 30.Madesh M, Hajnóczky G. VDAC-dependent permeabilization of the outer mitochondrial membrane by superoxide induces rapid and massive cytochrome c release. J Cell Biol. 2001;155:1003–1015. doi: 10.1083/jcb.200105057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin DP, Schmidt RE, DiStefano PS, Lowry OH, Carter JG, Johnson EM., Jr Inhibitors of protein synthesis and RNA synthesis prevent neuronal death caused by nerve growth factor deprivation. J Cell Biol. 1988;106:829–844. doi: 10.1083/jcb.106.3.829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinou I, Desagher S, Eskes R, Antonsson B, André E, Fakan S, Martinou JC. The release of cytochrome c from mitochondria during apoptosis of NGF-deprived sympathetic neurons is a reversible event. J Cell Biol. 1999;144:883–889. doi: 10.1083/jcb.144.5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miller TM, Johnson EM., Jr Metabolic and genetic analyses of apoptosis in potassium/serum-deprived rat cerebellar granule cells. J Neurosci. 1996;16:7487–7495. doi: 10.1523/JNEUROSCI.16-23-07487.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller TM, Moulder KL, Knudson CM, Creedon DJ, Deshmukh M, Korsmeyer SJ, Johnson EM., Jr Bax deletion further orders the cell death pathway in cerebellar granule cells and suggests a caspase-independent pathway to cell death. J Cell Biol. 1997;139:205–217. doi: 10.1083/jcb.139.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Neame SJ, Rubin LL, Philpott KL. Blocking cytochrome c activity within intact neurons inhibits apoptosis. J Cell Biol. 1998;142:1583–1593. doi: 10.1083/jcb.142.6.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Oppenheim RW. Cell death during development of the nervous system. Annu Rev Neurosci. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- 37.Pavlov EV, Priault M, Pietkiewicz D, Cheng EHY, Antonsson B, Manon S, Korsmeyer SJ, Mannella CA, Kinnally KW. A novel, high conductance channel of mitochondria linked to apoptosis in mammalian cells and Bax expression in yeast. J Cell Biol. 2001;155:725–731. doi: 10.1083/jcb.200107057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Putcha GV, Deshmukh M, Johnson EM., Jr Bax translocation is a critical event in neuronal apoptosis: regulation by neuroprotectants, Bcl-2, and caspases. J Neurosci. 1999;19:7476–7485. doi: 10.1523/JNEUROSCI.19-17-07476.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Putcha GV, Moulder KL, Golden JP, Bouillet P, Adams JA, Strasser A, Johnson EM., Jr Induction of BIM, a proapoptotic BH3-only Bcl-2 family member, is critical for neuronal apoptosis. Neuron. 2001;29:615–628. doi: 10.1016/s0896-6273(01)00238-0. [DOI] [PubMed] [Google Scholar]
- 40.Rabizadeh S, Gralla EB, Borchelt DR, Gwinn R, Valentine JS, Sisodia S, Wong P, Lee M, Hahn H, Bredesen DE. Mutations associated with amyotrophic lateral sclerosis convert superoxide dismutase from an antiapoptotic gene to a proapoptotic gene: studies in yeast and neural cells. Proc Natl Acad Sci USA. 1995;92:3024–3028. doi: 10.1073/pnas.92.7.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ratan RR, Murphy TH, Baraban JM. Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J Neurosci. 1994;14:4385–4392. doi: 10.1523/JNEUROSCI.14-07-04385.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rossé T, Olivier R, Monney L, Rager M, Conus S, Fellay I, Jansen B, Borner C. Bcl-2 prolongs cell survival after Bax-induced release of cytochrome c. Nature. 1998;391:496–499. doi: 10.1038/35160. [DOI] [PubMed] [Google Scholar]
- 43.Royall JA, Ischiropoulos H. Evaluation of 2′, 7′-dichlorofluorescein and dihydrorhodamine123 as fluorescent probes for intracellular H2O2 in cultured endothelial cells. Arch Biochem Biophys. 1993;302:348–355. doi: 10.1006/abbi.1993.1222. [DOI] [PubMed] [Google Scholar]
- 44.Schlesinger PH, Gross A, Yin X, Yamamoto K, Saito M, Waksman G, Korsmeyer SJ. Comparison of the ion channel characteristics of proapoptotic Bax and antiapoptotic BCL-2. Proc Natl Acad Sci USA. 1997;94:11357–11362. doi: 10.1073/pnas.94.21.11357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Schulz JB, Weller M, Klockgether T. Potassium deprivation-induced apoptosis of cerebellar granule neurons: a sequential requirement for new mRNA and protein synthesis, ICE-like protease activity, and reactive oxygen species. J Neurosci. 1996;15:4696–4706. doi: 10.1523/JNEUROSCI.16-15-04696.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shimizu S, Eguchi Y, Kosaka H, Kamiike W, Matsuda H, Tsujimoto Y. Prevention of hypoxia-induced cell death by Bcl-2 and Bcl-xL. Nature. 1995;374:811–813. doi: 10.1038/374811a0. [DOI] [PubMed] [Google Scholar]
- 47.Shimizu S, Ide T, Yanagida T, Tsujimoto Y. Electrophysiological study of a novel large pore formed by Bax and the voltage-dependent anion channel that is permeable to cytochrome c. J Biol Chem. 2000;275:12321–12325. doi: 10.1074/jbc.275.16.12321. [DOI] [PubMed] [Google Scholar]
- 48.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 49.Tan S, Sagara Y, Liu Y, Maher P, Schubert D. The regulation of reactive oxygen species production during programmed cell death. J Cell Biol. 1998;141:1423–1432. doi: 10.1083/jcb.141.6.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tsujimoto Y, Shimizu S. VDAC regulation by the Bcl-2 family of proteins. Cell Death Differ. 2000;7:1174–1181. doi: 10.1038/sj.cdd.4400780. [DOI] [PubMed] [Google Scholar]
- 51.Turrens JF. Superoxide production by the mitochondrial respiratory chain. Biosci Rep. 1997;17:3–8. doi: 10.1023/a:1027374931887. [DOI] [PubMed] [Google Scholar]
- 52.Von Ahsen O, Renken C, Perkins G, Kluck RM, Bossy-Wetzel E, Newmeyer DD. Preservation of mitochondrial structure and function after Bid- or Bax-mediated cyto c release. J Cell Biol. 2000;150:1027–1036. doi: 10.1083/jcb.150.5.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.White FA, Keller-Peck CR, Knudson CM, Korsmeyer SJ, Snider WD. Widespread elimination of naturally occurring neuronal death in Bax-deficient mice. J Neurosci. 1998;18:1428–1439. doi: 10.1523/JNEUROSCI.18-04-01428.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wolter KG, Hsu YT, Smith CL, Nechushtan A, Xi XG, Youle RJ. Movement of Bax from the cytosol to mitochondria during apoptosis. J Cell Biol. 1997;139:1281–1292. doi: 10.1083/jcb.139.5.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yuan J, Yanker BA. Apoptosis in the nervous system. Nature. 2001;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- 56.Zamzami N, Kroemer G. The mitochondrion in apoptosis: how Pandora's box opens. Nat Rev Mol Cell Biol. 2001;2:67–71. doi: 10.1038/35048073. [DOI] [PubMed] [Google Scholar]
- 57.Zou H, Henzel WJ, Liu X, Lutschg A, Wang X. Apaf-1, a human protein homologous to C. elegans Ced-4, participates in cytochrome c-dependent activation of caspase-3. Cell. 1997;90:405–413. doi: 10.1016/s0092-8674(00)80501-2. [DOI] [PubMed] [Google Scholar]