

Abstract
NPC-1 gene mutations cause Niemann–Pick type C (NPC), a neurodegenerative storage disease resulting in premature death in humans. Spontaneous mutation of the NPC-1 gene in mice generates a similar phenotype, usually with death ensuing by 12 weeks of age. Both human and murine NPC are characterized neuropathologically by ballooned neurons distended with lipid storage, axonal spheroid formation, demyelination, and widespread neuronal loss. To elucidate the biochemical mechanism underlying this neuropathology, we have investigated the phosphorylation of neuronal cytoskeletal proteins in the brains of npc-1 mice. A spectrum of antibodies against phosphorylated epitopes in neurofilaments (NFs) and MAP2 and tau were used in immunohistochemical and immunoblotting analyses of 4- to 12-week-old mice. Multiple sites in NFs, MAP2, and tau were hyperphosphorylated as early as 4 weeks of age and correlated with a significant increase in activity of the cyclin-dependent kinase 5 (cdk5) and accumulation of its more potent activator, p25, a proteolytic fragment of p35. At 5 weeks of age, the development of axonal spheroids was noted in the pons. p25 and cdk5 coaccumulated with hyperphosphorylated cytoskeletal proteins in axon spheroids. These various abnormalities escalated with each additional week of age, spreading to other regions of the brainstem, basal ganglia, cerebellum, and eventually, the cortex. Our data suggest that focal deregulation of cdk5/p25 in axons leads to cytoskeletal abnormalities and eventual neurodegeneration in NPC. The npc-1 mouse is a valuablein vivo model for determining how and when cdk5 becomes deregulated and whether cdk5 inhibitors would be useful in blocking NPC neurodegeneration.
Keywords: cdk5, p35, neurodegeneration, Niemann–Pick disease type C, cholesterol, axon spheroid, lipid rafts, caveolas, neurofilament phosphorylation, tau phosphorylation, cytoskeletal pathology
Niemann–Pick type C disease (NPC) is a rare, autosomal recessive, fatal, lysosomal lipidosis affecting multiple organs (Vanier et al., 1991a,b; Scriver et al., 2001). The disease is caused predominantly by mutations in the NPC-1gene and less frequently in the HE1 (also referred to asNPC-2) gene (Naureckiene et al., 2000; Millat et al., 2001;Scriver et al., 2001). The NPC-1 gene encodes for a cholesterol transporter in late endosomes, and the HE1 gene encodes for a lysosomal cholesterol-binding protein. Neuropathologically, NPC is characterized by neurons distended with lipid storage material having a foamy appearance, dendritic and axonal abnormalities, demyelination, and widespread neuronal loss (Elleder et al., 1985; Love et al., 1995; Suzuki et al., 1995). In addition, neurofibrillary tangles (NFTs), a diagnostic lesion of Alzheimer's disease (AD), are also a consistent finding, particularly in cases with a prolonged course of disease (Auer et al., 1995; Love et al., 1995;Suzuki et al., 1995) (H. H. Klünemann, B. Bu, J. Husseman, M. Elleder, K. Suzuki, S. Salamant, S. Love, H. Budka, C. Fligner, T. Bird, L.-W. Jin, D. Nochlin, and I. Vincent, unpublished observations). How these various neuropathologic features result from altered cholesterol metabolism in NPC is a mystery and a rather difficult one to resolve given the rarity of the disease.
A tremendous asset for unraveling the neuropathologic effects ofNPC-1 mutations is the BALB/cNpc-1nih mouse, which harbors a spontaneous mutation in its npc-1 gene (Loftus et al., 1997). Mice with homozygous npc-1 mutations (npc-1 mice) display extensive lipid storage accumulation, neuroaxonal dystrophy, and neuronal loss, similar to that of human NPC (Higashi et al., 1993; Suzuki et al., 1995; Sawamura et al., 2001). Cholesterol (Xie et al., 1999; Sawamura et al., 2001) and glycosphingolipids such as gangliosides GM2 and neutral glycolipids (Walkley, 1995; Zervas et al., 2001) are the predominant constituents of storage material in the npc-1 mouse brain. Curiously, however, neither alleviation of cholesterol (Patterson et al., 1993; Erickson et al., 2000; Camargo et al., 2001) nor ganglioside storage (Liu et al., 2000) ameliorate the neurological phenotype or progressive neuronal loss in npc-1 mice or feline NPC, although lipid storage was effectively reduced in neurons and other cells. Thus, it is yet unclear what mechanism underlies neuronal dysfunction and loss of neurons in NPC. A notable difference between the npc-1 mouse and human NPC is the absence of NFTs in the mouse (German et al., 2001a; Sawamura et al., 2001). However, in light of the conspicuous axonal abnormalities in human, murine, and feline NPC (Elleder et al., 1985; Higashi et al., 1993; Ong et al., 2001), we wondered whether cytoskeletal abnormalities contribute to neuronal dysfunction and degeneration in NPC. Therefore, we have undertaken a detailed characterization of cytoskeletal protein phosphorylation in the brains of npc-1 mice.
MATERIALS AND METHODS
All procedures in this study were approved by the Internal Review Board and Animal Use and Care Committee of the University of Washington.
Npc-1 mice. A breeding pair of heterozygousnpc-1 mice obtained from The Jackson Laboratory (Bar Harbor, ME) was bred to generate wild-type (+/+), heterozygous (npc-1 +/−), and homozygous (npc-1 −/−) mice, which were identified using an established PCR-based method (Loftus et al., 1997). Tail biopsies for genotyping were performed at the time of weaning (i.e., at ∼3 weeks). Only −/− mice have been reported to display pathology (Tanaka et al., 1988). In initial studies, we screened +/− mice and confirmed the absence of cytoskeletal pathology in this genotype. Hence, all further study concentrated on comparisons of −/− mice with +/+ siblings. Twenty-eight npc-1 −/− mice (4, 5, 7, and 9 weeks of age, n = 3; 6 and 8 weeks of age, n = 5; 10, 11, and 12 weeks of age,n = 2) and a minimum of two age-matched (for each week), wild-type littermates were analyzed by immunohistochemistry and immunoblotting.
Brain tissue. Mice were killed by carbon dioxide exposure followed by decapitation. The brains were removed quickly and divided sagittally into halves. The right halves were immersion fixed with 4% paraformaldehyde/PBS for 1 week and then embedded in paraffin. Where indicated, some mice were transcardially perfused with 4% paraformaldehyde/PBS, and the brain was then processed for paraffin embedding. The paraffin-embedded blocks were sectioned at 6 μm for histological analyses. The left halves were frozen at −80°C for biochemical study. In some cases, the forebrain, cerebellum, and brainstem were isolated and frozen separately for regional analysis.
Frozen hippocampus from a clinically and neuropathologically confirmed AD case was used in parallel with the mouse samples as a control for specificity of NFT antibodies.
Antibodies. The primary antibodies used in this study are summarized in Table 1.
Table 1.
Antibodies and specificities
| Antibody types | Specificity | Isotype | Source |
|---|---|---|---|
| Phosphotau | |||
| PHF-1 | Ser 396-404 | Mouse IgG1 | P. Davies |
| CP-13 | Ser 202 | Mouse IgG1 | P. Davies |
| CP-10 | Thr 231 | Mouse IgM | P. Davies |
| CP-22 | Ser 175 | Mouse IgM | P. Davies |
| MC-6 | Thr 235 | Mouse IgG1 | P. Davies |
| Sequence-tau | |||
| TG-5 | 220-240 | Mouse IgG1 | I. Vincent/P. Davies |
| ALZ-50 | 5-15, 312-322 | Mouse IgM | P. Davies |
| MC-1 | 5-15, 312-322 | Mouse IgG1 | P. Davies |
| Cytoskeletal | |||
| SMI 31 | Phospho-NF-H and -NF-M | Mouse IgG1 | Sternberger Monoclonals, Inc. (Lutherville, MD) |
| SMI 32 | Nonphospho-NF-H | Mouse IgG1 | Sternberger Monoclonals, Inc. |
| R39 | Total NF | Rabbit | H. C. Pant (National Institutes of Health/National Institute of Neurological Disorders and Stroke, Bethesda, MD) |
| AP-18 | Phospho-MAP2 | Mouse IgG1 | L. Binder (Northwestern University, Chicago, IL) |
| AP-20 | MAP2a + MAP2b + MAP2c | Mouse IgG1 | Sigma |
| Kinase | |||
| cdk5 (C-8) | cdk5 | Rabbit | Santa Cruz Biotechnologies (Santa Cruz, CA) |
| cdk5 (DC-17) | cdk5 | Mouse IgG1 | Santa Cruz Biotechnologies |
| p35 (C-19) | C-terminal p35 | Rabbit | Santa Cruz Biotechnologies |
| p35 (N-20) | N-terminal p35 | Rabbit | Santa Cruz Biotechnologies |
| Phospho-GSK-3β | phosphoSer9 of human GSK-3β | Rabbit | Cell Signaling Technology (Beverly, MA) |
| GSK-3β | N-end human GSK-3β | Rabbit | Cell Signaling Technology |
| Loading control | |||
| Anti-NeuN | NeuN | Mouse IgG1 | Chemicon International (Temecula, CA) |
Immunohistochemistry and immunofluorescent labeling.Immunohistochemical staining was performed on paraffin-embedded sections as described previously (Vincent et al., 1997, 1998) with modifications. Immersion-fixed sections were incubated with 88% formic acid for 7 min to enhance antigen recovery and washed three times for 15 min with Tris-buffered saline (in mm): 10 Tris-HCl, pH 7.4, and 150 NaCl, before the addition of primary antibody. Biotinylated, isotype-specific secondary antibodies followed by HRP-labeled streptavidin were used to detect primary antibody-specific binding, visualized using diaminobenzidine (DAB, brown). Sections were counterstained with hematoxylin (blue-purple).
Immunofluorescent labeling was conducted similarly (Vincent et al., 1998). For visualization of specific antibody binding, streptavidin-conjugated Cy-3 (red, 1:500) or Alexa Fluor 488 (green, 1:500) was used. Nuclei were counterstained in 1 μg/ml 4,6-diamino-2-phenyliodole (DAPI, blue) in distilled water for 30 sec at room temperature. Light and fluorescent micrographs were collected using a Nikon (Tokyo, Japan) Optiphot microscope connected to a computerized SPOT CCD camera (Diagnostic Instruments, Inc., Sterling Heights, MI).
Preparation of brain extracts. Frozen tissue was weighed and homogenized with a polytron in 10 volumes of ice-cold lysis buffer [in mm: 10 Tris-HCl, 150 NaCl, 20 NaF, 1 mm sodium vanadate, 2 ethylene glycol-bis (β-aminoethylether)-N,N,N′,N′-tetraacetic acid, 0.5% Triton X-100, and 0.1% SDS] and proteinase inhibitor cocktail (P-8340; Sigma, St. Louis, MO). The homogenates were aliquoted and stored at −80°C.
Immunoblotting analysis. Frozen aliquots were thawed and centrifuged at 12,000 × g at 4°C for 5 min, and the soluble fraction was used for immunoblotting. The protein content in supernatants was determined using a Bio-Rad (Hercules, CA) protein assay kit and a Microplate Reader (Molecular Devices, Sunnyvale, CA). The supernatants were then subjected to SDS-gel electrophoresis and immunoblotting analyses (Vincent et al., 1997). For the analyses of MAP2 and neurofilaments (NFs), 10 μg of protein was loaded per lane; for tau and cyclin-dependent kinase 5 (cdk5) studies, 40 μg of protein was loaded per lane. For heat-stable proteins, supernatants containing 30 μg of protein were boiled for 10 min at 100°C and then centrifuged. The supernatants containing the heat-soluble protein were resolved and immunoblotted according to routine procedures.
Immunoprecipitation. Protein (100 μg) from lysates in equal volume was immunoprecipitated with 2 μg of cdk5 polyclonal antibody as described previously (Vincent et al., 1997). The immunoprecipitates (IPs) were processed for immunoblotting (as above) or kinase assay as described below.
Kinase assay. The cdk5 IPs were washed twice with lysis buffer and once with kinase buffer (50 mm HEPES, pH 7.0, 10 mm MgCl2, 1 mm dithiothreitol, and 10 μm cold ATP), resuspended in 20 μl of kinase buffer containing 10 μg of histone H1 and 0.5 μCi of [γ-32P]ATP, and incubated at room temperature for 20 min. To stop the reaction, 5 μl of 5× sample buffer was added, and the samples were boiled for 5 min at 95°C. Samples were separated by SDS-PAGE. Resolved proteins in the gel were visualized with Coomassie blue dye, and the gels were then dried and exposed to film for autoradiography.
Densitometric analysis. ECL and autoradiographic films were scanned, the appropriate bands were outlined, and their densities were measured using NIH image software. Statistical differences were determined with Student's t test using Microsoft Excel (Seattle, WA)
RESULTS
Hyperphosphorylation and accumulation of neurofilament protein innpc-1 −/− mouse brain
The immunohistochemical staining with SMI 31 antibody was performed on immersion-fixed sagittal sections. Consistent with the expected localization of phosphorylated NFs (Julien and Mushynski, 1982), SMI 31 primarily stained axon tracts throughout the brain of wild-type (+/+) mice (Fig.1A,C,E). In npc-1 −/− mice, SMI 31 stained numerous spot-like structures in the same regions through which axons typically course (Fig.1B,D,F shown at higher magnification in Fig.1G,H, red, I,arrowhead). The abnormal spot-like structures were first noted in the pons at 5 weeks of age; they were present throughout the basal ganglia (Fig. 1B), brainstem (Fig.1D), and white matter of the cerebellum (data not shown) by 7–8 weeks of age but were rarely seen in the hippocampus (Fig. 1F, arrows). By 11–12 weeks of age, they were observed in small numbers in the cerebral cortex as well (data not shown). Although we did not directly analyze neuronal loss in this study, disruption of the continuity of the Purkinje neuron layer in the cerebellum was conspicuous at 6 weeks of age, and Purkinje cells were rarely present after 9–10 weeks of age (data not shown). To further characterize the spot-like structures, immunofluorescent labeling with SMI 31 (red) and nuclear counterstaining with DAPI (blue) was performed. The abnormally enlarged SMI 31-positive structures were devoid of nuclei (Fig. 1G). Occasional perikaryal staining was recognized by the presence of a nucleus within a larger-sized spot (Fig. 1H, arrow) relative to the more abundant smaller spot-like structures. Thus, the SMI 31-positive spot-like structures most likely represent the axonal spheroids described previously in human, murine, and feline NPC (Elleder et al., 1985; Higashi et al., 1993; Ong et al., 2001). These axonal spheroids often displayed a translucent core resembling lipid deposit (Fig.1I, arrowhead). In general, neuronal somata swollen with lipid-like material were unstained (Fig.1I, arrow). The SMI 32 antibody recognizing a nonphosphorylated epitope in 200 kDa high-molecular weight NF (NF-H) stained a few neuronal cell bodies in the brainstem and small fibers throughout the brain of +/+ mice (Fig.1J). In −/− mice, SMI 32 immunolabeled numerous axonal spheroids (Fig. 1K, arrows) with a regional distribution (shown for the pons only, Fig.1K) that was similar to those stained by SMI 31.
Fig. 1.
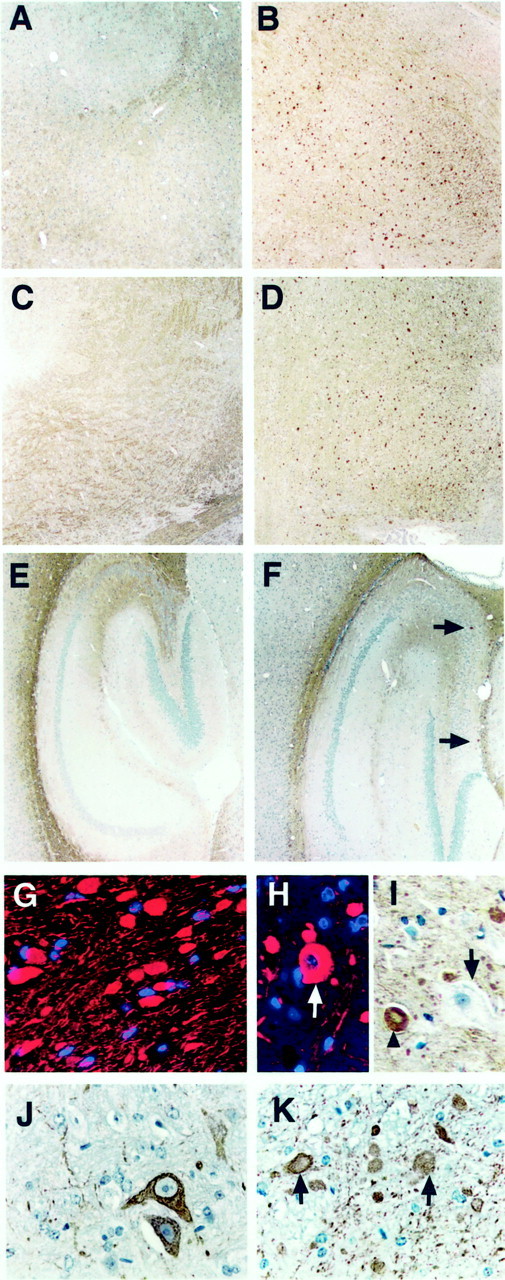
NF abnormalities in npc-1 −/− mice. Immersion-fixed, paraffin-embedded sagittal brain sections from 7-week-old +/+ (A, C, E, J) and −/− (B, D, F, G–I, K) mice were immunolabeled with antibodies SMI 31 (A–I) or SMI 32 (J, K). For some sections, antibody binding was visualized with DAB (brown) and hematoxylin counterstain for highlight ing the nuclei of all cells (blue-purple) or with Cy-3 (red) and DAPI counterstain for nuclei (blue). In the +/+ mouse, SMI 31 positively stained bundles of processes throughout the brain [shown for the basal ganglia (A), brainstem (C), and hippocampus (E)]. In sharp contrast, intense spheroid-like structures were observed in vast numbers in the basal ganglia (B) and brainstem (D) and sparsely in the hippocampus (F, arrows). The absence of nuclei within the spheroidal structures (G) and their size suggest that they are cross sections of swollen axons. Some rarer, larger spots containing a nucleus resembled perikarya (H, arrow in cortex). The gigantically enlarged neurons containing storage were not stained with SMI 31 (I, arrow), in contrast to axonal staining with a translucent core (I,arrowhead). SMI 32 stained a few large neurons and small fibers in the pons of the +/+ mouse (J) and axonal spheroids principally in the brainstem of −/− mice (K, arrows). Magnification:A–F, 4×; G–K, 40×.
To verify that the histological abnormalities were a result of increased phosphorylation and/or accumulation of NFs, immunoblotting analyses were conducted with whole-brain lysates from +/+ and −/− mice. Replicate blots were stained with SMI 31, SMI 32, and R39, a pan NF antibody recognizing all of the NF isoforms. The intensities of the NF-H, 160 kDa medium-molecular weight (NF-M), and 68 kDa low-molecular weight (NF-L) NF bands stained with R39 from four 8-week-old npc-1 mice and four controls were quantitated densitometrically, and the results indicated that the levels of the NF-H, NF-M, and NF-L were increased by 2.4-, 1.5-, and 1.17-fold (p < 0.03 for all), respectively, in thenpc-1 mice. These changes in NF levels were observed, whereas the levels of many other proteins, such as tau, MAP2, cdk5 (shown in Figs. 4 and 5), and neuronal-specific nuclear protein (NeuN) (Brazelton et al., 2000), in the same samples, were invariant. The results for NeuN are shown in Figure 2and indicate equal protein loading of all lanes. With respect to phosphorylation, a sixfold increase in SMI 31 immunoreactivity was observed with NF-M, but there was no significant increase in SMI31 immunoreactivity with NF-H (Fig. 2, SMI 31). These results suggest that NF-M is the only hyperphosphorylated NF isoform npc-1 mouse brain. They also imply that phosphorylation of NF-H did not increase in proportion to the increase in total NF-H levels, which corresponds to a net accumulation of nonphosphorylated NF-H in the diseased mice. This interpretation was supported by the marked increase in SMI 32 immunoreactivity with NF-H in the npc-2 mouse brain samples (Fig. 2, SMI 32) and the increased SMI 32 immunoreactivity with axonal spheroids (Fig. 1K). Neither SMI 31 nor SMI 32 stained any bands corresponding to tau, as has been observed in AD (Sternberger and Sternberger, 1983).
Fig. 4.
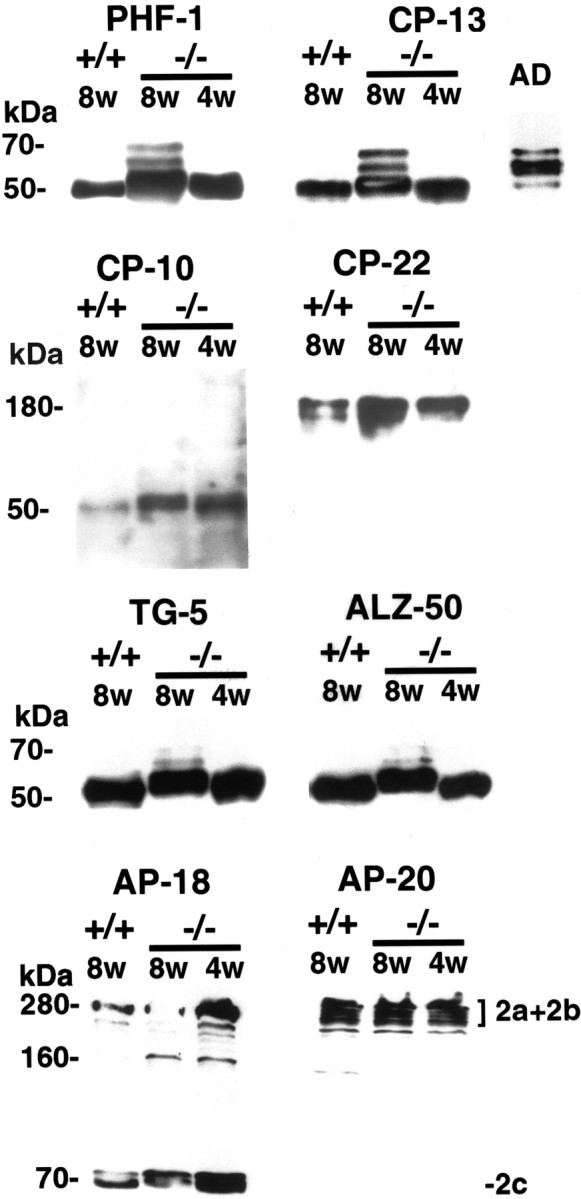
Increased phosphorylation of microtubule-associated proteins in npc-1 −/− mice. Ten micrograms of whole-brain, heat-stable protein from +/+ and −/− mice and a typical AD case were blotted with the indicated antibodies (supernatant only for AP-18). Ten and 8% gels were used for detection of tau and MAP2, respectively. An increase in intensity and in the apparent molecular weight of tau was observed with PHF-1 and CP-13 in 4-week-old and even more so in 8-week-old −/− mice, consistent with hyperphosphorylation of tau. The pattern of tau bands at 8 weeks of age resembled that of AD-tau. CP-10-detected phosphorylated tau was increased threefold in −/− mice. CP-22 raised against and shown to recognize phosphorylated tau did not react with tau in the BALB/c strain of mouse. However, the antibody detected a molecule of ∼180 kDa that was more highly phosphorylated in −/− mice. The tau sequence antibodies TG-5 and ALZ-50 displayed equivalent amounts of tau in alllanes. AP-18 immunoreactivity showed increased phosphorylation of the 280 kDa MAP2a and MAP2b and 70 kDa MAP2c protein −/− mice, particularly at 4 weeks of age. AP-20 did not recognize MAP2c but showed invariable amounts of total MAP2 (2a+2b) protein in all lanes.
Fig. 5.
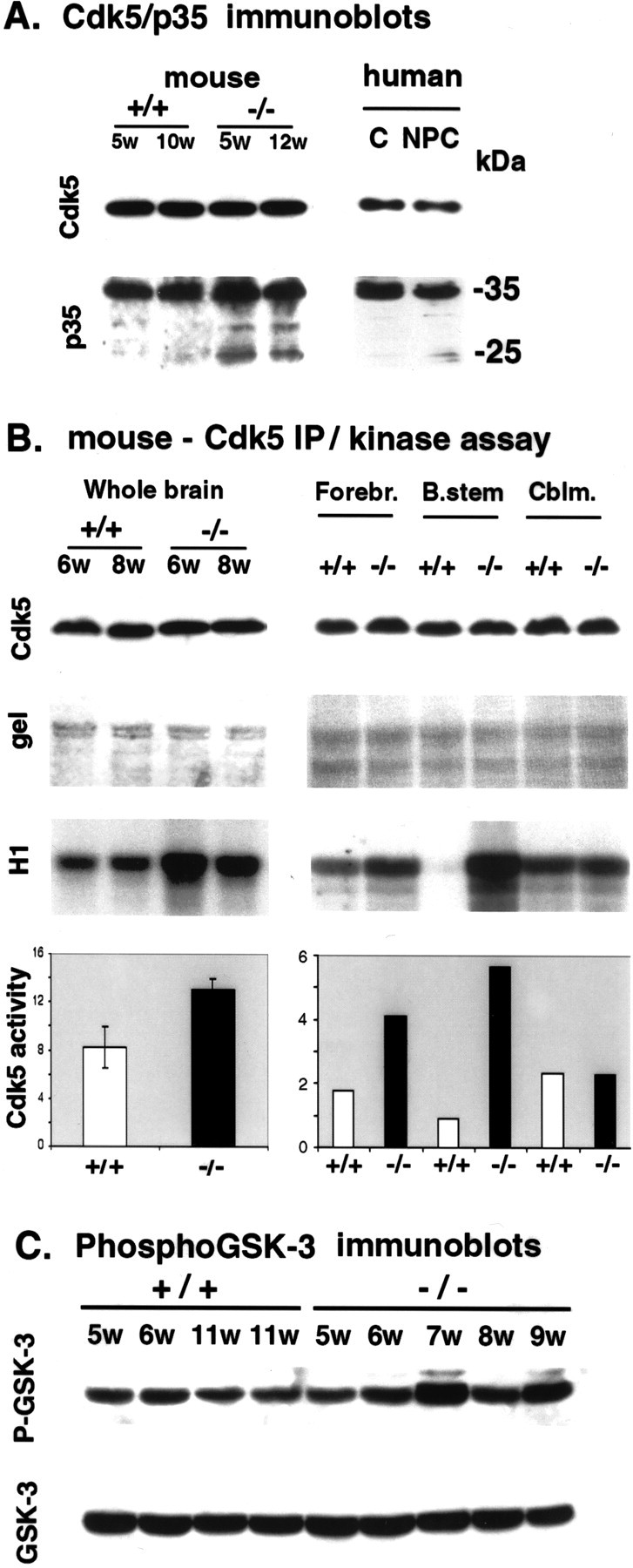
cdk5 and p25 abnormalities in thenpc-1 −/− mouse brain. A, cdk5/p35 immunoblots. Whole-brain lysates containing 40 μg of protein from +/+ and −/− mice were resolved on 12% gels and blotted with the indicated antibodies. cdk5 levels remained unchanged in −/− mice compared with controls. The p35 C-terminal antibody showed no difference in p35 levels but an enrichment of the 25 kDa p35 fragment (p25) in −/− mice (left panel, mouse). Similar preparations containing 100 μg of protein from a human control and an NPC hippocampus were immunoblotted with cdk5 and p35C antibodies. cdk5 and p35 levels were invariant, but p25 was elevated in NPC (right panel, human).B, cdk5 IP/kinase activity. Whole-brain lysates (left panel) from +/+ and −/− mice were immuno- precipitated with cdk5 (C-8) antibody. Immunoprecipitate (cdk5 IP) was immunoblotted with a different cdk5 antibody to show that the relative recoveries of enzyme in the samples were similar (Cdk5). Catalytic aliquots of cdk5 immunoprecipitate were incubated with kinase buffer containing histone H1 and [γ-32P]-ATP for 20 min at 30°C, and the reaction mixtures were resolved on 12% gels. The incorporation of [γ-32P]ATP into histone H1 (H1) was significantly higher with cdk5 IP from −/− mice compared with +/+ mice (left panel), on the basis of equivalent H1 loading demonstrated by staining of the gel with Coomassie blue (gel). The cdk5 activity in 5- to 10-week-old −/− mice was increased 1.5-fold over that seen in +/+ mice (p < 0.01; n = 12;left panel). Right panel, The increase in cdk5 activity in the forebrain (Forebr.) and brainstem (B.stem) but not in the cerebellum (Cblm.) of a 5-week-old −/− mouse compared with those of a 5-week-old +/+ mouse (all scales, ×1000). C, P-GSK-3β immunoblots. Whole-brain lysates containing 50 μg of protein from +/+ and −/− mice were resolved on 10% gels and blotted with P-GSK-3β antibody (P-GSK-3) or primary sequence-dependent antibody (GSK-3). The intensities of the resulting bands were quantitated densitometrically, and no significant differences (p = 0.11) were observed between the npc-1 and control mice.
Fig. 2.

Increased levels of NF-H and increased phosphorylation of NF-M in npc-1 −/− mice. Whole-brain lysates containing 10 μg of protein from 8-week-old +/+ and 4- and 8-week-old −/− mice were resolved on 8% gels, transferred to nitrocellulose, and blotted with the indicated antibodies. The pan NF antibody R39 identified the three major NF isoforms (marked NF-H, NF-M, and NF-L, respectively). The intensities of the bands visualized with SMI 31 and R39 were quantitated, and the results indicated a significant increase in the levels of all three isoforms and a marked increase in phosphorylation of the NF-M isoform. Equivalent loading of all samples is demonstrated with the NeuN antibody recognizing the 46–48 kDa neuronal-specific antigens.
Hyperphosphorylated tau and MAP2 accumulate in npc-1mouse brain
To determine whether other cytoskeletal proteins normally localized in axons and dendrites (i.e., tau and MAP2) (Binder et al., 1986; Matus, 1990) are also modified, their phosphorylation status was examined in npc-1 mice. A library of anti-tau antibodies [i.e., paired helical filament-1 (PHF-1), CP-13, CP-22, MC-6, MC-1, ALZ-50, and TG-5] raised against PHFs from AD brains (Table 1) was used. Overall, the pattern of staining obtained with the phosphotau antibodies was similar in that bundles of fibers were stained in +/+ mice (Fig.3A, PHF-1, D, CP-22) and numerous axon spheroids were detected in −/− mice (Fig.3B, PHF-1, E, CP-22). Classic NFTs were not observed with any of the anti-tau antibodies. Some perikarya of neurons (Fig. 3C, PHF-1) in the brainstem and basal ganglia was visible. Tau-positive axon spheroids were seen in the brainstem, basal ganglia, and white matter of −/− mice as early as 5 weeks of age and became more conspicuous over the following 7 weeks (shown only in 8-week-old mice) (Fig. 3B,C, PHF-1, E, CP-22). The pattern of staining with the CP-10 antibody recognizing phosphothreonine-231 in tau was somewhat different from that of the above phosphotau antibodies. CP-10 exhibited weak staining of neuronal cell bodies in +/+ mice (Fig. 3F), but in −/− mice there was a dramatic increase in the staining of neurons both in the cytoplasm and in the nucleus (Fig. 3G,arrow). Some neurons containing foamy material were conspicuously unstained (Fig. 3G, ∗), but others were stained (Fig. 3G, arrowhead). Curiously, the conformation- and sequence-dependent tau antibodies ALZ-50, MC-1, and TG-5 did not react with axonal spheroids or any other pathological feature in the npc-1 mice (data not shown). These antibodies stained axon fibers weakly in +/+ mice and fewer numbers of similar fibers in npc-1 −/− mice (data not shown).
Fig. 3.
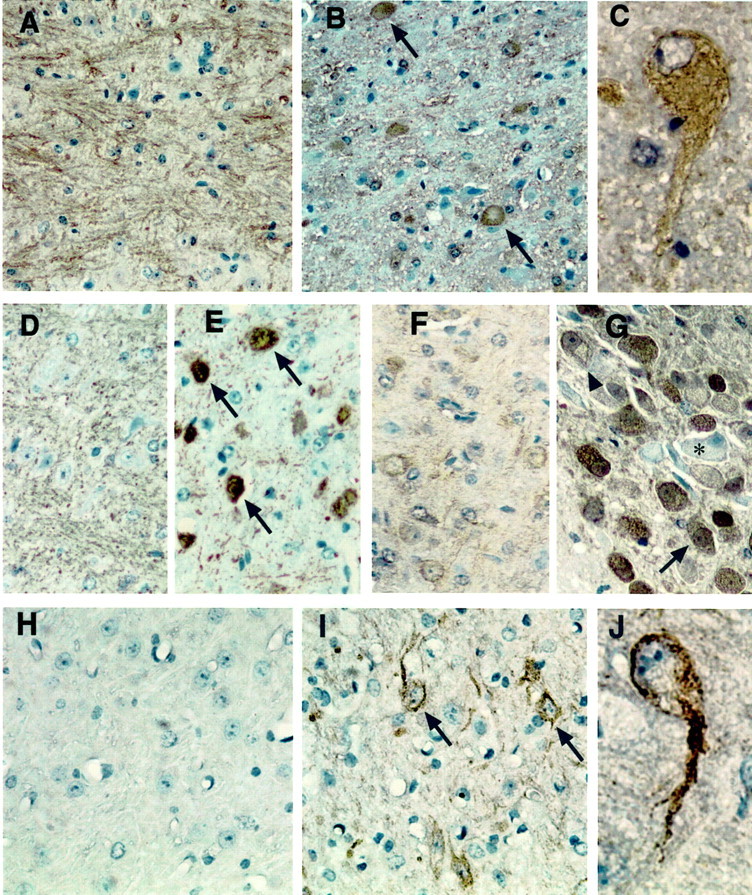
Tau and MAP2 abnormalities in thenpc-1 −/− mouse brain. Sagittal brain sections from +/+ (A, D, F, H) and 8-week-old −/− mice (B, C, E, G, I, J) were immunostained with PHF-1 (A–C), CP-22 (D, E), CP-10 (F, G), and AP-18 (H–J). PHF-1 stained bundles of processes in the brainstem and white matter of +/+ mice (A, pons) but numerous axonal spheroids (B, arrows) and some perikarya (C) of neurons in similar regions of the −/− mouse brain. CP-22 displayed much weaker staining of processes in +/+ mice (D) and a similar pattern of axonal spheroids (arrows) in the brainstem of −/− mice (E, pons). CP-10 exhibited weak staining of neuronal cell bodies in +/+ mice (F, pons), but in −/− mice there was a dramatic increase in the staining of neurons both in the cytoplasm and in the nucleus (G, large arrow). Some neurons containing foamy material were conspicuously unstained (G, asterisk), but others were CP-10 positive (arrowhead). AP-18 was primarily negative in the +/+ mouse brain (H) but labeled the soma and dendrites of several neurons (arrows) in the thalamus (I). J, One such neuron with prominent staining of the soma and apical dendrite. Magnification:A, B, D–G, I, J, 40×; C, H, 100×.
The AP-18 antibody recognizing a phosphoepitope in MAP2 gave positive results with perfusion-fixed sections only. In 7-week-old −/− mice, AP-18 displayed intense immunoreactivity in the somatodendritic compartment of large neurons of the thalamus (Fig.3I, arrows). The apical dendrite was prominently labeled in many of these cells (Fig. 3J). Axonal spheroids in the thalamus and brainstem were labeled to a lesser degree with AP-18 (data not shown) than with tau or NF antibodies. The antibody did not show any staining in +/+ brain sections (Fig.3H). AP-20, an antibody recognizing total MAP2 (i.e., the 240–280 kDa MAP2a and MAP2b isoforms), positively labeled the soma of many neurons throughout the brain, without any striking difference between the two groups of mice (data not shown).
Immunoblotting analyses of the microtubule-associated proteins was conducted with heat-stable supernatants from +/+ and −/− mice. An increase in tau phosphorylation was detected in npc-1 −/− mice at the earliest time point tested (i.e., 4 weeks of age) and increased further through 10 weeks of age (only data from the 4- and 8-week-old mice are shown) (Fig. 4). In 4-week-old npc-1 −/− mice, an increase in intensity of phosphotau antibody immunoreactivity with tau was seen compared with +/+ mice, although there was no striking change in electrophoretic mobility (Fig. 4, blots PHF-1, CP-13, and CP-10). In 8-week-oldnpc-1 −/− mice, the triplet pattern of hyperphosphorylated tau similar to that of AD became apparent with most of the antibodies (Fig. 4, compare bands stained with CP-13 in 8-week-oldnpc-1 −/− mice and AD). The average increase in immunoreactivity with phosphotau antibodies in 8-week-oldnpc-1 mice compared with age-matched +/+ mice was threefold for CP-10 (n = 4) and PHF-1 (n = 8) and twofold for CP-13 (n = 8). The tau sequence and conformation antibodies TG-5, ALZ-50, and MC-1 showed no difference in the intensity of the tau bands between the two groups of mice, indicating similar levels of tau protein in both (Fig. 4, only TG-5 and ALZ-50 shown). Together, these data support a net increase in the phosphorylation of tau in npc-1 mice.
Unexpectedly, CP-22 and MC-6, which were raised against purified PHF, did not react with tau in either +/+ or −/− mice. However, both antibodies recognized a protein of ∼180 kDa that was markedly elevated in heat-stable supernatants from npc-1 mice (Fig.4, shown for CP-22 only). The size of this band is larger than the 97–110 kDa high-molecular weight tau species (Georgieff et al., 1991), and we found that it was more abundant in the supernatant fraction without any heat treatment, suggesting that it is primarily heat labile (data not shown).
Consistent with the accumulation of phosphorylated MAP2 by immunohistochemistry, immunoblotting demonstrated that the phosphorylated high-molecular weight 240–280 kDa MAP2a and MAP2b isoforms of and the low-molecular weight 70 kDa MAP2c isoform were enriched in −/− mice, particularly at 4 weeks of age (Fig. 4, AP-18). At 8 weeks of age, no increase in phosphorylation of MAP2a and MAP2b was detected, but MAP2c appeared to be hyperphosphorylated. A duplicate blot stained with the AP-20 antibody recognizing the primary sequence in MAP2a and MAP2b showed similar amounts of these proteins in −/− mice relative to +/+ mice (Fig. 4, AP-20).
p25 levels and cdk5 activity are increased in npc-1 −/− mouse brain
Previous evidence has suggested that the PHF-1, CP-13, SMI 31, and AP-18 phosphoepitopes are generated by cdk5 kinase (Berling et al., 1994; Patrick et al., 1999; Nguyen et al., 2001) that is enriched in axons (Tsai et al., 1993). Therefore, we compared the levels of cdk5 and its activator, p35, and cdk5 activity in −/− and +/+ mice. Immunoblotting analyses showed that the cdk5 levels were invariable in −/− mice compared with +/+ mice (Fig.5A, cdk5, mouse). p35 levels were also unchanged, but an elevation in p25, a C-terminal truncated fragment of p35 (Patrick et al., 1999), was detected in −/− mouse brains from 5 to 12 weeks of age (Fig.5A, p35 mouse). A similar pattern of constant cdk5 levels but increased p25 levels is seen in human NPC (Fig. 5A, human). Elevated p25 correlates with an increase in cdk5 activity, because p25 has a longer half-life and is a better activator of cdk5 than p35 (Patrick et al., 1999). To see whether cdk5 activity is altered in −/− mice, the kinase was immunoprecipitated from whole-brain lysates and tested for its phosphorylation activity toward histone H1 in vitro (Fig. 5B, cdk5 IP/kinase assay). The recovery of cdk5 in all of the immunoprecipitates was similar as judged by immunoblotting with a cdk5 monoclonal antibody (Fig. 5B, whole brain, cdk5 blots in top row), and Coomassie blue staining of the gel showed equivalent amounts of H1 substrate in the kinase reaction in all lanes (Fig. 5, whole brain, gel shown in second row). However, incorporation of labeled phosphate from [γ-32P]ATP into H1 was significantly higher with cdk5 IPs from −/− mice than from +/+ mice (Fig. 5B, whole brain, H1 phosphorylation shown inthird row). The increase was most striking between 4 and 7 weeks of age and less so at ≥8 weeks of age. The lower increase in cdk5 activity in older mice may be a result of neuronal loss, which at 8 weeks of age is most prominent in the cerebellum but becomes more significant in other regions as well in later stages of the disease. The average increase in cdk5 activity in whole-brain lysates from 5- to 10-week-old −/− mice was 1.6-fold (Fig. 5B, whole brain, histogram; p < 0.01; n = 12).
Given the regional distribution of axonal spheroids in npc-1brain, we also performed a regional comparison of cdk5 activities. The cerebellum, brainstem, and remaining forebrain from 5-week-old mice were analyzed. As was done with cdk5 IPs from whole-brain lysates, equivalent recovery of cdk5 in the IP and the equivalent amount of H1 were verified with the samples from different brain regions (Fig.5B, Forebr., B.stem, Cblm., respectively, cdk5 blot shown in first row, H1 protein in gel shown insecond row). An increase in the amount of labeled phosphate incorporated into H1 was visible in the forebrain but was far more striking in the brainstem and barely noticeable in the cerebellum (Fig.5B, Forebr., B.stem, Cblm., H1 phosphorylation shown in third row). Quantitation of the data from two such determinations using samples from two different sets of mice revealed a sixfold increase in cdk5 activity in the brainstem and a twofold increase in the forebrain but no change in the cerebellum (Fig.5B, histogram).
To support the significance of these cdk5 activity changes in NPC, another proline-directed kinase [i.e., glycogen synthase kinase-3β (GSK-3β)] that has been shown to phosphorylate sites similar to those of cdk5 in the above cytoskeletal proteins (Julien and Mushynski, 1998; Mattson, 2001) was explored. The GSK-3β kinase is inactivated by phosphorylation of its ser9 residue (Grimes and Jope, 2001). The phospho-GSK-3β (P-GSK-3β) (ser9) antibody recognizing this phosphoepitope provides a convenient means of detecting changes in the activity of the kinase by tracing the phosphorylation status of this site. This P-GSK-3β antibody and another primary sequence-dependent GSK-3β antibody were used to explore the phosphorylation status and levels of GSK-3β in thenpc-1 mouse brain. Replicate blots were generated using brain lysates from five 5- to 9-week-old npc-1 mice and three controls of similar ages. One blot was stained with the P-GSK-3β antibody and the other with the sequence antibody (GSK-3); neither antibody revealed any consistent change in GSK-3β immunoreactivity in NPC (Fig. 5C). This experiment was run in duplicate, and the intensities of the bands obtained with each antibody in the npc-1 mice were compared with the corresponding bands in control mice. There was no significant difference (p = 0.11) in kinase levels or activity in the npc-1 mice (Fig. 5C). Reprobing the blots with NeuN antibody confirmed that all of the lanes were equally loaded (data not shown).
p25 accumulates and colocalizes with hyperphosphorylated NFs in axon spheroids of NPC −/− mouse brain
To further determine whether the cdk5 activity changes were relevant to the cytoskeletal pathology in npc-1 mice, immunohistochemical studies of cdk5 and p35 were conducted using perfusion-fixed brain sections. Despite the constant cdk5 levels in the immunoblots discussed above, cdk5 antibodies displayed an increase in immunoreactivity in some deformed neurons (Fig.6B, arrow) and axon speroids (arrowhead) in the brainstem and basal ganglia of −/− mice compared with +/+ mice of the same age (Fig.6A). An N-terminal p35 antibody recognizing only p35 but not p25 stained bundles of fibers in +/+ mice and fewer numbers of fibers in −/− mice but no axonal spheroids (data not shown). In contrast, a C-terminal p35 antibody recognizing both p35 and p25 exhibited intense and extensive staining of axonal spheroids in the brainstem, basal ganglia, and white matter of the cerebellum (Fig. 6, only brainstem shown, F, green), but in +/+ mice, its immunoreactivity was faint and localized in fibers and in soma of a few neurons (arrows in Fig. 6C,green). C-terminal p35 antibody-specific staining of NFTs suggested the accumulation of p25 in NFTs of the AD brain (Patrick et al., 1999). Thus, it appears that the C-terminal p35 antibody-specific staining of axonal spheroids may also suggest that increased p25 is localized to pathological structures in npc-1 mouse brain. To gather additional support for this idea, double labeling with C-terminal p35 antibody (green) and SMI 31 (red) was performed. Wild-type sections did not show significant colocalization of C-terminal p35 antibody immunoreactivity and SMI 31 (Fig.6C,D, respectively, and merged image inE). The micrograph shown from the brain of a 7-week-oldnpc-1 −/− mouse instead indicates considerable overlap in localization of the two antigens in axonal spheroids (arrows, Fig. 6, C-terminal p35 antibody, green,F and H, SMI 31, red, G andH) throughout the brain.
Fig. 6.
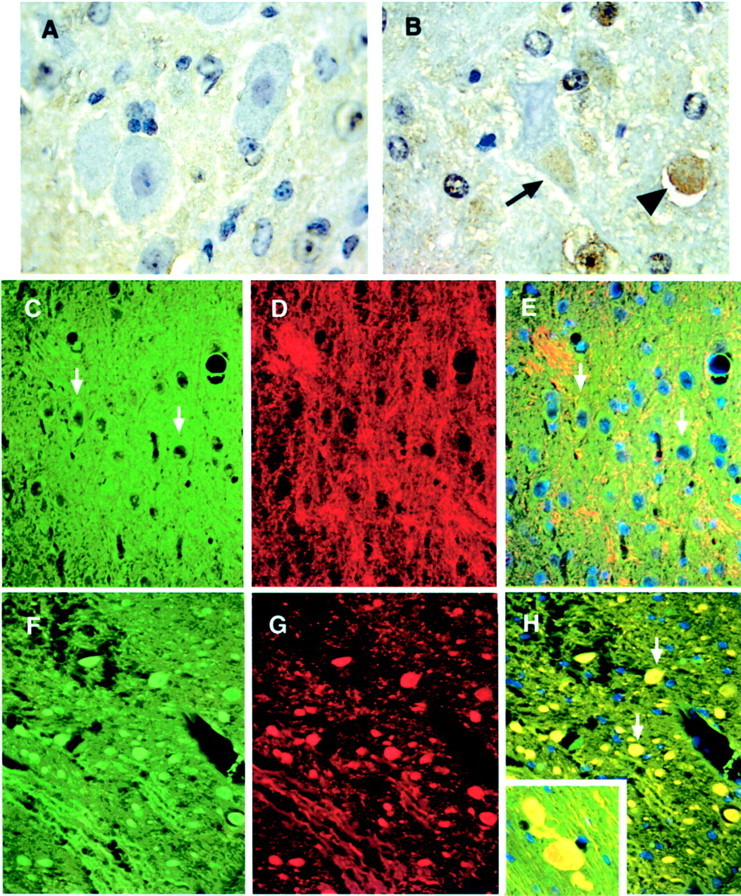
p35 accumulates and colocalizes with phosphorylated NF in axonal spheroids of npc-1 −/− mice. Perfusion-fixed, paraffin-embedded sections from 7-week-old +/+ (A, C–E) and −/− (B, F–H) mice were immunohistochemically labeled with cdk5 (brown) and counterstained with hematoxylin (blue-purple) or immunofluorescently labeled with p35 (Alexa 488, green) and SMI 31 (Cy-3, red) and counterstained with DAPI (blue). cdk5 barely stained neurons in +/+ mice (A, pontine nucleus). In contrast, cdk5 immunoreactivity was increased in the soma of some deformed neurons (arrow) and in the axonal spheroids (arrowhead) in the brainstem of −/− mice (B, pontine nucleus). p35 faintly labeled the soma of neurons in the brainstem of +/+ mice (arrows inC and E, pons) and displayed little colocalization with SMI 31 immunoreactivity (D andmerged image in E). In the −/− mouse brain, intense p35 immunoreactivity was seen in the axonal spheroids (F, pons), which perfectly matched the accumulation of SMI 31 immunoreactivity (G and arrows inmerged image in H).Inset in H, Gigantically enlarged axons containing p35 (green) and SMI 31 (red) immunoreactivity in the cerebral cortex. Magnification: A, B, 100×; C–H, 40×.
DISCUSSION
The cardinal findings of this study include elevated levels of NF-H and hyperphosphorylation of NF-M, tau, and MAP2 in thenpc-1 mouse brain. These changes coincide with deregulation of the neuronal cdk5/p35 kinase complex. Accelerated proteolytic cleavage of p35 to p25, a more powerful cdk5 activator, correlates with increased cdk5 activity, and p25 colocalizes with hyperphosphorylated cytoskeletal proteins in the most conspicuous neuropathological lesion, the axonal spheroid. These focally enlarged axonal abnormalities are concentrated in the brainstem, basal ganglia, and white matter of the cerebellum, regions containing long myelinated axons and having highNPC-1 gene expression levels (Prasad et al., 2000). Their abundance is directly proportional to the increase in cdk5 activity in each brain area. Chronologically, cdk5 activation and hyperphosphorylation are dramatic at 4 weeks of age, the earliest time point studied here, and axonal spheroids were first detected in the pons at 5 weeks of age. The severity of these changes escalated rapidly, reaching a maximum at 8–9 weeks of age, after which point marked brain atrophy became obvious. All of the biochemical changes detected here are observed in human NPC (Klünemann, Bu, Husseman, Elleder, Suzuki, Salamant, Love, Budka, Fligner, Bird, Jin, Nochlin, and Vincent, unpublished observations), but it is difficult to evaluate their temporal characteristics using autopsy brain tissue. The sequence of neuropathological events that we have delineated in thenpc-1 mouse highlights an opportunity to inhibit the NPC neurodegenerative cascade with cdk5 inhibitors. Our studies also emphasize the value of the npc-1 mouse model for testing and developing this therapeutic strategy and unraveling additional details of NPC neuropathogenesis.
NFTs were not detected in the npc-1 mouse brain using tau antibodies or Bielchowsky silver reagent (data not shown), confirming previous studies that used thioflavin S (German et al., 2001a) and in contrast to the widespread occurrence of NFTs in human NPC (Auer et al., 1995; Suzuki et al., 1995). Curiously, even with a threefold increase in the phosphorylation of tau at some sites, thenpc-1 mouse brain was devoid of TG-5, ALZ-50, and MC-1 immunoreactivity. Such sequence- and conformation-dependent tau antibodies rarely stain neurons of normal rodent or human brain; their epitopes are sensitive to fixation, but they react strongly with AD neurons because their fixation sensitivity is alleviated by phosphorylation and aggregation (Pollock and Wood, 1988; Papasozomenos, 1989; Weaver et al., 2000). Our data would argue that hyperphosphorylation of tau at sites 202 (CP-13) and 396–404 (PHF-1) in axons and of CP-10-positive tau in neuronal soma are inadequate for eliciting TG-5, ALZ-50, and MC-1 immunoreactivity in either neuronal compartment. Perhaps phosphorylation at additional sites or other modifications (Gonzalez et al., 1998; Takeda et al., 2000) is required to expose these epitopes in situ. The absence of such additional modifications in the npc-1 mouse brain might also be the key to their inability to form NFTs. The CP-22 and MC-6 antibodies recognize phosphothr-175 and phosphoser-235 in lysine–serine/threonine–proline motifs, respectively, in AD PHF (P. Davies, unpublished observations). Neither antibody reacted with tau in npc-1 mice, suggesting that these sites are not phosphorylated in NPC. Instead, both antibodies recognized a 180 kDa heat labile protein present in axon spheroids. The possibility of this protein being a tau aggregate is excluded by its negative staining with other tau antibodies. It could be a degradation product of a larger protein containing similar proline-directed phosphorylation sites (i.e., NF-H or MAP2). All three MAP2 isoforms are hyperphosphorylated in the neuronal somatodendritic compartment ofnpc-1 mice, but additional experiments are required to clarify their relationship with the 180 kDa protein.
Regarding the individual NF isoforms, the increase in NF-H levels was greater than the increase in SMI 31 immunoreactivity with NF-H; the increase in NF-M levels was less than the increased SMI 31 immunoreactivity with NF-M. These data translate into a selective increase in expression or reduced phosphorylation or degradation of NF-H and a selective increase in NF-M phosphorylation in NPC. The robust staining of axon spheroids with SMI 31 and SMI 32 suggests that a mixture of nonphosphorylated and hyperphosphorylated NF protein accumulates in the lesions. These results confirm earlier electron microscopic evidence for intermediate filaments in spheroids from human NPC (Elleder et al., 1985). However, axons normally contain hyperphosphorylated NFs, whereas perikarya contain hypophosphorylated NFs (Pant and Veeranna, 1995). Many neurodegenerative diseases, such as AD (Schmidt et al., 1991; Nixon, 1993), Parkinson's disease (Forno et al., 1986), and amyotrophic lateral sclerosis (ALS) (Julien, 1995), are characterized by perikaryal accumulations of hyperphosphorylated NFs. Axonal dilations have been documented in ALS and experimental models of axotomy (Stone et al., 2001) and in transgenic mice overexpressing four repeat human tau (Spittaels et al., 1999). These conditions are also associated with perikaryal NF or tau accumulation, implying disruption of proximal axonal transport. The absence of somatic accumulation of hyperphosphorylated tau and NFs in npc-1 mice suggests a defect downstream of the proximal axon in NPC. Using silver staining (Patel et al., 1999) and Golgi impregnation (Zervas et al., 2001), it was noted that terminal fields of axons and dendrites are the earliest sites of degeneration in npc-1 mice. Together, these data suggest that NPC neurodegeneration proceeds along a distal-to-proximal axis in long axons. The integrity of these axons has been thought to depend on the cycling of cholesterol between surrounding glial cells and the axons themselves (Xie et al., 1999), which may explain their special vulnerability to the lipid disturbances caused byNPC-1 mutations.
In accordance with this more “distal” origin of NPC axonopathy is the enrichment of cdk5 in distal axonal segments (Nikolic et al., 1996). cdk5 normally mediates phosphorylation of NFs, tau, and MAP2 and their detachment from microtubules in axons (Matsushita et al., 1996;Wada et al., 1998). In npc-1 mice, elevated p25 levels appear to be the trigger for cdk5 activation in distal axons, because p25 was enriched in cdk5 IPs from npc-1 brain and colocalized with hyperphosphorylated NFs in axonal spheroids. Moreover, the levels and activity of the GSK-3β kinase, another enzyme that has been implicated in NF and tau phosphorylation, were unaltered in thenpc-1 mice. In transgenic mice overexpressing p25, a twofold increase in cdk5 activity resulted in tau and NF hyperphosphorylation and cytoskeletal pathology (Ahlijanian et al., 2000). Similarly, increased p25 activity and a twofold increase in cdk5 activity correlated with tau and NF hyperphosphorylation and motor neuron degeneration in the SODG37R ALS mouse model (Nguyen et al., 2001). Innpc-1 mice, we measured as much as a sixfold increase in p25-associated cdk5 activity in the brainstem, where hyperphosphorylation and cytoskeletal abnormalities were intense. A causal role for cdk5 in NPC is also supported by evidence linking its activity to phosphorylation of serines 202 and 396–404 in tau, the SMI 31 epitope in NF, and the AP-18 epitope in MAP2 (Berling et al., 1994;Patrick et al., 1999; Ahlijanian et al., 2000; Grant et al., 2001;Nguyen et al., 2001). Presently, it is not possible to exclude the involvement of other kinases in NPC. Sawamura et al. (2001) reported upregulation of MAP kinase, and we (Bu, Klünemann, Suzuki, Husseman, Bird, Jin, and Vincent, unpublished observations) have observed aberrant expression of cell division cycle kinase (cdc2) and cyclin B1 in the npc-1 mouse cerebellum. Even with equivalent recovery of cdk5 in cdk5 IPs from the cerebellum and other brain regions, cdk5 activity and p25 were not elevated in the cerebellum, suggesting that other kinases mediate hyperphosphorylation in this region (Sawamura et al., 2001). The cerebellum has low p35 expression but is rich in p39 (Tang et al., 1995), which is degraded to a more effective activator, p29 (Patzke et al., 2002). We did not explore p39/p29 here, but if p29 was elevated in the npc-1cerebellum, an increase in cdk5 activity would have been detected using cdk5 IPs. p35 expression is induced by MAP kinase (Harada et al., 2001), which may explain the sustained p35 levels in npc-1mice that were observed even with its increased conversion to p25. The thr-231 tau epitope recognized by the CP-10 antibody when phosphorylated is a better acceptor site for cdc2 than cdk5. CP-10 immunoreactivity is produced abundantly in npc-1 mice and is the only tau phosphoepitope that we detected in neuronal soma. Thus, although cdk5 may be the principal effector of axonal tau and NF in NPC, a somatic pool of tau may be modified by a different proline-directed kinase.
Cholesterol, glycosphingolipids, and glycosyl-phosphatidylinositol-anchored proteins cluster in distinct cell membrane microdomains, called lipid rafts, and raft-enriched membrane invaginations called caveolas (Kurzchalia and Parton, 1999) constitute “signal transduction centers,” coordinating extracellular signals with neuronal function and stability (Brown and London, 1998; Masserini et al., 1999). cdk5 mediates such a link in axons (Maccioni et al., 2001), and it is intriguing that p35 through its interaction with Rac GTPase (Nikolic et al., 1998); calpain, the protease that converts p35 to cytosolic p25 (Kulkarni et al., 1999; Bialkowska et al., 2000; Kusakawa et al., 2000;Lee et al., 2000); and a fraction of cellular NPC-1 (Garver et al., 2000) are all present in caveolas. We propose that caveolas play a crucial role in NPC neuropathology. Alterations of the caveolar scaffold protein caveolin-1 and of annexin II innpc-1 mice (Garver et al., 1997a,b) support this idea. The demyelination induced by NPC-1 mutations (Elleder et al., 1985; German et al., 2002) may potentiate axonal pathology, because myelin regulates the levels and activity of calpain (Chakrabarti et al., 1990; Persson and Karlsson, 1991), the expression and phosphorylation of NFs (Starr et al., 1996; Gotow et al., 1999), and the density and stability of axonal microtubules (Sanchez et al., 2000;Kirkpatrick et al., 2001). It should be possible to unravel the temporal and spatial relationships of these events in thenpc-1 mouse model.
Footnotes
This work was supported by a grant from the Seattle Jim Lambright Medical Research Foundation (I.V.) and Grants AG12721 (I.V.), P50 AG 05136-16 (Alzheimer's Disease Research Center, M.R.), and MH38623 (P.D.) from the National Institute on Aging. We thank Drs. Harish C. Pant and Lester Binder for their gifts of antibodies and Drs. Kinuko Suzuki and Steven Walkely for providing us withnpc-1 mouse brain for preliminary studies. We also thank Dr. Jan Hallows for her reading of this manuscript and her suggestions.
Correspondence should be addressed to Inez Vincent, University of Washington, Department of Pathology, K072 HSB, Box 357705, 1959 Northeast Pacific Avenue, Seattle, WA 98195. E-mail:ivincent@u.washington.edu.
REFERENCES
- 1.Ahlijanian MK, Barrezueta NX, Williams RD, Jakowski A, Kowsz KP, McCarthy S, Coskran T, Carlo A, Seymour PA, Burkhardt JE, Nelson RB, McNeish JD. Hyperphosphorylated tau and neurofilament and cytoskeletal disruptions in mice overexpressing human p25, an activator of cdk5. Proc Natl Acad Sci USA. 2000;97:2910–2915. doi: 10.1073/pnas.040577797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Auer IA, Schmidt ML, Lee VM, Curry B, Suzuki K, Shin RW, Pentchev PG, Carstea ED, Trojanowski JQ. Paired helical filament tau (PHFtau) in Niemann-Pick type C disease is similar to PHFtau in Alzheimer's disease. Acta Neuropathol. 1995;90:547–551. doi: 10.1007/BF00318566. [DOI] [PubMed] [Google Scholar]
- 3.Berling B, Wille H, Roll B, Mandelkow EM, Garner C, Mandelkow E. Phosphorylation of microtubule-associated proteins MAP2a,b and MAP2c at Ser136 by proline-directed kinases in vivo and in vitro. Eur J Cell Biol. 1994;64:120–130. [PubMed] [Google Scholar]
- 4.Bialkowska K, Kulkarni S, Du X, Goll DE, Saido TC, Fox JE. Evidence that beta3 integrin-induced Rac activation involves the calpain-dependent formation of integrin clusters that are distinct from the focal complexes and focal adhesions that form as Rac and RhoA become active. J Cell Biol. 2000;151:685–696. doi: 10.1083/jcb.151.3.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Binder LI, Frankfurter A, Rebhun LI. Differential localization of MAP-2 and tau in mammalian neurons in situ. Ann NY Acad Sci. 1986;466:145–166. doi: 10.1111/j.1749-6632.1986.tb38392.x. [DOI] [PubMed] [Google Scholar]
- 6.Brazelton TR, Rossi FM, Keshet GI, Blau HM. From marrow to brain: expression of neuronal phenotypes in adult mice. Science. 2000;290:1775–1779. doi: 10.1126/science.290.5497.1775. [DOI] [PubMed] [Google Scholar]
- 7.Brown DA, London E. Functions of lipid rafts in biological membranes. Annu Rev Cell Dev Biol. 1998;14:111–136. doi: 10.1146/annurev.cellbio.14.1.111. [DOI] [PubMed] [Google Scholar]
- 8.Camargo F, Erickson RP, Garver WS, Hossain GS, Carbone PN, Heidenreich RA, Blanchard J. Cyclodextrins in the treatment of a mouse model of Niemann-Pick C disease. Life Sci. 2001;70:131–142. doi: 10.1016/s0024-3205(01)01384-4. [DOI] [PubMed] [Google Scholar]
- 9.Chakrabarti AK, Dasgupta S, Banik NL, Hogan EL. Regulation of the calcium-activated neutral proteinase (CANP) of bovine brain by myelin lipids. Biochim Biophys Acta. 1990;1038:195–198. doi: 10.1016/0167-4838(90)90204-s. [DOI] [PubMed] [Google Scholar]
- 10.Elleder M, Jirasek A, Smid F, Ledvinova J, Besley GT. Niemann-Pick disease type C. Study on the nature of the cerebral storage process. Acta Neuropathol. 1985;66:325–336. doi: 10.1007/BF00690966. [DOI] [PubMed] [Google Scholar]
- 11.Erickson RP, Garver WS, Camargo F, Hossain GS, Heidenreich RA. Pharmacological and genetic modifications of somatic cholesterol do not substantially alter the course of CNS disease in Niemann-Pick C mice. J Inherit Metab Dis. 2000;23:54–62. doi: 10.1023/a:1005650930330. [DOI] [PubMed] [Google Scholar]
- 12.Forno LS, Sternberger LA, Sternberger NH, Strefling AM, Swanson K, Eng LF. Reaction of Lewy bodies with antibodies to phosphorylated and non-phosphorylated neurofilaments. Neurosci Lett. 1986;64:253–258. doi: 10.1016/0304-3940(86)90337-x. [DOI] [PubMed] [Google Scholar]
- 13.Garver WS, Erickson RP, Wilson JM, Colton TL, Hossain GS, Kozloski MA, Heidenreich RA. Altered expression of caveolin-1 and increased cholesterol in detergent insoluble membrane fractions from liver in mice with Niemann-Pick disease type C. Biochim Biophys Acta. 1997a;1361:272–280. doi: 10.1016/s0925-4439(97)00047-1. [DOI] [PubMed] [Google Scholar]
- 14.Garver WS, Hsu SC, Erickson RP, Greer WL, Byers DM, Heidenreich RA. Increased expression of caveolin-1 in heterozygous Niemann-Pick type II human fibroblasts. Biochem Biophys Res Commun. 1997b;236:189–193. doi: 10.1006/bbrc.1997.6929. [DOI] [PubMed] [Google Scholar]
- 15.Garver WS, Heidenreich RA, Erickson RP, Thomas MA, Wilson JM. Localization of the murine Niemann-Pick C1 protein to two distinct intracellular compartments. J Lipid Res. 2000;41:673–687. [PubMed] [Google Scholar]
- 16.Georgieff IS, Liem RK, Mellado W, Nunez J, Shelanski ML. High molecular weight tau: preferential localization in the peripheral nervous system. J Cell Sci. 1991;100:55–60. doi: 10.1242/jcs.100.1.55. [DOI] [PubMed] [Google Scholar]
- 17.German DC, Quintero EM, Liang CL, Ng B, Punia S, Xie C, Dietschy JM. Selective neurodegeneration, without neurofibrillary tangles, in a mouse model of Niemann-Pick C disease. J Comp Neurol. 2001;433:415–425. doi: 10.1002/cne.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.German DC, Liang CL, Song T, Yazdani U, Xie C, Dietschy JM. Neurodegeneration in the Niemann-Pick C mouse: glial involvement. Neuroscience. 2002;109:437–450. doi: 10.1016/s0306-4522(01)00517-6. [DOI] [PubMed] [Google Scholar]
- 19.Gonzalez C, Farias G, Maccioni RB. Modification of tau to an Alzheimer's type protein interferes with its interaction with microtubules. Cell Mol Biol (Noisy-le-grand) 1998;44:1117–1127. [PubMed] [Google Scholar]
- 20.Gotow T, Leterrier JF, Ohsawa Y, Watanabe T, Isahara K, Shibata R, Ikenaka K, Uchiyama Y. Abnormal expression of neurofilament proteins in dysmyelinating axons located in the central nervous system of jimpy mutant mice. Eur J Neurosci. 1999;11:3893–3903. doi: 10.1046/j.1460-9568.1999.00820.x. [DOI] [PubMed] [Google Scholar]
- 21.Grant P, Sharma P, Pant HC. Cyclin-dependent protein kinase 5 (Cdk5) and the regulation of neurofilament metabolism. Eur J Biochem. 2001;268:1534–1546. [PubMed] [Google Scholar]
- 22.Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3beta in cellular signaling. Prog Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- 23.Harada T, Morooka T, Ogawa S, Nishida E. ERK induces p35, a neuron-specific activator of Cdk5, through induction of Egr1. Nat Cell Biol. 2001;3:453–459. doi: 10.1038/35074516. [DOI] [PubMed] [Google Scholar]
- 24.Higashi Y, Murayama S, Pentchev PG, Suzuki K. Cerebellar degeneration in the Niemann-Pick type C mouse. Acta Neuropathol. 1993;85:175–184. doi: 10.1007/BF00227765. [DOI] [PubMed] [Google Scholar]
- 25.Julien JP. A role for neurofilaments in the pathogenesis of amyotrophic lateral sclerosis. Biochem Cell Biol. 1995;73:593–597. doi: 10.1139/o95-064. [DOI] [PubMed] [Google Scholar]
- 26.Julien JP, Mushynski WE. Multiple phosphorylation sites in mammalian neurofilament polypeptides. J Biol Chem. 1982;257:10467–10470. [PubMed] [Google Scholar]
- 27.Julien JP, Mushynski WE. Neurofilaments in health and disease. Prog Nucleic Acid Res Mol Biol. 1998;61:1–23. doi: 10.1016/s0079-6603(08)60823-5. [DOI] [PubMed] [Google Scholar]
- 28.Kirkpatrick LL, Witt AS, Payne HR, Shine HD, Brady ST. Changes in microtubule stability and density in myelin-deficient shiverer mouse CNS axons. J Neurosci. 2001;21:2288–2297. doi: 10.1523/JNEUROSCI.21-07-02288.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kulkarni S, Saido TC, Suzuki K, Fox JE. Calpain mediates integrin-induced signaling at a point upstream of Rho family members. J Biol Chem. 1999;274:21265–21275. doi: 10.1074/jbc.274.30.21265. [DOI] [PubMed] [Google Scholar]
- 30.Kurzchalia TV, Parton RG. Membrane microdomains and caveolae. Curr Opin Cell Biol. 1999;11:424–431. doi: 10.1016/s0955-0674(99)80061-1. [DOI] [PubMed] [Google Scholar]
- 31.Kusakawa G, Saito T, Onuki R, Ishiguro K, Kishimoto T, Hisanaga S. Calpain-dependent proteolytic cleavage of the p35 cyclin-dependent kinase 5 activator to p25. J Biol Chem. 2000;275:17166–17172. doi: 10.1074/jbc.M907757199. [DOI] [PubMed] [Google Scholar]
- 32.Lee MS, Kwon YT, Li M, Peng J, Friedlander RM, Tsai LH. Neurotoxicity induces cleavage of p35 to p25 by calpain. Nature. 2000;405:360–364. doi: 10.1038/35012636. [DOI] [PubMed] [Google Scholar]
- 33.Liu Y, Wu YP, Wada R, Neufeld EB, Mullin KA, Howard AC, Pentchev PG, Vanier MT, Suzuki K, Proia RL. Alleviation of neuronal ganglioside storage does not improve the clinical course of the Niemann-Pick C disease mouse. Hum Mol Genet. 2000;9:1087–1092. doi: 10.1093/hmg/9.7.1087. [DOI] [PubMed] [Google Scholar]
- 34.Loftus SK, Morris JA, Carstea ED, Gu JZ, Cummings C, Brown A, Ellison J, Ohno K, Rosenfeld MA, Tagle DA, Pentchev PG, Pavan WJ. Murine model of Niemann-Pick C disease: mutation in a cholesterol homeostasis gene. Science. 1997;277:232–235. doi: 10.1126/science.277.5323.232. [DOI] [PubMed] [Google Scholar]
- 35.Love S, Bridges LR, Case CP. Neurofibrillary tangles in Niemann-Pick disease type C. Brain. 1995;118:119–129. doi: 10.1093/brain/118.1.119. [DOI] [PubMed] [Google Scholar]
- 36.Maccioni RB, Otth C, Concha II, Munoz JP. The protein kinase Cdk5. Structural aspects, roles in neurogenesis and involvement in Alzheimer's pathology. Eur J Biochem. 2001;268:1518–1527. doi: 10.1046/j.1432-1033.2001.02024.x. [DOI] [PubMed] [Google Scholar]
- 37.March PA, Thrall MA, Brown DE, Mitchell TW, Lowenthal AC, Walkley SU. GABAergic neuroaxonal dystrophy and other cytopathological alterations in feline Niemann-Pick disease type C. Acta Neuropathol (Berl) 1997;94:164–172. doi: 10.1007/s004010050689. [DOI] [PubMed] [Google Scholar]
- 38.Masserini M, Palestini P, Pitto M. Glycolipid-enriched caveolae and caveolae-like domains in the nervous system. J Neurochem. 1999;73:1–11. doi: 10.1046/j.1471-4159.1999.0730001.x. [DOI] [PubMed] [Google Scholar]
- 39.Matsushita M, Tomizawa K, Lu YF, Moriwaki A, Tokuda M, Itano T, Wang JH, Hatase O, Matsui H. Distinct cellular compartment of cyclin-dependent kinase 5 (Cdk5) and neuron-specific Cdk5 activator protein (p35nck5a) in the developing rat cerebellum. Brain Res. 1996;734:319–322. [PubMed] [Google Scholar]
- 40.Mattson MP. Neuronal death and GSK-3beta: a tau fetish? Trends Neurosci. 2001;24:255–256. doi: 10.1016/s0166-2236(00)01838-5. [DOI] [PubMed] [Google Scholar]
- 41.Matus A. Microtubule-associated proteins and the determination of neuronal form. J Physiol (Lond) 1990;84:134–137. [PubMed] [Google Scholar]
- 42.Millat G, Chikh K, Naureckiene S, Sleat DE, Fensom AH, Higaki K, Elleder M, Lobel P, Vanier MT. Niemann-pick disease type c: spectrum of he1 mutations and genotype/phenotype correlations in the npc2 group. Am J Hum Genet. 2001;69:1013–1021. doi: 10.1086/324068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Naureckiene S, Sleat DE, Lackland H, Fensom A, Vanier MT, Wattiaux R, Jadot M, Lobel P. Identification of HE1 as the second gene of Niemann-Pick C disease. Science. 2000;290:2298–2301. doi: 10.1126/science.290.5500.2298. [DOI] [PubMed] [Google Scholar]
- 44.Nguyen MD, Lariviere RC, Julien JP. Deregulation of Cdk5 in a mouse model of ALS: toxicity alleviated by perikaryal neurofilament inclusions. Neuron. 2001;30:135–147. doi: 10.1016/s0896-6273(01)00268-9. [DOI] [PubMed] [Google Scholar]
- 45.Nikolic M, Dudek H, Kwon YT, Ramos YF, Tsai LH. The cdk5/p35 kinase is essential for neurite outgrowth during neuronal differentiation. Genes Dev. 1996;10:816–825. doi: 10.1101/gad.10.7.816. [DOI] [PubMed] [Google Scholar]
- 46.Nikolic M, Chou MM, Lu W, Mayer BJ, Tsai LH. The p35/Cdk5 kinase is a neuron-specific Rac effector that inhibits Pak1 activity. Nature. 1998;395:194–198. doi: 10.1038/26034. [DOI] [PubMed] [Google Scholar]
- 47.Nixon RA. The regulation of neurofilament protein dynamics by phosphorylation: clues to neurofibrillary pathobiology. Brain Pathol. 1993;3:29–38. doi: 10.1111/j.1750-3639.1993.tb00723.x. [DOI] [PubMed] [Google Scholar]
- 48.Ong WY, Kumar U, Switzer RC, Sidhu A, Suresh G, Hu CY, Patel SC. Neurodegeneration in Niemann-Pick type C disease mice. Exp Brain Res. 2001;141:218–231. doi: 10.1007/s002210100870. [DOI] [PubMed] [Google Scholar]
- 49.Pant HC, Veeranna Neurofilament phosphorylation. Biochem Cell Biol. 1995;73:575–592. doi: 10.1139/o95-063. [DOI] [PubMed] [Google Scholar]
- 50.Papasozomenos SC. Tau protein immunoreactivity in dementia of the Alzheimer type: II. Electron microscopy and pathogenetic implications. Effects of fixation on the morphology of the Alzheimer's abnormal filaments. Lab Invest. 1989;60:375–389. [PubMed] [Google Scholar]
- 51.Patel SC, Suresh S, Kumar U, Hu CY, Cooney A, Blanchette-Mackie EJ, Neufeld EB, Patel RC, Brady RO, Patel YC, Pentchev PG, Ong WY. Localization of Niemann-Pick C1 protein in astrocytes: implications for neuronal degeneration in Niemann-Pick type C disease. Proc Natl Acad Sci USA. 1999;96:1657–1662. doi: 10.1073/pnas.96.4.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, Tsai LH. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature. 1999;402:615–622. doi: 10.1038/45159. [DOI] [PubMed] [Google Scholar]
- 53.Patterson MC, Di Bisceglie AM, Higgins JJ, Abel RB, Schiffmann R, Parker CC, Argoff CE, Grewal RP, Yu K, Pentchev PG, Brady RO, Barton NW. The effect of cholesterol-lowering agents on hepatic and plasma cholesterol in Niemann-Pick disease type C. Neurology. 1993;43:61–64. doi: 10.1212/wnl.43.1_part_1.61. [DOI] [PubMed] [Google Scholar]
- 54.Patzke H, Tsai LH. Calpain-mediated cleavage of the cyclin-dependent kinase 5 activator p39 to p29. J Biol Chem. 2002;277:8054–8060. doi: 10.1074/jbc.M109645200. [DOI] [PubMed] [Google Scholar]
- 55.Persson H, Karlsson JO. Calpain activity in a subcellular fraction enriched in partially degraded CNS myelin fragments compared with myelin. Neurosci Lett. 1991;130:81–84. doi: 10.1016/0304-3940(91)90232-i. [DOI] [PubMed] [Google Scholar]
- 56.Pollock NJ, Wood JG. Differential sensitivity of the microtubule-associated protein, tau, in Alzheimer's disease tissue to formalin fixation. J Histochem Cytochem. 1988;36:1117–1121. doi: 10.1177/36.9.2841371. [DOI] [PubMed] [Google Scholar]
- 57.Prasad A, Fischer WA, Maue RA, Henderson LP. Regional and developmental expression of the Npc1 mRNA in the mouse brain. J Neurochem. 2000;75:1250–1257. doi: 10.1046/j.1471-4159.2000.0751250.x. [DOI] [PubMed] [Google Scholar]
- 58.Sanchez I, Hassinger L, Sihag RK, Cleveland DW, Mohan P, Nixon RA. Local control of neurofilament accumulation during radial growth of myelinating axons in vivo. Selective role of site-specific phosphorylation. J Cell Biol. 2000;151:1013–1024. doi: 10.1083/jcb.151.5.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sawamura N, Gong JS, Garver WS, Heidenreich RA, Ninomiya H, Ohno K, Yanagisawa K, Michikawa M. Site-specific phosphorylation of tau accompanied by activation of mitogen-activated protein kinase (MAPK) in brains of Niemann-Pick type C mice. J Biol Chem. 2001;276:10314–10319. doi: 10.1074/jbc.M009733200. [DOI] [PubMed] [Google Scholar]
- 60.Schmidt ML, Lee VM, Trojanowski JQ. Comparative epitope analysis of neuronal cytoskeletal proteins in Alzheimer's disease senile plaque neurites and neuropil threads. Lab Invest. 1991;64:352–357. [PubMed] [Google Scholar]
- 61.Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B. The metabolic and molecular bases of inherited disease. McGraw-Hill; New York: 2001. [Google Scholar]
- 62.Spittaels K, Van den Haute C, Van Dorpe J, Bruynseels K, Vandezande K, Laenen I, Geerts H, Mercken M, Sciot R, Van Lommel A, Loos R, Van Leuven F. Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. Am J Pathol. 1999;155:2153–2165. doi: 10.1016/S0002-9440(10)65533-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Starr R, Attema B, DeVries GH, Monteiro MJ. Neurofilament phosphorylation is modulated by myelination. J Neurosci Res. 1996;44:328–337. doi: 10.1002/(SICI)1097-4547(19960515)44:4<328::AID-JNR3>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 64.Sternberger LA, Sternberger NH. Monoclonal antibodies distinguish phosphorylated and nonphosphorylated forms of neurofilaments in situ. Proc Natl Acad Sci USA. 1983;80:6126–6130. doi: 10.1073/pnas.80.19.6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stone JR, Singleton RH, Povlishock JT. Intra-axonal neurofilament compaction does not evoke local axonal swelling in all traumatically injured axons. Exp Neurol. 2001;172:320–331. doi: 10.1006/exnr.2001.7818. [DOI] [PubMed] [Google Scholar]
- 66.Suzuki K, Parker CC, Pentchev PG, Katz D, Ghetti B, D'Agostino AN, Carstea ED. Neurofibrillary tangles in Niemann-Pick disease type C. Acta Neuropathol. 1995;89:227–238. doi: 10.1007/BF00309338. [DOI] [PubMed] [Google Scholar]
- 67.Takeda A, Smith MA, Avila J, Nunomura A, Siedlak SL, Zhu X, Perry G, Sayre LM. In Alzheimer's disease, heme oxygenase is coincident with Alz50, an epitope of tau induced by 4-hydroxy-2-nonenal modification. J Neurochem. 2000;75:1234–1241. doi: 10.1046/j.1471-4159.2000.0751234.x. [DOI] [PubMed] [Google Scholar]
- 68.Tanaka J, Nakamura H, Miyawaki S. Cerebellar involvement in murine sphingomyelinosis: a new model of Niemann-Pick disease. J Neuropathol Exp Neurol. 1988;47:291–300. doi: 10.1097/00005072-198805000-00008. [DOI] [PubMed] [Google Scholar]
- 69.Tang D, Yeung J, Lee KY, Matsushita M, Matsui H, Tomizawa K, Hatase O, Wang JH. An isoform of the neuronal cyclin-dependent kinase 5 (Cdk5) activator. J Biol Chem. 1995;270:26897–26903. doi: 10.1074/jbc.270.45.26897. [DOI] [PubMed] [Google Scholar]
- 70.Tsai LH, Takahashi T, Caviness VS, Jr, Harlow E. Activity and expression pattern of cyclin-dependent kinase 5 in the embryonic mouse nervous system. Development. 1993;119:1029–1040. doi: 10.1242/dev.119.4.1029. [DOI] [PubMed] [Google Scholar]
- 71.Vanier MT, Rodriguez-Lafrasse C, Rousson R, Duthel S, Harzer K, Pentchev PG, Revol A, Louisot P. Type C Niemann-Pick disease: biochemical aspects and phenotypic heterogeneity. Dev Neurosci. 1991a;13:307–314. doi: 10.1159/000112178. [DOI] [PubMed] [Google Scholar]
- 72.Vanier MT, Rodriguez-Lafrasse C, Rousson R, Gazzah N, Juge MC, Pentchev PG, Revol A, Louisot P. Type C Niemann-Pick disease: spectrum of phenotypic variation in disruption of intracellular LDL-derived cholesterol processing. Biochim Biophys Acta. 1991b;1096:328–337. doi: 10.1016/0925-4439(91)90069-l. [DOI] [PubMed] [Google Scholar]
- 73.Vincent I, Jicha G, Rosado M, Dickson DW. Aberrant expression of mitotic cdc2/cyclin B1 kinase in degenerating neurons of Alzheimer's disease brain. J Neurosci. 1997;17:3588–3598. doi: 10.1523/JNEUROSCI.17-10-03588.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vincent I, Zheng JH, Dickson DW, Kress Y, Davies P. Mitotic phosphoepitopes precede paired helical filaments in Alzheimer's disease. Neurobiol Aging. 1998;19:287–296. doi: 10.1016/s0197-4580(98)00071-2. [DOI] [PubMed] [Google Scholar]
- 75.Wada Y, Ishiguro K, Itoh TJ, Uchida T, Hotani H, Saito T, Kishimoto T, Hisanaya S. Microtubule-stimulated phosphorylation of tau at Ser 202 and Thr 205 by cdk5 decreases its microtubule nucleation activity. J Biochem. 1998;124:738–746. doi: 10.1093/oxfordjournals.jbchem.a022174. [DOI] [PubMed] [Google Scholar]
- 76.Walkley SU. Pyramidal neurons with ectopic dendrites in storage diseases exhibit increased GM2 ganglioside immunoreactivity. Neuroscience. 1995;68:1027–1035. doi: 10.1016/0306-4522(95)00208-z. [DOI] [PubMed] [Google Scholar]
- 77.Weaver CL, Espinoza M, Kress Y, Davies P. Conformational change as one of the earliest alterations of tau in Alzheimer's disease. Neurobiol Aging. 2000;21:719–727. doi: 10.1016/s0197-4580(00)00157-3. [DOI] [PubMed] [Google Scholar]
- 78.Xie C, Turley SD, Pentchev PG, Dietschy JM. Cholesterol balance and metabolism in mice with loss of function of Niemann-Pick C protein. Am J Physiol. 1999;276:E336–E344. doi: 10.1152/ajpendo.1999.276.2.E336. [DOI] [PubMed] [Google Scholar]
- 79.Zervas M, Somers KL, Thrall MA, Walkley SU. Critical role for glycosphingolipids in Niemann-Pick disease type C. Curr Biol. 2001;11:1283–1287. doi: 10.1016/s0960-9822(01)00396-7. [DOI] [PubMed] [Google Scholar]