

Abstract
Mutations affecting the gating and channel properties of ionotropic neurotransmitter receptors in some hereditary epilepsies, in familial hyperekplexia, and the slow-channel congenital myasthenic syndrome (SCCMS) may perturb the kinetics of synaptic currents, leading to significant clinical consequences. Although at least 12 acetylcholine receptor (AChR) mutations have been identified in the SCCMS, the altered channel properties critical for disease pathogenesis in the SCCMS have not been identified. To approach this question, we investigated the effect of different AChR subunit mutations on muscle weakness and the function and viability of neuromuscular synapses in transgenic mice. Targeted expression of distinct mutant AChR subunits in skeletal muscle prolonged the decay phases of the miniature endplate currents (MEPCs) over a broad range. In addition, both muscle strength and the amplitude of MEPCs were lower in transgenic lines with greater MEPC duration. SCCMS is associated with calcium overload of the neuromuscular junctional sarcoplasm. We found that the extent of calcium overload of motor endplates in the panel of transgenic mice was influenced by the relative permeability of the mutant AChRs to calcium, on the duration of MEPCs, and on neuromuscular activity. Finally, severe degenerative changes at the motor endplate (endplate myopathy) were apparent by electron microscopy in transgenic lines that displayed the greatest activity-dependent calcium overload. These studies demonstrate the importance of control of the kinetics of AChR channel gating for the function and viability of the neuromuscular junction.
Keywords: synaptic currents, kinetics, degeneration, calcium, mutation, neuromuscular junction
The importance of the gating properties of the muscle and neuronal nicotinic acetylcholine receptor (AChR) ion channels has recently been highlighted by the description of several kinetic disorders of the AChR that comprise prominent subgroups of both the epilepsies and congenital myasthenic syndromes (Engel, 1994; Lerche et al., 2001; Steinlein, 2001; Muley and Gomez, 2002). The slow-channel congenital myasthenic syndrome (SCCMS) is characterized by weakness, impaired neuromuscular transmission, AChR loss, and progressive degeneration of the neuromuscular junction (NMJ) (endplate myopathy) associated with different missense mutations in the muscle AChR subunits. Although the pathogenesis of the SCCMS has not been fully explained, the common effect of point mutations in the SCCMS is to prolong AChR activation events and “slow” the channel closure rate. In neuromuscular preparations from SCCMS patients, endplate currents are prolonged 4- to >10-fold. Because the effect of prolonged AChR activation events is to increase the entry of Ca2+ and Na+into the junctional sarcoplasm through the endplate AChRs, it has been suggested that the disease results from a disturbance of intracellular ionic millieu. Ca2+ overload may have direct effects on AChR function (Siara et al., 1990) and may lead to focal muscle contractures or activation of calcium-activated proteases, DNases, and phospholipases resulting in subsynaptic vacuoles, degenerating junctional nuclei and mitochondria, and postsynaptic folds seen in the endplate myopathy (Engel et al., 1982; Choi, 1994). Elucidation of the pathogenesis of synaptic dysfunction in SCCMS will improve our understanding of kinetic disorders of the AChR, such as SCCMS, epilepsies, and other ion channelopathies.
Both the clinicopathological findings (severity of weakness and endplate myopathy) and the magnitude of change in synaptic currents in SCCMS vary significantly among individuals bearing different AChR mutations (Engel et al., 1996; Gomez et al., 1996b, 2002; Croxen et al., 1997). The ε subunit mutation, εL269F, has been identified in patients from at least two SCCMS kindreds and is associated with moderate to severe weakness (Gomez and Gammack, 1995; Engel et al., 1996). When expressed in vitro, this mutation causes a 9-fold increase in affinity for ACh and a 40-fold increase in AChR channel burst duration (Engel et al., 1996; Gomez et al., 1997). Single-channel currents recorded in εL269F patient endplates have burst durations similar to those recorded in endplates from SCCMS patients bearing other AChR mutations, but there is poor correlation between disease severity and the single AChR channel properties studied (Engel et al., 1996; Gomez et al., 2002). As with patients, transgenic mice expressing the εL269F mutation are weak, have prolonged synaptic currents, increased single channel open times, Ca2+ overload of endplate regions, and endplate myopathy (Gomez et al., 1997). Other SCCMS patients appear to have little or no endplate myopathy or severely perturbed ion channel kinetics, or both (Croxen et al., 1997; Gomez et al., 2002). Although the relationship is not yet clear, it is tempting to speculate that differences in kinetic properties of mutant AChRs in SCCMS are responsible for the different clinical and pathological picture. The determination of which critical changes in channel properties control the function and integrity of the neuromuscular synapse may have far-reaching implications for the diagnosis and treatment of ion channel disorders. In this study we used transgenic mice expressing different AChR subunit mutations to explore the relationship between the kinetics of endplate currents and weakness, impaired neuromuscular transmission, Ca2+ overload, and the degenerative changes at the synapse. We show that weakness, diminished miniature endplate current (MEPC) amplitudes, and endplate degeneration occur with increasing duration of endplate currents and synaptic Ca2+ overload.
MATERIALS AND METHODS
Oocyte studies. Characterization of AChR properties in Xenopus oocytes was performed as described previously (Charnet et al., 1992; Gomez et al., 1996a). Briefly, oocytes were injected with cRNA synthesized from linearized plasmids (pAlter,Promega, Madison WI) containing mutant or wild-type AChR subunit cDNAs using T7 polymerase and an in vitro transcription kit (mMessage mMachine, Ambion, Austin, TX). Fifty nanoliters of mRNA of mouse AChR subunit (at a concentration of 0.4 ng/nl) were injected into the cytoplasm of oocytes at an α/β/δ/ε ratio of 2:1:1:1. After 3 d, oocytes were placed individually in a 50 μl recording chamber. For recording, oocytes were kept under continuous gravity-driven perfusion. Signals were amplified using a Geneclamp 500 amplifier (Axon Instruments, Foster City, CA). Voltage-command, sampling, acquisition, and analysis were performed using a Digidata 1200 and the pClamp program (version 6.01, Axon Instruments). All experiments were performed at room temperature (20–25°C).
For macroscopic whole-cell recordings, voltage clamp of oocytes was performed using 3 m KCl-filled electrodes of 1–2 MΩ resistance. Junction potentials (typically <3–5 mV) were cancelled, and contaminating endogenous Ca2+-activated Cl− currents were suppressed by injecting BAPTA (in mm: BAPTA 100, HEPES 10, CsOH 10, pH 7.2) into oocytes by means of a third microelectrode and a pneumatic injector. The final intra-oocyte BAPTA concentration was estimated to be 2–5 mm. Current recording and analysis were performed using pClamp software.
The Ca2+/Na+permeability ratio for each mutant used to generate transgenic mice was measured by determining the change in reversal potential between Ca2+- and Na+- containing solutions. True ACh-induced currents were obtained by digital subtraction of the currents recorded during voltage ramps of −80 to +50 mV, both before and after ACh perfusion (ACh = 0.1–40 μm). Reversal potentials (Erev) were measured in solutions of the following composition (in mm): NaCl 100, HEPES 10, pH 7.2 with NaOH, or Ca50 (Ca2+Cl2 50, mannitol 50, HEPES 10, pH 7.2 with CaOH. Permeability ratios (PCa/PNa) were calculated as described (Mayer and Westbrook, 1987; Bertrand et al., 1993). Computations were done by the constant field theory using Goldman-Hodgkin-Katz equations for Na+, K+, and Ca2+permeabilities, yielding relative permeabilities. The activity coefficient for Ca2+ was 0.33, and calculations were performed using the Solver tool from Excel (Microsoft, Redmond, WA).
Single-channel recordings. Single-channel recordings were performed on both cell-attached and outside-out patches at room temperature (22–24°C) using the same extracellular and intrapipette solutions (100 mm KCl, 2 mmMgCl2, 10 mm HEPES, 10 mm EGTA, pH adjusted to 7.4 with KOH). In this solution, the oocyte resting membrane potential was close to 0 mV. ACh (2 μm) was included either in the bath solution (outside-out recordings) or in the pipette solution (cell-attached recordings). Pipettes made from KG33 borosilicate glass (Garner Glass, Claremont, CA), treated with Sylgard, and fire polished had a resistance of 10–15 MΩ. Data were recorded with a Geneclamp500 amplifier (Axon Instruments; 100 G-CV5 headstage), using Fetchex version 6.02 (Axon Instruments) in event-driven mode and digitized by a Labmaster interface. Single-channel open time and conductance were determined at different pipette voltages from −150 to +150 mV. Single-channel open times were measured from the time constant of exponential decays fitted to duration distributions of channel opening. Single-channel conductances were determined by fitting Gaussians to amplitude histograms.
AChR mutations and transgene constructs. We established previously that overexpression of a mutant AChR subunit in mouse muscle gives rise to mice with prolonged endplate currents (Gomez et al., 1996a, 1997). To develop a panel of transgenic mice with endplate responses that are prolonged over a wide range, we constructed transgenes from a series of distinct AChR subunit mutations. Site-directed mutagenesis of mouse α, δ, and ε subunit cDNAs (Lapolla et al., 1984; Boulter et al., 1985; Gardner, 1990) was performed using mutagenic oligonucleotide primers and single-stranded plasmid generated from the vector, pAlter, as per manufacturer's instructions (Promega). The mutations were confirmed by dideoxynucleotide sequence determination. The ε subunit mutation, εL269F, has been found in two families with moderately severe SCCMS (Gomez and Gammack, 1995; Engel et al., 1996). Overexpression of this mutation gave rise to several lines of mice that were found to develop progressive weakness and degeneration of the postsynaptic membrane, as is seen in the SCCMS (Gomez et al., 1997, 1998). The δ subunit mutation, δS262T, which is not known to be a spontaneously occurring mutation, was generated during structure-function studies of the M2 domain (Charnet et al., 1992). The kinetic properties of δS262T-AchRs and εL269F-AChRs in vitro and in vivo have been described previously (Engel et al., 1996; Gomez et al., 1996a, 1997). The α subunit mutations, αL251T and αC418W, were generated for this study but have been used by others in structure-function studies of the M2 (αL251T) and M4 (αC418W) domains (Filatov and White, 1995; Tamamizu et al., 1999). The properties of the AChRs expressing each of these mutations are described in Table 1.
Table 1.
Electrophysiological properties of oocytes and muscle expressing mutant AChRs
| Mutant | Study type | |||||||
|---|---|---|---|---|---|---|---|---|
| Single-channel oocytes | Voltage-clamp mouse diaphragm | |||||||
| τ11-a | τ2 | γ1-b | τs(n)1-c | τ1 | τ2 | Area (n)1-d | Amplitude (n)1-e | |
| Wild type | 0.8 ± 0.2 | ND1-f | 76 ± 6 | 1.31 ± 0.2 (5) | ND | ND | 6.8 ± 0.33 (18) | 2.72 ± 0.56 (5) |
| δS262T | 3.8 ± 0.7 | ND | 72 ± 1 | 2.6 ± 0.51 (4)* | 1.52 ± 0.25 | 5.2 ± 3.25** | 7.54 ± 0.83 (26) | 2.5 ± 0.55 (4) |
| αC418W | 0.5 ± 0.2 | 16 ± 3 | 82 ± 2 | 3.1 ± 0.69 (7)** | 1.64 ± 0.26 | 9.57 ± 3.0** | 11 ± 1.44 (17)* | 2.18 ± 0.32 (7) |
| αL251T | 2 ± 1 | 150 ± 13 | 81 ± 4 | 2.94 ± 0.89 (6)* | 1.22 ± 0.65 | 12.4 ± 3.3** | 8.43 ± 0.89 (17) | 2.12 ± 0.51 (6) |
| ɛL269F | 2.7 ± 3 | 39 ± 7 | 83 ± 1 | 10.1 ± 2.95 (8)** | 0.72 ± 0.26 | 25.8 ± 4.1** | 30.8 ± 3.8 (12)** | 2.04 ± 0.25 (8)*** |
All values are mean ± SEM (Weisberg, 1985). *p < 0.01, **p < 0.001, ***p < 0.04.
Time constants (milliseconds) derived from fitting of open duration histograms to mono- or bi-exponential equations (Gomez et al., 1996b, 1997; Tamamizu et al., 1999). n = 3-10 patches.
Single-channel conductance (picoSiemans).
Time constant (millisecond) of miniature endplate current decays fitted to one τs or two (τ1 and τ2) exponents (n= number of animals, where 4-18 endplates were studied per animal).
Mean area under curve described by MEPC (amperes per second).
Amplitude of MEPC (nA) (n = number of animals, where 4-18 endplates were studied per animal).
ND, No significant component detected.
The structure of the transgenes used to generate mice expressing each of these mutations was identical to those described previously (Gomez et al., 1996a, 1997). Each construct consisted of a minigene containing 3.3 kb of the promoter region from the mouse muscle creatine phosphokinase gene (Johnson et al., 1989) joined to the 5′ end of the mutant AChR subunit cDNA, which in turn is joined to the 5′ end of the 3′ UTR of neo and the SV 40 small-t intron from the construct pRSVNEO (Gorman et al., 1982).
Generation and characterization of slow-channel transgenic mice. Transgenic mice were generated by pronuclear injection followed by implantation into pseudopregnant females as described previously (Gomez et al., 1996a, 1997). Transgenic mice were derived on the FVB background (Taketo et al., 1991) with the exception of mice expressing δS262T, which were made on the C57BL/6 × DBA/2 F2 background (Gomez et al., 1996a). For each transgene, two to four founder lines, identified by analysis of tail DNA, were characterized initially. The expression levels of the transgenes were determined by RNA blotting using skeletal muscle total RNA separated by gel electrophoresis (Gomez et al., 1997). Blots were hybridized with labeled AChR subunit cDNA probes and, for standardization of levels among lines of mice expressing different mutations, to a universal transgene-specific probe consisting of a 1000 bp 3′ fragment from the common tail sequence.
Characterization of neuromuscular synaptic currents. MEPCs were recorded using the two-electrode voltage-clamp method as described previously (Maselli et al., 1989, 1991; Gomez et al., 1997). All microelectrode recordings were performed in the most posterior aspect of the left hemidiaphragm muscle. Muscle fibers were viewed using an upright microscope (Leitz Laborlux 12, Leitz Ltd., Heerbrugg, Switzerland) equipped with Hoffman interference contrast optics (magnification 250×). The preparations were bathed continuously in Tyrode's solution with the following ionic composition (in mm): NaCl 140, KCl 2.6, MgCl2 0.4, CaCl2 2.5, NaH2PO4 0.4, pH 7.4), bubbled with a 95% O2, 5% CO2 gas mixture at 26°C. Voltage and current electrodes were connected to an Axoclamp 2A amplifier (Axon Instruments), and the signals were sampled at 10 kHz by a 12-bit analog-to-digital converter (Data Translation 2818, Marlboro, MA). Exponential fits to the MEPC decays were performed using a nonlinear least square fitting routine. Because endplate currents in transgenic animals might reflect the activity of mixed populations of mutant and wild-type AChRs, leading to complex decay phases, both exponential decay time constants and the areas under the MEPCs were compared for each transgenic line. Grouped mean and SE were determined by the method of weighted least squares (Weisberg, 1985). The mean area under the MEPC is an estimate of the total amount of charge passing during receptor activation. Areas for each MEPC were obtained using a graphics program (Photoshop, Adobe, San Jose, CA). An average of 13 MEPCs were used to estimate the mean MEPC area for each transgene line.
Detection and quantitation of Ca2+-overloaded endplates. Mice were killed by anesthetic overdose either at rest or after an exercise protocol (see below). The number of Ca2+-overloaded neuromuscular junctions throughout the muscles of each transgenic line was estimated using a histological stain for motor endplates and accumulated Ca2+ on adjacent sections. Ca2+ accumulation (ionized Ca2+) at endplates was detected using the glyoxal bis 2 hydroxyanil (GBHA) method (Kashiwa, 1970; Evans, 1974;Bodensteiner and Engel, 1978). Motor endplates were localized using the stain for endplate-specific enzyme acetylcholinesterase (AChE) (Namba et al., 1967). Serial 8 μm frozen sections were taken from seven muscle groups, the muscular diaphragm, gastrocnemius, tibialis anterior, biceps, triceps, and the flexors and extensors of the forepaw. To facilitate counting and allow simultaneous labeling and examination of sections from each of these muscle groups, adjacent serial sections from each muscle group for each animal were arranged together on glass microscope slides. To prevent experimenter bias, the identities for each mouse were encoded. To estimate the number of motor endplates in these muscle sections, the first and third of three serial slides were stained for AChE. The brown-stained endplate regions in the seven sections of each first and third slide were counted by direct visualization, and the average number of endplates for the two slides was taken as the number of endplates present on the second slide. An average of 640 endplates was counted for each mouse. Slides with great differences in the endplate number were recounted and checked for loss of sections. Cholinesterase-reactive regions in muscle spindles, which were easily distinguished from normal muscle fibers, were not counted. To estimate the fraction of endplates that have become overloaded with Ca2+, the second slide of each series of three was stained using the GBHA procedure, and the number of red–black-stained, Ca2+-overloaded endplates was expressed as the ratio of the total number of cholinesterase-labeled endplates that were stained with GBHA.
Electron microscopy. For ultrastructural studies, mice were perfused transcardially with 2% glutaraldehyde. Forelimb flexor muscles were then removed and fixed overnight in fresh glutaraldehyde and rinsed in 0.1 m phosphate buffer. The muscle bellies were teased into 1 mm bundles, trimmed to lengths of 3 mm, osmicated, and embedded in Epon. Regions containing endplates were localized in toluidine blue-stained thick sections, and thin sections were obtained and viewed as described previously (Gomez et al., 1984,1997).
Exercise and measurement of strength. Muscle strength was assessed using a wire-hang test (Gomez et al., 1997), in which the ability of mice to grip and remain suspended from a wire was expressed as the mean of five trials. Briefly, mice were allowed to grip a taut wire suspended at a height of 35 cm by their front claws and were observed for periods of up to 60 sec. The possible outcomes consisted of escape or persistence on the wire for the entire period (score of 100%) or falling off before 1 min was reached (scored as the fraction of a minute). Five sets of three repetitions were conducted. Mice were tested with encoded identities and using littermate controls.
To increase neuromuscular synaptic activity, mice were subjected to a 30 min period of exercise that consisted of gently pulling the tail while the mouse gripped the bars of the cage top with forelimb and hindlimb claws. This led to a uniform routine of gentle exercise for limb muscle.
Quantitation of AChRs. The number of AChRs per endplate was estimated by determining the number of α bungarotoxin (α-BT) binding sites per cholinesterase-stained endplate region in teased muscle fibers segments using 125I-α-BT (Pestronk et al., 1985). Mice aged 16–21 months were killed by anesthetic overdose. The brachioradialis of the right forelimb was excised close to the origin. The excised muscle was (1) incubated at 37°C for 4 hr in Ham's F-12 culture medium containing 0.15 μg/ml125I-α-BT (specific activity 150–300 Ci/mmol), (2) rinsed 12 times at 5 min intervals with HBSS containing 0.5% BSA (fraction V), buffered to pH 7.2 with HEPES, (3) incubated at 4°C overnight, (4) washed five times at 5 min intervals, (5) pinned at resting length for fixation with 2.5% glutaraldehyde for 30 min, and (6) washed three times at 15 min intervals with 0.1m sodium acetate. The labeled, washed, and fixed muscle was gently teased to spread apart the muscle fibers and was stained for AChE to localize endplate regions (Namba et al., 1967). Sections of identical size with and without endplate regions were trimmed from each muscle belly. The number of endplates in the small bundles and the radioactivity were counted, and the specific125I-α-BT-binding to the endplates was determined by subtracting the background activity of the adjacent, endplate-free bundles.
Statistics. All statistical comparisons for single measurements were made using the Student's t test. Because strength testing and MEPC recordings were made on different animals in unpaired mouse groups for each transgene, a Matlab (The Mathworks, Natick, MA) routine was used to estimate a normally distributed set of paired variables to make possible a regression analysis. For this purpose, a weighted average of the normalized SDs (SD/mean) was made for the measurements for each transgenic mouse line. A normally distributed set of data points about the mean for eachx-y coordinate pair was calculated. It was assumed that the points were uniformly distributed in direction and normally distributed in distance from the mean.
RESULTS
In vitro characterization of mouse of AChR subunit mutations
To generate lines of transgenic mice with differing synaptic current properties, we chose AChR subunit mutations predicted to affect ion channel kinetics over a wide range. The mean single-channel open time and conductance recorded from expression studies inXenopus oocytes for the four mutations used in this study are compared with those of wild-type AChRs in Table1 and Figure1. The duration of the channel openings ranges from 0.8 msec in wild type to 150 msec in AChRs expressing the αL251T mutation. The single-channel conductance, determined from the current–voltage relationship with K+ as the predominant charge-carrying ion, varies little for the mutant AChRs (72–83 pS) (Table 1, Fig. 1B). The Ca2+/Na+relative permeabilities of the mutant AChRs are more variable than are the monovalent cation conductances, as shown on Figure 1C. The apparent increase in Ca2+ permeability for αL251T over wild type, i.e., 0.75 versus 0.61, for example, may relate to the addition of a polar residue in the ion permeation pathway (Imoto et al., 1991). On the other hand, the critical effect on Ca2+ permeability of the threonine at position 6′ in the M2 domain in the δS262T mutation suggests a previously unexpected effect of pore size on divalent cation conductance. Permeability to Ba2+ was similarly reduced for δS262T (data not shown). Thus, this panel of four AChR mutations with open times that vary >100-fold over that of wild type had a fourfold difference in relative Ca2+/Na+permeability and displayed minimal changes in conductance, allowing us to generate a panel of transgenic mice with widely differing synaptic currents.
Fig. 1.
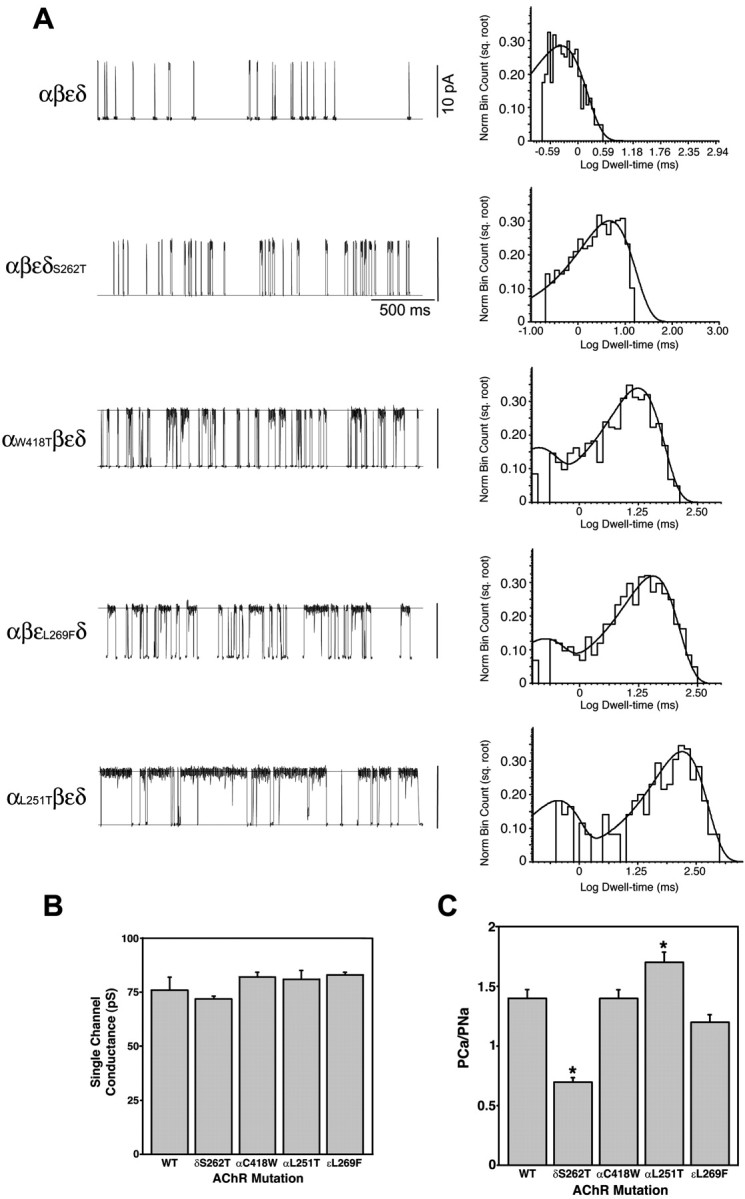
In vitro kinetic and permeation properties of a panel of mutant mouse AChRs used in transgenic mice.A, Single-channel recordings (left) and open duration histograms (right) for mouse wild-type and AChR mutants expressed in Xenopus oocytes. Recordings were performed at −100 mV and 2.0 μm ACh. Marquardt least squares fitting in pClamp 6 allowed resolution of two open components for each mutant (Table 1). B, Single-channel conductance obtained from oocyte patch-clamp recordings. There is no significant difference between the conductance for sodium among any of the mutants. C, Permeability of wild-type and mutant AChRs to calcium relative to sodium. The relative calcium permeability of αL251T (n = 17) AChRs is ∼33% greater than wild-type AChRs (n = 19;p < 0.005). The relative calcium permeability of δS262T AChRs (n = 20) is ∼67% less than wild type (p < 0.001).
A panel of mutant AChR subunit transgenes perturbs synaptic currents over a wide range
Using the mutant AChR subunit transgene constructs, we established at least three founder lines for each transgene (Gomez et al., 1996a,1997). The levels of expression of the mutant AChR subunit transgenes in skeletal muscle varied over a wide range, but at least two founder lines for each transgene had high levels of transgene expression. We selected lines with matched levels of high expression by hybridizing RNA blots with a common sequence probe present in all AChR subunit transgenes (Fig. 2A). To compare the levels of expression of the transgenes with those of endogenous AChR subunits, we hybridized RNA blots with a probe generated from α subunit cDNA (Fig. 2B). As noted previously, the levels of transgene expression greatly exceed those of endogenous AChR subunits (Merlie and Sanes, 1985; Gomez et al., 1996a). Although this degree of overexpression should lead, in theory, to complete substitution of the wild-type with mutant subunits (Bhattacharyya et al., 1997; Gomez et al., 1997), it is difficult to estimate the effect of the localized, subsynaptic expression of endogenous AChR subunits (Merlie and Sanes, 1985; Brenner et al., 1990).
Fig. 2.

Analysis of transgene mRNA expression. Expression of the mutant AChR mRNA in the panel of transgenic mice was comparable between several lines and well above that of endogenous mouse AChR subunit genes. A, Matched expression of AChR subunit transgenes. Top panel, Ten micrograms of total muscle RNA per mouse were hybridized with common sequence probe for all transgenes. Exposure was 4 d. Bottom panel, Same blot rehybridized with MCK cDNA to ensure uniform loading. Exposure was 4 hr. Lane 1, δS262T, line 40; lane 2, αC418W, line 42; lane 3, αL251T, line 39;lane 4, αL251T, line 36; lane 5, εL269F, line 14; lane 6, εL269F, line 5; lane 7, control muscle. B, Overexpression of mutant α subunits. Top panel, Total muscle RNA probed with mouse α subunit cDNA. Exposure was 5 d. Endogenous α subunit mRNA is undetectable at this explosure time. Lane 1, αC418W, line 42; lane 2, αL251T, line 39;lane 3, αL251T, line 36; lane 4, control muscle. Bottom panel, Same blot reprobed with MCK cDNA. Exposure was 4 hr. Size markers are in kilobases.
The duration of the MEPCs in mice expressing each mutant was significantly increased over control mice for each transgenic line, presumably reflecting the effects of the mutant AChRs on channel open time recorded in vitro (Table 1). The τs and τ2 time constants increased successively in transgenic lines δS262T, αC418W, αL251T, and εL269F. Figure3 compares the typical traces from control and transgenic mice expressing each mutation. In the αC418W, αL251T, and εL269F transgenic mice, the decay phases could be resolved well into two exponents (τ1 and τ2), with τ2proportions of ∼43, 17, and 45%, respectively. Based on the time constants of decay of wild-type MEPCs and single-channel data (Gomez et al., 1996a, 1997; Bhattacharyya et al., 1997), the τ1 may not necessarily reflect the open duration of a population of wild-type AChRs (Table 1). Nevertheless, in several cases the τ2 values, which varied nearly 20-fold over wild-type τs, are close to the long opening component of the corresponding mutant AChRs expressed in oocytes. The slow component, τ2, of the MEPC decay phase in αL251T mice, although increased to 12.4 msec, is smaller than the τ2 component of other lines of slow-channel mice and does not reflect the presence of the very long opening events (150 msec) resolved in oocyte expression studies. This may arise from the effect of coexpression of wild-type α subunits in muscle or a stabilizing effect of the mutant AChRs when associated with the subsynaptic apparatus compared with the oocyte membrane. Similar disparities have been seen between expression studies of recombinant AChRs bearing SCCMS mutations and single-channel studies using dissociated muscle fibers bearing the same mutation (Engel et al., 1996; Gomez et al., 1997). The value for MEPC area, which has the units of picocoulombs, is an estimate of amount of charge entering during synaptic activity for each transgenic line (Table 1). The mean MEPC area was significantly greater for αC418W and εL269F mice than for wild type (p < 0.01). The effect of increases in MEPC duration on MEPC area in αL251T mice was perhaps offset by the reduced MEPC amplitudes and low proportion of slow MEPC decay phase.
Fig. 3.
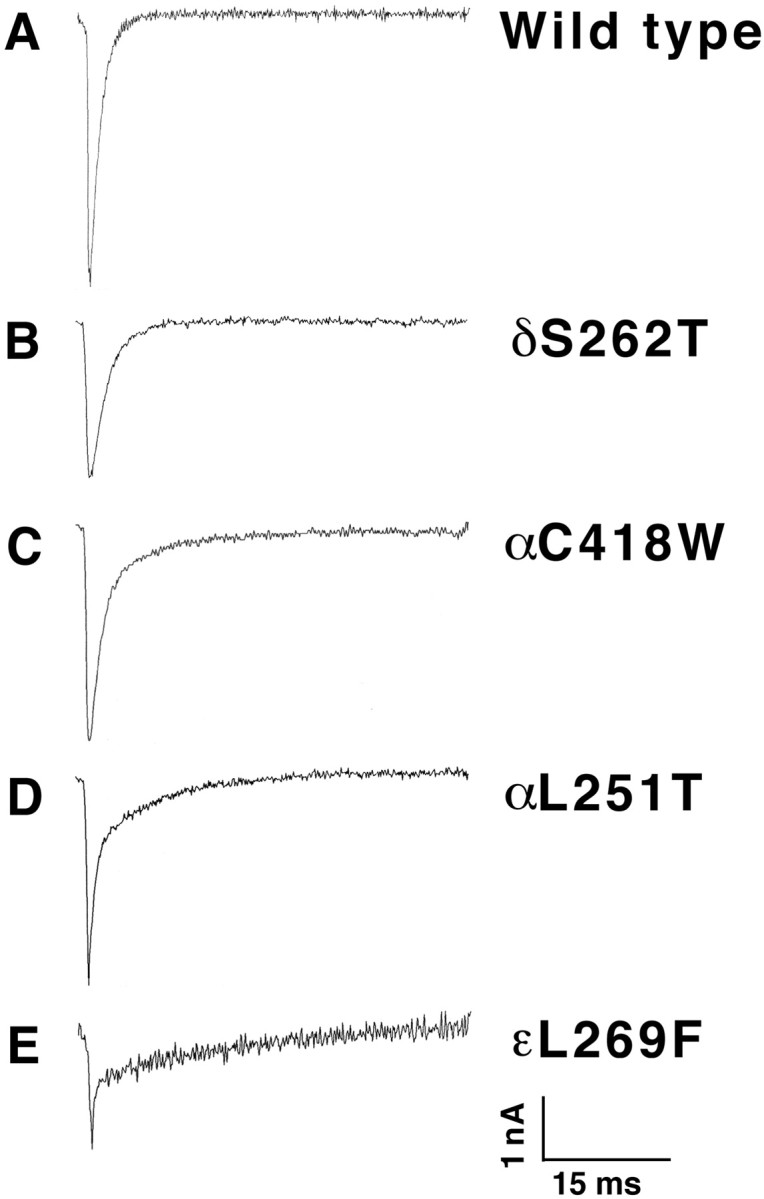
Miniature endplate current traces from individual diaphragm muscle fibers of wild type (A), δS262T (B), αC418W (C), αL251T (D), and εL269F (E). The prolonged and prominent bi-exponential decay phases correlate with the open times of the mutant channels expressed in the mice (Table 1). A, Average rise, 0.42 ± 0.47 msec; τs, 1.34 msec; amplitude (Amp), 4.15 ± 1.16 nA. B, Average rise, 0.32 ± 0.09 msec; τs = 2.34 msec; Amp, 2.69 ± 0.47 nA. C, Average rise, 0.25; τ1 = 1.52, τ2 = 9.89 msec; Amp, 3.48 nA. D, Average rise, 0.18 ± 0.14 msec; τ1 = 0.72 msec, τ2 = 8.37 msec; Amp, 3.31 ± 0.65 nA. E, Average rise, 0.37 ± 0.33 msec; τ1 = 0.17 msec, τ2 = 15.87 msec; Amp, 2.58 nA. Calibration: 1 nA, 15 msec.
Transgenic mouse strength varies with AChR mutation
The principal clinical features of impaired neuromuscular transmission in the SCCMS are weakness and fatigability (Engel et al., 1982; Gomez et al., 1996b). We demonstrated previously that εL269F mice have overt weakness (Gomez et al., 1997) and that δS262F mice are sensitive to low doses of curare (Gomez et al., 1996a). To compare the effect of the different AChR mutations on muscle strength we compared forelimb strength of mice from each transgenic line using a standardized, timed-grip strength test (Gomez et al., 1997). Weakness ranged from mild in the case of δS262F mice to severe in the case of the εL269F line (Fig.4A, left axis label, dark bars). There appeared to be a progression of increasingly poor performance in mice between lines in the order δS262F, αC418W, αL251T, and εL269F. Mice from three lines, αC418W (p < 0.025; n = 5), αL251T (p < 0.015; n = 4), and εL269F (p<<0.001; n = 8), performed significantly more poorly than control mice (n = 25).
Fig. 4.

Transgene-specific neuromuscular weakness correlates with MEPC current kinetics in slow-channel transgenic mice.A, Slow-channel mice exhibit a broad range of muscle weakness (left axis, dark bars). Strength testing was performed using a wire-hang paradigm (see Materials and Methods). Results are expressed as percentage of perfect score. The mean scores of αC418W (p < 0.025;n = 5), αL251T (p < 0.015; n = 4), and εL269F (p<<0.001;n = 8), but not that of δS262F (p = 0.5; n = 4), were significantly worse than age-matched control mice (n = 25). Performance by εL269F mice was also significantly poorer than αC418W mice (p< 0.02). Slow-channel mice have diminished miniature endplate current (MEPC) amplitudes (right axis,light bars). MEPC amplitudes (nanoamps) were recorded during voltage-clamp analysis of excised diaphragms of control (n = 5 mice) or δS262F (n = 4), αC418W (n = 7), αL251T (n = 6), or εL269F (n = 8) transgenic mice. The mean MEPC amplitude was significantly lower than control mice in εL269F transgenic mice (p< 0.025). B, εL269F (n = 7) transgenic mice have reduced endplate AChRs compared with control (p < 0.025; n = 4), as determined by 125I α−BT binding (see Materials and Methods). C, Plot of strength performance versus the slow decay time constant (τ2) of bi-exponential MEPCs for each transgenic mouse line. For this plot the value for τs was used for wild type because no τ2value was resolved for wild-type mice. D, Plot of strength performance versus the log of MEPC area as expressed in picocoulombs. Mouse lines for C and D:open circle, wild type; filled square, δS262; filled triangle, αC418W; filled diamond, αL251T; filled circle, εL269F.
The amplitude of the MEPC is a direct measure of synaptic strength and is a critical component of the safety factor of neuromuscular transmission. The MEPC amplitudes were diminished in each line, in parallel to the strength performance, although the reduction reached statistical significance only in εL269F mice (p < 0.025; n = 8) (Fig.4A, right axis label, light bars). The fact that strength performance was inversely proportional to MEPC amplitude for each transgenic line suggested that muscle weakness in these lines was predominantly of neuromuscular origin.
The postsynaptic causes of weakness and reduced MEPC amplitude include functional changes in AChR gating, reduced conductance of the AChR ion channel, reduced number and density of functional AChRs, and structural aspects of endplate degeneration (Engel et al., 1982;Bhattacharyya et al., 1997; Gomez et al., 1997, 1998, 2002; Milone et al., 1997). Figure 1B demonstrates that all the mutant AChRs used in this in vivo study have conductances similar to monovalent cations. We estimated the density of endplate AChRs by measuring the number of α-BT binding sites on muscle segments containing endplates. Endplate AChR numbers were significantly reduced to 50% of control (p < 0.025) in the εL269F transgenic mice, the line that had the greatest weakness and reduction of MEPC amplitudes, but were normal in other lines (Fig.4B). This suggests that reduction in AChR number plays a prominent role in neuromuscular weakness in εL269F mice. Additional mechanisms must contribute to the weakness in the other lines of slow-channel mice.
Changes in gating of AChRs bearing SCCMS mutations will predominantly affect the MEPC decay phase, although mutations with slowed activation rates that appear to reduce AChR responses (Gomez et al., 2002) have rarely been identified. To visualize how estimates of muscle strength for each transgenic line vary with line-specific differences in MEPC kinetics, we plotted the strength scores as a function of τ2 (Fig. 4C), τs (data not shown), and the mean MEPC area (Fig. 4D) for each transgenic line. τ2 and τs best reflect the duration of the endplate current, whereas MEPC area is an estimate of total charge entry during synaptic activity. The small number of mean data points and large variance for each value preclude an accurate prediction of a mathematical relationship. Nevertheless, it is clear that the strength for the mouse lines varies inversely with the duration of the MEPC and charge entry during synaptic activity. Although these relationships are based on unpaired transgene-specific measurements, we used weighted averages of the normalized SDs for the data from each mouse group and transgene to estimate a normal distribution of data points for a regression analysis. Of the three MEPC variables, τs, τ2, and area, the highest correlation coefficients were obtained for regressions between strength and MEPC area: r = 0.66 for a linear fit and r = 0.71 for an exponential fit. As is evident from the plot (Fig. 4D), if such a relationship exists between strength performance and MEPC area, the αL251T transgenic mice tend to perform somewhat more poorly than predicted. When the data points from the αL251T mice are removed, the correlation coefficients increase to r = 0.73 andr = 0.85 for linear and exponential fits, respectively. This suggests that AChRs bearing αL251T mutation may have a greater adverse effect on strength than predicted for estimates of MEPC duration, possibly because of the presence of additional properties.
Severe weakness is associated with active calcium overload of endplates
The greater adverse effect of the αL251T mutation on the strength and viability of the NMJ in slow-channel transgenic mice could be attributable to the fact that this mutation has additional properties not revealed in the voltage-clamp studies. Differences in Ca2+ permeability between SCCMS mutants is not readily apparent by comparing MEPCs or channel kinetics as they are routinely recorded. Figure 1C demonstrates that there are significant differences in relative Ca2+/Na+permeability between the same mutations as judged by reversal potential studies in oocytes. To explore the effects of different transgene mutations on Ca2+ entry into endplate regions, we used the histochemical stain GBHA as an indicator of gross Ca2+ overload. The GBHA stain is estimated to detect free Ca2+ in tissues at concentrations of 1–2 mm (Bodensteiner and Engel, 1978). It does not detect the Ca2+present in the sarcoplasmic reticulum or mitochondrial compartments, presumably because the Ca2+ is protein bound (Schwartz et al., 1967; Kashiwa, 1970; Bodensteiner and Engel, 1978). We determined the proportion of endplates that were overloaded with histochemically detectable Ca2+ for each transgenic line (Evans, 1974; Kawabuchi, 1982; Gomez et al., 1997) in a panel of seven muscle regions, using a histochemical stain for cholinesterase to localize endplates on adjacent sections. In muscle from normal mice or from mice bearing the δS262T mutation, there were no sites of staining with GBHA, either at endplate regions or elsewhere (Fig.5A,B). GBHA stain was present at some endplate regions in mice expressing the αC418W, αL251T, and εL269F mutant subunits, although in very different proportions (Fig.5A,B). Similar findings were obtained in mice from at least two founder lines for each transgene, indicating that the effect on Ca2+overload was not specific to transgene integration site. The proportion of Ca2+-overloaded endplates detected in resting αL251T mice (2.8%) was only slightly greater than that in αC418W mice (0.47%; p = 0.08). However, the proportion of GBHA-stained endplates in resting εL269F transgenic mice was significantly greater than that of the other resting mice (p ≪ 0.001).
Fig. 5.

Calcium accumulation at motor endplates differs with transgenic expression of distinct AChR mutations.A, Sections of forelimb flexor muscles from resting δS262T, αC418W, αL251T, and εL269F mice demonstrate differences in degree of GBHA staining for different transgenic lines. Sections in the left panels were stained for cholinesterase to localize endplates (cholinesterase is brown) (Namba et al., 1967) and counterstained with hematoxylin and eosin. The adjacent serial sections in the right panels were stained for glyoxal bis 2 hydroxyanil (Evans, 1974) (GBHA, dark red or black) and methylene blue. There is no GBHA staining at control (data not shown) or δS262T endplates. Scale bar, 25 μm. B, Calcium accumulation at motor endplates differs according to mutation and increases with exercise. The proportions of motor endplates stained histochemically with GBHA in αC418W, αL251T, and εL269F mice at rest or after exercise is displayed in vertical barslabeled below. Dark bars indicate the lines that develop endplate myopathy (Fig. 6). At rest, no GBHA stain was detected at control or δS262T endplates, whereas 0.47% were labeled in αC418W mice, 2.8% in αL251T, and 19% in εL269F mice. The proportion of Ca2+-overloaded endplates detected in resting αL251T mice (n = 6) was slightly greater than that in αC418W mice (0.47%; p = 0.08;n = 6). The proportion of GBHA-stained endplates in resting εL269F transgenic mice was significantly greater than that of the other resting mice (p ≪ 0.001;n = 5). After exercise (see Materials and Methods), there was still no GBHA staining at motor endplates or elsewhere in muscle fibers of wild-type or δS262T transgenic mice, whereas marked increases in GBHA staining were seen in the other three transgenic lines after exercise. In exercised αC418W transgenic mice (n = 5), the proportion of endplates stained for GBHA increased to 3.9%, an eightfold increase (p < 0.01). In αL251T transgenic mice (n = 4) and εL269F transgenic mice (n = 4), the proportion of GBHA-stained endplates increased to 26% (10-fold; p < 0.002) and 33% (1.7-fold; p < 0.04), respectively. The proportions of Ca2+-overloaded endplates after exercise in αL251T transgenic mice and εL269F transgenic mice were significantly greater than the Ca2+-overloaded endplates in exercised αC418W transgenic mice (p < 0.01).
To further explore the relationship between synaptic activity and Ca2+ overload in slow-channel transgenic mice, we investigated the effect of changes in neuromuscular activity on endplate Ca2+ overload. We found that exercise had a marked and selective effect on Ca2+ overload of transgenic endplates (Fig. 5B). Mice were given a 30 min standardized exercise protocol consisting of forced gripping of the cage top with all four paws. No GBHA staining was detected at motor endplates or elsewhere in muscle fibers of exercised wild-type or δS262T-transgenic mice. In contrast, in mice bearing the αC418W, αL251T, and εL269F mutations, exercise had a potent effect on the proportion of endplates overloaded with Ca2+. In αC418W transgenic mice, the proportion of endplates stained for GBHA increased to 3.9%, an eightfold increase (p < 0.01). In αL251T and εL269F transgenic mice, the proportion of GBHA-stained endplates increased to 26% (10-fold; p < 0.002) and 33% (1.7-fold; p < 0.04), respectively. Moreover, the proportions of Ca2+-overloaded endplates after exercise in αL251T transgenic mice and εL269F transgenic mice were significantly greater than the Ca2+-overloaded endplates in exercised αC418W transgenic mice (p < 0.01). Thus, in mice expressing mutant AChR subunits, Ca2+accumulation at motor endplates increased with neuromuscular activity.
Structural correlates of impaired neuromuscular transmission
Classical SCCMS is associated with striking degenerative changes of the postsynaptic membrane, referred to as endplate myopathy (Engel et al., 1982). We previously reported similar degenerative changes of the endplate region in εL269F mice (Gomez et al., 1997). Comparison of the NMJ at the ultrastructural level in the four transgenic lines studied here showed that δS262T and αC418W mice had essentially normal endplates (Fig.6A), although a few vacuoles were found in some αC418W endplates. In both αL251T (Fig.6B) and εL269F (Fig. 6C) transgenic mice, however, numerous endplates had marked ultrastructural changes. The most prominent abnormalities were collections of membrane-bound structures that in some cases filled the junctional sarcoplasm and appeared to arise from dilated sarcoplasmic reticulum. These vacuole-like structures were often small and uniform-sized in αL251T mice, but in εL269F mice and occasionally in αL251T mice these vacuoles were quite variable in size and filled with fluffy or granular material (Fig. 6C). In both transgenic lines, several other ultrastructural abnormalities typical of the endplate myopathy in the SCCMS were present and included degenerating nuclei, dilated or degenerating mitochondria, and membranous or granular debris. Although difficult to quantitate, these abnormalities, especially the nuclear degeneration, were qualitatively more abundant in εL269F transgenic mice. We found that 56% of endplates were abnormal in αL251T mice, compared with 66% of endplates in εL269F mice. Degenerating nuclei were seen in 25% of αL251T mice and in 55% of εL269F mice. The pronounced nuclear degeneration in εL269F mice most likely contributes to the marked reduction in endplate AChRs in εL269F transgenic mice because AChR subunit mRNA transcription is restricted to the subsynaptic nuclei (Merlie and Sanes, 1985; Brenner et al., 1990). These results support earlier findings which demonstrated that the severe ultrastructural changes in the NMJ seen in SCCMS can underlie progressive changes in muscle strength (Gomez et al., 2002). Moreover, the presence of severe endplate myopathy is consistent with the intense endplate calcium overload and the greater degree of weakness in αL251T and εL269F mice.
Fig. 6.

Endplate myopathy occurs in slow-channel transgenic mice with severe calcium overload. A, Neuromuscular junction from a 2-month-old αC418W mouse showing essentially normal ultrastructure of nerve terminus (white arrowheads), postsynaptic folds (black arrow), and junctional sarcoplasm (asterisk).B, Neuromuscular junction from 2-month-old αL251T mouse. The junctional sarcoplasm is filled with myriads of vacuole-like structures ranging in size from 0.05 to 1 μm (asterisk). Vacuoles are empty or filled with fluffy or granular material. Profiles of nuclei are present at either side of the accumulation of vacuoles. One nucleus appears severely degenerated (black arrow). In B and Cthe secondary synaptic folds and clefts are absent, and the nerve terminals (white arrowheads) are small and barely recognizable at the outer surface of the bulging postsynaptic regions.C, Neuromuscular junction from a εL269F mouse at 2 months. Vacuoles fill the junctional sarcoplasm and are present within the underlying sarcomeres (asterisk). The subsynaptic mitochondria are abnormally enlarged compared with those in the nerve terminus. Some show accumulations of dark, dense granules consistent with calcium (data not shown). Others contain multiple clear inclusions (black arrowhead). Some mitochondria are pathologically dilated (black arrow). One subsynaptic nucleus has normal ultrastructure. Scale bars: A, 0.9 μm;B, 4.1 μm; C, 1.5 μm.
DISCUSSION
The succinct termination of excitatory synaptic responses is important for reproducible activation and recovery of excitatory synapses. In the SCCMS, progressive weakness and degeneration of the postsynaptic portion of the neuromuscular junction is associated with missense mutations in the AChR that lead to prolonged opening of the AChR ion channel (Engel et al., 1982, 1996; Gomez et al., 1996b, 1997). This suggests that proper termination of synaptic responses is necessary to preserve the viability of the neuromuscular junction. The presence of deposits of Ca2+ at the neuromuscular junctions of these patients strongly suggests a role for Ca2+ overload in the pathogenesis of endplate degeneration (Engel et al., 1982).
The strength of neuromuscular transmission depends on several structural, cellular, and neurophysiological aspects of the neuromuscular junction, including cleft width, activity of AChE, and the density and channel properties of AChRs (Engel et al., 1982;Oosterhuis et al., 1987; Gomez et al., 1996b, 1997, 2002). Some of these properties are altered in the SCCMS and its animal model. In this study we have used transgenic mice expressing different AChR subunit mutations to determine the relationships between kinetic properties of synaptic currents and the strength of neuromuscular transmission, focal Ca2+ overload, and ultrastructural indicators of neuromuscular disease. The results are summarized in Table 2.
Table 2.
Summary of neuromuscular properties in transgenic mice2-a
| Mouse line | Strength testing | MEPC duration | MEPC area | MEPC amp | Calcium overload | |||
|---|---|---|---|---|---|---|---|---|
| Rest | Exercise | AChR loss | Endplate myopathy | |||||
| Wild type | − | − | − | − | − | − | − | − |
| δS262T | + | + | − | + | − | − | − | − |
| αC418W | ++ | ++ | + | + | + | + | − | +/− |
| αL251T | ++ | ++ | − | + | ++ | +++ | − | ++ |
| ɛL269F | +++ | +++ | ++ | ++ | +++ | +++ | ++ | ++ |
Assignments of + based on rank order and magnitude change in each parameter.
The limitations of this approach relate to issues of transgene expression. Expression of the transgenes derived by pronuclear injection occurs in combination with, rather than in place of, that of the endogenous subunits. The mutant phenotype relies on overexpression of the mutant gene. Although the bulk expression of these transgenes is considerably greater than that of endogenous genes, variations in expression between muscle fibers and even between myonuclei may arise because of position effects conferred by the sites of transgene integration. Such variations may affect a proportion of mutant AChRs in a given endplate and hence endplate kinetics. This is particularly the case for the expression of α subunit mutants, given the subunit stoichiometry of α2βδε. We believe the conclusions are valid for several reasons. (1) To minimize the effects of regional differences in expression, all comparisons between transgenes were made on measurements averaged over multiple mice, muscle groups, and muscle fibers. (2) There were no appreciable differences between lines bearing distinct integrations of the same transgene, suggesting that position effects could not explain the differences. (3) All transgenes were expressed at levels at least 100-fold above endogenous subunits (Gomez et al., 1996a). Moreover, there were no differences in electrophysiological abnormalities in mice expressing the δS262T transgene over a 50-fold range (Gomez et al., 1996a), indicating that the mutant phenotype was not sensitive to transgene expression level.
In the panel of slow-channel transgenic mice, neuromuscular weakness and reduction in MEPC amplitudes correlated well over a broad range of muscle strength. Milder weakness and reduced MEPC amplitudes were seen in δS262F and αC418W mice, which had kinetic changes but little or no Ca2+ overload. The mild impairment of neuromuscular transmission could not be attributed to structural changes or to AChR loss and is likely caused by additional functional changes arising at least in part from the kinetic disturbance (Milone et al., 1997; Gomez et al., 2002). Previous studies have shown that delayed AChR activation or increased desensitization may contribute significantly to reduced synaptic strength in some SCCMS patients or slow-channel mice (Bhattacharyya et al., 1997; Gomez et al., 2002). Functional changes are likely to include intrinsic properties of mutant AChRs, effects of smaller increases in Ca2+ entry on the rate of desensitization, blockade, and compensatory changes in the neuromuscular transmission (Ochoa et al., 1989; Siara et al., 1990; Nojima et al., 1994; Fenster et al., 1999).
Greater weakness and the most severe endplate myopathy were seen in the αL251T and εL269F transgenic mice. Intense overload of endplates with Ca2+, at rest or with exercise, attributable to the prolonged MEPC duration or a higher Ca2+ permeability overload, appear to be most closely associated with the typical changes in the SCCMS. Weakness was greatest in εL269F mice, the line that displayed the greatest perturbations in channel kinetics, active endplate Ca2+ overload, endplate myopathy, and endplate AChR loss. Thus, both channel gating activity and ion permeation are critical to maintain the ionic environment of the subsynaptic region and lead to active Ca2+overload, degeneration of the endplate, and impaired neuromuscular transmission when disrupted.
The findings in this study demonstrate that Ca2+ overload occurs as a direct result of mutant AChR channel activity. Ca2+overload in some slow-channel transgenic mice and patients is detectable using a relatively insensitive histochemical assay. The GBHA stain, which is the most sensitive for soluble Ca2+, has an estimated detection limit of 1–2 mm (Bodensteiner and Engel, 1978). Using this same assay we also detected a severalfold increase of Ca2+ overloaded endplates in transgenic mice after brief exercise. These results show that for some mutants, Ca2+ overload is governed by the Ca2+ permeability, open duration, and even frequency of opening of the AChR channel at the neuromuscular junction. The muscle AChR ion channel is highly permeable to Ca2+, which represents ∼2–3% of ions that comprise the adult endplate current (Decker and Dani, 1990). Nevertheless, Ca2+ does not accumulate at normal endplates unless AChE is inhibited pharmacologically (Evans, 1974; Kawabuchi, 1982). Changes in the kinetics of the channel opening affect the quantity of Ca2+ that enters the junctional sarcoplasm during synaptic activity. The findings in this study suggest that there are discrete thresholds for the duration of synaptic currents beyond which Ca2+ overload and endplate degeneration occur. The determinants of these thresholds are complex and related to the size of the neuromuscular junction and muscle fiber and Ca2+-buffering systems present in the junctional sarcoplasm (Kohr and Mody, 1991; Gunter and Gunter, 1994;Hartmann et al., 1994).
On the basis of the ultrastructural data, we predict that Ca2+ accumulates in the sarcoplasm, the sarcoplasmic reticulum, and the mitochondria (Fig. 6) (Gomez et al., 1997). The marked Ca2+ overload is associated with severe ultrastructural changes at the neuromuscular junction. The dilated vacuoles, localized contractures, and degenerating mitochondria and nuclei, termed endplate myopathy, which were seen in the αL251T and εL269F mice have been described previously in both mice and patients (Engel et al., 1982; Gomez et al., 1996b, 1997; Milone et al., 1997). Excessive intracellular Ca2+ levels might activate a number of harmful enzymatic or free radical degradative pathways. We have found evidence that the Ca2+-activated protease, calpain, contributes to impaired neuromuscular transmission in εL269F mice (J. Groshong, B. P. S. Vohra, M. J. Spencer, and C. M. Gomez, unpublished observations). Moreover, in mice and patients, multiple components of the caspase cascade appear to elicit a process of localized subcellular apoptosis (Vohra, R. Zayas, Groshong, R. A. Maselli, R. L. Wollmann, and Gomez, unpublished observations).
The findings reported here may have a number of important clinical implications. First, the capacity of some Ca2+ buffering systems, such as the mitochondria, may diminish with age (Martinez et al., 1992; Satrustegui et al., 1996), such that a given mutation might not lead to a phenotype until advanced age. This fact could provide an explanation for late onset or progression of some forms of the disease despite the lifelong presence of the mutation (Engel et al., 1982; Croxen et al., 1997). Second, deterioration of such buffering systems in excitatory pathways, for example, in mitochondrial disorders (Brini et al., 1999; Wasniewska et al., 2001), may be a route by which normal amounts of Ca2+ entry may lead to neurodegeneration. Third, because neuromuscular activity increases the extent of Ca2+ overload, the value of exercise in the long-term management of the SCCMS is uncertain. Finally, the biochemical processes coupling Ca2+overload to endplate degeneration are complex and may include activation of Ca2+-activated neutral protease, calpain, Dnase, or damage to cells through overproduction of free radicals (Choi, 1992). Collectively, these results suggest that agents that either serve to block Ca2+ entry through mutant ion channels or that enhance Ca2+ buffering would be additional neuroprotective strategies for this syndrome and those such as stroke and epilepsy, which are associated with excitotoxic neuronal stress.
Footnotes
This work was supported by National Institutes of Health Grants RO1 NS33202 and RO1 NS36809 and the Muscular Dystrophy Association to C.M.G., and by North Atlantic Treaty Organization Grant CRG 972059, Association Francaise contre les Myopathies, Association pour la Recherche contre le Cancer, and Ligue Nationale contre le Cancer to P.C. We thank Drs. John H. Anderson and Bruce Lynn for helpful discussions.
Correspondence should be addressed to Dr. Christopher M. Gomez, Box 295, Departments of Neuroscience and Neurology, 420 Delaware Street Southeast, Minneapolis, MN 55455. E-mail:gomez001@tc.umn.edu.
REFERENCES
- 1.Bertrand D, Galzi JL, Devillers TA, Bertrand S, Changeux JP. Mutations at two distinct sites within the channel domain M2 alter calcium permeability of neuronal alpha 7 nicotinic receptor. Proc Natl Acad Sci USA. 1993;90:6971–6975. doi: 10.1073/pnas.90.15.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhattacharyya BJ, Day J, Gundeck JE, Leonard S, Wollmann RW, Gomez C. Desensitization of mutant acetylcholine receptors in transgenic mice reduces the amplitude of synaptic currents. Synapse. 1997;27:367–377. doi: 10.1002/(SICI)1098-2396(199712)27:4<367::AID-SYN10>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 3.Bodensteiner JB, Engel AG. Intracellular calcium accumulation in Duchenne dystrophy and other myopathies: a study of 567,000 muscle fibers in 114 biopsies. Neurology. 1978;28:439–446. doi: 10.1212/wnl.28.5.439. [DOI] [PubMed] [Google Scholar]
- 4.Boulter J, Luyten W, Evans K, Mason P, Ballivet M, Goldman D, Stengelin S, Martin G, Heinemann S, Patrick J. Isolation of a clone coding for the α-subunit of a mouse acetylcholine receptor. J Neurosci. 1985;5:2545–2552. doi: 10.1523/JNEUROSCI.05-09-02545.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brenner HR, Witzemann V, Sakmann B. Imprinting of acetylcholine receptor messenger RNA accumulation in mammalian neuromuscular synapses. Nature. 1990;344:544–547. doi: 10.1038/344544a0. [DOI] [PubMed] [Google Scholar]
- 6.Brini M, Pinton P, King MP, Davidson M, Schon EA, Rizzuto R. A calcium signaling defect in the pathogenesis of a mitochondrial DNA inherited oxidative phosphorylation deficiency. Nat Med. 1999;5:951–954. doi: 10.1038/11396. [DOI] [PubMed] [Google Scholar]
- 7.Charnet P, Labarca C, Lester HA. Structure of the gamma-less nicotinic acetylcholine receptor: learning from omission. Mol Pharmacol. 1992;41:708–717. [PubMed] [Google Scholar]
- 8.Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- 9.Choi DW. Calcium and excitotoxic neuronal injury. Ann NY Acad Sci. 1994;747:162–171. doi: 10.1111/j.1749-6632.1994.tb44407.x. [DOI] [PubMed] [Google Scholar]
- 10.Croxen R, Newland C, Beeson D, Oosterhuis H, Chauplannaz G, Vincent A, Newsomdavis J. Mutations in different functional domains of the human muscle acetylcholine receptor alpha subunit in patients with the slow-channel congenital myasthenic syndrome. Hum Mol Genet. 1997;6:767–774. doi: 10.1093/hmg/6.5.767. [DOI] [PubMed] [Google Scholar]
- 11.Decker ER, Dani JA. Calcium permeability of the nicotinic acetylcholine receptor: the single-channel calcium influx is significant. J Neurosci. 1990;10:3413–3420. doi: 10.1523/JNEUROSCI.10-10-03413.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Engel AG. Myasthenic syndromes. In: Engel AG, Franzini-Armstrong C, editors. Myology. McGraw-Hill; New York: 1994. pp. 393–418. [Google Scholar]
- 13.Engel AG, Lambert EH, Mulder DM, Torres CF, Sahashi K, Bertorini TE, Whitaker JN. A newly recognized congenital myasthenic syndrome attributed to a prolonged open time of the acetylcholine-induced ion channel. Ann Neurol. 1982;11:553–569. doi: 10.1002/ana.410110603. [DOI] [PubMed] [Google Scholar]
- 14.Engel AG, Ohno K, Milone M, Wang HL, Nakano S, Bouzat C, Pruitt JN, Hutchinson DO, Brengman JM, Bren N, Sieb JP, Sine SM. New mutations in acetylcholine receptor subunit genes reveal heterogeneity in the slow-channel congenital myasthenic syndrome. Hum Mol Genet. 1996;5:1217–1227. doi: 10.1093/hmg/5.9.1217. [DOI] [PubMed] [Google Scholar]
- 15.Evans RH. The entry of labeled calcium into the innervated region of the mouse diaphragm. J Physiol (Lond) 1974;240:517–533. doi: 10.1113/jphysiol.1974.sp010621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fenster CP, Beckman ML, Parker JC, Sheffield EB, Whitworth TL, Quick MW, Lester RA. Regulation of alpha4beta2 nicotinic receptor desensitization by calcium and protein kinase C. Mol Pharmacol. 1999;55:432–443. [PubMed] [Google Scholar]
- 17.Filatov GN, White MM. The role of conserved leucines in the M2 domain of the acetylcholine receptor in channel gating. Mol Pharmacol. 1995;48:379–384. [PubMed] [Google Scholar]
- 18.Gardner PD. Nucleotide sequence of the epsilon-subunit of the mouse muscle nicotinic acetylcholine receptor. Nucleic Acids Res. 1990;18:6714. doi: 10.1093/nar/18.22.6714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gomez CM, Gammack JT. A leucine-to-phenylalanine substitution in the acetylcholine receptor ion channel. In: A family with the slow-channel syndrome. Neurology. 1995;45:982–985. doi: 10.1212/wnl.45.5.982. [DOI] [PubMed] [Google Scholar]
- 20.Gomez CM, Wollmann RL, Richman DP. Induction of the morphologic changes of both acute and chronic experimental myasthenia by monoclonal antibody directed against acetylcholine receptor. Acta Neuropathol. 1984;63:131–143. doi: 10.1007/BF00697195. [DOI] [PubMed] [Google Scholar]
- 21.Gomez CM, Bhattacharyya BB, Charnet P, Day JW, Labarca C, Wollmann RW, Lambert EH. A transgenic mouse model of the slow-channel syndrome. Muscle Nerve. 1996a;19:79–87. doi: 10.1002/(SICI)1097-4598(199601)19:1<79::AID-MUS11>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 22.Gomez CM, Ricardo Maselli BS, Lasalde J, Tamamizu S, Cornblath DR, Lehar M, McNamee M, Kuncl RW. A β subunit mutation in the acetylcholine receptor channel gate causes severe slow-channel syndrome. Ann Neurol. 1996b;39:717–723. doi: 10.1002/ana.410390607. [DOI] [PubMed] [Google Scholar]
- 23.Gomez CM, Maselli R, Gundeck JE, Chao M, Day JW, Tamamizu S, Lasalde JA, McNamee M, Wollmann RL. Slow-channel transgenic mice: a model of postsynaptic organellar degeneration at the neuromuscular junction. J Neurosci. 1997;17:4170–4179. doi: 10.1523/JNEUROSCI.17-11-04170.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gomez CM, Maselli R, Williams JM, Bhattacharyya BB, Wollmann RL, Day JW. Genetic manipulation of AChR responses in transgenic mice points to multiple causes of weakness in slow-channel syndrome. Ann NY Acad Sci. 1998;841:167–180. doi: 10.1111/j.1749-6632.1998.tb10923.x. [DOI] [PubMed] [Google Scholar]
- 25.Gomez C, Maselli R, Vohra B, Navedo M, Stiles J, Charnet P, Schott K, Rojas L, Keesey J, Verity A, Wollmann R, Lasalde-Dominicci J. Novel delta subunit mutation in slow-channel syndrome causes severe weakness by novel mechanisms. Ann Neurol. 2002;51:102–112. doi: 10.1002/ana.10077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gorman CM, Merlino GT, Willingham MC, Pastan I, Howard BH. The Rous sarcoma virus long terminal repeat is a strong promoter when introduced into a variety of eukaryotic cells by DNA-mediated transfection. Proc Natl Acad Sci USA. 1982;79:6777–6781. doi: 10.1073/pnas.79.22.6777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gunter KK, Gunter TE. Transport of calcium by mitochondria. J Bioenerg Biomembr. 1994;26:471–485. doi: 10.1007/BF00762732. [DOI] [PubMed] [Google Scholar]
- 28.Hartmann H, Eckert A, Muller WE. Disturbances of the neuronal calcium homeostasis in the aging nervous system. Life Sci. 1994;55:2011–2018. doi: 10.1016/0024-3205(94)00381-5. [DOI] [PubMed] [Google Scholar]
- 29.Imoto K, Konno T, Nakai J, Wang F, Mishina M, Numa S. A ring of uncharged polar amino acids as a component of channel constriction in the nicotinic acetylcholine receptor. FEBS Lett. 1991;289:193–200. doi: 10.1016/0014-5793(91)81068-j. [DOI] [PubMed] [Google Scholar]
- 30.Johnson JE, Wold BJ, Hauschka SD. Muscle creatine kinase sequence elements regulating skeletal and cardiac muscle expression in transgenic mice. Mol Cell Biol. 1989;9:3393–3399. doi: 10.1128/mcb.9.8.3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kashiwa HK. Calcium phosphate in osteogenic cells. A critique of the glyoxal bis(2-hydroxyanil) and the dilute silver acetate methods. Clin Orthop. 1970;70:200–211. [PubMed] [Google Scholar]
- 32.Kawabuchi M. Neostigmine myopathy is a calcium ion-mediated myopathy initially affecting the motor end-plate. J Neuropathol Exp Neurol. 1982;41:298–314. doi: 10.1097/00005072-198205000-00005. [DOI] [PubMed] [Google Scholar]
- 33.Kohr G, Mody I. Endogenous intracellular calcium buffering and the activation/inactivation of HVA calcium currents in rat dentate gyrus granule cells. J Gen Physiol. 1991;98:941–967. doi: 10.1085/jgp.98.5.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lapolla RJ, Mayne KM, Davidson N. Mouse muscle nicotinic acetylcholine receptor δ subunit: cDNA sequence and gene expression. Proc Natl Acad Sci USA. 1984;81:7970–7974. doi: 10.1073/pnas.81.24.7970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lerche H, Jurkat-Rott K, Lehmann-Horn F. Ion channels and epilepsy. Am J Med Genet. 2001;106:146–159. doi: 10.1002/ajmg.1582. [DOI] [PubMed] [Google Scholar]
- 36.Martinez SA, Blanco P, Satrustegui J. Calcium binding to the cytosol and calcium extrusion mechanisms in intact synaptosomes and their alterations with aging. J Biol Chem. 1992;267:4672–4679. [PubMed] [Google Scholar]
- 37.Maselli R, Nelson D, Richman D. Effects of a monoclonal anti-acetylcholine receptor antibody on the avian end-plate. J Physiol (Lond) 1989;411:271–283. doi: 10.1113/jphysiol.1989.sp017573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maselli R, Mass D, Distad B, Richman D. Anconeus muscle: a human preparation suitable for in-vitro microelectrode studies. Muscle Nerve. 1991;14:1189–1192. doi: 10.1002/mus.880141208. [DOI] [PubMed] [Google Scholar]
- 39.Mayer ML, Westbrook GL. The physiology of excitatory amino acids in the vertebrate central nervous system. Prog Neurobiol. 1987;28:197–276. doi: 10.1016/0301-0082(87)90011-6. [DOI] [PubMed] [Google Scholar]
- 40.Merlie JP, Sanes JR. Concentration of acetylcholine receptor mRNA in synaptic regions of adult muscle fibres. Nature. 1985;317:66–68. doi: 10.1038/317066a0. [DOI] [PubMed] [Google Scholar]
- 41.Milone M, Wang HL, Ohno K, Fukudome T, Pruitt JN, Bren N, Sine SM, Engel AG. Slow-channel myasthenic syndrome caused by enhanced activation, desensitization, and agonist binding affinity attributable to mutation in the m2 domain of the acetylcholine receptor α subunit. J Neurosci. 1997;17:5651–5665. doi: 10.1523/JNEUROSCI.17-15-05651.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muley S, Gomez C. Congenital myasthenic syndromes. In: Kaminiski H, editor. Mysathenia gravis and related disorders. Humana; Totowa, NJ: 2002. pp. 309–326. [Google Scholar]
- 43.Namba T, Nakamura T, Grob D. Staining for nerve fiber and cholinesterase activity in fresh frozen sections. Am J Clin Pathol. 1967;47:75–79. doi: 10.1093/ajcp/47.1.74. [DOI] [PubMed] [Google Scholar]
- 44.Nojima H, Kimura I, Kimura M. The evidence of accelerative interaction between cAMP-dependent protein kinase and external calcium for the desensitization of nicotinic acetylcholine receptor channel in mouse skeletal muscle cells. Neurosci Lett. 1994;167:113–116. doi: 10.1016/0304-3940(94)91040-5. [DOI] [PubMed] [Google Scholar]
- 45.Ochoa EL, Chattopadhyay A, McNamee MG. Desensitization of the nicotinic acetylcholine receptor: molecular mechanisms and effect of modulators. Cell Mol Neurobiol. 1989;9:141–178. doi: 10.1007/BF00713026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oosterhuis HJ, Newsom-Davis J, Wokke JH, Molenaar PC, Weerden TV, Oen BS, Jennekens FG, Veldman H, Vincent A, Wray DW, Prior C, Murray NMF. The slow channel syndrome. Two new cases. Brain. 1987;110:1061–1079. doi: 10.1093/brain/110.4.1061. [DOI] [PubMed] [Google Scholar]
- 47.Pestronk A, Drachman DB, Self SG. Measurement of junctional acetylcholine receptors in myasthenia gravis: clinical correlates. Muscle Nerve. 1985;8:245–251. doi: 10.1002/mus.880080311. [DOI] [PubMed] [Google Scholar]
- 48.Satrustegui J, Villalba M, Pereira R, Bogonez E, Martinez SA. Cytosolic and mitochondrial calcium in synaptosomes during aging. Life Sci. 1996;59:429–434. doi: 10.1016/0024-3205(96)00322-0. [DOI] [PubMed] [Google Scholar]
- 49.Schwartz M, Kashiwa HK, Jacobson A, Rehm WS. Concentration and localization of calcium in frog gastric mucosa. Am J Physiol. 1967;212:241–246. doi: 10.1152/ajplegacy.1967.212.2.241. [DOI] [PubMed] [Google Scholar]
- 50.Siara J, Ruppersberg JP, Rudel R. Human nicotinic acetylcholine receptor: the influence of second messengers on activation and desensitization. Pflügers Arch. 1990;415:701–706. doi: 10.1007/BF02584008. [DOI] [PubMed] [Google Scholar]
- 51.Steinlein OK. Genes and mutations in idiopathic epilepsy. Am J Med Genet. 2001;106:139–145. doi: 10.1002/ajmg.1571. [DOI] [PubMed] [Google Scholar]
- 52.Taketo M, Schroeder AC, Mobraaten LE, Gunning KB, Hanten G, Fox RR, Roderick TH, Stewart CL, Lilly F, Hansen CT. FVB/N: an inbred mouse strain preferable for transgenic analyses. Proc Natl Acad Sci USA. 1991;88:2065–2069. doi: 10.1073/pnas.88.6.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tamamizu S, Lee Y, Hung B, McNamee MG, Lasalde-Dominicci JA. Alteration in ion channel function of mouse nicotinic acetylcholine receptor by mutations in the M4 transmembrane domain. J Membr Biol. 1999;170:157–164. doi: 10.1007/s002329900545. [DOI] [PubMed] [Google Scholar]
- 54.Wasniewska M, Karczmarewicz E, Pronicki M, Piekutowska-Abramczuk D, Zablocki K, Popowska E, Pronicka E, Duszynski J. Abnormal calcium homeostasis in fibroblasts from patients with Leigh disease. Biochem Biophys Res Commun. 2001;283:687–693. doi: 10.1006/bbrc.2001.4834. [DOI] [PubMed] [Google Scholar]
- 55.Weisberg S. Applied linear regression, Ed 2. Wiley; New York: 1985. [Google Scholar]