

Abstract
Hypoxic preconditioning provides protection against ischemic brain lesions in animal models of cerebral ischemia–hypoxia. To analyze the underlying molecular mechanisms, we developed an invitro model of hypoxic neuroprotection in cerebellar granule neurons (CGN) by reducing the oxygen tension to 1–5% for 1–24 hr. Exposure to 5% O2 for 9 hr resulted in reduction of cell death after potassium deprivation, treatment with 100 μm glutamate, or 500 μm 3-nitroproprioninc acid (3-NP) by 46, 22, and 55%, respectively. Shorter (1 or 3 hr) or longer (>12 hr) intervals or pretreatment with lower oxygen tension failed to rescue CGN from death. In contrast, toxicity of four different chemotherapeutic drugs [1-(2-chloroethyl)-3-cyclohexyl-1-nitrosourea, cisplatine, topotecane, and vincristine] was unaffected by hypoxic preconditioning. The induction of protective effects was dependent on new protein synthesis. Protein levels of B-cell lymphoma protein-2 (BCL-2), BCL-xL/S, heat shock protein 70/90, and BCL-2-associated death protein remained unaltered. CGN incubated at 5% O2 for 9 hr showed increased levels of the vascular endothelial growth factor (VEGF), the VEGF receptor-2 (VEGFR-2), phosphorylated Akt/protein kinase B (PKB), and extracellular signal-regulated kinase 1 (ERK1). Incubation with a neutralizing anti-VEGF antibody, a monoclonal antibody to VEGFR-2, wortmannin, or antisense-Akt/PKB, but not treatment with U0126, an ERK-inhibitor, reverted the resistance acquired by hypoxic preconditioning. Inhibition of VEGFR-2 blocked the activation of Akt/PKB. Finally, pretreatment with recombinant VEGF resulted in a hypoxia-resistant phenotype in the absence of hypoxic preconditioning. Our data are indicating a sequential requirement for VEGF/VEGFR-2 activation and Akt/PKB phosphorylation for neuronal survival mediated by hypoxic preconditioning and propose VEGF as a hypoxia-induced neurotrophic factor.
Keywords: neuron, vascular endothelial growth factor, DC101, Akt, antisense oligonucleotide, glutamate, potassium, 3-nitropropionic acid
Preconditioning to ischemic tolerance is a phenomenon in which brief episodes of a subtoxic insult induce a robust protection against the deleterious effects of subsequent, prolonged ischemia. Hypoxic–ischemic preconditioning induces tolerance to focal ischemia (Miller et al., 2001), global ischemia (Kitagawa et al., 1990), and neonatal hypoxia (Jones and Bergeron, 2001). There are two temporally and mechanistically distinct types of protection afforded by preconditioning stimuli, acute and delayed preconditioning. Although they may share components of the signaling pathway, they differ in their requirement of new protein synthesis. The protective effects of acute preconditioning are protein synthesis independent, mediated by post-translational protein modifications, and are short lived. The effects of delayed preconditioning require new protein synthesis and are sustained for days and weeks (Nandagopal et al., 2001).
Descriptive studies have shown increased expression of heat shock proteins (HSPs) (Sharp et al., 1999), B-cell lymphoma protein-2 (BCL-2) (Shimazaki et al., 1994), and superoxide dismutase and decreased expression of NMDA receptor NR2A and NR2B subunits (Shamloo et al., 1999). NMDA receptor activation followed by nitric oxide (NO)-mediated activation of p21Rasand nuclear factor-κB (NF-κB) appears to be a crucial step in the generation of a signal transduction pathway resulting in hypoxic tolerance (Grabb and Choi, 1999; Gonzalez-Zulueta et al., 2000;Blondeau et al., 2001).
Recently, it became apparent that neurotrophic factors, such as nerve growth factor, brain-derived neurotrophic factor (BDNF), and insulin-like growth factor-1, protect cerebellar granule neurons (CGN), motor neurons, or hippocampal neurons from excitotoxic and ischemic insults (Segal and Greenberg, 1996; Han and Holtzman, 2000; Matsuzaki et al., 2001). These factors activate intracellular signal transduction pathways, including the phosphatidyl-inositol 3-kinase (PI3-K)/Akt and mitogen-activated protein (MAP) kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) pathways (Segal and Greenberg, 1996; Dudek et al., 1997). Vascular endothelial growth factor (VEGF) is an angiogenic peptide that is released in response to hypoxia in developing or neoplastic tissue; it acts on endothelial cells to promote the sprouting of blood vessels (Neufeld et al., 1999). The angiogenic action of VEGF involves an antiapoptotic effect that promotes endothelial cell survival and is mediated through the VEGF receptor-2 (VEGFR-2) and the PI3-K/Akt signaling pathway (Gerber et al., 1998). Systemic treatment of an experimental intracerebral glioma in mice with DC101, a monoclonal antibody to VEGFR-2, inhibited glioma angiogenesis and growth (Kunkel et al., 2001). The role for VEGF in neurons is less well defined. Deletion of the hypoxia-response element in the VEGF promoter in “knock-in” mice reduced the hypoxic VEGF expression in the spinal cord and caused motor neuron degeneration (Oosthuyse et al., 2001). In this study, it remained unclear whether VEGF was a survival factor for neurons through its effects on endothelial and/or neuronal cells. Others have demonstrated neurotrophic activity for VEGF on Schwann cells and survival-promoting effects on immortalized hippocampal neurons (Sondell et al., 1999; Jin et al., 2000).
These observations and the current interest in the therapeutic potential of VEGF for stroke treatment led us to investigate whether preconditioning with moderate hypoxia resulted in the induction of VEGF-dependent neuroprotective pathways. We used CGN to analyze the effects of hypoxic preconditioning without glucose deprivation on potassium deprivation-induced apoptosis, glutamate-induced excitotoxicity, and inhibition of oxidative phosphorylation by 3-nitroproprioninc acid (3-NP), an irreversible inhibitor of succinate dehydrogenase. Our results indicate that moderate hypoxia of 5% O2 applied for 3–9 hr protects CGN against toxic stimuli involving a pathway that sequentially requires the activation of VEGFR-2, PI3-K, and Akt.
MATERIALS AND METHODS
Cell culture. Cerebellar granule neurons were prepared from 7-d-old Sprague Dawley rats as described previously (Weller et al., 1994). In brief, freshly dissected cerebella were dissociated by mechanical disruption and by incubation at 37°C for 15 min in 0.3 mg/ml trypsin. Cells were plated in poly-l-lysine-precoated 60 mm culture plates or 24-well plates and seeded at a density of 1 × 106 cells/cm2in basal modified Eagle's medium supplemented with 10% fetal calf serum, 2 mm glutamine, 20 μg/ml gentamicin, and 25 mm KCl (high K+). Cytosine arabinoside was added at a concentration of 10 μm after 24 hr to prevent the growth of non-neuronal cells. Contamination with glial cells was <5% (Schulz et al., 1996).
Oxygen system. The standard oxygen level was defined as the pO2 that exists in a standard, conventional, humidified tissue culture incubator at 37°C (20%). The low (1–5%) oxygen systems were established in a humidified environmental chamber set at 37°C (C42; Labotect, Göttingen, Germany). This incubator used an oxygen analyzer to monitor and maintain the selected chamber oxygen concentration. This oxygen concentration was maintained with a calibrated gas mixture of 95% nitrogen and 5% carbon dioxide. The cells were incubated on day 8 after preparation for 1–12 hr at 1–5% pO2.
Treatment of cultures. In all experiments, neurons were cultured for 7 d in 25 mmK+ and 10% fetal calf serum before use. For studies of K+ deprivation, medium was replaced by serum-free basal modified Eagle's medium containing 5 mm potassium (low K+) or 25 mmpotassium (high K+) and supplemented with glutamine and gentamicin as indicated above. Glutamate (Invitrogen, Beverly, MA), 3-NP (Sigma, St. Louis, MO), 1-(2-chloroethyl)-3-cyclohexyl-1-nitrosourea (CCNU) (Bristol, Munich, Germany), cisplatine (Bristol), topotecane (GlaxoSmithKline Pharmaceuticals, Harlow, UK), or vincristine (Bristol) were added to neurons cultured in high K+ and serum on day 8 after preparation. Viability was measured at 24 hr after hypoxic preconditioning. To investigate whether protein synthesis is relevant, cycloheximide (CHX) (Sigma) was added to the cells. Wortmannin was purchased from Sigma. LY924002 was obtained from Biomol (Plymouth Meeting, PA), and U0126 (dissolved in DMSO from Sigma) was purchased from Calbiochem (La Jolla, CA).
Determination of viability. Neurons plated in 24-well plates were used for assessment of viability. Viability was measured by the capability of the cells to diesterify and retain fluorescein diacetate (FDA) in their cytoplasm. Medium was removed from neuronal cultures, and cells were incubated at 37°C for 5 min with Locke's solution (in mm: 154 NaCl, 5.6 KCl, 2.3 CaCl2, 1 MgCl2, 3.6 NaHCO3, 5 HEPES, and 20 glucose) containing 5 μg/ml FDA. Cultures were washed once with Locke's solution and examined under fluorescent light microscopy. Cell numbers were determined as described previously (Schulz et al., 1996). In brief, three random fields were chosen from each well and digitized by a CCD camera connected to an image processor (MCID-V; Imaging Research, St. Catharines, Ontario, Canada). Images were filtered, and the total number of stained cells was counted automatically by MCID-V computer software.
Immunoblot analysis. Blotting was performed essentially as described previously (Schulz et al., 1996). CGN were seeded on 60 mm dishes. After hypoxic preconditioning, medium was removed, and cultures were washed once in cold PBS before the cells were lysed for 15 min on ice in lysis buffer (1% Triton X-100 and 0.1% SDS with 10 μg/μl leupeptin and aprotinin). Cell debris was removed by high-speed centrifugation at 4°C. Samples containing 20 μg of protein were boiled in 1% SDS and 1% β-mercaptoethanol for 5 min, separated by 10–15% SDS-PAGE, and electrotransferred to a nitrocellulose membrane. Filters were blocked for 1 hr in blocking solution (10 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.1% Tween 20, and 5% skim milk), followed by incubation with primary antibodies overnight at 4°C and secondary, horseradish peroxidase-linked antibodies. Bound antibody was visualized using enhanced chemiluminescence (ECL kit; Amersham Pharmacia Biotech, Braunschweig, Germany). The following primary antibodies were used at a concentration of 2.5 μg/ml: rabbit polyclonal αphospho-Akt (Ser473) antibody (Cell Signaling Technology, Beverly, MA); α protein kinase B (PKB)/Akt antibody (Transduction Laboratories, Lexington, KY); α BCL-2-associated death protein (BAD) antibody (catalog #9292; Cell Signaling Technology); α-phospho-p44/42 MAP kinase antibody (E10 monoclonal; catalog #9106), αVEGF antibody (C-1; sc-7269), αVEGF antibody (147; sc-507), αVEGFR-1/Flt-1 antibody (A-3; sc-6251), αVEGFR-2/Flk-1 (C-17; sc-316), αHSP90 antibody (N-17; sc-1055), αERK1/2 antibody (1CK-23; sc-94), αBCL-2 antibody (N-19; sc-492), and αBCL-xL/s antibody (S-18; sc-634) (all from Santa Cruz Biotechnology, Santa Cruz, CA).
Akt antisense oligonucleotides. Transfection with an Akt antisense oligonucleotide (sequence, gct ggt ggt cct ggc cat ga; Interactiva Biotechnologie, Ulm, Germany) or a nonsense oligonucleotide (sequence, gac caa gat ccg acg gga tc; Interactiva Biotechnologie) at a DNA concentration of 1 μm was performed using Effectene transfection (Qiagen, Hilden, Germany).
DC101 antibody. The rat monoclonal antibody DC101 against VEGFR-2 was provided by ImClone Systems (New York, NY) and used at 10 μg/ml. The rat control IgG (R & D Systems, Minneapolis, MN) was used at 10 μg/ml.
Neutralizing VEGF antibody. The human neutralizing antibody against VEGF (10 μg/ml) (R & D Systems) and the human control IgG (R & D Systems) were applied at 10 μg/ml.
Recombinant VEGF. The rat protein was obtained from R & D Systems and used at 100 ng/ml.
Statistical analysis. Data are expressed as mean ± SEM. Statistical significance was assessed by one-way ANOVA, followed by Tukey's post hoc test. All experiments reported here represent at least three independent replications performed in triplicate. Values of p < 0.05 were considered significant.
RESULTS
Hypoxic preconditioning prevents apoptotic cell death in CGN
To establish an in vitro model of hypoxic preconditioning with chronic nonlethal hypoxia, we exposed CGN at dayin vitro 8 for 1–24 hr to a reduced oxygen tension of 1–10%. After reincubation in the standard oxygen tension of 21% for 24 hr, CGN that had been exposed to O2concentrations between 1 and 4% showed time-dependent cell death, whereas CGN that had been exposed to 5% O2remained fully viable (Fig. 1). We next asked whether this lowest nonlethal oxygen concentration of 5% was sufficient to alter the response of CGN to three established toxic stimuli and tried to determine the optimal exposure for preconditioning. We preexposed CGN to 5% O2 for 1, 3, 6, 9, 12, or 24 hr. Twenty-four hours after reexposure to 21% O2, we challenged CGN with potassium deprivation, with 100 μm glutamate, or with 500 μm 3-NP. Preconditioning by oxygen deprivation for 9 hr resulted in a reduction of cell death induced by potassium deprivation, glutamate, or 3-NP by 46, 22, and 55%, respectively (Fig.2A). Shorter (1 or 3 hr) or longer (>12 hr) intervals of low oxygen preconditioning failed to rescue the cells from death (data not shown). To determine whether this protective effect was a general principle for toxic stress in neurons, we challenged CGN with different chemotherapeutics (CCNU, cisplatine, topotecane, and vincristine) after hypoxic preconditioning. We showed recently that these chemotherapeutics induced apoptosis in CGN (A. Wick, W. Wick, R. Dringen, M. Weller, and J. B. Schulz, unpublished observations). However, there were no differential toxicities of the chemotherapeutics in preconditioned versus normoxic CGN (data not shown). Caspase-3 activity was required for cell death induced by potassium deprivation, glutamate, and 3-NP because caspase-3 was activated, and the panspecific caspase inhibitor zVAD-fmk (for N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone) inhibited cell death in these paradigms (data not shown). We next analyzed whether increased neuronal survival after hypoxic preconditioning was attributable to reduced caspase-3 activity in the different models of neuronal cell death. Preexposure of cerebellar granule neurons to 5% O2 for 9 hr led to a reduction of caspase-3 activity as determined by DEVD-amc (forN-acetyl-Asp-Glu-Val-Asp-amino-4-methylcoumarin) hydrolysis after potassium deprivation and glutamate treatment (Fig.2B). To determine whether the effect of hypoxic preconditioning depended on new protein synthesis, we exposed CGN to the protein synthesis inhibitor CHX during the 9 hr period of hypoxia and for another 15 hr under normoxic conditions before switching to parallel conditioned sister medium. At this time, cells were switched to conditioned sister culture medium to reestablish optimal culture conditions. The cells were treated with potassium deprivation, glutamate, and 3-NP at 24 hr after reexposure to 21% O2. Treatment with CHX abolished the death inhibitory effects of hypoxia-induced preconditioning (Fig.2C).
Fig. 1.

Oxygen deprivation-induced death of CGN. Neurons were incubated at different oxygen concentrations (1% O2, whitebars; 5% O2, black bars) for the indicated intervals. Viability was determined 24 hr after transfer to normoxic conditions. Data are expressed as mean ± SEM percentages of viability (n = 3; *p < 0.05; **p < 0.01; ANOVA, followed by Tukey'spost hoc test compared with normoxic controls).
Fig. 2.
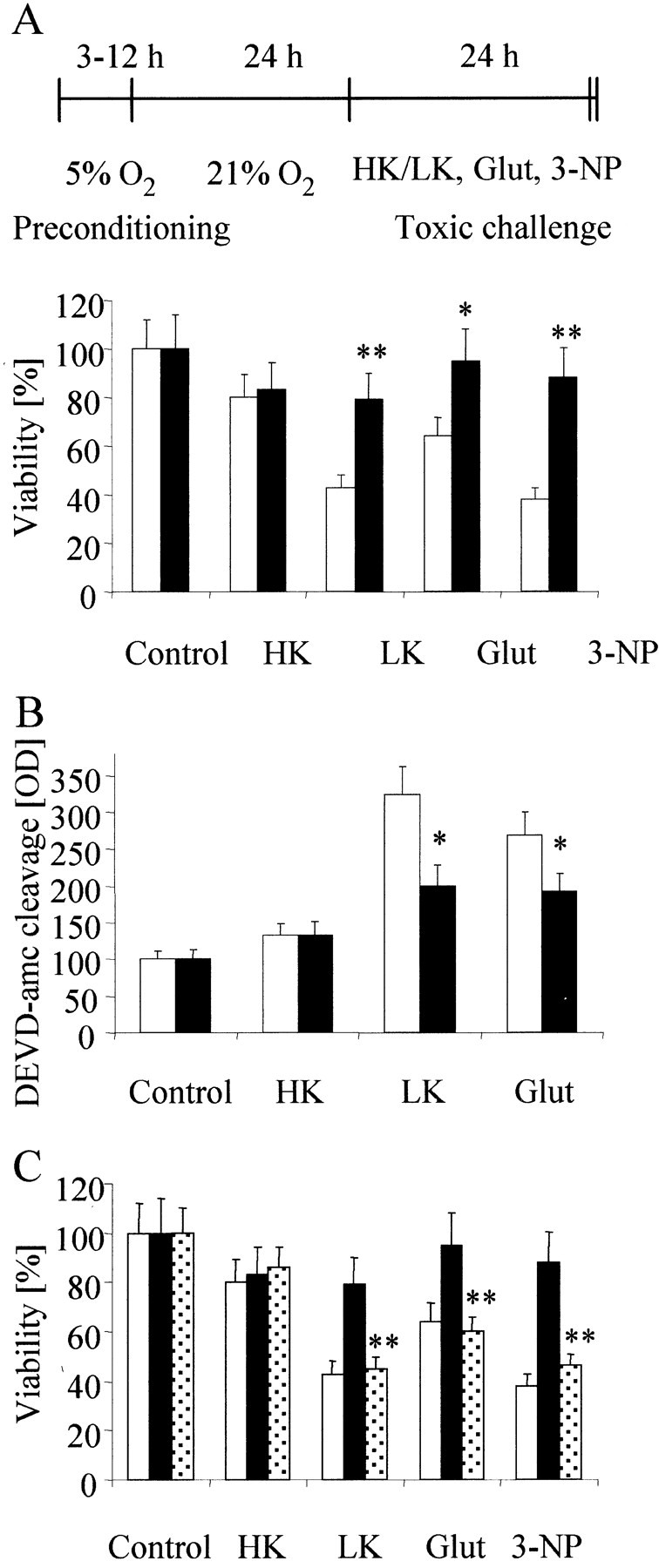
Hypoxic preconditioning attenuates cytotoxicity induced by potassium deprivation, glutamate, or 3-NP. A, CGN were exposed to 5% O2 for 9 hr (black bars) or not (white bars), reoxygenated for subsequent 24 hr, and challenged with potassium deprivation, 100 μm glutamate (Glut), or 500 μm 3-NP for 24 hr. Viability was assessed by FDA staining. B, CGN were treated as in A, and caspase-3 activity was measured by the cleavage of DEVD-amc (12.5 μm) at 6 hr after potassium deprivation or exposure to glutamate. Data are expressed as means ± SEM from three independent experiments performed in triplicate (*p< 0.05; **p < 0.01 compared with normoxic controls). C, CGN were treated according to the schedule displayed in A (black bars andwhite bars) or with CHX (10 μg/ml) in addition to incubation for 9 hr at 5% O2 and for another 15 hr before switching to regular parallel conditioned medium (dotted bars). Data are expressed as means ± SEM from three independent experiments performed in triplicate (*p< 0.05; **p < 0.01; ANOVA, followed by Tukey'spost hoc test compared with hypoxic but not with CHX-treated controls). HK, High K+;LK, low K+.
Neither BCL-2 family proteins nor HSP90 are induced by hypoxic preconditioning in CGN
Because induced expression of antiapoptotic proteins of the BCL-2 family has been described after hypoxia–reoxygenation treatment in hippocampal neurons (Tamatani et al., 2000), we examined whether a shift in the ratio of antiapoptotic and proapoptotic proteins of the BCL-2 family correlated with the observed preconditioning phenomenon. Protein levels of BAD, BCL-xL/S, or BCL-2 were not altered in response to hypoxic preconditioning. Similarly, protein levels of HSP90 that is regulated in neurons in response to different cellular stresses remained stable 24 hr after hypoxic challenging (5% O2) at different intervals (Fig.3).
Fig. 3.

No induction of BCL-2, BCL-xL/s, BAD, or HSP90 in hypoxic preconditioning. CGN were cultured at 5% O2 for the indicated intervals. The levels of BCL-2, BCL-xL/S, or HSP90 proteins were assessed by immunoblot. Co, Control.
Hypoxia-induced neuroprotection from death-inducing stimuli is mediated by VEGFR-2
Recent studies provided first evidences that VEGF, which was assumed to specifically affect endothelial cells, is a survival factor for CNS neurons (Matsuzaki et al., 2001; Oosthuyse et al., 2001). Transient hypoxia results in an increased expression of VEGF and its receptors (Marti and Risau, 1998). However, to date, a functional role of VEGF in ischemic preconditioning has not been defined. In our model, we found an increased expression of VEGF and VEGFR-1 and -2 after 1, 6, and 9 hr of 5% hypoxia (Fig.4A). To prove that induction of the VEGF/VEGFR system is indeed sufficient to protect from toxic stimuli, we used recombinant VEGF and a VEGF antibody with neutralizing function in CGN treated with potassium deprivation and glutamate excitotoxicity. Interestingly, VEGF mimicked whereas a neutralizing antibody against VEGF completely abolished the protective effects of hypoxic preconditioning (Fig.4B,C). To analyze the functional significance of the induction of VEGFR-2, we used a monoclonal antibody against VEGFR-2, DC101, that has been associated previously with inhibition of tumor angiogenesis (Rockwell et al., 1995; Prewett et al., 1999). DC101 alone was not toxic to control neurons. Treatment of CGN with 10 μg/ml DC101 in parallel to exposure to 5% hypoxia nullified the protective effects of hypoxic preconditioning against cell death induced by potassium deprivation, glutamate (Fig.5), or 3-NP (data not shown).
Fig. 4.
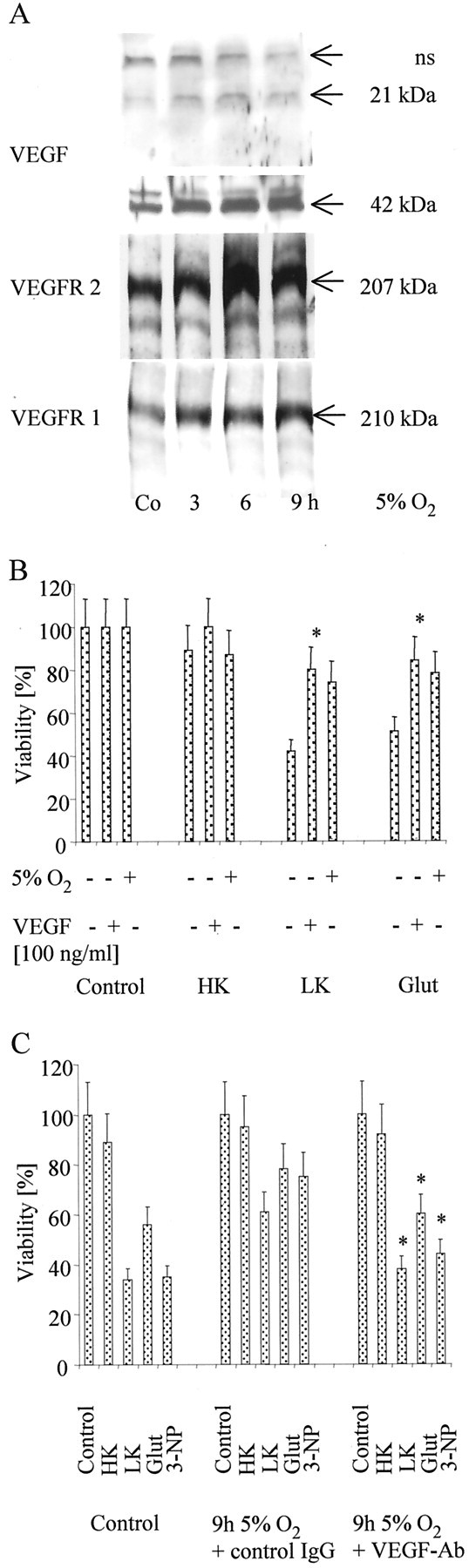
VEGF in hypoxia-induced neuroprotection.A, CGN were cultured at 5% O2 for the indicated intervals. VEGF (monomer at 21 kDa, dimer at 42 kDa; nonspecific bands, ns), VEGFR-2, or VEGFR-1 were assessed by immunoblot. B, CGN were exposed to 5% O2 for 9 hr, reoxygenated for subsequent 24 hr, or pretreated with recombinant VEGF (100 ng/ml) for 24 hr or left untreated, and, subsequently, they were challenged as in Figure2A. Viability was assessed by FDA staining. Data are expressed as means ± SEM from three independent experiments performed in triplicate (*p < 0.05, for the effect of VEGF; ANOVA, followed by Tukey's post hoc test).C, CGN were exposed to 5% O2 as detailed inA, challenged with potassium deprivation, glutamate, or 3-NP as displayed in Figure 2A, and were cotreated with an anti-human IgG control-antibody (10 μg/ml), or with a human neutralizing VEGF-antibody (10 μg/ml) (VEGF-Ab) as indicated. Viability was assessed by FDA staining. Data are expressed as means ± SEM from three independent experiments performed in triplicate (*p< 0.05, for the effect of the VEGF-antibody compared with the control-antibody; ANOVA, followed by Tukey's post hoctest). HK, High K+; LK, low K+; Glut, glutamate.
Fig. 5.

Inhibition of VEGFR-2 abolishes hypoxia-induced neuroprotection. CGN were treated as displayed in Figure2A or with DC101 or a control rat IgG alone, or cotreated with DC101 or rat IgG as labeled. Viability was assessed by FDA staining. Data are expressed as means ± SEM from three independent experiments performed in triplicate (*p< 0.05, for the effect of DC101 compared with rat IgG; ANOVA, followed by Tukey's post hoc test). HK, High K+; LK, low K+;Glut, glutamate.
Hypoxic preconditioning critically depends on Akt activation
The ERK and the PI3-K pathways have been implicated in paradigms of rescue from apoptosis in neurons. Because the PI3-K pathway has been implicated in VEGF-mediated protection (Gratton et al., 2001) and because BDNF protects the neonatal brain of Sprague Dawley rats via phosphorylation of ERK1/2 (Han and Holtzman, 2000), we investigated whether either pathway was involved in the paradigm of hypoxic preconditioning. Figure6A shows an increase in the phosphorylation of ERK, predominantly of the p44 form, 24 hr after exposure to a low oxygen tension of 5% for different time periods. To analyze whether this ERK activation was functionally relevant for the protective effects after hypoxic preconditioning, we treated CGN with the MEK1/2 inhibitor U0126. There was no influence of U0126 at up to 50 μm on the protective effects induced by hypoxic preconditioning, although the hypoxia-induced ERK phosphorylation was inhibited (data not shown). Similar to ERK, Akt phosphorylation, but not protein expression, was induced by hypoxic preconditioning for 9 hr (Fig. 6B). After exposure to the PI3-K inhibitor wortmannin, the phosphorylation of the serine 473 was completely blocked. In addition, both wortmannin (data not shown) and an Akt antisense oligonucleotide prevented the protective effects of hypoxic preconditioning on potassium deprivation and glutamate or 3-NP toxicity (Fig. 6C). The next experiment aimed at defining whether Akt is the key target for VEGF/VEGFR-2-controlled hypoxic neuroprotection in cerebellar granule neurons. Recombinant VEGF, similar to exposure to 5%O2 for 9 hr, lead to increased Akt phosphorylation. The blocking VEGFR-2 antibody DC101 abrogated the hypoxia-induced Akt phosphorylation (Fig. 6D), confirming that the pathway from hypoxia to enhanced survival of CGN involves VEGFR-2 and enhanced Akt activation.
Fig. 6.

Inhibition of the Akt pathway blocks hypoxia-induced neuroprotection. A, B, CGN were cultured at 5% O2 for the indicated intervals. ERK, phospho-ERK, Akt/PKB, or phospho-Akt levels in neurons treated with (+) or without (−) wortmannin (WM) were measured by immunoblot (nonspecific bands, ns).C, CGN were treated as displayed in Figure2A (white bars, normoxic neurons;black bars, hypoxic neurons) or treated with Akt antisense oligonucleotide or a nonsense control oligonucleotide in parallel to the induction of hypoxia. Viability was assessed by staining with FDA. Data are expressed as means ± SEM from three independent experiments performed in triplicate (*p< 0.05, for the effect of the Akt antisense oligonucleotide compared with nonsense controls; Student's t test). Immunoblot analysis for Akt protein was performed at the indicated times after addition of the Akt antisense (as) oligonucleotide, a nonsense (ns) control or vehicle (Co).D, The cells were cultured at 5% O2 for 9 hr. Phosphorylation of Akt at serine 473 in neurons treated with (+) or without (−) DC101 or VEGF (100 ng/ml) was assessed by immunoblot.NS, No switch; HK, high K+;LK, low K+; Glut, glutamate.
DISCUSSION
Although VEGF was identified as an angiogenic and vessel-permeability factor (Keck et al., 1989), recent studies have shown neurotrophic and neuroprotective functions of VEGF, as well as induction of VEGF in pathological states of the CNS (Sondell et al., 1999; Jin et al., 2000; Matsuzaki et al., 2001; Oosthuyse et al., 2001). Our results document an additional intrinsic neuroprotective function of VEGF via VEGFR-2 and Akt.
Our data are consistent with previous reports of preconditioning-induced tolerance in vivo orin vitro but make several novel points: (1) the induction of tolerance by moderate hypoxia of 5% O2 without glucose deprivation; (2) the wide range of different toxic stimuli attenuated by hypoxic preconditioning; and (3) the central role for VEGF/VEGFR-2 in the hypoxia-induced neuronal protection.
VEGF is induced in vivo by exposing mice to systemic hypoxia (Marti and Risau, 1998). VEGF induction was highest in the brain but also occurred in kidney, testis, lung, liver, and heart. In the brain, VEGF was expressed in neurons and in glial cells. Recent studies demonstrated that VEGF expression was significantly induced in the penumbra in cerebral ischemia (Leker et al., 2001). Although exogenous VEGF protected immortalized hippocampal neurons from hypoxia and glucose deprivation in vitro, VEGF, VEGFR-1 and VEGFR-2 expression were not induced in this paradigm (Jin et al., 2000). We established a paradigm of hypoxic preconditioning without the necessity of glucose deprivation or anoxia in CGN (Fig. 1). VEGF was induced by moderate hypoxia of 5% O2 over 3–9 hr (Fig.4A). These conditions did not result in immediate cell death or delayed reduction of cell survival (Fig. 1). Shorter periods of hypoxia did not lead to the induction of VEGF (data not shown). Our model circumvents the necessity for the combination of anoxia and glucose deprivation to induce hypoxic preconditioning, allowing to study the effects of pure hypoxia. With regard to hypoxia, our model may better reflect changes that occur in the penumbra. The penumbra is defined as a region in which cells only suffer functional but not structural impairments. The blood supply is decreased but not completely blocked. In the initial phase, energy metabolism is normal in the penumbra and deteriorates at a later phase (Hossmann, 1994). Therefore, we established a model that circumvented the induction of anoxia and complete glucose deprivation. Because CGN are the predominant cell type in our cultures, accounting for 95% of all cells, it appears likely that the VEGF that was detected was produced by CGN. This suggests an autocrine or paracrine mechanism. However, we cannot completely rule out that VEGF was derived from the 5% of astrocytes (Sinor et al., 1998) or oligodendrocytes in our culture.
CGN preconditioned by sublethal exposure to a reduced oxygen tension were rendered resistant to injuries induced by subsequent potassium deprivation or exposure to the excitotoxin glutamate or the mitochondrial toxin 3-NP (Fig.2A,B). In contrast, apoptosis induced by several chemotherapeutic drugs remained unaltered. Demonstration of such preconditioning-induced tolerance in vitro supports the view that toxin tolerance in vivomay be predominantly explained by intracellular alterations in neurons rather than by alterations in blood flow or systemic responses to the toxin. As in the initial gerbil studies (Kitagawa et al., 1990; Kato et al., 1992), we observed that tolerance developed slowly over many hours after the preconditioning episode, suggesting that changes in gene expression are involved. Supporting the hypothesis that long-term effects of ischemic preconditioning are depending on new protein synthesis treatment with the protein synthesis inhibitor CHX blocked the protective effects induced by 5% O2 (Fig.2C). We excluded certain BCL-2 family members (BCL-2, BCL-x, and BAD) as candidates mediating the protective effects of hypoxic preconditioning (Fig. 3). In contrast to these findings in CGN, VEGF has been demonstrated to induce BCL-2 expression in neoplastic cells and to block apoptosis (Pidgeon et al., 2001).
In addition to the induction of VEGF, we also found an increase in protein levels of VEGFR-1 and -2. VEGFR-1 apparently is primarily involved in endothelial morphogenesis, at least during embryonic development (Fong et al., 1995), but has no defined function in the activation of cell protection pathways (Matsuzaki et al., 2001). It has been shown that the VEGF-stimulated NO release is inhibited by blockade of VEGFR-1. Additional VEGFR-1 negatively regulates VEGFR-2-mediated proliferation via NO release and promotes the formation of capillary networks in human umbilical vein endothelial cells (Bussolati et al., 2001). The function of VEGFR-1 in neuronal cells, however, remains to be elucidated. In contrast, our data support and extent the finding that, in neurons, VEGFR-2 stimulation is linked to Akt activation and neuronal protection (Matsuzaki et al., 2001).
How might endogenous VEGF act to prevent or limit neuronal injury after hypoxic preconditioning? It has been suggested that VEGF signals neuroprotection through the VEGFR-2 receptor in association with both the PI3-K/Akt and the MEK/ERK pathways (Hetman et al., 1999; Matsuzaki et al., 2001). ERKs are activated by growth factors and G-protein-coupled receptors and are mostly involved in cell proliferation and survival. Both the stress activated and the ERK members of the MAP kinase family are activated by oxidative stress. As shown by increased phosphorylation of p44 (Fig. 6A), we detected ERK1 activation after hypoxic preconditioning. Because an MEK1/2 inhibitor failed to attenuate hypoxic preconditioning, this activation is unlikely to be of functional relevance in CGN. However, we found a functional involvement of the PI3-K/Akt pathway that was dependent on the activation of VEGFR-2. This result is in contrast to findings in primary cortical cell cultures in which preconditioning by oxygen–glucose deprivation requires the p21Ras/ERK cascade but not the PI3-kinase pathway (Gonzalez-Zulueta et al., 2000). The differences may be explained by the different cell type used (cortical neurons vs CGN) and by the different paradigm of hypoxic preconditioning (short but severe oxygen–glucose deprivation vs pure but extended hypoxia). For CGN, the PI3-K/Akt appears to be the predominant survival pathway (Datta et al., 1997; Dudek et al., 1997; Miller et al., 1997; Gleichmann et al., 2000). As shown by Hetman et al. (1999), not only the cell type but also the stress stimulus itself may determine the dominant signaling pathway for cellular survival. Using primary cortical neurons, these authors showed that BDNF-mediated protection against camptothecin toxicity through the ERK pathway, whereas it mediated protection against serum withdrawal through the PI3-K pathway. Crowder and Freeman (1999) demonstrated that the survival of sympathetic neurons promoted by potassium depolarization, but not by cAMP, required PI3-K and Akt. Vice versa, our findings that potassium withdrawal, glutamate, and 3-NP but not drug (CCNU, cisplatine, topotecan, and vincristine) toxicity are ameliorated by hypoxic preconditioning stress the specificity of the observed neuroprotection. Similarly, it has been shown for glioma cells that protection against death ligands (CD95L) and irradiation but not chemotherapeutics could be overcome by inhibition of PI3-K (Wick et al., 1999).
PI3-K is thought to regulate cell death by activating the serine–threonine protein kinase Akt, which enhances the activity of antiapoptotic proteins through the transcription factor NF-κB and inhibits proapoptotic signaling by BAD, caspase-9, and other effectors (Dudek et al., 1997). The serine 473 phosphorylation of Akt induced by 9 and 24 hr of hypoxia is prevented by the PI3-K inhibitor wortmannin. These effect of wortmannin on neurons is not specific for the hypoxic state but has been demonstrated to potentiate cell death in paradigms of glutamate-induced apoptosis (Gary and Mattson, 2001). Total protein levels of Akt were not affected (Fig. 6B). Both wortmannin and an Akt antisense oligonucleotide reverted the protective effects of hypoxic preconditioning (Fig. 6C). Treatment with the VEGFR-2 antagonist DC101 inhibited hypoxia-induced serine 473 phosphorylation of Akt (Fig. 6D). The hypoxia-induced protective effects mediated by VEGFR-2 require the secretion and binding of its ligand VEGF, because a VEGF antibody blocks the protective effects. Furthermore, the protective effects are reproduced by exogenous addition of VEGF. In summary, we provide a framework for the understanding of hypoxic preconditioning in CGN. The hypoxia-induced neuroprotective effects sequentially require the secretion of VEGF and the activation of VEGFR-2, PI3-K, and Akt.
Footnotes
This work was supported by a grant from the German Research Foundation (Sonderforschungsbereich 430, Teilprojekt B8) (J.B.S.).
Correspondence should be addressed to Dr. Jörg B. Schulz, Department of Neurology, University of Tübingen, School of Medicine, Hoppe-Seyler-Strasse 3, D-72076 Tübingen, Germany. E-mail: joerg.b.schulz@uni-tuebingen.de.
REFERENCES
- 1.Blondeau N, Widmann C, Lazdunski M, Heurteaux C. Activation of the nuclear factor-κB is a key event in brain tolerance. J Neurosci. 2001;21:4668–4677. doi: 10.1523/JNEUROSCI.21-13-04668.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bussolati B, Dunk C, Grohman M, Kontos CD, Mason J, Ahmed A. Vascular endothelial growth factor receptor-1 modulates vascular endothelial growth factor-mediated angiogenesis via nitric oxide. Am J Pathol. 2001;159:993–1008. doi: 10.1016/S0002-9440(10)61775-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crowder RJ, Freeman RS. The survival of sympathetic neurons promoted by potassium depolarization, but not by cyclic AMP, requires phosphatidylinositol 3-kinase and Akt. J Neurochem. 1999;73:466–475. [PubMed] [Google Scholar]
- 4.Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell. 1997;91:231–241. doi: 10.1016/s0092-8674(00)80405-5. [DOI] [PubMed] [Google Scholar]
- 5.Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 6.Fong GH, Rossant J, Gertsenstein M, Breitman ML. Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376:66–70. doi: 10.1038/376066a0. [DOI] [PubMed] [Google Scholar]
- 7.Gary DS, Mattson MP. Integrin signaling via the PI3-kinase-Akt pathway increases neuronal resistance to glutamate-induced apoptosis. J Neurochem. 2001;76:1485–1496. doi: 10.1046/j.1471-4159.2001.00173.x. [DOI] [PubMed] [Google Scholar]
- 8.Gerber HP, McMurtrey A, Kowalski J, Yan M, Keyt BA, Dixit V, Ferrara N. Vascular endothelial growth factor regulates endothelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Requirement for Flk-1/KDR activation. J Biol Chem. 1998;273:30336–30343. doi: 10.1074/jbc.273.46.30336. [DOI] [PubMed] [Google Scholar]
- 9.Gleichmann M, Weller M, Schulz JB. Insulin-like growth factor-1-mediated protection from neuronal apoptosis is linked to phosphorylation of the pro-apoptotic protein BAD but not to inhibition of cytochrome c translocation in rat cerebellar neurons. Neurosci Lett. 2000;282:69–72. doi: 10.1016/s0304-3940(00)00857-0. [DOI] [PubMed] [Google Scholar]
- 10.Gonzalez-Zulueta M, Feldmann AB, Klesse LJ, Kalb RG, Dillmann JF, Parada LF, Dawson TM, Dawsen VL. Requirement for nitric oxide activation of p21(ras)/extracellular regulated kinase in the neuronal ischemic preconditioning. Proc Natl Acad Sci USA. 2000;97:436–441. doi: 10.1073/pnas.97.1.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grabb MC, Choi DW. Ischemic tolerance in murine cortical cell culture: critical role for NMDA receptors. J Neurosci. 1999;19:1657–1662. doi: 10.1523/JNEUROSCI.19-05-01657.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gratton J-P, Morales-Ruiz M, Kureishi Y, Fulton D, Walsh K, Sessa WC. Akt down-regulation of p38 signaling provides a novel mechanism of vascular endothelial growth factor-mediated cytoprotection in endothelial cells. J Biol Chem. 2001;276:30359–30365. doi: 10.1074/jbc.M009698200. [DOI] [PubMed] [Google Scholar]
- 13.Han BH, Holtzman DM. BDNF protects the neonatal brain from hypoxic-ischemic injury in vivo via the ERK pathway. J Neurosci. 2000;20:5775–5781. doi: 10.1523/JNEUROSCI.20-15-05775.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hetman M, Kanning K, Cavanaugh JE, Xia Z. Neuroprotection by brain-derived neurotrophic factor is mediated by extracellular signal-regulated kinase and phosphatidylinositol 3-kinase. J Biol Chem. 1999;274:22569–22580. doi: 10.1074/jbc.274.32.22569. [DOI] [PubMed] [Google Scholar]
- 15.Hossmann KA. Viability thresholds and the penumbra of focal ischemia. Ann Neurol. 1994;36:557–565. doi: 10.1002/ana.410360404. [DOI] [PubMed] [Google Scholar]
- 16.Jin KL, Mao XO, Greenberg DA. Vascular endothelial growth factor: direct neuroprotective effect in in vitro ischemia. Proc Natl Acad Sci USA. 2000;97:10242–10247. doi: 10.1073/pnas.97.18.10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jones NM, Bergeron M. Hypoxic preconditioning induces changes in HIF-1 target genes in neonatal rat brain. J Cereb Blood Flow Metab. 2001;21:1105–1114. doi: 10.1097/00004647-200109000-00008. [DOI] [PubMed] [Google Scholar]
- 18.Kato H, Liu Y, Araki T, Kogure K. MK-801, but not anisomycin, inhibits the induction of tolerance to ischemia in the gerbil hippocampus. Neurosci Lett. 1992;139:118–121. doi: 10.1016/0304-3940(92)90871-4. [DOI] [PubMed] [Google Scholar]
- 19.Keck PJ, Hauser SD, Krivi G, Sanzo K, Warren T, Feder J, Connolly DT. Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science. 1989;246:1309–1312. doi: 10.1126/science.2479987. [DOI] [PubMed] [Google Scholar]
- 20.Kitagawa K, Matsumoto M, Tagaya M, Hata R, Ueda H, Niinobe M, Handa N, Fukunaga R, Kimura K, Mikoshiba K, Kamada T. “Ischemic tolerance” phenomenon found in the brain. Brain Res. 1990;528:21–24. doi: 10.1016/0006-8993(90)90189-i. [DOI] [PubMed] [Google Scholar]
- 21.Kunkel P, Ulbricht U, Bohlen P, Brockmann MA, Fillbrandt R, Stavrou D, Westphal M, Lamszus K. Inhibition of glioma angiogenesis and growth in vivo by systemic treatment with a monoclonal antibody against vascular endothelial growth factor receptor-2. Cancer Res. 2001;61:6624–6628. [PubMed] [Google Scholar]
- 22.Leker RR, Teichner A, Ovadia H, Keshet E, Reinherz E, Ben Hur T. Expression of endothelial nitric oxide synthase in the ischemic penumbra: relationship to expression of neuronal nitric oxide synthase and vascular endothelial growth factor. Brain Res. 2001;909:1–7. doi: 10.1016/s0006-8993(01)02561-6. [DOI] [PubMed] [Google Scholar]
- 23.Marti HH, Risau W. Systemic hypoxia changes the organ-specific distribution of vascular endothelial growth factor and its receptors. Proc Natl Acad Sci USA. 1998;95:15809–15814. doi: 10.1073/pnas.95.26.15809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsuzaki H, Tamatani M, Yamaguchi A, Namikawa K, Kiyama H, Vitek MP, Mitsuda N, Tohyama M. Vascular endothelial growth factor rescues hippocampal neurons from glutamate-induced toxicity: signal transduction cascades. FASEB J. 2001;15:1218–1220. [PubMed] [Google Scholar]
- 25.Miller BA, Perez RS, Shah AR, Gonzales ER, Park TS, Gidday JM. Cerebral protection by hypoxic preconditioning in a murine model of focal ischemia-reperfusion. NeuroReport. 2001;12:1663–1669. doi: 10.1097/00001756-200106130-00030. [DOI] [PubMed] [Google Scholar]
- 26.Miller TM, Tansey MG, Johnson EM, Creedon DJ. Inhibition of phosphatidylinositol 3-kinase activity blocks depolarization- and insulin-like growth factor I-mediated survival of cerebellar granule cells. J Biol Chem. 1997;272:9847–9853. doi: 10.1074/jbc.272.15.9847. [DOI] [PubMed] [Google Scholar]
- 27.Nandagopal K, Dawson TM, Dawson VL. Critical role for nitric oxide signaling in cardiac and neuronal ischemic preconditioning and tolerance. J Pharmacol Exp Ther. 2001;297:474–478. [PubMed] [Google Scholar]
- 28.Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999;13:9–22. [PubMed] [Google Scholar]
- 29.Oosthuyse B, Moons L, Storkebaum E, Beck H, Nuyens D, Brusselmans K, Van Dorpe J, Hellings P, Gorselink M, Heymans S, Theilmeier G, Dewerchin M, Laudenbach V, Vermylen P, Raat H, Acker T, Vleminckx V, Van Den Bosch L, Cashman N, Fujisawa H. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat Genet. 2001;28:131–138. doi: 10.1038/88842. [DOI] [PubMed] [Google Scholar]
- 30.Pidgeon GP, Barr MP, Harmey JH, Foley DA, Bouchier-Hayes DJ. Vascular endothelial growth factor (VEGF) upregulates BCL-2 and inhibits apoptosis in human and murine mammary adenocarcinoma cells. Br J Cancer. 2001;85:273–278. doi: 10.1054/bjoc.2001.1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Prewett M, Huber J, Li Y, Santiago A, O'Connor W, King K, Overholser J, Hooper A, Pytowski B, Witte L, Bohlen P, Hicklin D. Antivascular endothelial growth factor receptor (fetal liver kinase 1) monoclonal antibody inhibits tumor angiogenesis and growth of several mouse and human tumors. Cancer Res. 1999;59:5209–5218. [PubMed] [Google Scholar]
- 32.Rockwell P, Neufeld G, Glassman A, Caron D, Goldstein N. In vitro neutralization of vascular endothelial growth factor activation of FLK-1 by a monoclonal antibody. Mol Cell Differ. 1995;3:91–109. [Google Scholar]
- 33.Schulz JB, Weller M, Klockgether T. Potassium deprivation-induced apoptosis of cerebellar granule neurons: a sequential requirement for new mRNA and protein synthesis, ICE-like protease activity, and reactive oxygen species. J Neurosci. 1996;16:4696–4706. doi: 10.1523/JNEUROSCI.16-15-04696.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Segal RA, Greenberg ME. Intracellular signaling pathways activated by neurotrophic factors. Annu Rev Neurosci. 1996;19:463–489. doi: 10.1146/annurev.ne.19.030196.002335. [DOI] [PubMed] [Google Scholar]
- 35.Shamloo M, Rytter A, Wieloch T. Activation of the extracellular signal-regulated protein kinase cascade in the hippocampal CA1 region in a rat model of global cerebral ischemic preconditioning. Neuroscience. 1999;93:81–88. doi: 10.1016/s0306-4522(99)00137-2. [DOI] [PubMed] [Google Scholar]
- 36.Sharp FR, Massa SM, Swanson RA. Heat-shock protein protection. Trends Neurosci. 1999;22:97–99. doi: 10.1016/s0166-2236(98)01392-7. [DOI] [PubMed] [Google Scholar]
- 37.Shimazaki K, Ishida A, Kawai N. Increase in bcl-2 oncoprotein and the tolerance to ischemia-induced neuronal death in the gerbil hippocampus. Neurosci Res. 1994;20:95–99. doi: 10.1016/0168-0102(94)90026-4. [DOI] [PubMed] [Google Scholar]
- 38.Sinor AD, Irvin SM, Cobbs CS, Chen J, Graham SH, Greenberg DA. Hypoxic induction of vascular endothelial growth factor (VEGF) protein in astroglial cultures. Brain Res. 1998;812:289–291. doi: 10.1016/s0006-8993(98)00976-7. [DOI] [PubMed] [Google Scholar]
- 39.Sondell M, Lundborg G, Kanje M. Vascular endothelial growth factor has neurotrophic activity and stimulates axonal outgrowth, enhancing cell survival and Schwann cell proliferation in the peripheral nervous system. J Neurosci. 1999;19:5731–5740. doi: 10.1523/JNEUROSCI.19-14-05731.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tamatani M, Mitsuda N, Matsuzaki H, Okada H, Miyake S, Vitek MP, Yamaguchi A, Tohyama M. A pathway of neuronal apoptosis induced by hypoxia/reoxygenation: roles of nuclear factor-κB and BCL-2. J Neurochem. 2000;75:683–693. doi: 10.1046/j.1471-4159.2000.0750683.x. [DOI] [PubMed] [Google Scholar]
- 41.Weller M, Montpied P, Paul SM. NMDA receptor-mediated excitoprotection of cultured cerebellar granule neurons fails to alter glutamate-induced expression of c-fos and c-jun mRNAs. Mol Brain Res. 1994;22:227–235. doi: 10.1016/0169-328x(94)90051-5. [DOI] [PubMed] [Google Scholar]
- 42.Wick W, Furnari FB, Naumann U, Cavenee WK, Weller M. PTEN gene transfer in human malignant glioma: sensitization to irradiation and CD95L-induced apoptosis. Oncogene. 1999;18:3936–3943. doi: 10.1038/sj.onc.1202774. [DOI] [PubMed] [Google Scholar]