

Abstract
The winged helix gene Brain factor-1 (BF1) has a pleiotropic role in the development of the cerebral hemispheres of the brain. Mice lacking BF1 have defects in the morphogenesis of the structures of the dorsal telencephalon (e.g., neocortex) and the ventral telencephalon (e.g., the basal ganglia). This study focuses on the functions of BF1 in the dorsal telencephalon. We showed previously that telencephalic progenitor cells lacking BF1 differentiate into neurons prematurely. Here, we demonstrate that the loss of BF1 also results in an early lengthening of the cell cycle in neocortical progenitors. To investigate the mechanisms by which BF1 regulates progenitor cell proliferation and differentiation in the developing brain, we have replaced the endogenous BF1 protein with a DNA binding defective form of BF1 in mice, BF1NHAA. The BF1NHAA protein restores the growth of the dorsal telencephalon, by improving the proliferation of progenitor cells. However, the BF1NHAA protein does not correct the early neuronal differentiation associated with the loss of BF1. In contrast, replacement of endogenous BF1 with wild-type BF1 corrects the defects in both the proliferation and differentiation of neocortical progenitors. These results demonstrate that BF1 controls progenitor cell proliferation and differentiation in the neocortex through distinct DNA binding-independent and binding-dependent mechanisms.
Keywords: BF1, foxg1, winged helix, neocortex, brain development, telencephalon, neurogenesis
The neocortex is the brain structure associated with higher cognitive functions. Evolution of the mammalian brain is characterized by a disproportionate expansion of the neocortex relative to other structures (Clark et al., 2001). Neocortical neurons are generated from a sheet of neuroepithelial cells of the dorsal telencephalon, during the period of neurogenesis. The length of the cell cycle in progenitor cells and the duration of the neurogenetic interval primarily determine the total number of neurons that will comprise the neocortex (Takahashi et al., 1993, 1996a; Caviness et al., 1995). Between embryonic day 11 (E11) and E18 in the mouse, the G1 phase of the cell cycle becomes progressively longer, resulting in a slower rate of cell division (Takahashi et al., 1993, 1996b). Concomitantly, a steadily increasing fraction of cells withdraw from the cell cycle and ventricular zone to differentiate, leading to the depletion of the progenitor cell population and the cessation of neurogenesis. To understand how the growth of neocortex is regulated, we are examining the mechanisms that control the neurogenetic program in the telencephalic neuroepithelium.
Expression of the Winged Helix gene Brain factor-1(BF1/foxg1) within the neural tube is limited to the progenitor population that comprises the telencephalon (Tao and Lai, 1992). BF1 is an early marker of cells specified as telencephalic neuroepithelium. It continues to be expressed in neurons and glia derived from the telencephalon in the adult brain (Dou et al., 1999). BF1 plays a critical role in the development of the cerebral hemispheres. Embryos lacking BF1 die at birth with hypoplasia of the cerebral hemispheres (Xuan et al., 1995). In the dorsal telencephalon, the major anomaly is an accelerated rate of neuronal differentiation within the population of progenitors of the neocortical neuroepithelium. This results in the premature depletion of the progenitor cell pool and ultimately, in the generation of fewer neurons. BF1(−/−) mutants also have reduced progenitor cell proliferation in the anterior third of the dorsal telencephalon.
Members of the Winged Helix (WH) family are characterized by a highly conserved DNA binding domain, functioning both as transcriptional activators and repressors. BF1 has been shown to act primarily as a transcriptional repressor (Li et al., 1995; Bourguignon et al., 1998). This activity requires an intact WH domain and the ability to bind to DNA. Recently, however, we have obtained evidence that BF1 may also function through additional mechanisms that are independent of DNA binding (Dou et al., 2000). In vitro studies showed that a DNA binding defective mutant BF1 protein (BF1NHAA) could antagonize the antiproliferative activity of TGFβ, as well as transcriptional activation by TGFβ. These results led us to postulate that BF1 may control the development of the neocortex through both DNA binding dependent and DNA binding independent mechanisms. To test this possibility in vivo and to dissect the molecular pathways controlled by BF1 in the developing brain, we examined the consequences of replacing the endogenous BF1 protein in mice with a DNA binding defective form.
MATERIALS AND METHODS
Targeting of the tetracycline transactivator gene to the BF1 locus. The targeting construct was made by replacing thelacZ sequence in the BF1 targeting vector previously described (Xuan et al., 1995) with the sequence encoding the tetracycline transactivator (tTA) from pUHD 15–1 (Gossen and Bujard, 1992). The SalI site in the tTA sequence was eliminated by mutagenesis. The tTA sequence was inserted between aSalI–ApaI fragment containing the BF1promoter and an EcoRI–KpnI fragment containing an SV40 intron and poly(A) sequence (Hebert and McConnell, 2000). TheSalI–BamHI fragment was then inserted into theSalI–BamHI sites in the pHBL3 plasmid. Linearized targeting vector was electroporated into W9.5 embryonic stem (ES) cells. Targeted ES clones were identified by PCR and Southern blot as previously described. Three correctly targeted clones were injected into blastocysts to generate chimeric mice that were bred with C57/BL6 mice.
Generation and screening of tetO transgenic lines.IRES3lacZ was generated by inserting a lacZ cassette (Xuan et al., 1995) into the Nco–Sma sites of the IRES-poly plasmid (generously provided by T. Jessell). AnXba–Xho fragment of IRES3lacZ was ligated to the 3′ end of Xba site of pUHD 10.3 plasmid to generate a plasmid containing the multiple tet-Operator sites and promoter, the IRES lacZ cassette, with an intervening Xba site. The BF-1NHAA mutant sequence (Dou et al., 2000) was cloned into this Xba site. Linearized plasmid was injected into oocytes. Transgenic offspring were identified by Southern analysis. Twelve transgenic lines were examined for the responsiveness of the tetO promoter to activation by tTA. Males from each line were mated with females heterozygous for the tTA gene at the BF1 locus (BF1 (+/−tTA)). Embryos were obtained from timed pregnancies, with noon of the plug date defined as E0.5. Embryos were fixed in 4% paraformaldehyde and embedded in OCT for frozen sections. β-gal staining in 10 μm frozen sections was performed as previously described (Xuan et al., 1995). Embryos were examined at E13.5 by staining for β-galactosidase activity. Nine lines with β-gal activity were studied further.
Immunohistochemistry and in situ hybridization.Immunohistochemical detection of BrdU and MAP2 were performed as described previously, (Xuan et al., 1995) except for the following changes. For BrdU, frozen sections (10 μm) were treated with 1.25 μg/ml proteinase K at 37°C for 17 min for E13.5 embryos. Shorter treatment was performed for younger embryo tissues. Sections were then postfixed with 4% paraformaldehyde for 10 min. For the secondary antibody, biotinylated rabbit anti-rat antibodies were diluted 1:1000 in PBS with 1.5% normal rabbit serum. For MAP2, goat anti-mouse antibodies conjugated with peroxidase were diluted 1:100 in PBS containing 0.5% Triton X-100 and 1% normal goat serum. Immunohistochemical localization of phosphorylated histone H3 was performed with mouse antibody from Upstate Biotechnology (Lake Placid, NY), followed by detection with the ABC system (Vector Laboratories, Burlingame, CA).
In situ hybridization using digoxigenin-labeled probes was performed as previously described (Yang et al., 1999) with the following modifications. Endogenous peroxidase activity was quenched with 1.5% H2O2 in methanol for 15 min. Digoxigenin-labeled cRNA probes (100 ng/ml) were hybridized at 60°C. Sections were incubated with anti-DIG-POD antibody (Ab) (1:500) overnight in a humidified chamber at 4°C. Biotinyl-tyramide was applied at 37°C for 15 min. Bone morphogenetic protein (Bmp) probe templates were kindly provided by B. Hogan (Vanderbilt University) (Bmp2,4,6,7). The BF1Sfi–Sma probe (nucleotide 1391–1850) was used to detect transcripts in the NHAA rescue and wild-type (WT) rescue lines.
Timed BrdU labeling. The quantitation of BrdU-labeled nuclei after a pulse of BrdU is a useful tool to monitor the proliferative activity of a population of cells. However, it is a relatively insensitive method to detect changes in the rate of cell proliferation. In the neocortical neuroepithelium, a significant increase in the length of the cell cycle from 12 to 15 hr would result only in a 20% reduction in the fraction of labeled nuclei and be difficult to detect. We took advantage of the fact that nuclei of neural progenitor cells move within the ventricular zone in a regulated manner during the cell cycle. The nuclei in cells undergoing mitosis are positioned next to the apical (ventricular) surface. During G1, the nuclei migrate to the basal surface, where they initiate DNA replication in the S phase. Subsequently, the nuclei migrate back toward the apical surface during G2. Thus, the position of the nucleus in the ventricular zone reflects its position in the cell cycle. By monitoring the number of BrdU-labeled nuclei at the apical surface, we can more readily detect changes in cell cycle length between two populations of cells.
Previous studies have established that the length of the cell cycle in neocortical progenitors increases progressively between E11 and E17, from 8 to ∼18 hr. Nearly all of this increase is attributable to the prolongation of the G1 phase of the cell cycle (Takahashi et al., 1993). The S phase is relatively constant at 4 hr in length, whereas G2 + M is ∼2 hr long. BrdU given intraperitoneally at 25 μg/gm to a pregnant mouse will result in a pulse of labeling for ∼4 hr in developing embryos (Takahashi et al., 1992) because BrdU is rapidly cleared. A single injection will label a cohort of nuclei that are in the S phase of the cell cycle during the labeling period. These nuclei will span an 8 hr window (4 hr labeling period plus the 4 hr length of the S phase) within the cell cycle. By examining embryos at various time points after BrdU labeling, we will be able to follow this cohort of labeled nuclei as they progress through the cell cycle. We selected the 15 hr time point because it is approximately the length of the cell cycle at E14.5 (Takahashi et al., 1993).
Quantitation of BrdU-labeled nuclei. BrdU-labeled nuclei in frozen sections were identified by immunohistochemistry using a monoclonal rat anti-BrdU antibody (Harlan Bioproducts for Science, Indianapolis, IN). Both the total number of nuclei and the number of BrdU-labeled nuclei along the apical surface of the ventricular zone were counted. Nuclei were counted under a Nikon light microscope in 10 μm sections, 40–60 μm apart, with 6–10 sections counted from each embryo (spacing and number of sections increased with embryo age). For the 15 hr labeled sections in Figure 5, data were collected from four to six embryos at each age [two from each of the genotypes ((BF1(+/−), BF1(−/−), NHAA rescue)]. We counted >950 nuclei for each genotype at E11.5, and >1100 nuclei at E12.5, E13.5, and E14.5. Nuclei were counted from the posterolateral region, as shown in Figure 5. For the 2 hr pulse labeling in Figure 6, data were collected from six embryos [three fromBF1(+/−), three from BF1(−/−)]. A minimum of 2800 nuclei was counted from each region of the telencephalon (anterior and posterior as shown in Fig. 6) for each genotype. Data from each set is reported as the mean of the calculated percentage of BrdU-labeled nuclei, ±SEM.
Fig. 5.
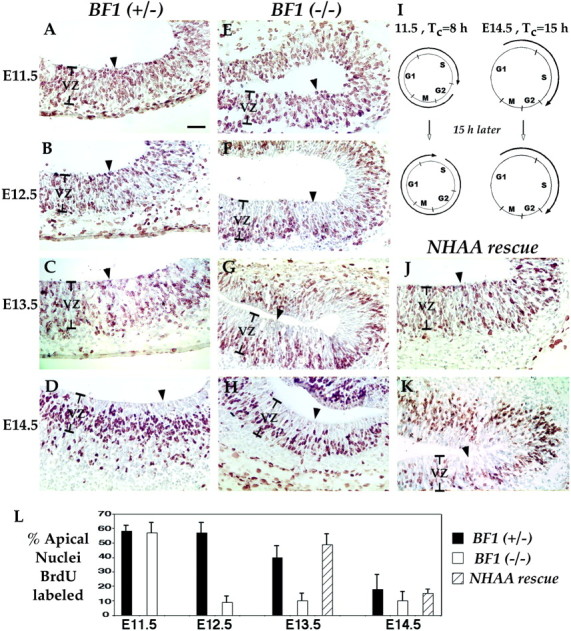
Aberrant pattern of BrdU labeling in the ventricular zone of the BF1 (−/−) mutant and restoration of normal patterns in the NHAA rescue. Diagram of the cell cycle in neocortical progenitor cells at E11.5 and E14.5 (I). Arrows depict the approximate position in the cycle of cells that will be labeled with BrdU after a single intraperitoneal injection of BrdU to the pregnant mouse. The position of this cohort of BrdU-labeled nuclei is also depicted 15 hr later, when the embryos are examined. When the cell cycle length is the same as the labeling period or slightly longer, labeled nuclei will be present in all phases of the cell cycle. Under these conditions, BrdU-labeled nuclei are distributed throughout the ventricular zone. This is observed from E11.5 to E13.5 in WT embryos (A–C) and at E11.5 in BF1 (−/−) mutants (E). When the cell cycle length increases to ∼15 hr or greater, labeled nuclei will be absent from the G2 and M phase of the cell cycle (I). This will result in a marked reduction in the number of BrdU-labeled nuclei at the apical surface of the ventricular zone, as seen at E14.5 in WT embryos (D) and at E12.5 to E14.5 inBF1 (−/−) mutants (F–H). In NHAA rescue embryos, 15 hr labeling at E13.5 (J) shows BrdU-labeled nuclei distributed throughout the ventricular zone, as found in normal embryos. BrdU-labeled nuclei are not excluded from the apical surface until E14.5 (K).Arrowheads point to the apical surface of the ventricular zone. Shown in this figure are high magnification views of the posterolateral region of the telencephalic neuroepithelium approximately at the positions indicated by thearrowheads in the horizontal sections of Figure 3. The posterior telencephalon is on the right side of each section.L, Quantitation of the percentage of BrdU-labeled nuclei at the apical surface of the ventricular zone in the posterolateral telencephalon, 15 hr following a single injection of BrdU from E11.5 to E14.5. Nuclei lining the ventricular surface were counted. The percentages of BrdU-labeled nuclei are plotted as the mean ± SEM. Scale bar, 50 μm.
Fig. 6.
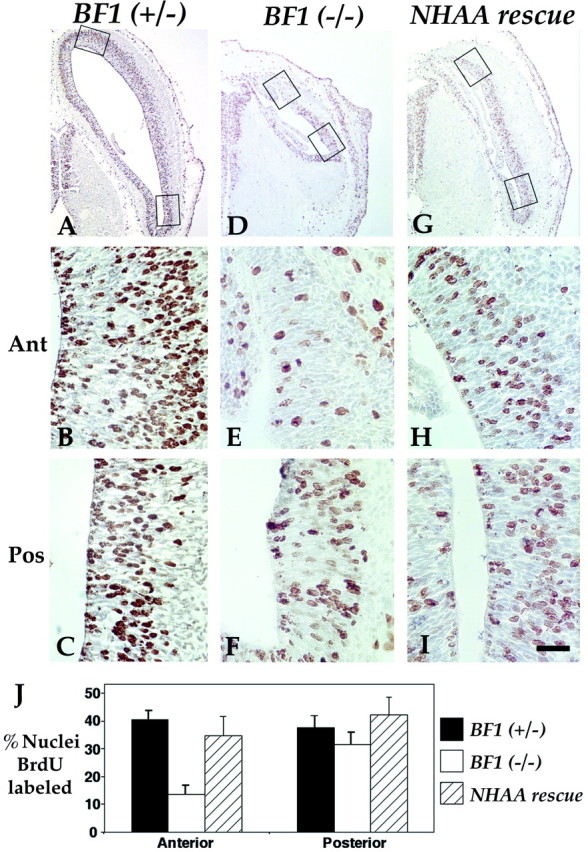
The BF1NHAA protein improves the proliferation of telencephalic progenitors in the anterior telencephalon. Horizontal sections of E13.5 embryos fromBF1(+/−) heterozygotes (A–C),BF1(−/−) mutants (D–F), and NHAA rescue (G–I). The positions of higher magnification views of the anterior and posterior regions of the telencephalic neuroepithelium telencephalon are indicated withrectangles. J, The fraction of BrdU-positive nuclei after a 2 hr pulse of BrdU was determined in the anterior and posterior regions. The reduction in BrdU labeling in the anterior telencephalon of the BF1(−/−) mutant is corrected to near normal levels in the NHAA rescue. Scale bar:A, D, G, 400 μm;B, C, E, F,H, I, 50 μm.
Quantitation of BF1 proteins in the embryo. The telencephalic vesicles from each E12.5 embryo were obtained by dissection of the head. Surface ectoderm was removed. The tissue was homogenized by sonication in 100 μl of 50 mmTris buffer, pH 8, 50 mm NaCl, 1% Triton-X 100, 1% sodium deoxycholate, 0.1% SDS, and protease inhibitors (P8340; Sigma, St. Louis, MO). Ten micrograms of protein lysate were loaded on an SDS gel. Immunoblotting was performed with affinity-purified rabbit anti-BF1 polyclonal antibody at 1:1000 dilution (Yao et al., 2001). This antibody was raised to the C-terminal peptide of BF1, and therefore should recognize WT BF1 and BF1NHAA equally well.
RESULTS
Targeting of the tetracycline transactivator gene to the BF1 locus
To examine the molecular pathways regulated by BF1 in the developing embryo, we developed a transgenic mouse model to direct the expression of a mutant form of BF1 in the developing telencephalon. This model uses the tetracycline-regulated system developed by Gossen and Bujard (1992) and Kistner et al. (1996). We first generated a mouse line expressing the tetracycline transactivator in BF1-expressing cells by targeting the tTA gene to the BF1 locus, through homologous recombination in ES cells (Fig.1A). Germline transmission of this ES cell line generates mice in which one copy of the BF1 gene is replaced with the tTA gene [BF1(+/−tTA) line]. In situ hybridization studies demonstrate that the tTA gene is expressed in a pattern that closely matches that of the BF1gene. The expression pattern of tTA is identical to that ofBF1 in the telencephalic neuroepithelium, the olfactory epithelium, in the pharyngeal pouches and ectodermal placodes (Fig.1B–E) (data not shown). Expression of tTA is not detectable in the optic vesicle. Because BF1 expression levels in the optic vesicle are lower than at other sites and the tTA probe is less sensitive than the BF1 probe, it is possible that tTA is expressed in the optic vesicle of theBF1(+/−tTA) mice at levels below the sensitivity of our assay.BF1(+/−tTA) mice have a normal life span and are fertile. They have no observed anomalies in brain development as assessed by light microscopy of embryos from E12.5 to postnatal day 0.
Fig. 1.

Targeting of the tetracycline transactivator (tTA) to the BF1 locus. A, Map of the targeting vector used to replace the BF1 gene in ES cells by homologous recombination. In the BF1 (+/− tTA) heterozygote at E13.5, the expression pattern of the tTA gene matches that of BF1 in the telencephalon (B, C) and olfactory epithelium (D, E), as detected by in situ hybridization. Mating of theBF1(+/−tTA) mice with a transgenic animal with a target gene under the control of multiple tetO sites results in offspring that express the target gene in BF1-expressing cells. An IRES–lacZ cassette placed downstream of the target gene directs translation of the β-galactosidase gene, which serves a reporter of target gene expression (F). Staining for β-gal activity in a BF1(+/−tTA):tetO-BF1NHAA IRES lacZ embryo at E13.5 demonstrates the expression of the target gene in the telencephalic neuroepithelium (H). No β-gal activity is detected in embryos lacking the tTA (G) or in BF1(+/−tTA): tetO- BF1NHAA IRES lacZ embryos harvested from pregnant dams that were fed doxycycline (20 μg/ml) in the drinking water (I). Scale bar, 200 μm.
Directed expression of target genes in the telencephalic neuroepithelium
A DNA binding defective form of BF1 (BF1NHAA) was previously generated by site-directed mutagenesis of the Winged Helix domain (Dou et al., 2000). The three-dimensional structure of the winged helix identifies two amino acids in Helix 3 that make critical base contacts with the DNA. The corresponding two residues in BF1, N(165) and H(169), were mutated to alanines to generate the BF1NHAA protein. We have shown that this mutant protein (BF1NHAA) is unable to bind to high-affinity BF1 binding sites and is inactive in a transcriptional repression assay. Mutation of the corresponding two amino acids in Qin, the chick homolog of BF1, disrupt its ability to transform chick embryo fibroblasts, a function that is mediated through a transcriptional repression mechanism (Ma et al., 2000).
To express BF1NHAA in the developing cerebral hemispheres under the control of the tTA protein, we generated several transgenic lines with this target gene fused to a promoter containing multiple tet-Operator sites. An IRES lacZ cassette was placed downstream of the target gene to facilitate the detection of cells expressing the target gene under the control of the tet-transactivator (Fig. 1F) (Mountford and Smith, 1995). Twelve transgenic lines were generated, of which nine exhibited lacZ expression in the telencephalon when mated with theBF1(+/−tTA) line to generate BF1(+/−tTA): tetO BF1NHAA embryos. LacZ expression in these embryos indicates that the target gene,BF1NHAA, is transcribed under the control of the tTA protein (Fig. 1H). No lacZ expression is detected in the absence of the tTA protein (Fig.1G). The level of lacZ expression varies between transgenic lines, most likely reflecting the different response of the tetO promoter at different integration sites. However, the pattern of expression is constant among the different transgenic lines, matching that of the tTA gene. In some of the high expresser lines, we observe ectopic expression in scattered cells of the diencephalon and midbrain. Based on the level of lacZ expression, we have grouped the transgenic lines into high (three lines), medium (three lines), and low (three lines) level expressers. In this report we will focus on representative lines of the medium (#25, 77) and high expresser class (#83, 99). The level of BF1NHAA protein expression in these lines is described below. Expression of the lacZ reporter in embryos is abolished when pregnant mice are given doxycycline in the drinking water beginning on the day the coital plug is identified (Fig.1I), providing additional evidence that the target gene is under the control of the tTA protein. Doxycycline concentrations of 2 μg/ml are sufficient to repress lacZ expression to undetectable levels.
Replacement of endogenous BF1 with tTA-directed expression of BF1NHAA
To examine the activity of the BF1NHAA protein in a BF1 null background, we generated mice in which this mutant protein replaces the endogenous BF1 protein. We first crossed the tetOBF1NHAA line with theBF1(+/−lacZ) line (Xuan et al., 1995), to generate animals with the tetOBF1NHAA transgene and with one copy of BF1 replaced with the lacZ gene. These mice are phenotypically normal, and express lacZ under the control of the BF1 promoter. These males were then mated with femaleBF1(+/−tTA) mice (Fig.2E). One quarter of the offspring will have no copies of the BF1 gene [BF1(-tTA/-lacZ)], and one half of these will also be hemizygous for the tetOBF1NHAA gene. These embryos will be referred to as BF1 mutant (BF1(-tTA/-lacZ)) and NHAA rescue (BF1 mutant +BF1NHAA), respectively.BF1(-tTA/-lacZ) mutants are indistinguishable from the previously describedBF1(-lacZ/-lacZ) mutants (Xuan et al., 1995). Newborn mice die within minutes of birth, have a flattened forehead, and irregularly shaped small eyes. NHAA rescue animals also die within minutes of birth and have eye anomalies. However, the forehead of these mice is notably more rounded in shape (data not shown).
Fig. 2.

Replacement of endogenous BF1 with BF1NHAA. Mating ofBF1(+/−lacZ):tetOBF1NHAA IRES lacZ males withBF1 (+/−tTA) females will yield offspring, one quarter of which will lack BF1. One half of these (or one eighth of the total) will also have the tetO BF1NHAAIRES lacZ construct. We refer to these as “NHAA rescue” embryos (E). Expression of theBF1NHAA transcript in these embryos was monitored by β-gal staining and by in situhybridization (B,#77; C, #99;D, #83). This was compared with the level of theBF1 transcript in BF1 (+/−) heterozygotes (A). The level of BF1NHAAprotein was determined by immunoblotting extracts from dissected telencephalon (#99 line) at E12.5, and compared with that in WT embryos, heterozygotes, and BF1 (−/−) mutants (F). Scale bar, 200 μm.
To compare the approximate expression level of the tTA-directedBF1NHAA transcript with that of endogenous BF1, we performed in situhybridization with a probe for BF1 that is within the protein coding sequence (BF1 Sfi–Sma fragment). Hybridization signals were compared between BF1heterozygotes and NHAA rescue embryos at E12.5. Sections shown were processed simultaneously under identical conditions. For transgenic lines in the moderate expresser group, expression levels of theBF1NHAA transcript in a mutant background are slightly lower than endogenous levels ofBF1 transcripts in BF1 (+/−) heterozygotes (Fig.2B). High expresser lines have levels three to four times higher (Fig. 2C,D). Among the different tetOBF1NHAA lines, the relative levels of BF1NHAA expression byin situ hybridization correlates well with the relative level of lacZ expression detected by β-gal staining. However, β-gal activity in high expresser tetOBF1NHAA lines is severalfold lower than in BF1(+/−lacZ) heterozygotes, indicating that translation of the lacZ gene from the IRES is much less efficient than translation initiated from the 5′ end of the transcript.
To determine the level of BF1NHAA protein expression, we immunoblotted extracts from the forebrain of E12.5 embryos with an anti-BF1 antibody. We find that expression of the BF1NHAA protein in a high expresser line (#99) is higher than levels of endogenous BF1 in WT embryos. Expression levels in the medium expresser group (#25, 77) are threefold to fourfold lower and are slightly lower than the level of BF1 inBF1(+/−) heterozygous embryos (data not shown). Thus, there is a good correlation between BF1 mRNA and protein levels expressed under the control of the tTA protein. NHAA rescue embryos from both the high and medium expresser lines have very similar phenotypes.
BF1NHAA protein improves the growth of the dorsal telencephalon
We found that the cerebral hemispheres of the NHAA rescue embryos were intermediate in size between that of normal mice andBF1 mutants. The basal ganglia, which are derived from the basal telencephalon, were absent in the NHAA rescue embryos, a result similar to that observed in the BF1 (−/−) mutant. However, a marked improvement in the growth of the dorsal telencephalon was observed in the NHAA rescue. In the BF1 mutant, the dorsal telencephalon is much smaller than normal (Fig.3D,H). By contrast, the dorsal telencephalon of the NHAA rescue, is nearly equal in size to the dorsal telencephalon of normal embryos (Fig. 3, compare A,B). The neocortex of the BF1 mutant also has an irregular contour (Fig. 3H), reflecting variations in the rate of progenitor cell proliferation within the anterior telencephalic neuroepithelium. This anomaly is corrected in the NHAA rescue. The neocortex of NHAA rescue embryos has a smooth contour resembling that of normal embryos. These results show that the BF1NHAA protein can substitute for the WT protein to enhance the growth of the dorsal telencephalon. However, the development of the dorsal telencephalon is not completely normalized by the BF1NHAA protein. Anomalies remain, notably in the morphology of the dorsomedial telencephalon (Fig.3F, arrows).
Fig. 3.

Improved growth of the dorsal telencephalon in NHAA and WT BF1 rescue embryos at E13.5. Coronal (A–D) and horizontal (E–H) sections show that the size of the dorsal telencephalon in the NHAA rescue is similar to that of the dorsal telencephalon in BF1 (+/−) heterozygotes and in the BF1 WT rescue. The NHAA rescue has a thicker cortical plate than that found inBF1(+/−) heterozygotes or in the BF1 WT rescue.Asterisks indicate the position of the cortical plate, which expresses β-galactosidase at lower levels than in the ventricular zone and contains the differentiated neurons of the dorsal telencephalon (F, H). dt, Dorsal telencephalon; bt, basal telencephalon. Scale bar, 200 μm.
Aberrant cell cycle progression throughout the dorsal telencephalon of the BF1 mutant
We have previously described anomalies in progenitor cell proliferation in the anterior third of the dorsal telencephalon of mice lacking BF1. We detected a significant reduction in the fraction of BrdU-labeled nuclei in this region of the mutant telencephalon (Xuan et al., 1995). To determine whether cell proliferation was altered in the remainder of the dorsal telencephalon of BF1 (−/−) mutants, we devised a method to monitor the lengthening of the cell cycle during the period of neurogenesis. We examined the fraction of BrdU-labeled nuclei at the apical surface of the ventricular zone 15 hr after a single injection of BrdU. This method is based on interkinetic nuclear migration of the progenitor cells of the neuroepithelium (Takahashi et al., 1996b). Nuclei move from the apical surface to the basal surface and back during every cell cycle (Fig. 4A). To determine the position of mitotic nuclei in the telencephalic neuroepithelium of BF1(−/−) mutants, we localized the phosphorylated form of histone H3 by immunohistochemistry. We show that mitotic nuclei are restricted to the apical surface of the ventricular zone in the BF1(−/−) mutant as well as in their normal littermates at E13.5 (Fig. 4B,C). After a short BrdU labeling period of 2 hr, we found that BrdU-positive cells are present throughout the apical and basal regions of the ventricular zone, in both BF1 mutants and their normal littermates (Xuan et al., 1995) (see Fig. 6). This suggests that BrdU-labeled nuclei are able to move from the apical to the basal region in the BF1 mutant. Taken together, these results indicate that interkinetic nuclear migration is not disrupted by the loss of BF1.
Fig. 4.
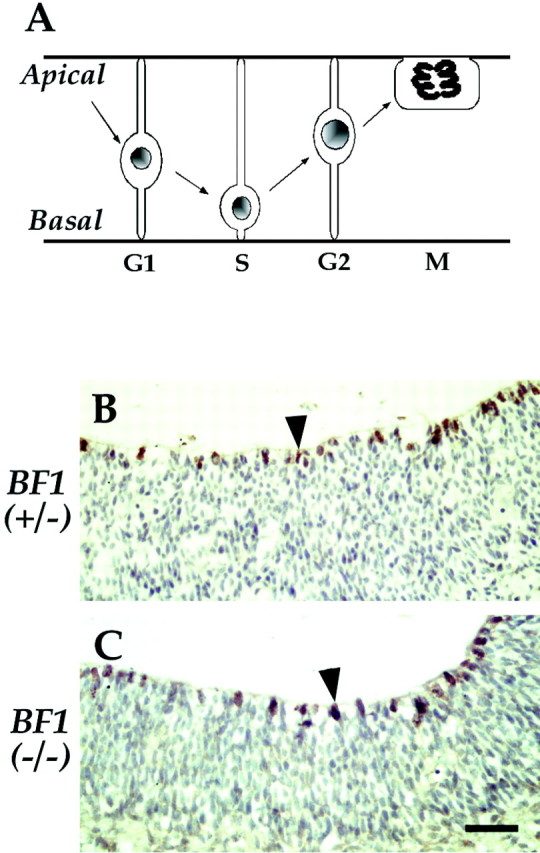
Interkinetic nuclear migration of neocortical progenitors is not altered in the BF1 (−/−) mutant. Mitosis takes place at the apical surface, adjacent to the ventricle. Immunohistochemical localization of phosphorylated histone H3 demonstrates mitotic nuclei at the apical surface of the ventricular zone in the telencephalon of both normal BF1(+/−) heterozygotes and BF1(−/−) mutants at E13.5.
We estimated that a single injection of BrdU in a pregnant mouse labels a cohort of nuclei spanning an 8 hr interval in the cell cycle, based on the length of the S phase and the clearance rate of BrdU (Takahashi et al., 1992; Caviness et al., 1995). When the length of the cell cycle is 8 hr (at E11.5), this cohort spans the entire cell cycle (Fig.5I), and BrdU-labeled nuclei are distributed throughout the ventricular zone (Fig.5A). As the cell cycle length increases, the labeled cohort represents a progressively smaller fraction of the total interval. When the average cell cycle length increases to ∼15 hr, few labeled nuclei will be in the G2 and M phase of the cell cycle when this cohort is examined 15 hr after labeling (Fig. 5I). This will result in a marked reduction of BrdU-labeled nuclei at the apical surface of the ventricular zone (Fig. 5D). Thus, the fraction of BrdU-labeled nuclei at the apical surface in this assay correlates with the fraction of progenitor cells with a cell cycle length of ∼15 hr or longer.
We examined the BrdU labeling pattern in BF1 (+/−) embryos from E11.5 through E14.5, after an injection of BrdU given 15 hr earlier. We focused on the posterolateral telencephalon, as indicated by the arrowheads in Figure 3. From E11.5 through E13.5, we observe BrdU-labeled nuclei throughout the ventricular zone (Fig.5A–C). However, at E14.5, BrdU-labeled nuclei are absent from the apical surface of the neocortical neuroepithelium (Fig.5D). The cell cycle length at E14.5 has been previously estimated to be 15.1 hr (Takahashi et al., 1993). We next examined the pattern of BrdU labeling in the BF1(−/−) mutant embryos. At 11.5, BrdU-labeled nuclei are present throughout the ventricular zone (Fig. 5E). However, at E12.5 and later, BrdU-labeled nuclei are sharply reduced in number at the apical surface of the neocortical neuroepithelium in the lateral and posterior telencephalon (Fig. 5F–H). This reduction was quantified by counting BrdU-labeled and unlabeled nuclei at the apical surface of the ventricular zone (Fig. 5L). The fraction of BrdU labeled nuclei at the apical surface begins to decline at E13.5 and is reduced to 18% at E14.5 in the BF1 (+/−) heterozygotes. In contrast, this fraction is reduced to ∼10% by E12.5 in theBF1(−/−) mutant. We interpret these results to suggest that the cell cycle length increases at an earlier stage in development than normal in the absence of BF1. At E12.5, the fraction of cells with an elongated cell cycle in the neocortical neuroepithelium of theBF1(−/−) mutant is comparable to that found in normal E14.5 embryos.
BF1NHAA improves the proliferation of telencephalic progenitor cells
To determine the basis for the increased size of the neocortex in mice expressing BF1NHAA, we examined the proliferative behavior of telencephalic progenitor cells by BrdU labeling. We find that BF1NHAA improves progenitor cell proliferation in the rostral region of the dorsal telencephalon by two criteria. We compared the pattern of BrdU-labeled nuclei after 15 hr, with that observed in BF1 mutants and their normal littermates (Fig. 5). In the NHAA rescue, we find that the fraction of BrdU-labeled nuclei at the apical surface of the ventricular zone of the telencephalon is higher than in the mutant and comparable to that of normal embryos at E12.5 and E13.5 (Fig.5J,L). The fraction of BrdU-labeled nuclei at apical surface decreases at E14.5 to the level found in normal embryos (Fig.5K,L).
We also determined the BrdU labeling index following a 2 hr labeling period in both the anterior third (anterior) and the posterolateral (posterior) region of the dorsal telencephalon in horizontal sections (Fig. 6J). TheBF1 mutant has marked reduction in the fraction of BrdU-labeled nuclei in the anterior dorsal telencephalic neuroepithelium compared to the posterior dorsal telencephalon (Xuan et al., 1995) (Fig. 6D–F), in contrast to a more uniform fraction of BrdU-labeled nuclei throughout the neocortical neuroepithelium of the normal BF1(+/−) heterozygote (Fig.6A–C). NHAA rescue embryos have a significant improvement in the BrdU labeling index in the anterior telencephalon, which is similar to that observed in normal embryos (Fig.6G–J). Thus, by two assays, we find that the BF1NHAA protein restores the proliferation of progenitor cells throughout the dorsal telencephalon to near normal levels.
BF1NHAA does not correct the premature neuronal differentiation associated with the loss of BF1
In the BF1 mutant, neuronal differentiation is initiated at the usual time, E11.5. However, by E12.5 we detect a large increase in the number of MAP2-positive cells that demarcate a thicker mantle zone (preplate and cortical plate) in the BF1 mutant compared with normal littermates. This is especially prominent in the lateral and posterior regions of the dorsal telencephalon (Xuan et al., 1995). These findings are consistent with a role for BF1 in reducing the rate of neuronal differentiation in the population of cortical progenitor cells. Although accelerated neuronal differentiation must be associated with an increased rate of cell cycle exit, we do not observe a significant reduction in the fraction of BrdU-labeled nuclei in the posterolateral telencephalon of the BF1 mutant. We postulate that this is the result of the rapid migration out of the ventricular zone to the cortical plate of progenitor cells that withdraw from the cell cycle. The observation that the neuroepithelium is thinner in this region is consistent with this hypothesis.
We compared the thickness of the mantle zone to the thickness of the ventricular zone (VZ) as an indicator of the extent of neuronal differentiation (mantle:VZ ratio). In the dorsal telencephalon of normal embryos, neuronal differentiation begins earlier in the posterior region, resulting in a higher mantle:VZ ratio posteriorly. At E12.5, the mantle:VZ ratio ranges from 0.1 anteriorly to 0.4 posteriorly. In the BF1 mutant at E12.5, accelerated neuronal differentiation results in a marked increase in the thickness of the mantle layer. The mantle:VZ ratio in the BF1 mutant ranges from 0.5 anteriorly to 4 posteriorly (Xuan et al., 1995). Expression of the BF1NHAA protein does not change this ratio significantly. The mantle:VZ ratio in the NHAA rescue embryo at E12.5 ranges from 0.5 anteriorly to 3 posteriorly. In the lateral telencephalon, shown in a coronal section at the level of the eye, the mantle:VZ ratio is 0.15 in the BF1 (+/−) heterozygote (Fig. 7A,D) and 1.4 in the NHAA rescue (Fig. 7B,E). The abnormally thick mantle zone in NHAA rescue embryos can also be seen in Figures 2 and 3. Thus, the accelerated neuronal differentiation associated with the loss of BF1 is not corrected by the expression of BF1NHAA.
Fig. 7.
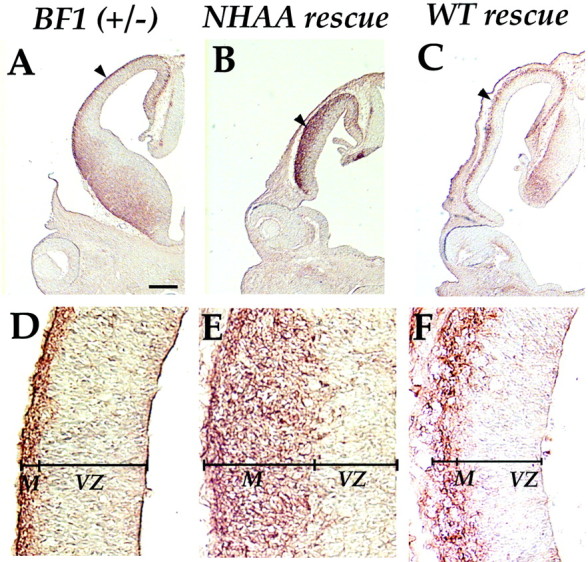
Expression of the neuronal marker, MAP2, in the dorsal telencephalon at E12.5. MAP2 expression in coronal sections inBF1 (+/−) heterozygotes (A, D), NHAA rescue embryos (B, E), and BF1 WT rescue embryos (C, F) identifies differentiated cells of the preplate and cortical plate (mantle zone). The thickness of the mantle zone in the NHAA rescue is much greater than in the BF1heterozygote or in the BF1 WT rescue. The mantle zone:ventricular zone ratio is 0.15 in D, 1.4 inE, and 0.3 in F. The mantle zone is thinnest anteriorly and thicker posteriorly within the telencephalon. Therefore we show coronal sections from comparable planes at the level of the eye, in the heterozygote, the NHAA rescue, and theBF1 WT rescue. Scale bar: A–C, 400 μm;D–F, 50 μm.
The accelerated neuronal differentiation in the neocortical neuroepithelium of the NHAA rescue suggests that the regulation of neuronal differentiation requires the DNA binding activity of BF1. However, it remained possible that this deficit is the result of an imperfect replacement of BF1 activity with the tTA system rather than to the loss of DNA binding activity in the BF1NHAA protein. To address this issue, we replaced the endogenous BF1 gene with that of WT BF1 under the control of the tTA protein. We find that “WT BF1 rescue” embryos exhibit normal growth of the dorsal telencephalon (Fig.3C,G). In two lines that express WT BF1 at levels lower than that of moderate BF1NHAA expresser lines, the growth of the dorsal telencephalon is comparable with that in normal embryos. Both the rate of progenitor proliferation as assessed by BrdU incorporation studies, and the timing of neuronal differentiation are corrected (data not shown). The thickness of the mantle zone in the WT rescue is comparable to that in WT embryos and significantly thinner than in the NHAA rescue (Fig.7C,F). These results support the conclusion that BF1 controls the timing of neuronal differentiation in the neocortex through a DNA binding-dependent mechanism.
Ectopic expression of Bmp genes in the dorsal telencephalon of the BF1 (−/−) mutant and the NHAA rescue
We have shown that Bmp4 is ectopically expressed in the telencephalic neuroepithelium of the BF1(−/−) mutant (Dou et al., 1999). Here we report thatBmp6, Bmp7, andBmp2 (Fig. 8) (data not shown) are also ectopically expressed in the telencephalon of embryos lacking BF1. Instead of being restricted to the dorsomedial telencephalon and the medial wall of the telencephalic vesicles, in a pattern complementary to that of BF1 (Furuta et al., 1997), Bmps 2, 4, 6, and7 are expressed broadly in the telencephalic neuroepithelium. BMP expression is expanded dorsolaterally and ventrolaterally from its normal domain in the medial wall. Because the ventral telencephalon is almost entirely missing in the BF1(−/−) mutant, the lateral dorsal telencephalon assumes a ventral position in the embryo. Coronal sections through the anterior telencephalon show that ectopic Bmp expression is detectable throughout the mutant neuroepithelium (Dou et al., 1999) (data not shown). In more posterior sections of the telencephalon, ectopic Bmp expression is not present uniformly throughout the neuroepithelium, but is highest in the dorsolateral and ventrolateral telencephalon (Fig. 8B,E). This ectopic expression overlaps the expression domain of the lacZ reporter (Dou et al., 1999) (data not shown).
Fig. 8.

Ectopic expression of Bmps is not corrected in the NHAA rescue. Bmp6(A–C) and Bmp7(D–F) expression is restricted to the dorsomedial telencephalic neuroepithelium in the BF1(+/−) heterozygote at E11.5 (arrows in A,D). In the BF1 (−/−) mutant and the NHAA rescue at E11.5, ectopic Bmp6 andBmp7 is detected in the lateral telencephalon. The level of Bmp6 andBmp7 is highest dorsolaterally and ventrolaterally (arrowheads). Scale bar, 200 μm.
To determine the potential role of increased BMP activity in regulating the proliferation and differentiation of telencephalic progenitor cells in the BF1 (−/−) mutant, we examined the expression ofBmps in NHAA rescue embryos. We find thatBmps 2, 4, 6, and 7 are ectopically expressed in the telencephalic neuroepithelium in a pattern similar to that found in the BF1 (−/−) mutant (Fig.8C,F) (data not shown). These findings show that DNA binding activity is required for BF1 to repress the expression ofBmp genes. They also suggest that increased BMP activity in the telencephalic neuroepithelium does not result in the reduced progenitor cell proliferation rate observed in theBF1 (−/−) mutant.
DISCUSSION
Replacement of endogenous BF1 with a DNA binding defective form
We have adapted the tetracycline-regulated system developed byGossen and Bujard (1992) and Kistner et al. (1996) to control the expression of genes within the telencephalic neuroepithelium. A mouse line, BF1(+/−tTA), was generated that expresses the tTA protein under the control of the BF1 promoter by targeting the tTA sequence to the BF1 locus by homologous recombination. We also generated transgenic lines that contain constructs expressing either WT BF1 or the DNA binding defective form,BF1NHAA, under the control of the tet-Operator. These new lines and the previously describedBF1(+/−lacZ) line, were used to generate embryos that lack endogenous BF1 and express either WT BF1 or BF1NHAA under the control of the tTA protein in the telencephalic neuroepithelium. Examination of these embryos reveals that some critical functions of BF1 in the developing brain are mediated through DNA binding-independent mechanisms, whereas others do require the ability to bind to DNA.
The utility of the BF1/tTA system is supported by the following results: (1) The BF1NHAAtranscript is expressed in a pattern that closely matches that of the endogenous BF1 protein. (2) We show that protein levels of BF1NHAA in embryos that demonstrate a phenotypic rescue, range from just below the level of BF1 inBF1(+/−) heterozygotes to slightly higher than the level of BF1 in wild-type embryos. Thus, the BF1NHAA protein functions at protein levels in the physiological range. (3) The phenotypic changes observed in the NHAA rescue are dependent on the expression of adequate levels of the BF1NHAA protein. Three low expresser lines have a phenotype that is indistinguishable from that of the BF1 (−/−) mutant. Furthermore, when pregnant dams of high expresser lines are given doxycycline to shut down the activity of the tTA protein, the BF1NHAAtranscript is not expressed (Fig. 1I), and NHAA rescue embryos are phenotypically indistinguishable from BF1(−/−) mutants (data not shown). (4) Replacement of endogenous BF1 with WT BF1 using the tTA system rescues the growth and development of the dorsal telencephalon. This occurs despite the relatively low expression levels in the two transgenic lines that we have generated. In contrast, rescue of ventral telencephalon development is poor. Further studies will be required to determine whether higher levels of WT BF1 will restore the development of the ventral telencephalon with the tTA system.
BF1 maintains the proliferation rate of telencephalic progenitor cells during the neurogenetic period
We provide evidence that BF1 regulates the length of the cell cycle throughout the neocortical neuroepithelium after E11.5. These findings extend our previous work that showed that BF1 is required for normal progenitor cell proliferation in the anterior third of the dorsal telencephalon. The requirement for BF1 in progenitor cell proliferation is not observed at earlier stages in development. Cell proliferation in the neocortical neuroepithelium is unaffected by the loss of BF1 from E8.5 when BF1 is first detectable in the neural plate, to E11.5, a period of rapid cell proliferation (Wilson, 1982). Loss of BF1 results in anomalies in the cell cycle only after E11.5, when the cell cycle length begins to increase and neuronal differentiation is initiated. Instead of a gradual increase in the cell cycle length between E11.5 and E17.5, neocortical progenitors of mice lacking BF1 display a rapid increase in cell cycle length. This result suggests that one function of BF1 may be to regulate the response of telencephalic neuroepithelial cells to mitogenic or antiproliferative signals. We have reported evidence that supports the latter possibility. Telencephalic neuroepithelial cells lacking BF1 proliferate in culture at a rate similar to that of WT cells in response to the growth factor, FGF-2. In contrast, primary cultures of cortical progenitor cells lacking BF1 are more sensitive to the antiproliferative activity of TGFβ and activin (Dou et al., 2000).
BF1 regulates neocortical progenitor cell proliferation through a DNA binding-independent mechanism
By examining the consequences of replacing endogenous BF1 with the DNA binding defective BF1NHAA protein, we show that the maintenance of progenitor cell proliferation in the developing telencephalon by BF1 is mediated through a DNA binding-independent mechanism. BF1NHAAenhances the growth of the telencephalic vesicles, compared with embryos lacking BF1. The size of the neocortex in the NHAA rescue embryos is restored to that of normal embryos. We show that the BF1NHAA protein is able to substitute for the WT BF1 protein to maintain the proliferation of neocortical progenitors, to normalize BrdU incorporation in the anterior dorsal telencephalon and to reverse the accelerated lengthening of the cell cycle in the dorsal telencephalon, associated with the loss ofBF1. Misexpression studies in Xenopus have led to the proposal that repression of the cell cycle inhibitor p27 is an important mechanism by which BF1 affects cell proliferation (Hardcastle and Papalopulu, 2000). We find that mice lacking both BF1and p27 have reduced telencephalic progenitor cell proliferation as found in the BF1 mutant (C. Hanashima and E. Lai, unpublished observations). This result together with the findings in the NHAA rescue, suggest that repression of p27expression may not be an essential mediator of the control of cell proliferation by BF1. Our new findings support previous in vitro studies that demonstrated that BF1 antagonizes the antiproliferative activity of TGFβ through a DNA binding-independent mechanism (Dou et al., 2000). We speculate that antagonism of the antiproliferative activity of TGFβ family members may be an important mechanism by which BF1 controls neocortical neurogenesis.
Control of neuronal differentiation in the neocortex: the role of transcriptional repression by BF1
In contrast with the regulation of progenitor cell proliferation, the inhibition of neuronal differentiation by BF1 is dependent on its DNA binding activity. NHAA rescue embryos have a thicker mantle zone in the dorsal telencephalon, compared with normal embryos of the same age. The BF1NHAA protein does not inhibit the withdrawal of neocortical progenitors from the cell cycle and delay their differentiation into neurons. Our results suggest that regulation of gene expression is the primary mechanism by which BF1 controls neuronal differentiation. Because transcriptional repression has been shown to be the predominant DNA binding-dependent activity of BF1, we investigated potential regulatory targets that could mediate its ability to inhibit neuronal differentiation.
We present evidence that multiple Bmp genes are targets of repression by BF1. Bmps 2, 4, 6, and 7 are ectopically expressed in the telencephalic neuroepithelium of mouse embryos lacking BF1. The BF1NHAA protein does not repress ectopic expression of Bmps associated with the absence of BF1. This is consistent with a requirement for DNA binding activity for BF1 to repress Bmpexpression. Bmp4 has been reported to inhibit the proliferation of neocortical progenitor cells as well as to promote their differentiation into neurons and glia (Li et al., 1998; Mabie et al., 1999). Our results suggest that repression ofBmp expression by BF1 is not required for normal proliferation rates of telencephalic progenitor cells. NHAA rescue embryos exhibit normal growth of the dorsolateral telencephalon despite having ectopic Bmp expression. The inability of the BF1NHAA protein to correct the ectopic expression of Bmps may contribute to the anomalies of the dorsomedial telencephalon in the NHAA rescue embryos. Studies with additional regional markers are in progress to delineate the mechanisms by which BF1 regulates dorsoventral patterning of the telencephalon.
Bmps exert their effects by binding to and activating a complex of cell surface receptors. The type I receptors, BmpR-1A and BmpR-1B, are expressed in the neuroepithelium (Dewulf et al., 1995) and are believed to be the primary transducers of Bmp signals in the developing brain. Expression of a constitutively activated Bmp receptor (caBmpr-1b) in the neuroepithelium results in premature neuronal differentiation in mid-gestation embryos, supporting a role for this receptor in mediating a terminal response to Bmps, resulting in apoptosis or differentiation of neural progenitors (Panchision et al., 2001). Antagonism of Bmp activity in neocortical progenitor cells with a dominant-negative Bmptype I receptor or with noggin, results in the inhibition of neuronal differentiation, demonstrating that endogenous Bmp signals trigger the differentiation of neocortical neurons (Li et al., 1998; Li and LoTurco, 2000). Taken together with our studies, we infer that ectopic Bmp expression resulting from the absence of BF1, promotes the differentiation of telencephalic progenitors into neurons and that repression ofBmp gene expression by BF1 mediates its ability to inhibit neuronal differentiation.
The key finding in this study is that the control of progenitor cell proliferation and differentiation in the neocortex by BF1 are mediated by distinct mechanisms. BF1 promotes the proliferation of telencephalic neuroepithelial cells through a DNA binding-independent mechanism, whereas BF1 delays neuronal differentiation in the neocortex through a DNA binding-dependent mechanism. This study also provides the first genetic evidence that a WH gene has essential functions that do not require the DNA binding activity of the WH domain. Finally, the targeting of the tetracycline transactivator to the BF1 locus creates a mouse line that will be a useful tool for the study of forebrain development.
C.H. and L.S. contributed equally to this work.
This work was supported by National Institutes of Health (NIH) Grants HD29584 and EY11124 (E.L.), a grant from the Muscular Dystrophy Association (S.L.), and a Cancer Center Support Grant from the NIH. We thank Leah Brunk, Marissa DeVito, and Dina Lomonte for excellent technical support and Willie Mark and the members of the Transgenic Mouse Core Facility for help with the mice. We are grateful to H. Bujard, S. McConnell, B. Hogan, and T. Jessell for providing plasmids and Lorenz Studer for critically reviewing this manuscript.
Correspondence should be addressed to Eseng Lai, Cell Biology Program, Memorial Sloan-Kettering Cancer Center, 1275 York Avenue, New York, NY 10021. E-mail: lai@ski.mskcc.org.
REFERENCES
- 1.Bourguignon C, Li J, Papalopulu N. XBF-1, a winged helix transcription factor with dual activity, has a role in positioning neurogenesis in Xenopus competent ectoderm. Development. 1998;125:4889–4900. doi: 10.1242/dev.125.24.4889. [DOI] [PubMed] [Google Scholar]
- 2.Caviness VJ, Takahashi T, Nowakowski R. Numbers, time and neocortical neuronogenesis: a general developmental and evolutionary model. Trends Neurosci. 1995;18:379–383. doi: 10.1016/0166-2236(95)93933-o. [DOI] [PubMed] [Google Scholar]
- 3.Clark D, Mitra P, Wang S. Scalable architecture in mammalian brains. Nature. 2001;411:189–193. doi: 10.1038/35075564. [DOI] [PubMed] [Google Scholar]
- 4.Dewulf N, Verschueren K, Lonnoy O, Moren A, Grimsby S, Vande Spiegle K, Miyazono K, Hulebroeck D, ten Dijke P. Distinct spatial and temporal expression patterns of two type I receptors for bone morphogenetic proteins during mouse embryogenesis. Endocrinology. 1995;136:2652–2663. doi: 10.1210/endo.136.6.7750489. [DOI] [PubMed] [Google Scholar]
- 5.Dou C, Li S, Lai E. Dual role of brain factor-1 in regulating growth and patterning of the cerebral hemispheres. Cereb Cortex. 1999;9:543–550. doi: 10.1093/cercor/9.6.543. [DOI] [PubMed] [Google Scholar]
- 6.Dou C, Lee J, Liu B, Massague J, Xuan S, Lai E. BF-1 interferes with transforming growth factor beta signaling by associating with smad partners. Mol Cell Biol. 2000;20:6201–6211. doi: 10.1128/mcb.20.17.6201-6211.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Furuta Y, Piston DW, Hogan BLM. Bone morphogenetic proteins (Bmps) as regulators of dorsal forebrain development. Development. 1997;124:2203–2212. doi: 10.1242/dev.124.11.2203. [DOI] [PubMed] [Google Scholar]
- 8.Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-reponsive promoters. Proc Natl Acad Sci USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hardcastle Z, Papalopulu N. Distinct effects of XBF-1 in regulating the cell cycle inhibitor p27xici and imparting a neutral fate. Development. 2000;127:1303–1314. doi: 10.1242/dev.127.6.1303. [DOI] [PubMed] [Google Scholar]
- 10.Hebert J, McConnell S. Targeting of cre to the Foxg1 (BF1) locus mediates loxP recombination in the telencephalon and other developing head structures. Dev Biol. 2000;222:296–306. doi: 10.1006/dbio.2000.9732. [DOI] [PubMed] [Google Scholar]
- 11.Kistner A, Gossen M, Zimmermann F, Jerecic J, Ullmer C, Lubbert H, Bujard H. Doxycycline-mediated quantitative and tissue-specific control of gene expression in transgenic mice. Proc Natl Acad Sci USA. 1996;93:10933–10938. doi: 10.1073/pnas.93.20.10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li J, Chang H, Lai E, Parker E, Vogt P. The oncogene qin codes for a transcriptional repressor. Cancer Res. 1995;55:5540–5544. [PubMed] [Google Scholar]
- 13.Li W, LoTurco J. Noggin is a negative regulator of neuronal differentiation in developing neocortex. Dev Neurosci. 2000;22:68–73. doi: 10.1159/000017428. [DOI] [PubMed] [Google Scholar]
- 14.Li W, Cogswell C, LoTurco J. Neuronal differentiation of precursors in the neocortical ventricular zone is triggered by Bmp. J Neurosci. 1998;18:8853–8862. doi: 10.1523/JNEUROSCI.18-21-08853.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma Y, Geerdes D, Vogt P. Oncogenic transformation by the FOX protein Qin requires DNA binding. Oncogene. 2000;19:4815–4821. doi: 10.1038/sj.onc.1203834. [DOI] [PubMed] [Google Scholar]
- 16.Mabie P, Mehler M, Kessler J. Multiple roles of bone morphogenetic protein signaling in the regulation of cortical cell number and phenotype. J Neurosci. 1999;19:7077–7088. doi: 10.1523/JNEUROSCI.19-16-07077.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mountford P, Smith A. Internal ribosome entry sites and dicistronic RNA's in mammalian transgenesis. Trends Genet. 1995;11:179–184. doi: 10.1016/S0168-9525(00)89040-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Panchision D, Pickel J, Studer L, Lee S, Turner P, Hazel T, McKay R. Sequential actions of Bmp receptors control neural precursor cell production and fate. Genes Dev. 2001;15:2094–2110. doi: 10.1101/gad.894701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takahashi T, Nowakowski R, Caviness VJ. BUdR as an S-phase marker for quantitative studies of cytokinetic behavior in the murine cerebral ventricular zone. J Neurocytol. 1992;21:185–197. doi: 10.1007/BF01194977. [DOI] [PubMed] [Google Scholar]
- 20.Takahashi T, Nowakowski S, Caviness V. Cell cycle parameters and patterns of nuclear movement in the neocortical proliferative zone on the fetal mouse. J Neurosci. 1993;13:820–833. doi: 10.1523/JNEUROSCI.13-02-00820.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takahashi T, Nowakowski R, Caviness VJ. The leaving or Q fraction of the murine cerebral proliferative epithelium: a general model of neocortical neuronogenesis. J Neurosci. 1996a;16:6183–6196. doi: 10.1523/JNEUROSCI.16-19-06183.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takahashi T, Nowakowski R, Caviness VJ. Interkinetic and migratory behavior of a cohort of neocortical neurons arising in the early embryonic murine cerebral wall. J Neurosci. 1996b;16:5762–5776. doi: 10.1523/JNEUROSCI.16-18-05762.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tao W, Lai E. Telencephalon-restricted expression of BF-1, a new member of the HNF-3/fork head gene family, in the developing rat brain. Neuron. 1992;8:957–966. doi: 10.1016/0896-6273(92)90210-5. [DOI] [PubMed] [Google Scholar]
- 24.Wilson D. The cell cycle during closure of the neural folds in the C57BL mouse. Dev Brain Res. 1982;2:420–424. doi: 10.1016/0165-3806(81)90049-3. [DOI] [PubMed] [Google Scholar]
- 25.Xuan S, Baptista C, Balas G, Tao W, Soares V, Lai E. Winged helix transcription factor BF-1 is essential for the development of the cerebral hemispheres. Neuron. 1995;14:1141–1152. doi: 10.1016/0896-6273(95)90262-7. [DOI] [PubMed] [Google Scholar]
- 26.Yang H, Wanner I, Roper S, Chaudhari N. An optimized method for in situ hybridization with signal amplification that allows the detection of rare mRNAs. J Histochem Cytochem. 1999;47:431–445. doi: 10.1177/002215549904700402. [DOI] [PubMed] [Google Scholar]
- 27.Yao J, Lai E, Stifani S. The winged-helix protein brain factor 1 interacts with groucho and hes proteins to repress transcription. Mol Cell Biol. 2001;21:1962–1972. doi: 10.1128/MCB.21.6.1962-1972.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]