

Abstract
The physiological activity of hypothalamic arcuate nucleus (ARC) neurons is critical for dynamic maintenance of body energy homeostasis, and its malfunction can result in common metabolic disorders, such as obesity. It is therefore of interest to determine which set of ion channels shapes electrical activity in the ARC. Whole-cell patch clamp of ARC neurons in mouse brain slices identified three electrophysiologically distinct types of neurons. These were distinguished by their rebound “signatures” after hyperpolarizing current injection in current clamp and by the presence of transient inward (Type-B neurons) or outward (Type-A and Type-C neurons) subthreshold voltage-gated currents in voltage-clamp recordings. In turn, the transient outward current (A-current) of Type-C neurons had a lower activation threshold and different time and voltage dependence of inactivation than that of Type-A neurons. The brain–gut peptide cholecystokinin (CCK) has long been recognized to control food intake, but how endogenous CCK modulates the activity of central appetite-regulating networks remains unresolved. Here, we show that low (picomolar) concentrations of CCK rapidly and reversibly slow the firing of ARC Type-C neurons. This effect is mediated by postsynaptic CCK-B receptors and is attributable to potentiation of the A-current. Our study thus identifies several fundamental biophysical mechanisms underlying the physiological activity of ARC neurons and suggests a novel mechanism by which endogenous CCK may control appetite.
Keywords: hypothalamus, arcuate nucleus, appetite, energy homeostasis, electrophysiology, CCK, A-currents, CCK-B receptor
The hypothalamus maintains body energy homeostasis by integrating neural, endocrine, and metabolic information to elicit behavioral, autonomic, and endocrine responses (Elmquist et al., 1999; Schwarz et al., 2000). Among the hypothalamic regions involved, the arcuate nucleus (ARC) has recently gained much prominence because of several findings. First, ARC neurons are the source of several powerful neuropeptide modulators of food intake and metabolic rate, such as α-melanocyte-stimulating hormone, cocaine- and amphetamine-regulated transcript, neuropeptide Y, and agouti protein (Elias et al., 1998; Hahn et al., 1998). Second, mRNA levels of these peptides in the ARC are changed by perturbations of body energy balance, such as fasting, and by changes in blood levels of insulin and leptin (Schwarz et al., 2000). Third, ARC neurons are strategically located near a “window” in the blood–brain barrier at the median eminence (Elmquist et al., 1999; Ganong, 2000), which allows them to directly translate blood-borne signals of body energy status, such as glucose, insulin, and leptin (Spanswick et al., 1997, 2000; Cowley et al., 2001) into neuronal activity.
Together, these findings indicate that ARC neurons act as key integrators of peripheral and central signals of body energy status and are critical for central control of body energy balance (Schwarz et al., 2000). Furthermore, defects in ARC function have been linked to obesity (Barsch et al., 2000). It is therefore important to determine the biophysical mechanisms that control the electrical activity of ARC neurons and thereby the release of the neuropeptides they contain. Within the hypothalamus, the region best characterized electrophysiologically is the ventromedial nucleus. In neurons from this region, spontaneous firing and responses to perturbations of membrane potential are probably determined by currents with activation thresholds below that of the voltage-gated Na+ channel (subthreshold currents) (Minami et al., 1986; Miki et al., 2001). Some ARC neurons are also spontaneously active (Rauch et al., 2000), suggesting that subthreshold currents may shape their electrical activity. Of particular interest are the currents that directly tune neuronal firing and are under control of neuromodulators, such as hyperpolarization-activated currents (Lüthi and McCormick, 1998) and A-type K+ currents (Connor and Stevens, 1971;Liss et al., 2001; Yang et al., 2001).
Cholesystokinin (CCK), which functions as both a hormone and a neurotransmitter (Crawley and Corwin, 1994), has long been recognized to play a key role in the short-term control of food intake by inducing satiety (Gibbs et al., 1973; Moran, 2000). However, the mechanism by which endogenous CCK controls appetite is still only poorly understood, and both the site of CCK action (central or peripheral) and the type of receptor involved (CCK-A or CCK-B) remain controversial (Baldwin et al., 1998). The ARC is a target for both synaptic (Siegel et al., 1987;Ciofi and Tramu, 1990; Baldwin et al., 1998) and probably also blood-borne (Ganong, 2000) CCK. It is therefore possible that endogenous CCK may modulate feeding by interacting with ARC neurons.
We used the whole-cell patch-clamp technique to investigate the intrinsic biophysical properties and CCK responsiveness of ARC neurons in mouse brain slices.
MATERIALS AND METHODS
Brain slice preparation. Procedures involving animals were performed in accordance with the Animals (Scientific Procedures) Act, 1986 (United Kingdom). C57BL/6 mice (12–16 d postnatal) were killed by cervical dislocation. Brains were removed quickly, immersed in ice-cold solution, and then blocked for slicing. Coronal slices (250- to 300-μm-thick) containing the arcuate nucleus were cut with a Vibroslice (Campden Instruments, London, UK), while bathed in ice-cold artificial CSF (ACSF), containing (in mm) the following: 118 NaCl, 25 NaHCO3, 3 KCl, 1.2 NaH2PO4, 1.5 CaCl2, 1 MgCl2, and 10 glucose (bubbled with a mixture of 95% O2 and 5% CO2). After sectioning, slices were allowed to recover for >20 min in a chamber filled with gassed ACSF at room temperature (22–24°C) before the experiment.
Electrophysiology and data analysis. For whole-cell patch-clamp recordings, slices were continuously perfused at 2–4 ml/min with ACSF bubbled with a mixture of 95% O2 and 5% CO2. Patch pipettes were pulled from borosilicate glass (GC150TF/F; Clark, Reading, UK) and had tip resistances of 3–5 MΩ when filled with internal solution, which contained (in mm) the following: 130 K-gluconate, 1 NaCl, 0.1 EGTA, 1 MgCl2, 10 HEPES, and 5 K2-ATP, pH 7.3 adjusted with KOH. A low concentration of EGTA was used to mimic physiological cytosolic Ca2+ buffering and to preserve Ca2+-dependent K+ currents, which can affect the spontaneous firing rates of neurons (Wolfart et al., 2001). Liquid junction potentials were measured in separate experiments, in which the bath (connected to the reference electrode by a 3m KCl agar bridge) was perfused alternately with ACSF or the internal solution, and the zero-current pipette potentials in the two solutions were compared. Junction potentials were <8 mV and were not corrected for. The arcuate nucleus was located using a mouse brain stereotaxic atlas (Franklin and Paxinos, 1997). Neurons were visualized by infrared interference contrast video microscopy with a Newvicon camera (Hamamatsu, Hamamatsu City, Japan), mounted to an upright microscope (Axioscop FS; Zeiss, Oberkochen, Germany) (Stuart et al., 1993). Whole-cell recordings were performed in current- or voltage-clamp mode using an EPC-9 patch-clamp amplifier (Heka Elektronik, Lambrecht, Germany). Only cells with an input resistance of >0.8 GΩ were used. The series resistance was <15 MΩ and was not compensated. Data was sampled (at 3 kHz) and analyzed using the program package Pulse+pulsefit (Heka Elektronik). The voltage dependence of A-current inactivation (see Figs. 4B, 5B) was fitted by the Boltzman equation, using Microcal Software (Northampton, MA) Origin software. Here,Vm is the membrane potential,V0.5 is the membrane voltage at which the normalized current amplitude (I/Imax) is half-maximal, and k is the slope factor. Concentration–response data for CCK were fitted according to the Hill equation, ΔI = ΔImax − ΔImax/{1 + ([CCK]/EC50)h}, where ΔI is the increase in current produced by CCK, ΔImax is the maximum current increase, [CCK] is the concentration of CCK, and h is the Hill coefficient. Statistical significance was tested using a two-tailed unpaired t test (Origin software; Microcal Software). Data are given as mean ± SEM unless stated otherwise.
Fig. 4.
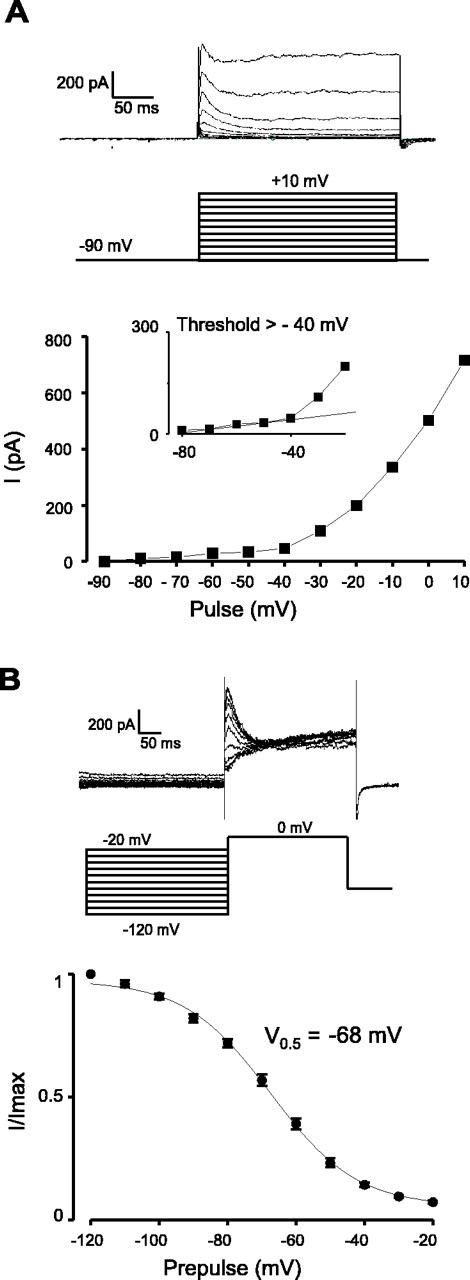
Voltage dependence of A-current activation and inactivation in ARC Type-A neurons. TTX (700 nm) was added to block Na+ currents. A , Current–voltage relationship. The protocol is the same as in Figure 3. The data are representative of 12 cells. The inset is an expansion of the threshold region and shows that the A-current diverges from the linear leak current at potentials positive to −40 mV.B, Voltage dependence of inactivation. Themiddle and top parts show the voltage-clamp protocol and an example of the data obtained, respectively. The protocol consisted of a 300 msec prepulse to between −120 and −20 mV (in 10 mV increments), followed by a 250 msec test pulse to 0 mV. The peak currents during the test pulse were expressed as a fraction of the maximum and plotted against the prepulse potential (bottom; n = 7). The line is the best fit of the data to the Boltzman equation (see Materials and Methods).
Fig. 5.
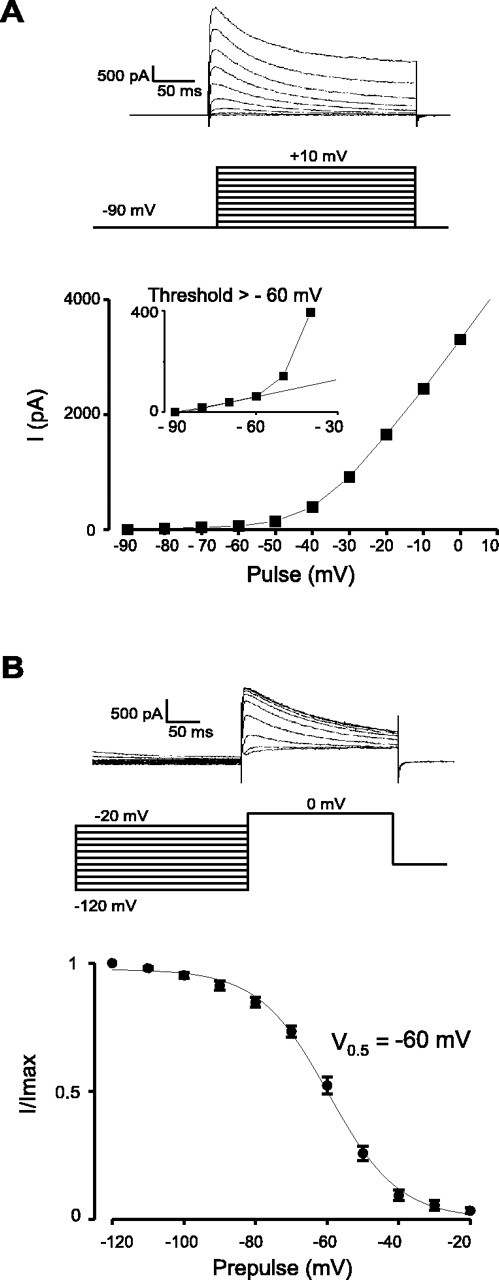
Voltage dependence of A-current activation and inactivation in ARC Type-C neurons. TTX (700 nm) was added to block Na+ currents. A, Whole-cell currents (top), voltage protocol (middle), and current–voltage relationship (bottom). The data are representative of 12 cells. Theinset is an expansion of the threshold region and shows that the A-current diverges from the linear leak current at potentials positive to −60 mV. B, Voltage dependence of inactivation (n = 6). The line is the best fit of the data to the Boltzman equation. All protocols are the same as in Figure 4.
Chemicals. CCK-8S (the main molecular form in both the CNS and the periphery) (Dockray 1978; Baldwin et al., 1998) was obtained from Calbiochem (La Jolla, CA). CCK-8S, gastrin (Calbiochem), or 4-aminopyridine (4-AP) (Sigma, St. Louis, MO) were applied locally at a rate of 35 μl/min (controlled by a syringe pump system) through a quartz pipette (0.25 mm inner diameter) placed over the ARC using a second manipulator. Tetrodotoxin (TTX) (Sigma) was included in the extracellular solution in some experiments, as indicated.
RESULTS
Three distinct electrophysiological phenotypes of ARC neurons
Current-clamp analysis of ARC neurons revealed that they fall into three electrically distinct types. “Type-A” neurons (membrane capacitance, 7–16 pF; n = 80) fired spontaneously at 5–10 Hz and immediately resumed normal firing after hyperpolarizing current injections (Fig.1A). “Type-B” neurons (membrane capacitance, 10–25 pF; n = 16) fired spontaneously at 2–8 Hz and showed a slow rebound depolarization spike at the end of a hyperpolarizing current pulse (Fig.1B). “Type-C” neurons (membrane capacitance, 15–35 pF; n = 34) exhibited spontaneous activity at 1–5 Hz (n = 27) or were electrically silent with resting membrane potentials between −45 and −50 mV (n= 7); on recovery from hyperpolarizing pulses, these cells displayed a rebound hyperpolarization, which caused them to resume firing with a prominent delay (Fig. 1C). After a hyperpolarizing pulse to −80 mV, the latency to the first spike was 51 ± 6 msec in Type-A cells (n = 80), 4.7 ± 3.5 msec in Type-B cells (n = 16), and 648 ± 83 msec in Type-C cells (n = 27). The latency was significantly longer in Type-C cells than in either Type-A or Type-B cells (p < 0.00001 in both cases).
Fig. 1.

Three distinct electrophysiological phenotypes of ARC neurons. These phenotypes were distinguished by their recovery from hyperpolarizing current injections (30–80 pA) in current clamp (left) and by the currents elicited by a voltage step from −90 to −40 mV in voltage clamp (right). Current- and voltage-clamp protocols are displayed schematically below.A, Type-A ARC cells show no rebound potential (left) and a small, rapidly inactivating outward current (right) in response to repolarization from hyperpolarized potentials. B, Type-B ARC neurons exhibit rebound depolarization (left) and a prominent subthreshold inward current (right). C, Type-C ARC neurons display rebound hyperpolarization (left) and a large outward current that inactivates slowly relative to that of Type-A cells (right). Horizontal lines on thecurrent-clamptraces(left) show the zero-current potential.
To visualize the currents that underlie these three types of rebound potential, we used a voltage-clamp protocol in which cells were held at −90 mV for 300 msec and then stepped to −40 mV. This protocol revealed fundamental differences in subthreshold currents in the three cell types. In Type-A cells, a voltage step from −90 to −40 mV evoked a small (44 ± 3 pA), fast-inactivating outward current (n = 80) (Fig. 1A), whereas in Type-C cells, it induced a large (248 ± 14 pA), slowly activating and inactivating outward current (n = 34) (Fig.1C). The amplitudes of these currents were significantly different (p < 0.00001). In Type-B cells, the same voltage step evoked a transient inward current (−34 ± 3 pA;n = 16) (Fig. 1B).
The differences in the recovery from hyperpolarization and the properties of voltage-gated currents were sufficient to identify unambiguously the type of each ARC neuron.
Absence of hyperpolarization-activated current in ARC neurons
ARC cells often exhibit regular spontaneous firing (Fig. 1). In extracellular recordings, this activity is not abolished by blockade of synaptic transmission (Rauch et al., 2000), which suggests that ARC neurons may have intrinsic pacemaker activity. We therefore investigated whether the hyperpolarization-activated current (Ih), which can be critical for neuronal pacemaking (Lüthi and McCormick, 1998), is present in ARC neurons.
Hyperpolarizing current injections failed to elicit the typical “sag” in membrane potential characteristic ofIh activation in all three types of ARC neurons (Fig.2A–C), in contrast to neurons from the neighboring dorsomedial nucleus (n = 4) (Fig. 2D). A voltage-clamp step from −40 to −120 mV confirmed that Ih is elicited by hyperpolarization in dorsomedial nucleus cells (n = 3) (Fig. 2D) but not in ARC neurons (Fig.2A–C). Thus, our results suggest thatIh channels with fast activation kinetics (i.e., those containing HCN1 subunits; Franz et al., 2000) are not present in all three types of ARC cells. We were unable to determine whether Ih currents with slower activation kinetics (channels lacking HCN1; Franz et al., 2000) are present in the ARC, because this requires holding cells at voltages negative to −90 mV for >2 sec (Franz et al., 2000), which was not well tolerated by ARC neurons in our experiments. However, because ARC cells fire at 2–10 Hz (Fig. 1), it seems unlikely that a slowly activating Ih current would contribute significantly to spike-to-spike pacemaking in the ARC.
Fig. 2.

Absence of fast Ihcurrents in ARC neurons. Overlay of membrane potential responses to hyperpolarizing current injections of increasing intensity (left) and currents recorded in response to hyperpolarization from −40 to −120 mV (right) in ARC (A–C) or dorsomedial nucleus (D) neurons. The prominentIh-mediated sag in potential and rapidly activating inward current found for dorsomedial nucleus neurons (D) are not observed for ARC neurons (A–C). Current- and voltage-clamp protocols are shown schematically below representative traces.Horizontal lines on the current-clamp traces (left) indicate the zero potential. Note the difference in current scale in A–C(right).
Type-B ARC neurons express a low-threshold Ca2+ current
The transient inward current and rebound depolarization in Type-B neurons (Fig. 1B) were significantly reduced or abolished by switching to ACSF containing low (0.3 mm) Ca2+ and high (9 mm) Mg2+(n = 3), suggesting that they were attributable to activation of a Ca2+ current. Voltage steps to between −80 and +10 mV from a holding potential of −90 mV revealed that this current activated between −60 and −50 mV (n = 10) (Fig. 3). This activation threshold is characteristic of neuronal T-type Ca2+ channels (Tsien et al., 1988).
Fig. 3.
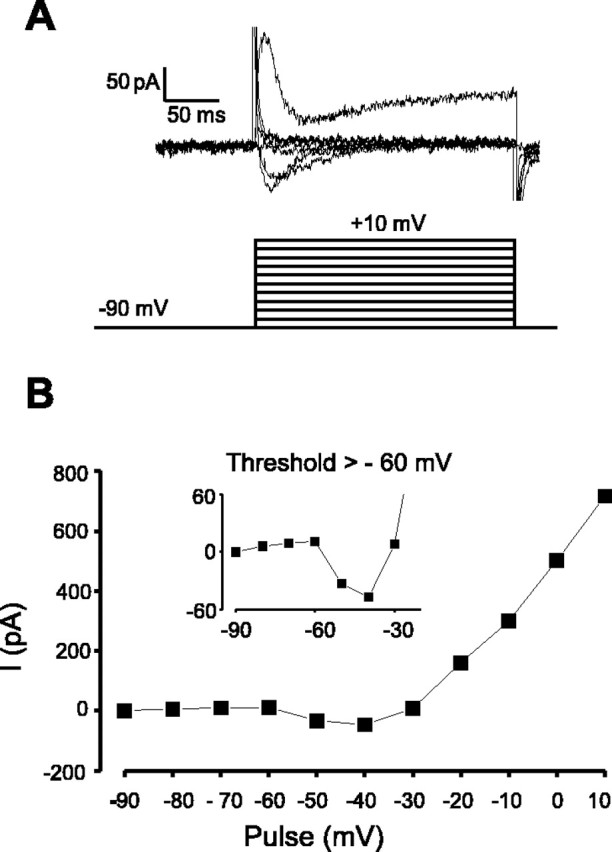
Voltage dependence of activation of Ca2+ current in Type-B cells. TTX (700 nm) was added to block Na+ currents.A, Representative example of membrane currents (top) evoked by the voltage protocol below. The cells were held at −90 mV for 300 msec and then stepped to +10 mV in 10 mV increments for 250–300 msec (interpulse interval of 2 sec). B, Peak amplitudes of resulting currents were measured relative to the baseline at −90 mV and plotted against the pulse potential. The data are representative of 10 cells. Theinset is an expansion of the threshold region and shows that the inward current activates between −60 and −50 mV.
A-Currents in Type-A and Type-C ARC neurons are different
The rapid activation and inactivation of the outward current observed in Type-A and Type-C ARC neurons is typical of A-type potassium channels. Furthermore, the transient outward current was completely eliminated by the A-current blocker 4-aminopyridine (10 mm; n = 4) (see Fig. 7B). A-Type potassium currents are key determinants of neuronal firing frequency (Rudy, 1988; Liss et al., 2001) and are modulated by neurotransmitters (Aghajanian, 1985) and neurotrophic factors (Yang et al., 2001). We therefore analyzed the properties of the A-current in Type-A and Type-C ARC neurons in detail. Type-B neurons appeared to express a fast transient outward current similar to the A-current in Type-A cells (Fig. 3); we did not analyze this current further because of the low numbers of Type-B cells in the ARC and because it was partially shunted by inward currents activated in the same voltage range (Fig. 3).
Fig. 7.
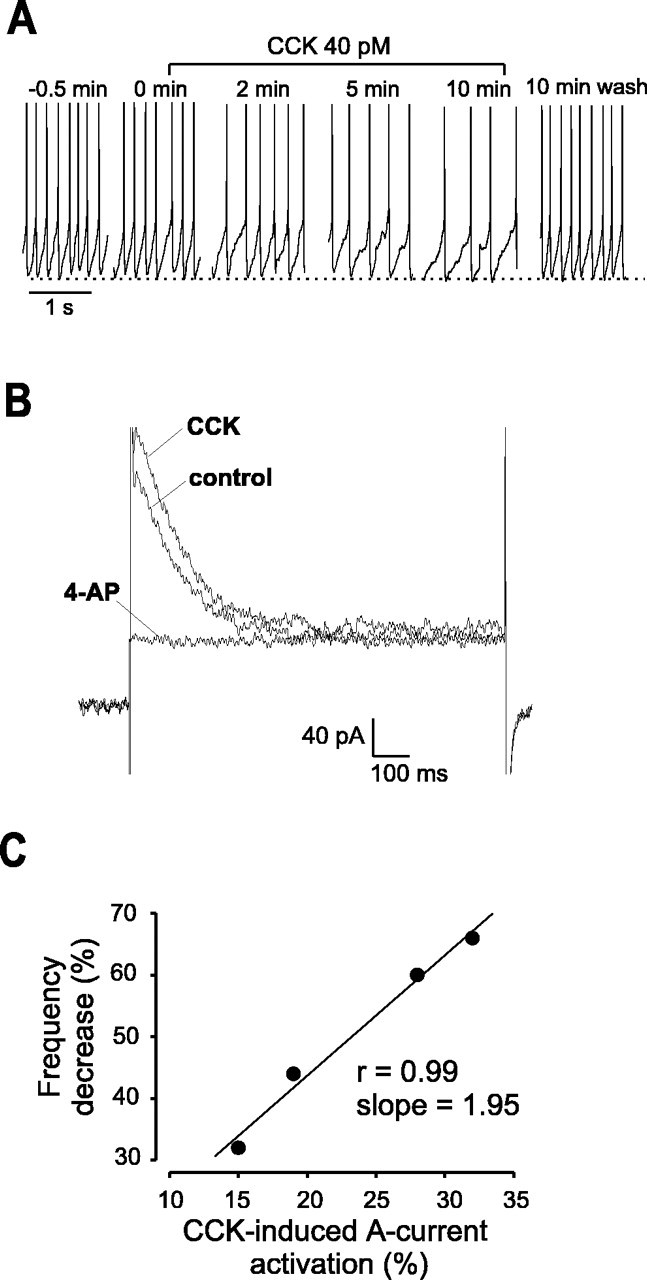
CCK slows firing and activates A-currents in ARC Type-C neurons. A, Membrane potential recordings of a Type-C neuron before, during, and after CCK application. Action potentials are truncated at 0 mV, and the dotted lineindicates −50 mV. B, Voltage-clamp recordings of the corresponding A-currents taken from the same cell. The cell was held at −90 mV for 300 msec and then stepped to −40 mV for 800 msec to elicit the A-current. Note that the peak A-current is greater in the presence CCK, but the steady-state current amplitude is unchanged (see Results). 4-AP at 10 mm abolished the A-current. Thetraces in A and B are representative of four cells. C, CCK-induced decrease in firing frequency plotted against the percentage of activation of the A-current for four different neurons. There is a strong linear correlation (r = 0.99; slope of 1.95).
Voltage steps to between −80 and +10 mV from a holding potential of −90 mV revealed that, in Type-A cells, the A-current activated between −40 and −30 mV (n = 12) (Fig.4A), whereas in Type-C cells, it activated at a much lower threshold, between −60 and −50 mV (n = 12) (Fig.5A). The voltage dependence of A-current inactivation was also significantly different in the two cell types: half-maximal inactivation occurred at −68 ± 1 mV in Type-A cells (Fig. 4B) and at −60 ± 1 mV in Type-C cells (p < 0.0001) (Fig. 5B). In addition, the time course of A-current inactivation in Type-C cells was substantially slower than that in Type-A cells (Fig.6), consistent with the differences in rebound potentials in the two cell types (Fig.1A,C). The rate of A-current inactivation was essentially voltage independent in both cell types (Fig. 6), as found for other neurons (Liss et al., 2001).
Fig. 6.
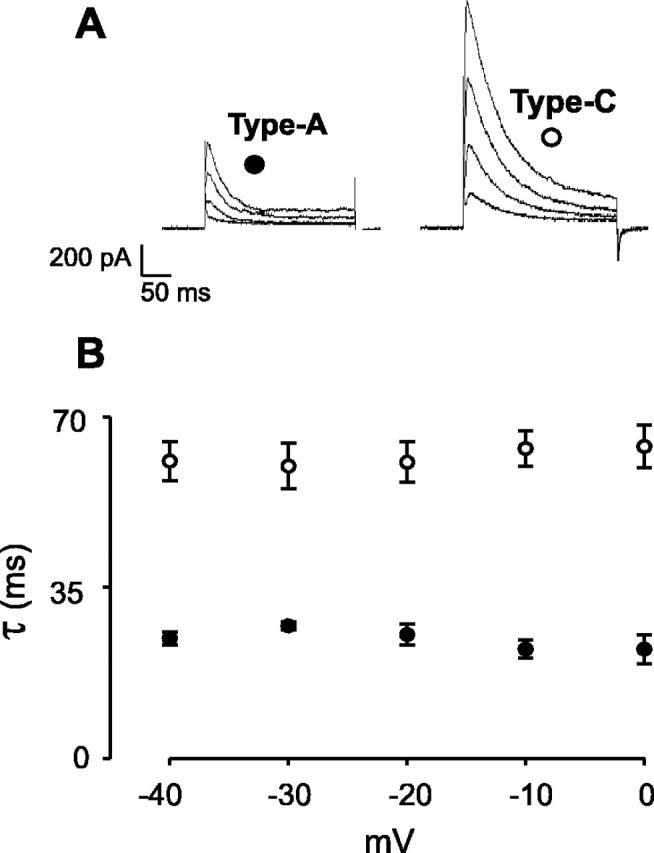
A-Current inactivation is slower in Type-C than in Type-A neurons. TTX (700 nm) was added to block Na+ currents. A, Examples of A-currents elicited by steps from −40 to 0 mV from a holding potential of −90 mV. A monoexponential fit (I =I0 + Aexp{−(t −t0)/τ}) to the A-current decay was used to calculate the inactivation time constant, τ.B, Voltage dependence of τ in Type-C cells (open circles; n = 5) and Type-A cells (filled circles; n = 5).
CCK acutely slows firing and potentiates A-currents in ARC Type-C neurons
We next examined whether CCK modulates the electrical activity of ARC neurons. The firing frequency of Type-A (n = 5) and Type-B (n = 4) neurons was unaffected by 40 pm CCK. However, in Type-C ARC neurons, CCK (40 pm) slowed firing within 0.5–5 min of application, without inducing detectable hyperpolarization (n = 4) (Fig.7A). This type of behavior can be caused by an increase in A-current activity (Liss et al., 2001). To address this possibility, we monitored A-currents and firing frequency before and after stimulation with CCK in the same Type-C neurons. Because no detectable activation of delayed outward current was seen at −40 mV in Type-C cells (n = 10) (Fig. 7B), we measured A-current amplitude at this potential. We found that the magnitude of the A-current, but not of the steady-state current, was greater after CCK application (n = 4) (Fig.7B). Furthermore, the current activated by CCK was abolished by 10 mm 4-AP. This suggests that CCK selectively affects the A-current. Plotting the decrease in firing frequency against the corresponding increase in A-current amplitude revealed a strong linear correlation (r = 0.99) (Fig.7C), as would be expected if CCK-induced slowing of firing was attributable to an increase in the A-current.
The mechanism of CCK modulation of A-currents in ARC Type-C neurons
We next explored the mechanism underlying the activation of A-currents in ARC Type-C cells by CCK (Fig.8). Neurons were synaptically isolated by including 1 μm TTX in the extracellular solution and voltage clamped at −60 mV to prevent membrane potential changes from influencing A-current activity. A-Current amplitude was measured every 30 sec using the protocol shown in Figure 8A. Under these conditions, CCK (2–40 pm) induced a reversible (Fig. 8A) and dose-dependent (Fig.8B) increase in A-current amplitude. Above 40 pm, CCK was less effective at increasing A-currents (n = 4), probably because of the rapid desensitization that is typical of high-affinity CCK receptors (Burdakov and Galione, 2000). The best fit of the Hill equation to the concentration–response data gave a maximum response of 31% and an apparent EC50 of 18.7 pm(n = 12).
Fig. 8.
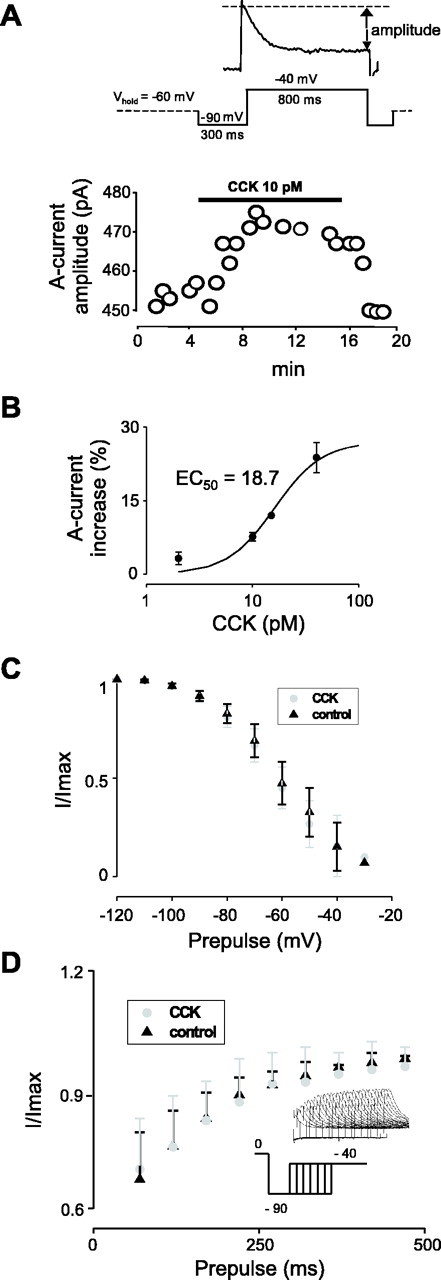
Pharmacological and kinetic analysis of CCK modulation of A-currents in ARC Type-C neurons. TTX (1 μm) was present to block Na+ currents.A, Time course of A-current increase by CCK. Theinset shows the voltage-clamp protocol used to monitor the A-current amplitude. The figure is representative of responses in eight cells. B, Relationship between CCK concentration and the increase in A-current ampli- tude. The line is drawn to the Hill equation with EC50 of 18.7 and h = 1.32 (n = 12). For details, see Results.C, The voltage dependence of A-current inactivation (measured as in Fig. 5B) was not significantly shifted by CCK (n = 5; control,V0.5 = −58 ± 2 mV; CCK,V0.5 = −60 ± 1 mV;p > 0.1). D, Time dependence of A-current recovery from inactivation. Cells were held at 0 mV for 500 msec to inactivate A-currents and then stepped to −90 mV for varying durations to remove inactivation, after which the A-current amplitude was measured at −40 mV. The inset shows the protocol and an example of the data obtained. CCK did not significantly change the time dependence of recovery from inactivation (n = 3).
The potentiation of the A-current by CCK was specific to ARC Type-C cells: kinetically similar A-currents in neurons of the neighboring premammillary nucleus were not modulated by the peptide (n = 4). The voltage and time dependence of A-current inactivation were not significantly different before and after application of CCK (Fig. 8C,D), suggesting that CCK enhances the activation of the A-current without affecting its inactivation.
Two CCK receptor subtypes are known to exist. The affinity of the CCK-A receptor for CCK-8S is 1000-fold higher than that for gastrin, whereas the CCK-B receptor has similar affinities for gastrin and CCK-8S (Crawley and Corwin, 1994). Application of 40 pm gastrin mimicked the effects of 40 pm CCK on firing and A-currents in ARC Type-C cells (n = 4) (Fig.9). These results suggest that CCK slows the firing and potentiates the A-current in ARC Type-C cells by activation of postsynaptic CCK-B receptors.
Fig. 9.

CCK slows firing and activates A-currents in ARC Type-C cells by acting on CCK-B receptors. Top trace, Expansions of membrane potential recordings before, during, and after application of 40 pm gastrin. Action potentials are truncated at 0 mV. Bottom traces, Corresponding A-currents, measured using the same protocol as in Figure8A. The data are representative of four cells.
DISCUSSION
In this study, we show that three biophysically distinct types of neurons are found in the ARC, which can be readily identified on the basis of their firing patterns and membrane currents. Each cell type has a unique recovery behavior after a hyperpolarizing current injection in current clamp (Fig. 1). Furthermore, voltage-clamp analysis shows that Type-B cells differ from Type-A and Type-C cells in that they exhibit a pronounced low-threshold Ca2+ current (Fig. 3), whereas Type-A and Type-C cells can be readily distinguished by differences in their A-currents (Figs. 4-6). The A-current of Type-C cells (Fig.5B) is much less inactivated in the resting potential region (−40 to −50 mV) than that of Type-A cells (Fig.4B), and it inactivates much more slowly (Fig. 6). These differences probably account for the finding that Type-C cells fire at lower rates than Type-A cells.
Modulation of Ih by neurotransmitter-generated intracellular second messengers such as cAMP can influence cell firing (Brown et al., 1979; Lüthi and McCormick, 1998). Our demonstration that ARC cells do not possesIh currents (Fig. 2) suggests that such mechanisms probably do not play a role in modulating the electrical activity of the ARC.
We found that the relatively small increases in A-current amplitude produced by CCK in ARC Type-C cells led to large changes in neuronal firing frequency (1.95% change in frequency per 1% A-current change) (Fig. 7C). A similar steep linear dependence of firing frequency on A-current amplitude was reported recently for dopaminergic midbrain neurons (1.78% change in frequency per 1% A-current change;Liss et al., 2001). Modulating A-current activity therefore seems to be a powerful mechanism for controlling neuronal firing rates in several brain regions.
Our finding that CCK acutely regulates firing frequency in ARC neurons may help to explain how endogenous CCK controls appetite. There is considerable evidence that CCK modulates food intake via both hormonal (Lieverse et al., 1995) and synaptic (McLaughlin et al., 1985; Scallet et al., 1985; Schick et al., 1986a, 1987; Sodersten and Linden, 1992) modes of action. ARC neurons are likely to be exposed to CCK from both of these sources. The absence of a blood–brain barrier at the median eminence (Broadwell and Brightman, 1976; Ganong, 2000) implies that they can directly sense circulating CCK. In this respect, it is significant that, in our study, CCK modulated firing of ARC Type-C cells in the same concentration range (low picomolar) (Fig.8B) as that which is found physiologically in the blood (Liddle et al., 1984) and within which the classical targets of blood-borne CCK, such as the exocrine pancreatic cells, are activated by the peptide (Petersen et al., 1991; Burdakov and Galione, 2000). It is therefore possible that modulation of ARC neurons by CCK may be involved in the negative feedback control of appetite by circulating CCK, which is considered to be responsible for meal termination (Crawley and Corwin, 1994). In turn, the presence of CCK-immunoreactive nerve fibers and terminals in the ARC (Ciofi and Tramu, 1990) suggests that ARC neurons are the target of synaptically released CCK. Release of CCK within the ARC is triggered by conditions such as stress (Siegel et al., 1987), and thus modulation of ARC neurons by synaptically released CCK may be involved in stress–appetite interactions.
Both CCK-A and CCK-B receptors are found in the hypothalamus, but their relative importance in appetite regulation by endogenous CCK is a matter of considerable debate (for review, see Baldwin et al., 1998). Our data suggest that CCK modulates the activity of the ARC appetite center via CCK-B receptors (Fig. 9). The apparent EC50 of CCK-8S for increasing A-current amplitude (Fig. 8B) may be an underestimate because of desensitization. However, it is noteworthy that it is in reasonable agreement with CCK-8S IC50 values determined for the CCK-B receptor in radioligand displacement studies (Kopin et al., 1994). The importance of the CCK-B receptor in appetite regulation is supported by a number of other pharmacological studies. For example, intracerebroventricularly administered CCK-8S and gastrin-17II were equally potent in reducing food intake (Schick et al., 1986b), as would be expected if CCK-B receptors are involved (Jensen et al., 1989). Studies with the CCK-B-selective antagonist L365,260 (Dourish et al., 1989; Schick et al., 1991; Corp et al., 1997) also suggest that CCK-B receptors are involved in modulation of food intake.
CCK increased A-currents and slowed firing within 0.5–5 min of application (Figs. 7, 8). The variability in the latency of this response probably reflects differences in drug accessibility, because different neurons were located at different depths within the slice. However, no detectable response was observed within 0.5 min of application, even in neurons lying on the slice surface. This suggests that CCK action involves the generation of intracellular second messenger(s). In some non-excitable cells, it has been shown that activation of CCK-B receptors stimulates the phospholipase C/inositol trisphosphate pathway and thereby mobilizes intracellular Ca2+ (Noble and Roques, 1999). Whether a similar transduction pathway exists in ARC Type-C cells remains to be resolved.
In summary, our results provide a biophysical framework for understanding the physiological and pathological activity of the ARC. In particular, we show for the first time that circulating concentrations of CCK modulate the activity of a specific subset of ARC neurons. This novel mechanism is likely to be involved in the regulation of appetite by this important brain region.
Footnotes
We thank Dr. Stefan Trapp for instruction on working with brain slices and the Wellcome Trust for support. D.B. holds a Wellcome Prize Studentship, and F.M.A. holds the Royal Society GlaxoSmithKline Research Professorship.
Correspondence should be addressed to Prof. F. M. Ashcroft, University Laboratory of Physiology, Parks Road, Oxford, OX1 3PT, UK. E-mail: frances.ashcroft@physiol.ox.ac.uk.
REFERENCES
- 1.Aghajanian GK. Modulation of a transient outward current in serotonergic neurons by α1-adrenoceptors. Nature. 1985;315:501–503. doi: 10.1038/315501a0. [DOI] [PubMed] [Google Scholar]
- 2.Baldwin BA, Parrot RF, Ebenezer IS. Food for thought: a critique on the hypothesis that endogenous cholecystokinin acts as a physiological satiety factor. Prog Neurobiol. 1998;55:477–507. doi: 10.1016/s0301-0082(98)00005-7. [DOI] [PubMed] [Google Scholar]
- 3.Barsch GS, Farooqi IS, O'Rahilly SO. Genetics of body weight regulation. Nature. 2000;404:644–651. doi: 10.1038/35007519. [DOI] [PubMed] [Google Scholar]
- 4.Broadwell RD, Brightman MW. Entry of peroxidase into neurons of the central and peripheral nervous systems from extracerebral and cerebral blood. J Comp Neurol. 1976;166:257–283. doi: 10.1002/cne.901660302. [DOI] [PubMed] [Google Scholar]
- 5.Brown HF, DiFrancesco D, Noble SJ. How does adrenaline accelerate the heart? Nature. 1979;280:235–236. doi: 10.1038/280235a0. [DOI] [PubMed] [Google Scholar]
- 6.Burdakov D, Galione A. Two neuropeptides recruit different messenger pathways to evoke Ca2+ signals in the same cell. Curr Biol. 2000;10:993–996. doi: 10.1016/s0960-9822(00)00649-7. [DOI] [PubMed] [Google Scholar]
- 7.Ciofi P, Tramu G. Distribution of cholecystokinin-like immunoreactive neurons in the guinea pig forebrain. J Comp Neurol. 1990;300:82–112. doi: 10.1002/cne.903000107. [DOI] [PubMed] [Google Scholar]
- 8.Connor JA, Stevens CF. Predictions of repetitive firing behaviour from voltage-clamp data on an isolated neuronal soma. J Physiol (Lond) 1971;213:31–53. doi: 10.1113/jphysiol.1971.sp009366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corp ES, Curcio M, Gibbs J, Smith GP. The effect of centrally administered CCK-receptor antagonists on food intake in rats. Physiol Behav. 1997;61:823–827. doi: 10.1016/s0031-9384(96)00561-6. [DOI] [PubMed] [Google Scholar]
- 10.Cowley MA, Smart JL, Rubinstein M, Cerdan MG, Diano S, Horvath TL, Vone RD, Low MJ. Leptin activates anorexigenic POMC neurons through a neural network in the arcuate nucleus. Nature. 2001;411:480–484. doi: 10.1038/35078085. [DOI] [PubMed] [Google Scholar]
- 11.Crawley JN, Corwin RL. Biological actions of cholecystokinin. Peptides. 1994;4:731–755. doi: 10.1016/0196-9781(94)90104-x. [DOI] [PubMed] [Google Scholar]
- 12.Dockray GJ. Isolation, structure and biological activity of two cholecystokinin octapeptides from sheep brain. Nature. 1978;274:711–713. doi: 10.1038/274711a0. [DOI] [PubMed] [Google Scholar]
- 13.Dourish CT, Rycroft W, Iversen SD. Postponement of satiety by blockade of brain cholecystokinin (CCK-B) receptors. Science. 1989;245:1509–1511. doi: 10.1126/science.2781294. [DOI] [PubMed] [Google Scholar]
- 14.Elias CF, Lee C, Kelly J, Aschkenasi C, Ahima RS, Couceyro PR, Kuhar MJ, Saper CB, Elmquist JK. Leptin activates hypothalamic CART neurons projecting to the spinal cord. Neuron. 1998;21:1375–1385. doi: 10.1016/s0896-6273(00)80656-x. [DOI] [PubMed] [Google Scholar]
- 15.Elmquist JK, Elias CF, Saper CB. From lesions to leptin: hypothalamic control of food intake and body weight. Neuron. 1999;22:221–232. doi: 10.1016/s0896-6273(00)81084-3. [DOI] [PubMed] [Google Scholar]
- 16.Franklin K, Paxinos G. The mouse brain in stereotaxic coordinates. Academic; San Diego: 1997. [Google Scholar]
- 17.Franz O, Liss B, Neu A, Roeper J. Single-cell mRNA expression of HCN1 correlates with a fast gating phenotype of hyperpolarisation-activated cyclic nucleotide-gated ion channels (Ih) in central neurons. Eur J Neurosci. 2000;12:2685–2693. doi: 10.1046/j.1460-9568.2000.00151.x. [DOI] [PubMed] [Google Scholar]
- 18.Ganong WF. Circumventricular organs: definition and role in the regulation of endocrine and autonomic function. Clin Exp Pharm Physiol. 2000;27:422–427. doi: 10.1046/j.1440-1681.2000.03259.x. [DOI] [PubMed] [Google Scholar]
- 19.Gibbs J, Young RC, Smith GP. Cholecystokinin decreases food intake in rats. J Comp Physiol Psychol. 1973;84:488–495. doi: 10.1037/h0034870. [DOI] [PubMed] [Google Scholar]
- 20.Hahn TM, Breininger JF, Baskin DG, Schwartz MW. Co-expression of Agrp and NPY in fasting activated hypothalamic neurons. Nat Neurosci. 1998;1:271–272. doi: 10.1038/1082. [DOI] [PubMed] [Google Scholar]
- 21.Jensen RT, Wank SA, Rowley WH, Sato S, Gardner JD. Interaction of CCK with pancreatic acinar cells. Trends Pharmacol Sci. 1989;10:418–423. doi: 10.1016/0165-6147(89)90192-2. [DOI] [PubMed] [Google Scholar]
- 22.Kopin AS, Beinborn M, Lee Y-M, McBridge EW, Quinn SM. The CCK-B/gastrin receptor. Ann NY Acad Sci. 1994;713:67–78. doi: 10.1111/j.1749-6632.1994.tb44053.x. [DOI] [PubMed] [Google Scholar]
- 23.Liddle RA, Goldfine ID, Williams JA. Bioassay of plasma cholecystokinin in rats: effects of food, trypsin inhibitor and alcohol. Gastroentereology. 1984;87:542–549. [PubMed] [Google Scholar]
- 24.Lieverse RJ, Jansen JBMJ, Masclee AAM, Lamers CBHW. Satiety effects of a physiological dose of cholecystokinin in humans. Gut. 1995;36:176–179. doi: 10.1136/gut.36.2.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liss B, Franz O, Sewing S, Bruns R, Neuhoff H, Roeper J. Tuning pacemaker frequency of individual dopaminergic neurons by Kv4.3L and Kchip3.1 transcription. EMBO J. 2001;20:5715–5724. doi: 10.1093/emboj/20.20.5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lüthi A, McCormick DA. H-current: properties of a neuronal and network pacemaker. Neuron. 1998;21:9–12. doi: 10.1016/s0896-6273(00)80509-7. [DOI] [PubMed] [Google Scholar]
- 27.McLaughlin A, Baile CA, Buonomo FC. Meal-stimulated increased concentrations of CCK in the hypothalamus of Zucker obese and lean rats. Physiol Behav. 1985;35:215–220. doi: 10.1016/0031-9384(85)90339-7. [DOI] [PubMed] [Google Scholar]
- 28.Miki T, Liss B, Minami K, Shiuchi T, Saraya A, Kashima Y, Horiuchi M, Ashcroft F, Minokoshi Y, Roeper J, Seino S. ATP-sensitive K channels in the hypothalamus are essential for the maintenance of glucose homeostasis. Nat Neurosci. 2001;4:507–512. doi: 10.1038/87455. [DOI] [PubMed] [Google Scholar]
- 29.Minami T, Oomura Y, Sugimori M. Electrophysiological properties and glucose responsiveness of guinea-pig ventromedial hypothalamic neurons in vitro. J Physiol (Lond) 1986;380:127–143. doi: 10.1113/jphysiol.1986.sp016276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moran TH. Cholecystokinin and satiety: current perspectives. Nutrition. 2000;16:858–865. doi: 10.1016/s0899-9007(00)00419-6. [DOI] [PubMed] [Google Scholar]
- 31.Noble F, Roques BP. CCK-B receptor: chemistry, molecular biology, biochemistry and pharmacology. Prog Neurobiol. 1999;58:349–379. doi: 10.1016/s0301-0082(98)00090-2. [DOI] [PubMed] [Google Scholar]
- 32.Petersen CCH, Toescu EC, Petersen OH. Different patterns of receptor-activated cytoplasmic Ca2+ oscillations in single pancreatic acinar cells: dependence on receptor type, agonist concentration and intracellular Ca2+ buffering. EMBO J. 1991;10:527–533. doi: 10.1002/j.1460-2075.1991.tb07979.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rauch M, Riediger T, Schmid HA, Simon E. Orexin A activates leptin-responsive neurons in the arcuate nucleus. Pflügers Arch. 2000;440:699–703. doi: 10.1007/s004240000342. [DOI] [PubMed] [Google Scholar]
- 34.Rudy B. Diversity and ubiquity of K channels. Neuroscience. 1988;25:729–749. doi: 10.1016/0306-4522(88)90033-4. [DOI] [PubMed] [Google Scholar]
- 35.Scallet AC, Della-Fera MA, Baile CA. Satiety, hunger and regional brain content of cholecystokinin/gastrin and met-enkephalin immunoreactivity in the sheep. Peptides. 1985;6:937–943. doi: 10.1016/0196-9781(85)90325-0. [DOI] [PubMed] [Google Scholar]
- 36.Schick RR, Yaksh TL, Go VLW. An intragastric meal releases the putative satiety factor cholecystokinin from hypothalamic neurons in cats. Brain Res. 1986a;370:349–353. doi: 10.1016/0006-8993(86)90492-0. [DOI] [PubMed] [Google Scholar]
- 37.Schick RR, Yaksh TL, Go VLM. Intracerebroventricular injections of cholecystokinin octapeptide suppress feeding in rats: pharmacological characterisation of this action. Regul Peptides. 1986b;14:277–292. doi: 10.1016/0167-0115(86)90170-9. [DOI] [PubMed] [Google Scholar]
- 38.Schick RR, Reilly WM, Roddy DR, Yaksh TL, Go VLW. Neuronal cholecystokinin-like immunoreactivity is postprandially released from primate hypothalamus. Brain Res. 1987;418:20–26. doi: 10.1016/0006-8993(87)90957-7. [DOI] [PubMed] [Google Scholar]
- 39.Schick RR, Schusdziarra V, Enders C, Eberl T, Classen M. Putative sleep factors and satiety in rats: role of cholecystokinin (CCK) and delta sleep inducing peptide (DSIP). Neuropeptides. 1991;10:41–48. [Google Scholar]
- 40.Schwarz MW, Woods SC, Porte D, Jr, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature. 2000;404:661–671. doi: 10.1038/35007534. [DOI] [PubMed] [Google Scholar]
- 41.Siegel RA, Dueker EM, Pahnke U, Wuttke W. Stress-induced changes in cholecystokinin and substance P in discrete regions of rat hypothalamus. Neuroendocrinology. 1987;46:74–81. doi: 10.1159/000124799. [DOI] [PubMed] [Google Scholar]
- 42.Sodersten P, Linden A. Plasma and cerebrospinal fluid concentrations of CCK and satiety. Multiple cholecystokinin receptors in the CNS (Dourish CT, Cooper CJ, Iversen SD, Iversen LL, eds), pp 222–233. Oxford UP; Oxford: 1992. [Google Scholar]
- 43.Spanswick D, Smith MA, Groppi VE, Logan SD, Ashford MLJ. Leptin inhibits hypothalamic neurons by activation of ATP-sensitive potassium channels. Nature. 1997;390:521–525. doi: 10.1038/37379. [DOI] [PubMed] [Google Scholar]
- 44.Spanswick D, Smith MA, Mirshamsi S, Routh VH, Ashford MLJ. Insulin activates ATP-sensitive K channels in hypothalamic neurons of lean, but not obese rats. Nat Neurosci. 2000;3:757–758. doi: 10.1038/77660. [DOI] [PubMed] [Google Scholar]
- 45.Stuart G, Dodt H-U, Sakmann B. Patch-clamp recordings from the soma and dendrites of neurons in brain slices using infrared video microscopy. Pflügers Arch. 1993;423:511–518. doi: 10.1007/BF00374949. [DOI] [PubMed] [Google Scholar]
- 46.Tsien RW, Lipscombe D, Madison DV, Bley KR, Fox AP. Multiple types of neuronal calcium channels and their selective modulation. Trends Neurosci. 1988;11:431–438. doi: 10.1016/0166-2236(88)90194-4. [DOI] [PubMed] [Google Scholar]
- 47.Wolfart J, Neuhoff H, Franz O, Roeper J. Differential expression of the small conductance, calcium-activated potassium channel SK3 is critical for pacemaker control of dopaminergic midbrain neurons. J Neurosci. 2001;21:3443–3456. doi: 10.1523/JNEUROSCI.21-10-03443.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang F, Feng L, Zheng F, Johnson SW, Du J, Shen L, Wu C-P, Lu B. GNDF acutely modulates excitability and A-type K channels in midbrain dopaminergic neurons. Nat Neurosci. 2001;4:1071–1078. doi: 10.1038/nn734. [DOI] [PubMed] [Google Scholar]