

Abstract
Small conductance Ca2+-activated K+ (SK) channels open in response to increased cytosolic Ca2+ and contribute to the afterhyperpolarization in many excitable cell types. Opening of SK channels is initiated by Ca2+ binding to calmodulin that is bound to the C terminus of the channel. Based on structural information, a chemomechanical gating model has been proposed in which the chemical energy derived from Ca2+ binding is transduced into a mechanical force that restructures the protein to allow K+ ion conduction through the pore. However, the residues that comprise the physical gate of the SK channels have not been identified. In voltage-gated K+ (Kv) channels, access to the inner vestibule is controlled by a bundle crossing formed by the intracellular end of the sixth transmembrane domain (S6) of each of the four channel subunits. Probing SK channels with internally applied quaternary amines suggests that the inner vestibules of Kv and SK channels share structural similarity. Using substituted cysteine accessibility mutagenesis, the relatively large molecule [2-(trimethylammonium)] methanethiosulfonate accessed positions near the putative bundle crossing more rapidly in the open than the closed state but did not modify S6 positions closer to the selectivity filter. In contrast, the smaller compound, 2-(aminoethyl) methanethiosulfonate (MTSEA), modified a position predicted to lie in the lumen immediately intracellular to the selectivity filter equivalently in the open and closed states. The pore blocker tetrabutylammonium impeded MTSEA access to this position in both open and closed channels. The results suggest that the SK channel gate is not formed by the cytoplasmic end of S6 but resides deep in the channel pore in or near the selectivity filter.
Keywords: SK channel, pore, activation gate, cysteine scanning, gating, inner vestibule
Three highly homologous small conductance Ca2+-activated K+ (SK) channel subunits have been cloned (SK1, SK2, and SK3), each containing six putative transmembrane segments with predicted topologies similar to voltage-gated K+ (Kv) channels (Kohler et al., 1996). The fourth transmembrane domain, S4, contains positively charged residues, but SK channels are not gated by voltage. Rather, they are gated solely by intracellular Ca2+ ions. SK channels function as heteromeric complexes with calmodulin (CaM) that is constitutively attached to a binding domain, the CaMBD, in the membrane-proximal region of the intracellular C terminus of the channel. CaM functions as the Ca2+ sensor for SK channels, transducing the Ca2+-gating signal through the CaMBD to a yet unidentified activation gate (Xia et al., 1998; Keen et al., 1999;Schumacher et al., 2001).
Crystal structure and electron paramagnetic resonance (EPR) studies of the bacterial K+ channel, KcsA, and structure–function studies of Kv and cyclic nucleotide-gated (CNG) cation channels have given rise to mechanistic models for the coupling of the gating cue to channel opening (Perozo et al., 1999; del Camino et al., 2000; Flynn et al., 2001). Within the KcsA channel, the four M2 segments form a vestibule in the shape of an inverted teepee (Doyle et al., 1998). The bundle crossing near the membrane interface with the cytoplasm forms a structure like the “smokehole” of a teepee that constitutes the physical gate of the channel. Rearrangement of the helices in response to the gating cue opens or closes the smokehole. This model is supported by EPR measurements that detected relatively large movements near this region of KcsA during gating and smaller movements on the intracellular side of the selectivity filter (Perozo et al., 1999). Further investigation of KcsA has led to the hypothesis that selectivity filter conformational changes during gating are coupled to the rearrangement of the channel activation gate (Perozo et al., 1999; Liu et al., 2001; Zhou et al., 2001b).
For Shaker Kv channels, the substituted cysteine accessibility method (SCAM) (Liu et al., 1997; del Camino et al., 2000, 2001; Yellen, 2001) and cross-linking experiments (Holmgren et al., 1998) strongly suggest that the S6 bundle crossing forms the activation gate. Thus, for both KcsA and Kv channels, the residues that form the activation gate are believed to reside at the cytoplasmic end of the inner vestibule. In contrast, studies of the CNG channel suggest that the selectivity filter of the CNG1 channel functions as the channel gate (Sun et al., 1996; Becchetti et al., 1999; Flynn et al., 2001). This is particularly relevant to SK channels, because both CNG and SK channels are voltage independent, relying on the binding of an intracellular ligand to a C-terminal domain for channel gating (Zagotta and Siegelbaum, 1996; Xia et al., 1998; Keen et al., 1999).
In this study, block by intracellular quaternary amines and SCAM were used to probe the topology of the SK2 channel inner vestibule, examine conformational changes during SK channel gating, and test whether the distal domain of S6 forms an SK activation gate.
MATERIALS AND METHODS
Molecular biology. Constructs were subcloned either into pJPA5 for expression in COSm6 cells or into the oocyte expression vector pBF. In vitro mRNA was synthesized from pBF constructs using SP6 polymerase (Invitrogen, Gaithersburg, MD). Site-directed mutagenesis was performed using pfu polymerase (Stratagene, La Jolla, CA) and the overlap PCR technique (Ho et al., 1989). The complete nucleotide sequences of the coding regions were verified before expression studies.
Electrophysiology. For COSm6 experiments, cells were transfected using Polyfect reagent (Qiagen, Valencia, CA) as per the manufacturer's instructions and plated on Fisherbrand Growth coverslips (Fisher Scientific, Houston, TX); currents were recorded 24–48 hr after transfection. For oocyte experiments, 50 nl of mRNA (20–50 ng) per oocyte was microinjected as described previously, and currents were recorded 2–7 d after injection. Bath solution contained either 150 mm (COSm6) or 120 mm (oocyte) potassium methanesulfonate, 10 mm HEPES, 1 mm EGTA, and CaOH to yield the desired free Ca2+concentration and was adjusted to a pH of 7.2 with methanesulfonic acid (Fabiato and Fabiato, 1979). Free Ca2+ for all open-state experiments was adjusted to 1 μmto maximally activate the channels, except for A392C, which was adjusted to 10 μm. For closed-state experiments, no Ca2+ was added (0 Ca2+), yielding a free Ca2+ level of <2 nm. Inside-out patches were pulled using borosilicate glass patch electrodes filled with 10 μm Ca2+ solution and pulled to resistances between 1 and 3 MΩ. Leak and background currents were measured by changing the bath solution on the inside face of the patch to 0 Ca2+ to close SK2 channels. Rapid solution changes were performed using an RSC-200 (Molecular Kinetics, Pullman, WA). Currents were measured and digitized with an EPC9 (Heka Elektronik, Lambrecht/Pfalz, Germany), currents were sampled and filtered at 1 kHz, and analysis was performed using Pulse (Heka Elektronik) and Igor (Wavemetrics, Lake Oswego, OR) software. No differences in Ca2+sensitivity, gating kinetics, or methanethiosulfonate (MTS) reactivity were observed between patches from oocytes or COSm6 cells, and in some cases, results obtained from both expression systems were combined. All reagents were diluted from concentrated stocks prepared the day of the experiment, and MTS compounds were used within 30 min of mixing into solution.
Data analysis. All values are reported as the mean ± SEM of n experiments. Statistical significance was evaluated using a Student's t test, and a p value of ≤0.05 was considered significant. Modification rates were related to the time constant (τ) of single exponential fits to plots of current amplitude versus MTS exposure duration by the reciprocal of τ × MTS, where MTS was the concentration of [2-(trimethylammonium)] methanethiosulfonate (MTSET) or 2-(aminoethyl) methanethiosulfonate (MTSEA) used in the experiment. Dose–response relationships for tetraethylammonium (TEA), triethylhexylammonium (C6-TEA), tetrabutylammonium (TBuA), and tetrahexylammonium (THexA) were fit with a single binding isotherm: Icontrol ×X/(X + IC50), whereX is the concentration of blocker,Icontrol is the current amplitude before application of blocker, and IC50represents the concentration at which macroscopic current is half blocked.
RESULTS
At least four gating models may be postulated for SK channels (Fig. 1A). Channel opening and closing could occur by: (1) a smokehole at the S6 bundle crossing similar to the KcsA and Kv channel-gating models (model a), (2) an expansion or collapse of the inner vestibule (model b), (3) a “ball-and-chain” mechanism analogous to that which underlies N-type inactivation of Kv channels, perhaps with CaM as the blocking particle (model c), and (4) a rearrangement of the selectivity filter (model d). The different models were tested by examining the topology and gating-dependent conformational changes in the inner vestibule using excised membrane patches containing heterologously expressed SK2 channels.
Fig. 1.

Possible SK channel-gating mechanisms.A, SK channel-gating models. In each model, two of four subunits are depicted in cross section embedded in the membrane with CaM (dumbbells) associated with the intracellular C-terminal domain. For the open state, the SK–CaM complex is modeled as a dimer-of-dimers, as indicated by the CaMBD/Ca2–CaM crystal structure. In the four closed-state models (a–d), the CaMBD/CaM complex is monomeric (Schumacher et al., 2001). a, The S6 bundle crossing acts as the gate preventing access to the lumen and selectivity filter, as suggested for Kv and KcsA channels.b, Collapse of the inner vestibule, including the aqueous lumen, closes SK channels. c, CaM may act as a blocking particle in a ball-and-chain type mechanism. d, Selectivity filter rearrangement prevents ion permeation in closed SK channels, as suggested for ligand-gated CNG channels. B, Sequence alignments of S6 and proximal cytoplasmic residues of Shaker B, a cyclic nucleotide-gated channel (CNG1), and the proton-gated bacterial K+ channel KcsA. Boxed region, Amino acids 386–403 examined with the SCAM technique in this study. Numbered arrows, Residues of particular importance (see Results). Shaded residues, Residues conserved in all SK family members.
A conserved inner vestibule topology between SK and Kv channels
Small quaternary amines, such as TEA, have been used to probe the architecture of the inner pore region of Kv channels, providing evidence for an inner vestibule within the membrane field that is accessed only in the open state (Armstrong and Hille, 1972; Choi et al., 1993; Holmgren et al., 1997; Liu et al., 1997). Furthermore, these experiments showed that increasing the length of one or more of the four hydrophobic side chains of TEA increases the potency of block, suggesting that a hydrophobic pocket stabilizes the interaction between channel and pore blocker (Choi et al., 1991, 1993). Despite limited primary sequence homology between SK and Kv channels in the inner vestibule region, both channel types have highly hydrophobic S6 domains (Fig. 1B).
Dose–response relationships for block of SK2 by TEA and three of its derivatives, TBuA, THexA, and C6-TEA, were obtained by applying compounds to the intracellular face of inside-out patches (data not shown). TEA only weakly blocked SK2 (IC50 = 46.8 ± 2.2 mm; n = 8). However, increasing one of the four side chains from two to six carbons (C6-TEA) decreased the IC50 more than fivefold (8.6 ± 0.4 mm; n = 3). When all four side chains were increased from two to four carbons (TBuA), the IC50 decreased ∼26-fold (1.8 ± 0.2 mm; n = 6). Increasing all of the side chains to six carbons (THexA) increased blocking potency >3000-fold (0.014 ± 0.002 mm;n = 5). Therefore, the rank order of potency for block of SK2 channels by these compounds, THexA > TBuA > C6-TEA > TEA, is similar to that determined for Kv channels, although in general, the IC50 was lower for SK2 than for Shaker channels (e.g., ∼120 times lower for TEA and 86 times lower for C6-TEA) (Choi et al., 1993). For both channel types, the potency of block increased with increased side-chain length. Moreover, as for Kv channels, SK channel block by all of the compounds was voltage dependent. Ratios of mean IC50 at 80 mV to mean IC50 at −80 mV were 0.16, 0.04, 0.08, and 0.17 for TEA, C6-TEA, TBuA, and THexA, respectively. The results suggest open-state access by TEA derivatives to an inner vestibule within the SK2 membrane field.
State-dependent conformational changes in the S6 helix
State-dependent accessibility to the inner vestibule of SK2 channels was explored using SCAM and cysteine-reactive reagents. Each of nine residues in S6 and eight residues C-terminal to S6 were individually mutated to cysteine (Fig. 1B) and assayed for reactivity with MTS compounds when the channels were either open or closed. If the side chain of the introduced cysteine residue resides in the conduction pathway and is available to MTS compounds, then disulfide bond formation may irreversibly reduce K+ permeation either through electrostatic or steric hindrance (Karlin and Akabas, 1998). If the cysteine residue points away from the pore, buried in the lipid bilayer or surrounding protein, or if MTS reagents do not attach or attach only very slowly, little if any current reduction will be observed. Thus, the availability and orientation of specific residues can be tested in the open and closed state.
Inside-out patches containing heterologously expressed wild-type (WT) or cysteine-substituted SK2 channels were exposed to 1 mmMTSET for 8 sec in either the open or closed state. This concentration and duration of exposure were sufficient to reveal sites that reacted quickly with MTSET and to reveal strong state dependence while avoiding the complications of channel rundown observed in some of the mutant channels. Representative traces showing the effects of MTSET exposure in the open and closed states are presented in Figure2 for WT and two different cysteine-substituted sites in SK2, A392C, and R396C. For open-state experiments, channels were first closed and opened, confirming rapid solution exchange (typical time constants of 20 and 50 msec for activation and deactivation using 1 μm and 0 Ca2+ solutions, respectively). Channels were then exposed to 1 mm MTSET for 8 sec, and the fraction of irreversibly blocked current was measured after MTSET washout (Fig.2A, left). WT SK2 contains nine native cysteine residues, including one at position 386 in S6 (Fig.1B). As shown in Figure 2A(left), WT SK2 channels were rapidly and reversibly blocked by ∼14% in the open state, as current recovered to ∼97% of the control level after MTSET washout. This suggests that MTSET does not react with endogenous cysteines to block ion permeation but rather behaves as a reversible pore blocker. To evaluate reactivity in the closed state, channels were closed and then exposed to MTSET for 8 sec, washed in 0 Ca2+ solution for 2 sec, and reopened to determine the amount of irreversible block. Figure2A (right) shows that for WT channels, the current after closed-state MTSET exposure was ∼96% of the control current. These results demonstrate that MTSET did not irreversibly react with WT SK2 in either the open or closed states (open-state current reduced by 3.6 ± 1.1%, n = 8; closed-state current reduced by 3.8 ± 1.3%, n = 6). In contrast, R396C, which is located just inside the cytoplasm beneath S6, demonstrated irreversible reactivity with MTSET in both states (open-state current reduced by 44.8 ± 3.7%,n = 5; closed-state current reduced by 49.7 ± 4.5%, n = 3) (Fig. 2C), suggesting that this site is available to MTSET regardless of whether channels are open or closed. A392C, which is located between the selectivity filter and putative bundle crossing, showed state-dependent modification by MTSET, reacting rapidly and irreversibly in the open state and not in the closed state (Fig. 2B). The current was reduced by 69.4 ± 5.4% (n = 8) and 1.6 ± 6.6% (n = 9) in the open and closed states, respectively, suggesting that conformational changes occur at or near position 392 during gating.
Fig. 2.

MTSET reactivity in WT and cysteine-substituted SK2 channels. A–C, Representative traces showing open- and closed-state MTSET reactivity for WT, A392C, and R396C, respectively. For open-state experiments (left), channels were first closed and then reopened using saturating Ca2+ solutions to verify adequate exchange rates. Next, MTSET (1 mm) was applied to open channels for 8 sec, MTSET was washed out, and current amplitudes before and after MTSET application were compared. For closed-state experiments (right), MTSET (1 mm) was applied to closed channels (0 Ca2+) for 8 sec and washed out with 0 Ca2+ solution for 2 sec, channels were reopened, and current amplitudes were compared before and after closed-state application. Bars above the traces indicate when MTSET was applied, the dashed line indicates 0 current level, and the solid lineabove the trace indicates Ca2+ steps from saturating (1 μm; O, open state) to 0 Ca2+ (<2 nm; C, closed state) solution.
To obtain more information about accessibility of introduced cysteines, the modification rates by MTSET were determined for both open and closed states. The open-state modification rate was defined by the inverse exponential constant of a single exponential fit to the decay of the current amplitude plotted as a function of the product of cumulative exposure time and MTSET concentration (Fig.3B,D). For the representative traces shown in Figure 3B,D, open-state exponential constants were 0.2 and 0.1 mmsec and rates were 4.2 × 103 and 1.1 × 104M−1sec−1for A392C and R396C, respectively. Closed-state modification rates were determined by repeating the closed-state protocol (Fig. 2, right panels) several times. Current amplitudes after MTSET washout and channel reopening were measured after each 8 sec exposure (Fig.3A,C) and plotted as a function of the product of cumulative exposure time and MTSET concentration (Fig. 3B,D, ●). The data points were fitted by a single exponential, and the inverse of the exponential constant defined the closed-state modification rate. For these A392C and R396C examples, closed-state exponential constants were 65.9 and 0.2 mmsec and rates were 15 and 5944 M−1sec−1, respectively. For comparison, the open- and closed-state modification time courses were overlaid for A392C in Figure 3B and for R396C in Figure 3D.
Fig. 3.

Determination of MTSET modification rates.A, Repeated closed-state 1 mm MTSET application to A392C channels using the protocol of Figure 2.B, Open-state (O, black trace) and closed-state (C, ●) decay time courses for A392C normalized to control current and plotted versus the product of cumulative MTSET exposure and MTSET concentration (mmsec). The open-state trace is taken from Figure2B, and the closed-state decay is determined by measuring current amplitude after each of the 10 applications of MTSET shown in A. A single exponential was fit to the data yielding an exponential constant of 0.2 and 65.9 mmsec for the open and closed state, respectively. C, Closed-state modification at position R396C. Channels were repeatedly exposed to 10 μm MTSET according to the closed-state protocol shown in Figure 2. D, Open-state (O, solid trace) and closed-state (C, ●) decay time courses of R396C normalized as in B. The open-state trace is taken from Figure 2C, and the closed-state rate was determined by measuring the current amplitude after each of the 10 applications of MTSET shown in A. A single exponential was fit to the data yielding an exponential constant of 0.09 and 0.17 mm for open and closed states, respectively.
A summary of the MTSET results for the different cysteine-substituted channels is presented in Figure 4, showing the percentage of inhibition after a single 8 sec exposure to MTSET for each position in the open and closed states (Fig.4A) and the modification rate for open and closed channels (Fig. 4B). Positions without bars either did not yield current, or current amplitudes were too small for reliable measurements (Fig. 4A). MTSET modification rates were determined as shown in Figure 3 for two positions deep in the inner vestibule (386C and T387C) and four positions in the putative bundle-crossing region (391, 392, 395, and 396). Current through WT or T387C channels was not affected by MTSET exposure in either the open or the closed state (modification rates < 2 M−1sec−1). Positions V391C, A392C, and V393C within S6 showed significant state dependence after a single MTSET exposure, and V391C and A392C were modified significantly faster in open than in closed channels (V391C: 47.9 ± 11.4 M−1sec−1,n = 4; 6.7 ± 2.1 M−1sec−1,n = 5; p < 0.05; A392C: 1.5 ± 0.4 × 103M−1sec−1,n = 8; 33.6 ± 6.3 M−1sec−1,n = 6; p < 0.05 for open and closed states, respectively). Modification of position 395 resulted in significant block in either state with channels in the closed state being more rapidly modified than those in the open state (open state: 9.7 ± 1.6 × 102M−1sec−1,n = 4; closed state: 3.9 ± 2.4 × 103M−1sec−1,n = 5; p < 0.0001). State-independent modification was clearly observed for position 396 that resides on the S6-cytoplasm interface (open state: 7.4 ± 2.4 × 103M−1sec−1,n = 3; closed state: 5.9 ± 0.6 × 103M−1sec−1,n = 6; p > 0.5). Further into the cytoplasmic domain, at positions 398 and 399, the percentage of closed-state inhibition was greater than open-state inhibition (Fig.4A). Significant MTSET reactivity was not detected at positions N-terminal to 391 in the open or closed states. It is important to note the differences between the percentage of inhibition after an 8 sec exposure to MTSET and the modification rate. For example, Figure 4A shows a much higher percentage block for A392C in the open than in the closed state after an 8 sec exposure. However, the steady-state percentage block by MTSET during repeated application to closed A392C channels was similar to that in the open state (Fig. 3A). This is because access to position 392 is strongly state dependent, and therefore the modification rate is considerably slower in the closed state than in the open state, although the block eventually reaches the same level. In contrast, an 8 sec MTSET application to R396C channels produced equivalent current reductions, and the rates of modification in the open or closed states were similar (Figs. 3D, 4B). Together, the results suggest that the S6 region of SK2 channels undergoes conformational changes during gating that alter MTS access to several positions.
Fig. 4.
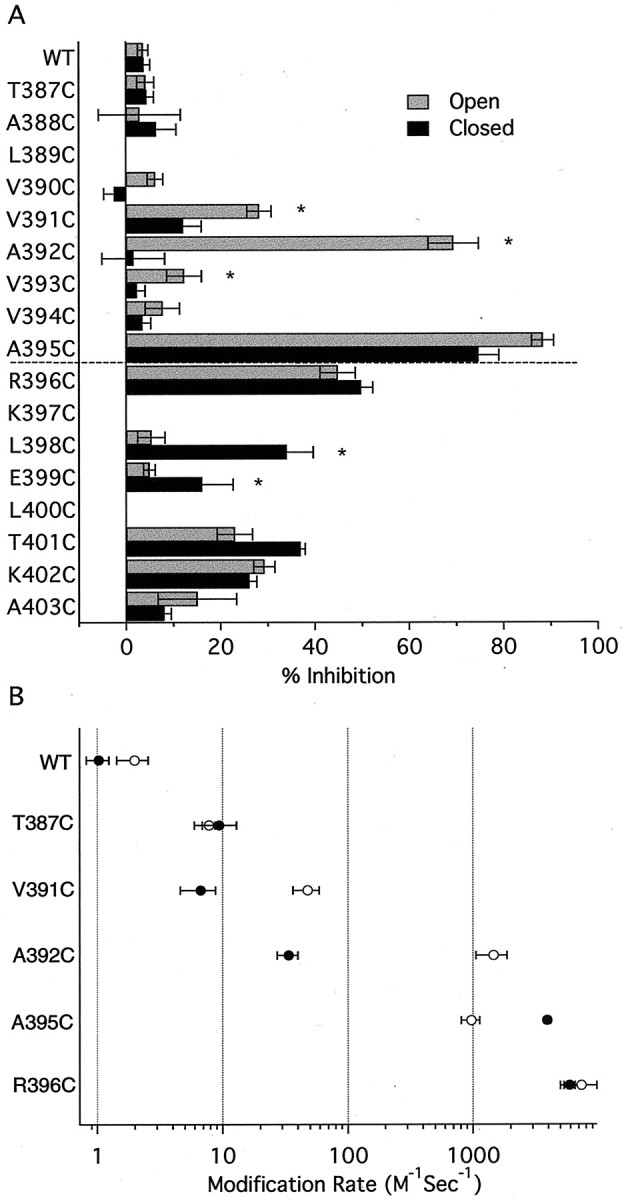
Summary of MTSET reactivity for WT and cysteine-substituted channels. A, Reactivity was measured for open (gray bars) or closed (black bars) channels, and the dashed line indicates the predicted boundary between the cytoplasm and membrane. Data from n ≥ 3 patches were normalized and averaged and plotted as the percentage of inhibition ±SEM after an 8 sec exposure to 1 mm MTSET. Note that positions 391, 392, and 393 show stronger open- than closed-state reactivity, whereas positions at the predicted membrane/cytoplasm boundary (395 and 396) show equal open- and closed-state reactivity. Cytoplasmic positions (398 and 399) show stronger closed- than open-state reactivity. ∗Statistically significant differences between open- and closed-state reactivity (p < 0.05). Reactivity at position 390 could not be reliably measured (<10% current reduction).B, Open-state (○) and closed-state (●) modification rates for WT, T387C, and four residues in the putative bundle-crossing region. Rates were determined from single exponential fits to current decay (see Results) and are presented as mean ± SEM fromn ≥ 3 patches.
Localization of the SK2 activation gate
The open-state-dependent access to positions 391–393 can be interpreted in three ways: (1) these residues rotate away from the pore lumen during channel closing, (2) a gate C-terminal to 393 occludes MTSET access when channels are closed, and (3) the inner vestibule narrows when channels close, and this constriction restricts access to these positions.
Compared with a K+ ion (Pauling radius of 1.33 Å), MTSET is a relatively large molecule that may be modeled to fit in a cylinder ∼5.8 Å in diameter and 10 Å long (Karlin and Akabas, 1998; Flynn and Zagotta, 2001) and so may be sterically hindered from reporting the availability of residues that K+ ions might access, such as those N-terminal to position 391. MTSEA is a smaller, positively charged compound (head group diameter of ∼3.6 Å) that has been used to reveal state-dependent access to sites in the inner vestibule of Shaker channels and in the pore of CNG channels (Liu et al., 1997; Liu and Siegelbaum, 2000). Modification by MTSEA of residues examined in Figure4 yielded results similar to those for MTSET. One important difference was that MTSEA could additionally access T387C, a site deeper in the SK2 inner vestibule that was not modified by MTSET (Fig.5). Application of MTSEA (2.5 mm) to open or closed T387C channels resulted in complete block within 8 sec (open-state inhibition, 99.9 ± 1.2%,n = 4; closed-state inhibition, 94.2 ± 7.2%,n = 6). WT channels containing 386C were not significantly altered by MTSEA in either the open state (inhibition of 2.5 ± 3.3%; n = 7) or closed state (increase of 1.3 ± 2.1%; n = 3), and the double mutation C386S and T387C was completely blocked by MTSEA in both the open (99.6 ± 6%; n = 5) and closed (97.9 ± 4%;n = 3) states, demonstrating that MTSEA reacts specifically at position T387C. The rate of modification of T387C by MTSEA was determined as described for MTSET, except that the exposure time during repeated MTSEA application in the closed state was decreased from 8 to 1 sec to accurately record channel modification (Fig. 5B). The open-state and closed-state modification rates of T387C by MTSEA were not significantly different (open state, 379 ± 109 M−1sec−1,n = 6; closed state, 286 ± 26 M−1sec−1,n = 3; p > 0.4). Modification rates for WT channels were <1 M−1sec−1 in either the open or closed states.
Fig. 5.

MTSEA reactivity for WT, T387C, and T387C/C386S channels. A, Current reduction was determined for the three channels using 8 sec applications of 2.5 mm MTSEA, as described for MTSET in Figure 2. ∗T387C, T387C in the C386S background. B, Open-state (O,black trace) and closed-state (C, ▪) modification of T387C by MTSEA. Data are plotted and fitted as for MTSET in Figure 3. Single exponential fits to the data yielded exponential constants of 1.7 and 3.5 mmsec for open and closed states, respectively.
One concern when using MTSEA is that it may cross the membrane, in this case, accessing T387C from the external vestibule and causingtrans modification. This possibility was addressed by including 10 mm cysteine in the external patch pipette solution (Holmgren et al., 1996), which had no effect on the extent or rate of modification (data not shown). It is also possible that MTSEA in an uncharged form might access position 387 by partitioning into the membrane or hydrophobic portions of the protein. If MTSEA accessed T387C only through the pore, then preapplication of TBuA, a pore blocker, should restrict MTSEA access to the site. Therefore, open or closed T387C channels were first exposed to 10 mm TBuA (∼97% block of open-state current). Then, in the continued presence of TBuA, 2.5 mmMTSEA was applied for 8 sec. After washout of both compounds, current amplitudes before and after exposure were compared (Fig.6A, inset). The protocol was repeated several times, and the data were plotted and fitted as described for Figure 3A. As shown in Figure 6, TBuA protected T387C from MTSEA modification in the open and closed states. Rates were ∼10-fold slower in the open state when channels were preblocked with TBuA (36.7 ± 7.5 M−1sec−1;n = 5) and ∼18-fold slower in the closed state when TBuA was present (16.2 ± 3.6 M−1sec−1;n = 4). Together, the data suggest that MTSEA gains access to T387C by fitting into the pore in either the open or closed states.
Fig. 6.

TBuA protects T387C from MTSEA modification.A, TBuA protects T387C from modification by MTSEA in the open state. The open-state modification time course without TBuA (black trace) is the same as in Figure5B. The open-state modification time course in the presence of 10 mm TBuA (∼97% block, ▵) was determined by repeatedly applying 2.5 mm MTSEA for 8 sec to blocked channels (inset) and measuring the fractional current remaining after each exposure (inset shows an example of a single exposure; dashed line represents zero current). Calibration (inset): 50 pA, 2 sec; barsindicate when TBuA and MTSEA were applied to open (O) channels. Single exponential fits to the data yielded exponential constants of 1.7 and 34.9 mmsec in the absence and presence of blocker, respectively. B, TBuA protects T387C from modification by MTSEA in the closed state. The closed-state modification time course with no blocker present (▪) is taken from Figure 5B. The closed-state modification time course in the presence of TBuA (▴) was determined by repeatedly applying 2.5 mm MTSEA for 8 sec to closed channels in the presence of TBuA and measuring the fractional current remaining after each exposure. Single exponential fits to the data yielded exponential constants of 3.7 and 77.5 mmsec in the absence and presence of blocker, respectively. C, The WT channel MTSEA modification rate in the open state (■) and closed state (▪) was <1 M−1sec−1. The open-state MTSEA modification rate of T387C without (■) and with (▵) 10 mm TBuA present and the closed-state MTSEA modification rate of T387C without (▪) and with (▴) 10 mm TBuA are shown. Data are presented as mean ± SEM fromn ≥ 3 patches.
DISCUSSION
The molecular details of Ca2+ gating of SK channels have begun to emerge from biochemical, electrophysiological, and crystallographic studies (Xia et al., 1998;Schumacher et al., 2001). The results suggest that SK channel opening may involve a large-scale reorganization in which four monomeric CaMBD/CaM complexes transition into two dimers of CaMBD/Ca2+–CaM. This rearrangement of the proximal C terminus may impose a conformational alteration on the associated S6 helices that opens the SK channel gate (Schumacher et al., 2001). The structural identity of the gate, the positions that occlude ion permeation in the closed state, and the mechanism by which the conformational change is transduced from the CaMBD–CaM complex to the channel gate are not known.
SK2 channel block by TEA and longer side-chain derivatives suggests that the inner vestibules of SK and Kv channels are conserved. SCAM results with the largest cysteine-specific probe used in this study, MTSET, revealed open-state-dependent access to three positions just internal to the putative S6 bundle crossing (391–393) (Figs. 3, 4). This was in contrast to more distal positions, such as A395C and R396C, that rapidly reacted with MTSET in both the open and closed states of the channel and to positions closer to the selectivity filter (386–390) that were not affected by MTSET exposure in either the open or closed state of the channel. These data are similar to the reactivity profile observed at analogous positions in Shaker Kv channels (Liu et al., 1997) and are consistent with a SK channel gate formed at the S6 bundle crossing between positions 392 and 395 (Fig.1A, model a).
The KcsA crystal structure reveals a wide aqueous lumen immediately cytoplasmic to the selectivity filter that is believed to be essential for rapid K+ conduction (Doyle et al., 1998; Roux and MacKinnon, 1999; Zhou et al., 2001b). This lumen is ∼10 Å wide and accommodates a fully hydrated K+ ion, thereby maintaining K+ in a low-energy state near the middle of the cell membrane (Doyle et al., 1998). Functional data from Kv channels and EPR data from KcsA support the maintenance of the lumen even in closed channels. For KcsA, the width of the lumen as measured by EPR does not markedly change when pH favors channel opening versus channel closing (Perozo et al., 1999; Liu et al., 2001). For Kv channels, a long-chain TEA derivative can be trapped between the selectivity filter and the gate formed by S6, presumably in an analogous aqueous lumen (Holmgren et al., 1997). Thus, in Kv and KcsA channels, access to the lumen is regulated by an intracellular gate that prevents ion exchange with cytoplasmic K+ in the closed state.
Two experiments presented here are consistent with the existence of a lumen, but not an intracellular gate, in closed SK channels. First, position 387 was rapidly modified by MTSEA even in closed channels, suggesting that there must be a lumen large enough to accommodate MTSEA even in closed channels (Figs. 5, 6). Second, a pore blocker with a fixed charge, TBuA, protected closed T387C channels from modification by MTSEA. The protection afforded was even greater in the closed than the open state, suggesting that the TBuA-binding site is preserved in closed SK channels (Fig. 6). These data are not consistent with a gate formed by the S6 bundle crossing between positions 392 and 395.
State-independent access to residue T387C deep in the vestibule raises the question of why MTSET access to the more distal position 392 is state dependent (Fig. 4A,B). The simplest interpretation of this result is that during channel closing, position 392 rotates away from the conduction pathway, perhaps into the channel protein surrounding the S6 helices. In this way, an unobstructed path to position 387 is maintained in the closed channel, and state-dependent modification of position 392 is achieved. It is also possible that 392 does not move but that surrounding protein moves to protect the site in the closed state. Although this possibility cannot be ruled out, state-dependent MTSET access to V391C and V393C, immediately adjacent to A392C, and to the more distal L398C and E399C, is also consistent with a rotation of the S6 helix during Ca2+ gating (Fig. 4).
To model the position of 387 and the other sites examined, SK2 residues were substituted for their counterparts in the KcsA crystal structure (Fig. 7). Despite the sequence divergence between KcsA and SK2, the orientations predicted from the substituted structure are consistent with results from the SCAM experiments in the inner vestibule. Residues that are predicted to reside in the inner vestibule, facing the permeation pathway (387, 391, and 392), reacted with MTS compounds in open SK channels to reduce current amplitude, and residues that point away from the permeation pathway (386 and 390), buried in surrounding protein or membrane, were not affected by MTS exposure. Although this model adequately predicts the reactivity of residues relatively deep in the inner vestibule (∼386–392), more cytoplasmic residues are less well depicted. For example, position 396 appears to point away from the permeation pathway, but MTSET reactivity at this site significantly decreases current amplitude, and state-dependent access to positions 398 and 399 would not necessarily be predicted based on the model. This divergence from the data at the cytoplasmic end of the model is not surprising; not only is the gating mechanism fundamentally different between SK and KcsA channels, but MTSEA access to T387C suggests that, at least in the closed state, the bundle-crossing region of SK is wider than in KcsA.
Fig. 7.
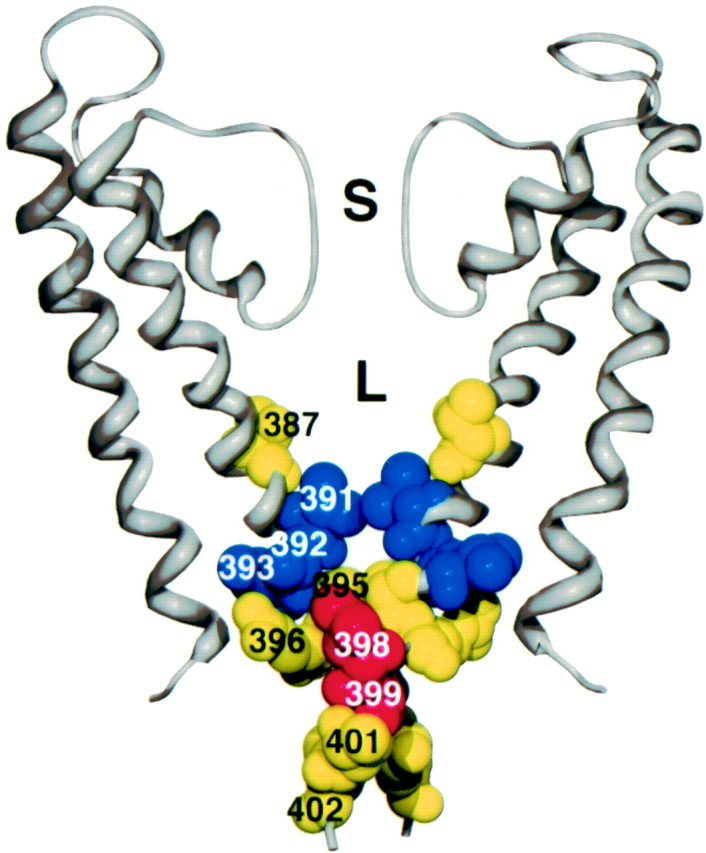
Model of SK2 residues substituted into the KcsA crystal structure. Two of four subunits are shown (gray ribbon). For each subunit, S5, the pore region, S6, and the S6 extension into the cytoplasm are shown. For reference, the selectivity filter at the top (S) is ∼3.3 Å wide.L, The lumen immediately internal to the selectivity filter. Amino acids in yellow (387,395, 396, 401, and402) show approximately equal open- and closed-state reactivity, blue residues (391,392, and 393) show more open-state than closed-state reactivity, and red (398 and399) shows more closed-state than open-state reactivity. Reactivity to MTSET is shown for all residues except T387C, which depicts MTSEA reactivity.
Using the model of the deep inner vestibule and pore region as an approximation of the SK structure, position 387 projects into the lumen, residing ∼8 Å from the selectivity filter (Fig. 7). Because MTSEA would fit in a cylinder ∼6 Å wide by 10 Å long, with a head group ∼3.6 Å in diameter (Karlin and Akabas, 1998; Flynn and Zagotta, 2001), a vestibule of at least these dimensions is predicted near position 387 even in closed SK channels. The closed-state vestibule may be even larger than this, particularly if TBuA protects T387C from MTSEA by binding to its site close to the selectivity filter, as demonstrated for Shaker and KcsA channels (Choi et al., 1993; Zhou et al., 2001a), and suggested by the voltage dependence of the open-state block in SK channels. If the smokehole (Fig.1A, model a), the collapsing vestibule (Fig. 1A, model b), or the ball-and-chain (Fig. 1A, model c) mechanism accounts for SK2 channel gating, MTSEA access to T387C should be occluded in the closed state. Because open- and closed-state modification rates do not differ (Figs. 5B, 6C), these models cannot accurately describe SK2 channel gating. Rather, rapid access by MTSEA to the region immediately cytoplasmic to the selectivity filter even in the closed state suggests that the SK channel activation gate lies at or external to the selectivity filter (Fig. 1A,model d). The difference between Kv and SK channels is clearly reflected by the residue analogous to 387 in the Shaker Kv channel (I470C) that is rapidly modified by MTSEA in the open but not closed state (Liu et al., 1997).
Recent evidence in other K+ channels suggests that structural changes may occur not only in the inner vestibule but also in the selectivity filter during gating. When pH is altered to favor gating of KcsA, EPR measurements detected small movements at the intracellular end of the selectivity filter (Perozo et al., 1999). Furthermore, KcsA crystal structures solved in high and low concentrations of K+ show two distinct selectivity filter conformations, one presumably conductive and the other nonconductive (Zhou et al., 2001b). In an inward rectifier type K+ channel, altering the electronegativity of the pore alters gating at hyperpolarized potentials (Lu et al., 2001). Finally, in Shaker Kv channels, some mutations in the selectivity filter lead to subconductance states with ion selectivities that differ from the selectivity profile of fully open channels (Zheng and Sigworth, 1997, 1998).
The data presented here support a model for SK channel gating in which Ca2+-induced rearrangements of the CaMBD/CaM complex are transduced through S6 to the channel activation gate, which lies in or near the channel selectivity filter. This is similar to the gating model for CNG channels, in which cyclic nucleotide binding to the channel C terminus is transduced to a gate that presumably resides in the channel selectivity filter (Sun et al., 1996; Flynn et al., 2001). Together, these studies suggest that there may be a conserved selectivity filter-based gating mechanism for many channels gated by intracellular ligands that is distinctly different from the activation gating mechanism proposed for voltage-gated channels.
Footnotes
This work was supported by National Institutes of Health grants (J.P.A. and J.M.). We thank Chris Bond, Dr. Paco Herson, and Dr. Aaron Gerlach for stimulating discussions.
Correspondence should be addressed to James Maylie, Department of Obstetrics and Gynecology, Oregon Health and Sciences University, Portland, OR 97201. E-mail: mayliej@ohsu.edu.
REFERENCES
- 1.Armstrong CM, Hille B. The inner quaternary ammonium ion receptor in potassium channels of the node of Ranvier. J Gen Physiol. 1972;59:388–400. doi: 10.1085/jgp.59.4.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Becchetti A, Gamel K, Torre V. Cyclic nucleotide-gated channels. Pore topology studied through the accessibility of reporter cysteines. J Gen Physiol. 1999;114:377–392. doi: 10.1085/jgp.114.3.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choi KL, Aldrich RW, Yellen G. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+ channels. Proc Natl Acad Sci USA. 1991;88:5092–5095. doi: 10.1073/pnas.88.12.5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Choi KL, Mossman C, Aube J, Yellen G. The internal quaternary ammonium receptor site of Shaker potassium channels. Neuron. 1993;10:533–541. doi: 10.1016/0896-6273(93)90340-w. [DOI] [PubMed] [Google Scholar]
- 5.del Camino D, Yellen G. Tight steric closure at the intracellular activation gate of a voltage-gated K+ channel. Neuron. 2001;32:649–656. doi: 10.1016/s0896-6273(01)00487-1. [DOI] [PubMed] [Google Scholar]
- 6.del Camino D, Holmgren M, Liu Y, Yellen G. Blocker protection in the pore of a voltage-gated K+ channel and its structural implications. Nature. 2000;403:321–325. doi: 10.1038/35002099. [DOI] [PubMed] [Google Scholar]
- 7.Doyle DA, Morais Cabral J, Pfuetzner RA, Kuo A, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 8.Fabiato A, Fabiato F. Calculator programs for computing the composition of the solutions containing multiple metals and ligands used for experiments in skinned muscle cells. J Physiol (Lond) 1979;75:463–505. [PubMed] [Google Scholar]
- 9.Flynn GE, Zagotta WN. Conformational changes in S6 coupled to the opening of cyclic nucleotide-gated channels. Neuron. 2001;30:689–698. doi: 10.1016/s0896-6273(01)00324-5. [DOI] [PubMed] [Google Scholar]
- 10.Flynn GE, Johnson JP, Jr, Zagotta WN. Cyclic nucleotide-gated channels: shedding light on the opening of a channel pore. Nat Rev Neurosci. 2001;2:643–651. doi: 10.1038/35090015. [DOI] [PubMed] [Google Scholar]
- 11.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 12.Holmgren M, Liu Y, Xu Y, Yellen G. On the use of thiol-modifying agents to determine channel topology. Neuropharmacology. 1996;35:797–804. doi: 10.1016/0028-3908(96)00129-3. [DOI] [PubMed] [Google Scholar]
- 13.Holmgren M, Smith PL, Yellen G. Trapping of organic blockers by closing of voltage-dependent K+ channels: evidence for a trap door mechanism of activation gating. J Gen Physiol. 1997;109:527–535. doi: 10.1085/jgp.109.5.527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holmgren M, Shin KS, Yellen G. The activation gate of a voltage-gated K+ channel can be trapped in the open state by an intersubunit metal bridge. Neuron. 1998;21:617–621. doi: 10.1016/s0896-6273(00)80571-1. [DOI] [PubMed] [Google Scholar]
- 15.Karlin A, Akabas MH. Substituted-cysteine accessibility method. Methods Enzymol. 1998;293:123–145. doi: 10.1016/s0076-6879(98)93011-7. [DOI] [PubMed] [Google Scholar]
- 16.Keen JE, Khawaled R, Farrens DL, Neelands T, Rivard A, Bond CT, Janowsky A, Fakler B, Adelman JP, Maylie J. Domains responsible for constitutive and Ca2+-dependent interactions between calmodulin and small conductance Ca2+-activated potassium channels. J Neurosci. 1999;19:8830–8838. doi: 10.1523/JNEUROSCI.19-20-08830.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kohler M, Hirschberg B, Bond CT, Kinzie JM, Marrion NV, Maylie J, Adelman JP. Small-conductance, calcium-activated potassium channels from mammalian brain. Science. 1996;273:1709–1714. doi: 10.1126/science.273.5282.1709. [DOI] [PubMed] [Google Scholar]
- 18.Liu J, Siegelbaum SA. Change of pore helix conformational state upon opening of cyclic nucleotide-gated channels. Neuron. 2000;28:899–909. doi: 10.1016/s0896-6273(00)00162-8. [DOI] [PubMed] [Google Scholar]
- 19.Liu Y, Holmgren M, Jurman ME, Yellen G. Gated access to the pore of a voltage-dependent K+ channel. Neuron. 1997;19:175–184. doi: 10.1016/s0896-6273(00)80357-8. [DOI] [PubMed] [Google Scholar]
- 20.Liu YS, Sompornpisut P, Perozo E. Structure of the KcsA channel intracellular gate in the open state. Nat Struct Biol. 2001;8:883–887. doi: 10.1038/nsb1001-883. [DOI] [PubMed] [Google Scholar]
- 21.Lu T, Ting AY, Mainland J, Jan LY, Schultz PG, Yang J. Probing ion permeation and gating in a K+ channel with backbone mutations in the selectivity filter. Nat Neurosci. 2001;4:239–246. doi: 10.1038/85080. [DOI] [PubMed] [Google Scholar]
- 22.Perozo E, Cortes DM, Cuello LG. Structural rearrangements underlying K+-channel activation gating. Science. 1999;285:73–78. doi: 10.1126/science.285.5424.73. [DOI] [PubMed] [Google Scholar]
- 23.Roux B, MacKinnon R. The cavity and pore helices in the KcsA K+ channel: electrostatic stabilization of monovalent cations. Science. 1999;285:100–102. doi: 10.1126/science.285.5424.100. [DOI] [PubMed] [Google Scholar]
- 24.Schumacher MA, Rivard AF, Bachinger HP, Adelman JP. Structure of the gating domain of a Ca2+-activated K+ channel complexed with Ca2+/calmodulin. Nature. 2001;410:1120–1124. doi: 10.1038/35074145. [DOI] [PubMed] [Google Scholar]
- 25.Sun ZP, Akabas MH, Goulding EH, Karlin A, Siegelbaum SA. Exposure of residues in the cyclic nucleotide-gated channel pore: P region structure and function in gating. Neuron. 1996;16:141–149. doi: 10.1016/s0896-6273(00)80031-8. [DOI] [PubMed] [Google Scholar]
- 26.Xia XM, Fakler B, Rivard A, Wayman G, Johnson-Pais T, Keen JE, Ishii T, Hirschberg B, Bond CT, Lutsenko S, Maylie J, Adelman JP. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature. 1998;395:503–507. doi: 10.1038/26758. [DOI] [PubMed] [Google Scholar]
- 27.Zagotta WN, Siegelbaum SA. Structure and function of cyclic nucleotide-gated channels. Annu Rev Neurosci. 1996;19:235–263. doi: 10.1146/annurev.ne.19.030196.001315. [DOI] [PubMed] [Google Scholar]
- 28.Zheng J, Sigworth FJ. Selectivity changes during activation of mutant Shaker potassium channels. J Gen Physiol. 1997;110:101–117. doi: 10.1085/jgp.110.2.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zheng J, Sigworth FJ. Intermediate conductances during deactivation of heteromultimeric Shaker potassium channels. J Gen Physiol. 1998;112:457–474. doi: 10.1085/jgp.112.4.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhou M, Morais-Cabral JH, Mann S, MacKinnon R. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 2001a;411:657–661. doi: 10.1038/35079500. [DOI] [PubMed] [Google Scholar]
- 31.Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature. 2001b;414:43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]