

Abstract
Pain can be effectively controlled by endogenous mechanisms based on neuroimmune interactions. In inflamed tissue immune cell-derived opioid peptides activate opioid receptors on peripheral sensory nerves leading to potent analgesia. This is brought about by a release of opioids from inflammatory cells after stimulation by stress or corticotropin-releasing hormone (CRH). Immunocytes migrate from the circulation to inflamed tissue in multiple steps, including their rolling, adhesion, and transmigration through the vessel wall. This is orchestrated by adhesion molecules on leukocytes and vascular endothelium. Intercellular adhesion molecule-1 [ICAM-1 (or CD54)] is expressed by endothelium and mediates adhesion and extravasation of leukocytes. The goal of this study was to show that ICAM-1 regulates the homing of opioid-producing cells and the subsequent generation of analgesia within sites of painful inflammation. This was accomplished using immunofluorescence, flow cytometry, and behavioral (paw pressure) testing. We found that ICAM-1 is upregulated on the vascular endothelium, simultaneously with an enhanced immigration of opioid-containing immune cells into inflamed paw tissue. The intravenous administration of a monoclonal antibody against ICAM-1 markedly decreased the migration of opioid-containing leukocytes and of granulocytes, monocytes–macrophages, and T cells to the inflamed tissue. At the same time, circulating immunocytes increased in numbers, and macroscopic inflammation (hyperalgesia, paw volume, and paw temperature) remained primarily unchanged. Most importantly, peripheral opioid analgesia elicited either by cold water swim stress or by intraplantar administration of CRH was dramatically reduced. Together, these findings indicate that ICAM-1 expressed on vascular endothelium recruits immunocytes containing opioids to promote the local control of inflammatory pain.
Keywords: adhesion molecules, intercellular adhesion molecule-1, opioids, endogenous, stress, analgesia, pain, inflammation
It is now recognized that pain and pain modulation is not solely mediated by neurons but can involve neuroimmune interactions (Machelska and Stein, 2000; Watkins and Maier, 2000; DeLeo and Yezierski, 2001). In inflammation, circulating immunocytes containing opioid peptides (predominantly β-endorphin) migrate to the injured tissue, and, concurrently, opioid receptors are upregulated on peripheral endings of primary afferents (Cabot et al., 1997, 2001; Mousa et al., 2001; Rittner et al., 2001). During stressful stimulation (cold water swim or postoperative stress), these cells locally secrete opioids to elicit potent and clinically relevant opioid receptor-specific analgesia devoid of central side effects (Stein et al., 1990a, 1993; Rittner et al., 2001). An important endogenous trigger for stress-induced analgesia is corticotropin-releasing hormone (CRH) produced locally in inflammatory cells. CRH activates its receptors on leukocytes, leading to release of opioid peptides, which then activate peripheral opioid receptors to relieve pain (Schäfer et al., 1994, 1996; Cabot et al., 1997, 2001). Immunosuppression abolishes stress- and CRH-induced analgesia, demonstrating the functional relevance of immunocytes (Stein et al., 1990b; Przewlocki et al., 1992; Schäfer et al., 1994).
The recruitment of leukocytes from the circulation to inflammatory foci involves interactions with vascular endothelium, which are orchestrated by adhesion molecules. Initially, selectins expressed on leukocytes and endothelial cells mediate leukocyte capture and rolling along the vessel wall. Subsequently, interactions between integrins and Ig-like members arrest the rolling cells and mediate firm adhesion, leading to their migration into sites of injury (Springer, 1990; Butcher and Picker, 1996; Petruzzelli et al., 1999). Intercellular adhesion molecule-1 [ICAM-1 (or CD54)] is one of the major molecules required primarily for the firm adhesion and diapedesis of leukocytes (Springer, 1990; Butcher and Picker, 1996; Petruzzelli et al., 1999). In addition, it is the most extensively studied adhesion molecule in anti-inflammatory therapy. ICAM-1 is constitutively expressed by endothelium, leukocytes, and synovial lining cells in animal models and in patients with inflammatory diseases (Szekanecz et al., 1994; Bennett et al., 1997; Salmi et al., 1997; Veihelmann et al., 1999). Blockade of ICAM-1 by monoclonal antibodies (mAbs), antisense oligonucleotides, or deletion of the ICAM-1 gene was shown to impair the migration of immunocytes to inflamed tissue and to decrease inflammation in animals and humans (Sligh et al., 1993; Kavanaugh et al., 1994; Bullard et al., 1996; Bennett et al., 1997). More recently, we showed that adhesion mechanisms are not exclusively involved in mounting an immune response to pathogens but also in pain control. Thus, interruption of rolling by selectin blockade attenuated intrinsic opioid analgesia within injured tissue (Machelska et al., 1998).
In this study, we sought to determine whether interfering with later steps in the adhesion cascade, i.e., with ICAM-1-dependent adhesion and transendothelial migration of opioid-expressing leukocytes, will affect opioid-mediated pain inhibition. To this end, we investigated the cellular coexpression of ICAM-1 and β-endorphin, and we evaluated the effects of an mAb against ICAM-1 (anti-ICAM-1) on the trafficking of opioid-containing immune cells and on peripheral stress- and CRH-induced opioid analgesia in a rat model of inflammatory pain.
MATERIALS AND METHODS
Animals. Experiments were performed on male Wistar rats (140–150 gm) (Freie Universität, Berlin, Germany) in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by the local animal care committee. Rats were housed individually in cages and maintained on a 12 hr light/dark schedule, with food pellets and waterad libitum. Room temperature was maintained at 22 ± 0.5°C and a relative humidity between 60 and 65%.
Antibodies. For immunofluorescence the following antibodies (Abs) were used: polyclonal rabbit anti-rat β-endorphin (Peninsula Laboratories, Belmont, CA), monoclonal mouse anti-rat ICAM-1 (clone number 1A29; Seikagaku, Tokyo, Japan), and secondary Abs: goat anti-rabbit Texas Red-conjugated (anti-rabbit–Texas Red) and horse anti-mouse– fluorescein isocyanate (FITC) (Vector Laboratories, Burlingame, CA). For flow cytometry, the following Abs were used: mouse IgG2a, rat anti-mouse IgG2a+b-phytoerythrin (PE), mouse anti-rat: CD3-PE (T cell marker), CD4-PE, CD11b-FITC (PharMingen/Becton Dickinson, Heidelberg, Germany), CD45-CyChrome (CyC) (all hematopoetic cell marker), RP-1-PE (granulocyte marker), ED1-FITC (monocyte–macrophage marker) (Serotec, Oxford, UK), and mouse 3E7 (opioid peptides) (subtype IgG2a; Gramsch Laboratories, Schwabhausen, Germany). 3E7 was generated against β-endorphin and recognizes the pan-opioid sequence Tyr-Gly-Gly-Phe-Met at the N terminus of opioid peptides (Gramsch et al., 1983). All Abs used in flow cytometry are monoclonal. For behavioral experiments monoclonal mouse anti-rat ICAM-1 (Ab clone number 1A29; PharMingen/Becton Dickinson) (Tamatani and Miyasaka, 1990) and mouse IgG (Sigma, Deisenhofen, Germany) were used.
Induction and evaluation of inflammation. Rats received an intraplantar injection of 0.15 ml of Freund's complete adjuvant (Calbiochem, La Jolla, CA) into the right hindpaw under brief halothane (Willy Rüsch GmbH, Böblingen, Germany) anesthesia. Paw volume and dorsal surface temperature were measured with a plethysmometer (Ugo Basile, Comerio, Italy) and a contact thermometer (Cooper Instrument, Middlefield, CT), respectively, and they were determined by averaging two consecutive trials (Stein et al., 1990a). Nociceptive thresholds were assessed using the paw pressure algesiometer (modified Randall-Selitto test; Ugo Basile). The pressure required to elicit paw withdrawal, the paw pressure threshold (PPT) (cutoff at 250 gm), was determined by averaging three consecutive trials separated by 10 sec (Stein et al., 1990a). All experiments were performed at 6 hr after induction of inflammation.
Immunofluorescence. The expression of ICAM-1 and β-endorphin in noninflamed and inflamed paw tissue was analyzed in rats (n = 5–6 per group) that did not receive any intravenous injections and in rats that received intravenous anti-ICAM-1 or mouse IgG (both at 4 mg/kg in 0.6 ml of sterile water) under brief halothane anesthesia immediately before induction of inflammation. Six hours later, rats were deeply anesthetized with halothane and perfused transcardially (0.1 m PBS, followed by fixative solution: PBS containing 4% paraformaldehyde, pH 7.4). The skin with adjacent subcutaneous tissue was removed from both hindpaws, postfixed for 30 min at 4°C in the fixative solution, and cryoprotected overnight at 4°C in PBS containing 10% sucrose. The tissue was embedded in Tissue Tek compound (OCT; Miles, Elkhart, IN), frozen, cut into 7 μm sections, mounted onto gelatin-coated slides, and processed for immunofluorescence (Mousa et al., 2000). The sections were incubated with anti-ICAM-1 (1:200) alone or in combination with anti-β-endorphin (1:1000) and then with secondary Abs. The sections were washed with PBS, mounted in Vectashield (Vector Laboratories), and viewed under a fluorescence microscope (Zeiss, Jena, Germany) with appropriate filters. The following control experiments were included: (1) preabsorption of anti-β-endorphin with β-endorphin (Peninsula Laboratories), and (2) omission of either the primary or the secondary Abs (Mousa et al., 2000).
The expression of ICAM-1 and β-endorphin was quantified by an observer blinded to the experimental protocol, using a Zeiss microscope (objective, 20×; eyepiece, 10×). The mean number of ICAM-1-expressing blood vessels and β-endorphin-expressing cells in three sections per animal and five squares (384 mm2 for ICAM-1; 384 μm2 for β-endorphin) per section was calculated. The percentage of ICAM-1-stained vessels was determined according to the following formula: number of ICAM-1-stained vessels/number of all vessels × 100 (Mousa et al., 2000).
Flow cytometry. To assess the contribution of ICAM-1 in trafficking of opioid-containing immune cells, rats (n = 6–12 per group) received intravenously anti-ICAM-1 or mouse IgG (both at 4 mg/kg in 0.6 ml of sterile water) under brief halothane anesthesia immediately before induction of inflammation. To evaluate the involvement of ICAM-1 in immune cell migration under normal conditions, rats received the same intravenous treatment but without induction of inflammation. Six hours after treatments, blood samples (1 ml) were obtained by direct parasternal cardiac puncture under halothane anesthesia. Rats were then killed, and subcutaneous tissue was dissected from inflamed paws. To obtain a single cell suspension, the tissue was cut into 1–2 mm pieces and digested (Rittner et al., 2001). Because no immune cells were found in noninflamed paws, these were not processed for flow cytometry.
For surface staining, cells were incubated for 15 min at room temperature with anti-CD3-PE (4 μg/ml) and anti-CD45-CyC (2 μg/ml), washed, and fixed with PBS containing 1% paraformaldehyde. Intracellular staining was performed as described previously (Rittner et al., 2001). Cells were permeabilized in PBS containing 0.5% saponin and 0.5% bovine serum albumin (Sigma) and subsequently incubated for 30 min at room temperature with the intracellular Abs (RP-1-PE, 6 μg/ml; ED1-FITC, 2 μg/ml; or 3E7, 20 μg/ml). For 3E7 staining, cell suspensions were stained with the secondary anti-mouse IgG2a+b-PE (1.5 μg/ml) for 30 min at room temperature. Negative controls included the replacement of the primary Ab with an isotype-matched irrelevant Ab (mouse IgG2a).
Aliquots of 100 μl of blood were left unstained or incubated with two Abs: anti-CD4-PE (4 μg/ml) and anti-CD11b-FITC (10 μg/ml) for 15 min at room temperature. The leukocyte subpopulations were identified as follows: granulocytes, CD4−/CD11b+; monocytes–macrophages, CD4+/CD11b+; and T cells, CD4+/CD11b−. Cells were lysed and fixed with fluorescence activated cell staining (FACS) lysing solution (PharMingen/Becton Dickinson) according to the instructions of the manufacturer. Granulocytes, monocytes–macrophages, and T cells were analyzed by forward–sideward scatter characteristics, as well as by expression of CD4 and CD11b.
To calculate absolute numbers of cells per paw and in circulating blood, the stained cell suspensions were analyzed together with a known number of fluorescent beads in a TRUCOUNT tube (PharMingen/Becton Dickinson). Numbers of cells per tube were calculated in relation to the known number of fluorescent TRUCOUNT beads and extrapolated for the whole paw. At least 10,000 FACS events were collected in FACScan and analyzed using CellQuest software (PharMingen/Becton Dickinson). To exclude nonviable and nonhematopoetic cells in the paw, only CD45+ cells were analyzed.
Behavioral experiments. To determine the contribution of peripheral opioid receptors in endogenous and CRH-induced analgesia, naloxone hydrochloride (NLX) (both from Sigma) was tested. Rats received an intraplantar injection of NLX (1.125 μg) into the inflamed paw, and, 5 min later, they were subjected to cold (2–4°C) water swim stress for 1 min in a metal container (Stein et al., 1990a). PPTs were reevaluated at the time of their maximum elevation, i.e., 1 min after swim stress (Stein et al., 1990a). In separate experiments, rats received bilateral intraplantar injections of CRH (4 ng), an agent triggering the release of opioid peptides from immune cells (Schäfer et al., 1994, 1996; Cabot et al., 1997, 2001). PPTs were reevaluated at the time of their maximum elevation, i.e., 5 min after CRH (Schäfer et al., 1994). Because CRH produced analgesia only in inflamed paws, the subsequent groups of rats received NLX (140 ng) concomitantly with CRH (4 ng) only into inflamed paws, and PPTs were measured 5 min later. Baseline PPTs were taken before all treatments. The doses of NLX and CRH chosen were the most effective in pilot experiments. Control rats were injected with an equivalent volume of vehicle and tested analogously. To confirm a peripheral site of action CRH (8 ng) or NLX (125 μg in case of swim stress- and 140 ng in case of CRH-induced analgesia) were injected subcutaneously (volume, 0.2 ml) into an animal back and tested like after intraplantar injections.
To evaluate the involvement of ICAM-1 in peripheral opioid analgesia, rats received intravenous anti-ICAM-1 or mouse IgG (both at 2–8 mg/kg in 0.6 ml of sterile water) immediately before induction of inflammation. Six hours later, paw temperature was measured, baseline PPTs were taken, and rats were subjected to swim stress. PPTs were reevaluated 1 min later, and paw volume was measured thereafter. In separate experiments, rats received CRH (4 ng) into inflamed paws (instead of swim stress), and PPTs were reevaluated 5 min later.
Intraplantar (volume, 0.1 ml) injections were done under brief halothane anesthesia. NLX was dissolved in 0.9% NaCl and CRH in sterile water. The number of animals was six to eight per group. The experimenter was blinded to the treatments.
Statistical analysis. Data are presented as means ± SEM and are expressed in raw values, except paw volume and paw temperature, which are expressed as a percentage of control in graphs and in raw values in the text. Data were analyzed using the pairedt test for dependent data, t test for independent normally distributed data, and Mann–Whitney test for independent not normally distributed data. Differences were considered significant ifp ≤ 0.05.
RESULTS
ICAM-1 is upregulated simultaneously with an enhanced immigration of β-endorphin-containing cells to inflamed paw tissue
Immunofluorescence revealed ICAM-1 on the vascular endothelium of some small vessels in noninflamed subcutaneous tissue (Fig.1A,D). β-Endorphin-positive cells were extremely scarce (Fig.1D). In inflamed tissue, ICAM-1 was strongly expressed in the endothelium of small and large vessels (Fig.1B,C,E,F). Inflammation significantly increased the total number and the percentage of ICAM-1-positive blood vessels (p< 0.001 and p < 0.01, respectively; pairedt test) (Table 1). There were also numerous cells expressing β-endorphin in the vicinity of ICAM-1-positive vessels (Fig. 1E). Intravenous pretreatment with IgG or anti-ICAM-1 (both at 4 mg/kg) did not significantly change the total number or the percentage of ICAM-1-positive blood vessels in inflamed or in noninflamed paws (p > 0.05; ANOVA) (Fig.1C,F; Table 1). However, anti-ICAM-1 significantly decreased the number of β-endorphin-expressing cells in inflamed paws (10.4 ± 0.59 vs 5.6 ± 0.45 cells, IgG vs anti-ICAM-1; p < 0.001; t test). Control experiments using omission of anti-ICAM-1 or preabsorption of anti-β-endorphin with β-endorphin revealed no immunoreactivity (data not shown).
Fig. 1.

Expression of ICAM-1 and β-endorphin in subcutaneous paw tissue. Immunofluorescent single staining of ICAM-1 (green; A–C) and double staining of ICAM-1 and β-endorphin (red;D–F). The expression of ICAM-1 and the infiltration of β-endorphin-containing immune cells is markedly increased in inflamed (B, E) compared with non-inflamed tissue (A, D). Pretreatment with anti-ICAM-1 (4 mg/kg, i.v.) does not change the expression of ICAM-1; however, it decreases the number of β-endorphin-containing cells in inflamed tissue (C,F). Scale bar, 20 μm.
Table 1.
Effects of anti-ICAM-1 on the number of ICAM-1-expressing blood vessels in the paw
| Treatment | Absolute number | Percentage | ||
|---|---|---|---|---|
| Inflamed paw | Noninflamed paw | Inflamed paw | Noninflamed paw | |
| No intravenous injection | 9.4 ± 0.5* | 3.1 ± 0.2 | 45 ± 2.71-160 | 28 ± 0.4 |
| Control (IgG, 4 mg/kg, i.v.) | 9.7 ± 0.6* | 2.7 ± 0.1 | 46 ± 2.51-160 | 24 ± 1.6 |
| Anti-ICAM-1 (4 mg/kg, i.v.) | 9.8 ± 0.4* | 2.6 ± 0.1 | 47 ± 1.71-160 | 24 ± 1.0 |
Data are expressed as means ± SEM.
p < 0.001 and
F1-160: p< 0.01 (paired t test) indicate statistically significant differences compared with respective noninflamed paws. There are no significant differences between different treatments in both inflamed and noninflamed paws (p > 0.05; ANOVA).
Anti-ICAM-1 impairs the trafficking of opioid-containing immune cells to inflamed paw tissue
For analysis by flow cytometry, immune cells isolated from the inflamed paws were pregated by staining with anti-CD45 to exclude debris and nonhematopoetic cells, such as fibroblasts or lipocytes (Fig. 2A). CD45+ cells were further characterized into granulocytes, monocytes–macrophages and T cells, as described in Materials and Methods and by Rittner et al. (2001). An intracellular stain for opioid peptides was then established: CD45+ cells were stained with 3E7 or an isotype-matched control IgG2a mAb and quantified (Fig. 2B). Twenty percent of CD45+ cells were 3E7 positive (Fig.2B, bottom panel), whereas only 0.53% of these cells were stained with IgG2a(Fig. 2B, top panel), demonstrating the specificity of the opioid staining. Corresponding results were obtained in another experiment in which 16% of CD45+ cells contained opioid peptides (Fig. 3, compare control groups inC, E). Specificity of the opioid staining was additionally demonstrated by dose-dependent and significant inhibition of 3E7 labeling by preincubation with β-endorphin but not with a control peptide (adrenocorticotropic hormone), as described by Rittner et al. (2001). Because no immune cells were found in noninflamed paws, these were not processed for flow cytometry.
Fig. 2.
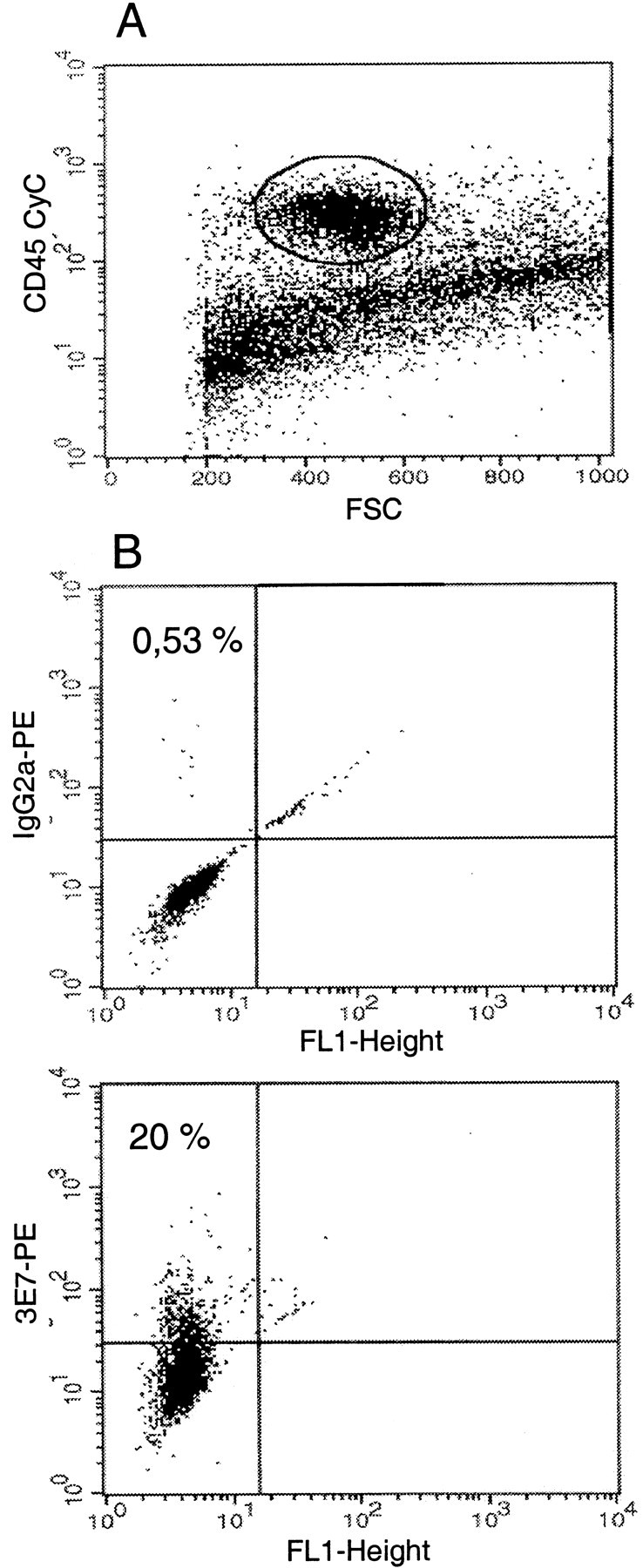
Fluorescence activated cell staining for intracellular opioid peptides in hematopoetic (CD45+) cells in the inflamed paw tissue. Single cell suspensions from the paws were stained with CD45 CyChrome (A). CD45+ cells were pregated and examined for expression of 3E7 (opioid peptides) (B). CD45+ cells were stained with a control mAb (anti-mouse IgG2a;B, top) or anti-pan-opioid 3E7 (B, bottom), and both were subsequently incubated with PE-conjugated anti-mouse IgG2a. Twenty percent of the CD45+ cells contain opioid peptides (B, bottom). Because no immune cells were found in noninflamed paws, these were not processed for flow cytometry.FSC, Forward scatter characteristic; FL1, flow laser 1.
Fig. 3.

Effects of anti-ICAM-1 (4 mg/kg, i.v.) on trafficking of opioid-containing immune cells. In the blood (A, B), anti-ICAM-1 significantly increases the total number of all leukocytes (A), granulocytes, monocytes–macrophages, and T cells (B) (p < 0.05;t test). In inflamed paws (C–E), anti-ICAM-1 significantly decreases the total number of all leukocytes (C), granulocytes, monocytes–macrophages (p < 0.01; Mann–Whitney test), T cells (p < 0.001; t test) (D), and opioid-containing leukocytes (p < 0.01;t test) (E). Opioid-containing leukocytes represent 16% (compare control group in Cwith control group in E) to 20% of all leukocytes (CD45+) (Fig. 2B,bottom). Control (mouse IgG; 4 mg/kg, i.v.),white bars; 4 mg/kg anti-ICAM-1, black bars. Data are expressed as means ± SEM. * indicates a statistically significant difference compared with respective controls. Because no immune cells were found in noninflamed paws, these were not processed for flow cytometry.
In the absence of inflammation, anti-ICAM-1 (4 mg/kg, i.v.) significantly increased the number of all leukocytes in the circulation (234 ± 20 vs 365 ± 35 × 10 cells/μl, control vs anti-ICAM-1; p < 0.01; t test) and of granulocytes (106 ± 12 vs 194 ± 23 × 10 cells/μl, control vs anti-ICAM-1; p < 0.01; Mann–Whitney test) and T cells (58 ± 4.9 vs 87 ± 6.0 × 10 cells/μl, control vs anti-ICAM-1; p < 0.01; t test). The number of monocytes–macrophages was slightly but significantly decreased by anti-ICAM-1 (12 ± 1.4 vs 6.9 ± 1.1 × 10 cells/μl, control vs anti-ICAM-1; p < 0.01;t test).
Inflammation increased the migration of immune cells to affected paws (Fig. 3C, control group). The majority of these cells were granulocytes, followed by monocytes–macrophages and T cells (Fig.3D, control groups). Sixteen to 20% of immune cells contained opioid peptides (Figs. 2B, bottom panel, 3E, control group). In the blood, anti-ICAM-1 (4 mg/kg, i.v.) significantly increased the total number of leukocytes, including all subpopulations, i.e., granulocytes, monocytes–macrophages, and T cells (p < 0.05;t test) (Fig. 3A,B). Consistently, anti-ICAM-1 significantly decreased the total number of immunocytes, as well as granulocytes, monocytes–macrophages (p < 0.01; Mann–Whitney test), and T cells (p < 0.001; t test) in the inflamed paws (Fig. 3C,D). Anti-ICAM-1 substantially decreased the number of leukocytes containing opioids in the inflamed paws (p < 0.01; t test) (Fig.3E).
Anti-ICAM-1 decreases peripheral opioid analgesia
Six hours after intraplantar injection of Freund's adjuvant, rats developed inflammation, confined to the inoculated paw and characterized by hyperalgesia (decreased PPT) (Fig.4, top panel, Table2, inflamed compared with noninflamed paws of control group), swelling (increased paw volume, 1.6 ± 0.04 vs 0.97 ± 0.01 ml, inflamed vs noninflamed paw), and hyperthermia (elevated paw temperature, 36 ± 0.2 vs 33.6 ± 0.4°C, inflamed vs noninflamed paw) (p < 0.001; paired t test).
Fig. 4.

Effects of anti-ICAM-1 (2–8 mg/kg, i.v.) on macroscopic inflammation. Anti-ICAM-1 does not significantly change hyperalgesia (p > 0.05; ttest) (top). Anti-ICAM-1 (4 mg/kg) decreases paw volume (p < 0.001; t test) (middle), and anti-ICAM-1 (8 mg/kg) decreases paw temperature (p < 0.001; Mann–Whitney test) (bottom) of the inflamed paws. No significant changes are observed in noninflamed paws (p > 0.05;t test). Control (mouse IgG; 2–8 mg/kg, i.v.),white bars; 2 mg/kg anti-ICAM-1, cross-hatched bars; 4 mg/kg anti-ICAM-1, black bars; 8 mg/kg anti-ICAM-1, diagonal bars. The dashed lines represent controls (100%). Data are expressed as means ± SEM. * indicates a statistically significant difference compared with respective controls.
Table 2.
Effects of swim stress and intraplantar CRH on PPT (gm) and reversibility of these effects by intraplantar NLX
| Treatment | Inflamed paw | Noninflamed paw |
|---|---|---|
| Swim stress | ||
| Control | ||
| Baseline | 22 ± 1.3 | 57 ± 1.3 |
| 1 min | 116 ± 6.1* | 60 ± 2.0 |
| NLX (1.125 μg) | ||
| 1 min | 61 ± 4.72-160 | 62 ± 2.0 |
| CRH (4 ng) | ||
| Control | ||
| Baseline | 30 ± 2.4 | 62 ± 4.5 |
| 5 min | 76 ± 8.3* | 58 ± 2.9 |
| NLX (140 ng) | ||
| 5 min | 27 ± 3.72-165 | 65 ± 2.8 |
Data are expressed as means ± SEM.
indicates a statistically significant difference compared with respective baseline (p < 0.001; paired t test).
F2-160: indicates a statistically significant difference compared with 1 min after swim stress of control (intraplantar 0.9% NaCl).
F2-165: indicates a statistically significant difference compared with 5 min after intraplantar CRH of control (intraplantar 0.9% NaCl and 4 ng of CRH) (p < 0.001; t test).
Anti-ICAM-1 (2–8 mg/kg, i.v.) did not cause any significant changes in hyperalgesia (baseline PPT) (p > 0.05;t test) (Fig. 4, top panel). This treatment significantly decreased paw volume (1.8 ± 0.02 vs 1.6 ± 0.05 ml, control vs 4 mg/kg anti-ICAM-1; p< 0.001; t test) (Fig. 4, middle panel) and paw temperature (35 ± 0.1 vs 34 ± 0.03°C, control vs 8 mg/kg anti-ICAM-1; p < 0.001; Mann–Whitney test) (Fig. 4, bottom panel). No significant changes were observed in noninflamed paws (p > 0.05;t test) (Fig. 4).
Exposure of rats to swim stress produced potent analgesia in inflamed but not in noninflamed paws (p < 0.001 andp > 0.05, respectively; paired t test) (Table 2). NLX (1.125 μg) injected into inflamed paws significantly decreased stress-induced analgesia (p < 0.001;t test) (Table 2). No significant changes were observed in noninflamed paws (p > 0.05; Mann–Whitney test) (Table 2). Subcutaneous NLX (1.125 μg) did not significantly change stress-induced analgesia (p > 0.05;t test; data not shown), demonstrating a peripheral site of action.
Anti-ICAM-1 (4 mg/kg, i.v.) strongly decreased stress-induced analgesia (p = 0.001; t test) (Fig.5, top panel). This effect was slightly but significantly less compared with the effect of NLX (1.125 μg) on stress-induced analgesia (p< 0.05; t test) (Fig. 5, top panel, 4 mg of anti-ICAM-1 compared with Table 2, 1.125 μg of NLX). No significant changes were observed in inflamed paws after anti-ICAM-1 (2 and 8 mg/kg) or in noninflamed paws after any treatment (p > 0.05; t test) (Fig. 5,top panel; Table 2).
Fig. 5.

Effects of anti-ICAM-1 on stress- (top) and CRH- (bottom) induced analgesia. Anti-ICAM-1 (4 mg/kg, i.v.) significantly decreases stress- and CRH (4 ng)-induced analgesia (p = 0.001 and p = 0.01, respectively; t test). Two and 8 mg/kg anti-ICAM-1 does not significantly change stress-induced analgesia (p > 0.05;t test). No significant changes are observed in noninflamed paws (p > 0.05;t test). Control (mouse IgG; 2–8 mg/kg, i.v.),white bars; 2 mg/kg anti-ICAM-1, cross-hatched bars; 4 mg/kg anti-ICAM-1, black bars; 8 mg/kg anti-ICAM-1, diagonal bars. Data are expressed as means ± SEM. * indicates a statistically significant difference compared with respective controls.
Bilateral intraplantar CRH (4 ng) produced analgesia in inflamed but not in noninflamed paws (p < 0.001 andp > 0.05, respectively; paired t test) (Table 2). CRH-induced analgesia was blocked by intraplantar NLX (140 ng); no significant changes were observed in noninflamed paws (p < 0.001 and p > 0.05, respectively; t test) (Table 2), after subcutaneous NLX (140 ng) or after subcutaneous CRH (8 ng) (p > 0.05;t test; data not shown), demonstrating a peripheral site of action.
Anti-ICAM-1 (4 mg/kg, i.v.) substantially reduced CRH (4 ng)-induced analgesia (p = 0.01; t test) (Fig. 5,bottom panel). This effect was slightly but significantly less compared with the effect of NLX (140 ng) on CRH (4 ng)-induced analgesia (p < 0.001; ttest) (Fig. 5, bottom panel, 4 mg of anti-ICAM-1 compared with Table 2, 140 ng of NLX). No significant changes were observed in noninflamed paws (p > 0.05; t test) (Fig. 5, bottom panel; Table 2).
DISCUSSION
The major finding of this study is that blocking of ICAM-1 dramatically decreases opioid-mediated analgesia and the extravasation of opioid-containing cells in inflammation, indicating that ICAM-1 is an important molecule governing intrinsic pain control in injured tissue. In the present and previous studies, we showed that our stress paradigm (cold water swim) and CRH produce opioid receptor-specific peripheral analgesia because the effects were dose dependently abolished by local (intraplantar) but not by systemic (subcutaneous and intravenous) treatment with opioid receptor antagonists and Abs against opioid peptides (Stein et al., 1990a; Schäfer et al., 1994). Furthermore, this analgesia can be blocked by immunosuppression with cyclosporine or whole-body irradiation (Stein et al., 1990b; Przewlocki et al., 1992; Schäfer et al., 1994), indicating that immune cells are the source of opioid peptides. Consistently, using flow cytometry, we showed here that a substantial portion of opioid-containing leukocytes migrate to inflamed paws. The majority of immune cells were granulocytes, followed by monocytes–macrophages. T cells represented the smallest population at this early stage of inflammation. The present immunofluorescence experiments also revealed numerous cells expressing β-endorphin in inflammation, whereas in noninflamed tissue, they were virtually absent. These findings are in line with our previous studies (Przewlocki et al., 1992; Cabot et al., 1997; Rittner et al., 2001). Previously, we showed that the predominant endogenous stimulus for the release of opioid peptides at later stages of inflammation is CRH. This release is CRH receptor specific and calcium dependent (Schäfer et al., 1994, 1996; Cabot et al., 1997, 2001). Here we found that the local application of CRH elicits potent peripheral opioid receptor-specific analgesia also at this early stage and, importantly, that CRH-induced analgesia is substantially diminished by anti-ICAM-1.
The most likely mechanism of the anti-ICAM-1-mediated decrease of peripheral analgesia is an impaired migration of opioid-containing immune cells. We found constitutive expression of ICAM-1 in noninflamed tissue and ICAM-1 upregulation on endothelium of small and large vessels in inflammation. Similar findings were reported for other inflammatory diseases in animals and humans (Szekanecz et al., 1994;Bennett et al., 1997; Salmi et al., 1997; Veihelmann et al., 1999). ICAM-1 has been proposed to be primarily involved in firm adhesion of leukocytes, which carry its major ligands β2(CD18) integrins, lymphocyte-function associated antigen-1 (CD11a/CD18), and Mac-1 (CD11b/CD18). As a consequence of the interactions between ICAM-1 and its ligands, leukocytes leave the circulation and migrate to inflammatory foci (Beekhuizen et al., 1993). In agreement with this concept, our flow cytometry experiments show that, in both the absence and presence of inflammation, anti-ICAM-1 increased the number of circulating leukocytes. The anti-ICAM-1-induced drop of circulating monocytes–macrophages in the absence of inflammation could be attributable to sequestering effects of intact Abs [in contrast to F(ab′)2 fragments] (Mulligan et al., 1994). Such effects are apparently overcompensated in inflammation. Importantly, anti-ICAM-1 significantly reduced the immigration of granulocytes, monocytes–macrophages, and T cells into the inflamed tissue. Impaired immune cell extravasation resulting from ICAM-1 inactivation was also shown by others (Sligh et al., 1993;Bullard et al., 1996; Bennett et al., 1997). Our study extends these observations in that anti-ICAM-1 substantially decreased the migration of opioid-containing leukocytes to the inflamed paws. This is shown by both our flow cytometry and immunofluorescence experiments. Anti-ICAM-1 did not significantly change the vascular expression of ICAM-1 but apparently impaired its function because less immunocytes extravasated into the inflamed tissue. Together, our findings strongly suggest that blockade of ICAM-1 on vascular inflamed endothelium reduces endogenous analgesia that is normally generated by immune cell-derived opioids.
The effects of anti-ICAM-1 were potent and similar to the effects of NLX on stress- and CRH-induced analgesia. Our finding that a high dose of anti-ICAM-1 tended to loose its blocking effect is in line with other studies reporting stimulatory effects of mAbs (Clayton et al., 1998; Vuorte et al., 1999) and with therapeutic effects of mAbs that strongly vary with their dosage (Bach et al., 1993; Willenborg et al., 1993). Notwithstanding, anti-ICAM-1 is very effective in attenuating intrinsic opioid analgesia in our model. In contrast, to achieve an optimal effect on inflammation, ICAM-1 had to be blocked simultaneously with other adhesion molecules (Kakimoto et al., 1992; Issekutz, 1998;McCafferty et al., 1999). Thus, our findings point to a specific role of ICAM-1 in intrinsic pain control. This is further supported by our observation that macroscopic inflammatory parameters, such as paw edema and temperature, were essentially unchanged. This may be explained by a lack of influence of anti-ICAM-1 on the vascular component of Freund's complete adjuvant-induced paw inflammation. Other studies have also shown a lack of correlation between cellular (leukocyte accumulation) and vascular (edema, plasma extravasation, and vasodilatation) components of inflammation after anti-adhesion treatments (Nwariaku et al., 1996; Taylor et al., 1997; De Mora et al., 1998). Furthermore, hyperalgesia (i.e., baseline PPT) was not significantly changed by anti-ICAM-1. This could be explained by a lack of the influence of ICAM-1 on the migration of cells containing hyperalgesic cytokines or transmitters (e.g., 5-hydroxytryptamine, histamine, nerve growth factor, substance P, or calcitonin gene-related peptide) (Dray et al., 1994; Shu and Mendell, 1999; Machelska et al., 2001; Cunha and Ferreira, 2002). In addition, anti-ICAM-1 is unlikely to block the release of nerve-derived substance P and/or calcitonin gene-related peptide. On the other hand, although anti-ICAM-1 diminished the immigration of opioid-containing leukocytes to inflamed paws, their nociceptive baseline thresholds were not decreased. This may be attributable to a lack of sensitivity of our paw pressure assay. It is also possible that the endogenous opioid system is not essential for tonic pain control in our model. This would be consistent with several studies using opioid receptor antagonists or mice lacking either opioid receptors or opioid peptides, which have shown no changes in basal nociceptive transmission in acute and inflammatory pain (Roques et al., 1993; Mogil and Grisel, 1998; Qiu et al., 2000). Furthermore, we showed previously that immune cells need to be stimulated by stress or CRH to release opioid peptides and produce analgesia (Schäfer et al., 1994, 1996; Cabot et al., 1997, 2001). Our present observations also suggest that ICAM-1 has no major influence on other immune cell-derived analgesic substances, such as acetylcholine or analgesic cytokines (interleukin-4, -10, and -13 and interleukin-1 receptor antagonist) (Machelska et al., 2001; Cunha and Ferreira, 2002). In summary, it seems that, in our model, ICAM-1 plays a specific role in the recruitment of opioid-containing cells and comes into play when this intrinsic system is activated to cope with inflammatory pain.
What is the clinical significance of our findings? ICAM-1 blockade has been proposed as a novel anti-inflammatory strategy (Iigo et al., 1991;Kakimoto et al., 1992; Kavanaugh et al., 1994; Bullard et al., 1996;Bennett et al., 1997; Bendjelloul et al., 2000). Our studies indicate that ICAM-1 blockade can result in severely impaired endogenous pain inhibition. Thus, in addition to other reported detrimental effects (Marvin et al., 1998; Samoilova et al., 1998; McCafferty et al., 1999), pain may be exacerbated after anti-ICAM-1 treatments.
In conclusion, ICAM-1 is an important molecule promoting the endogenous control of inflammatory pain (Fig. 6). Blocking of ICAM-1 expressed on vascular endothelium strongly reduces local intrinsic and CRH-induced opioid analgesia. This results from a decreased extravasation of opioid-containing immune cells infiltrating the inflamed tissue. Because analgesic effects of endogenous immune-derived opioids are potent in the clinical setting (Stein et al., 1993), it is important that future therapeutic strategies aimed at limiting the adhesion of harmful cells in inflammatory diseases (e.g., rheumatoid arthritis) do not interfere with the migration of opioid-containing cells promoting pain control.
Fig. 6.

Schema of immune mechanisms in peripheral opioid analgesia. Subcutaneous inflammation leads to an increased centrifugal transport of opioid receptors (OR) from dorsal root ganglia (DRG) and to their consequent upregulation on peripheral terminals of sensory neurons (left). P-Selectin and ICAM-1 are upregulated on vascular endothelium, and L-selectin is coexpressed by immune cells producing opioid peptides (EOP). L- and P-Selectin mediate rolling of opioid-containing immunocytes along the vessel wall. ICAM-1 mediates adhesion and diapedesis of these leukocytes from the circulation to inflamed tissue. Selectins and ICAM-1 interact with their respective ligands. In response to stress (cold water swim) or CRH, immune cells secrete opioid peptides. CRH is produced by local inflammatory cells and releases opioids by interacting with CRH receptors (CRHR). Opioid peptides bind to peripheral opioid receptors and interrupt nociceptive transmission. Blockade of selectins (Machelska et al., 1998) or ICAM-1 (present study) diminishes the migration of opioid-containing immunocytes to inflamed tissue, resulting in severely impaired endogenous pain inhibition.
Footnotes
This study was supported by the Deutsche Forschungsgemeinschaft (Sonderforschungsbereich 507/B8 and Klinische Forschergruppe 100/1) and by the International Anesthesia Research Society.
Correspondence should be addressed to Dr. Halina Machelska, Klinik für Anaesthesiologie und operative Intensivmedizin, Klinikum Benjamin Franklin, Freie Universität Berlin, Hindenburgdamm 30, D-12200 Berlin, Germany. E-mail:machelska@zop-admin.ukbf.fu-berlin.de.
REFERENCES
- 1.Bach JF, Fracchia GN, Chatenoud L. Safety and efficacy of therapeutic monoclonal antibodies in clinical therapy. Immunol Today. 1993;14:421–425. doi: 10.1016/0167-5699(93)90243-E. [DOI] [PubMed] [Google Scholar]
- 2.Beekhuizen H, Blokland I, van Furth R. Cross-linking of CD14 molecules on monocytes results in a CD11/CD18- and ICAM-1-dependent adherence to cytokine-stimulated human endothelial cells. J Immunol. 1993;150:950–959. [PubMed] [Google Scholar]
- 3.Bendjelloul F, Maly P, Mandys V, Jirkovska M, Prokesova L, Tuckova L, Tlaskalova H. Intercellular adhesion molecule-1 (ICAM-1) deficiency protects mice against severe forms of experimentally induced colitis. Clin Exp Immunol. 2000;119:57–63. doi: 10.1046/j.1365-2249.2000.01090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bennett CF, Kornbrust D, Henry S, Stecker K, Howard R, Cooper S, Dutson S, Hall W, Jacoby HI. An ICAM-1 antisense oligonucleotide prevents and reverses dextran sulfate sodium-induced colitis in mice. J Pharmacol Exp Ther. 1997;280:988–1000. [PubMed] [Google Scholar]
- 5.Bullard DC, Hurley LA, Lorenzo I, Sly LM, Beaudet AL, Staite ND. Reduced susceptibility to collagen-induced arthritis in mice deficient in intercellular adhesion molecule-1. J Immunol. 1996;157:3153–3158. [PubMed] [Google Scholar]
- 6.Butcher EC, Picker LJ. Lymphocyte homing and homeostasis. Science. 1996;272:60–66. doi: 10.1126/science.272.5258.60. [DOI] [PubMed] [Google Scholar]
- 7.Cabot PJ, Carter L, Gaiddon C, Zhang Q, Schäfer M, Loeffler JP, Stein C. Immune cell-derived β-endorphin: production, release and control of inflammatory pain in rats. J Clin Invest. 1997;100:142–148. doi: 10.1172/JCI119506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cabot PJ, Carter L, Schäfer M, Stein C. Methionine-enkephalin- and Dynorphin A-release from immune cells and control of inflammatory pain. Pain. 2001;93:207–212. doi: 10.1016/S0304-3959(01)00322-0. [DOI] [PubMed] [Google Scholar]
- 9.Clayton A, Evans RA, Pettit E, Hallett M, Williams JD, Steadman R. Cellular activation through the ligation of intercellular adhesion molecule-1. J Cell Sci. 1998;111:443–453. doi: 10.1242/jcs.111.4.443. [DOI] [PubMed] [Google Scholar]
- 10. Cunha FQ, Ferreira SH. Peripheral hyperalgesic cytokines. Immune mechanisms of pain and analgesia Machelska H, Stein C. 2002. Landes Bioscience; Georgetown, Washington, DC, in press. [Google Scholar]
- 11.DeLeo JA, Yezierski RP. The role of neuroinflammation and neuroimmune activation in persistent pain. Pain. 2001;90:1–6. doi: 10.1016/s0304-3959(00)00490-5. [DOI] [PubMed] [Google Scholar]
- 12.De Mora F, Williams CM, Frenette PS, Wagner DD, Hynes RO, Galli SJ. P- and E-selectins are required for the leukocyte recruitment, but not the tissue swelling, associated with IgE- and mast cell-dependent inflammation in mouse skin. Lab Invest. 1998;78:497–505. [PubMed] [Google Scholar]
- 13.Dray A, Urban L, Dickenson A. Pharmacology of chronic pain. Trends Pharmacol Sci. 1994;15:190–197. doi: 10.1016/0165-6147(94)90147-3. [DOI] [PubMed] [Google Scholar]
- 14.Gramsch C, Meo T, Reithmuller G, Herz A. Binding characteristics of a monoclonal beta-endorphin antibody recognizing the N-terminus of opioid peptides. J Neurochem. 1983;40:1220–1226. doi: 10.1111/j.1471-4159.1983.tb13560.x. [DOI] [PubMed] [Google Scholar]
- 15.Iigo Y, Takashi T, Tamatani T, Miyasaka M, Higashida T, Yagita H, Okumura K, Tsukada W. ICAM-1-dependent pathway is critically involved in the pathogenesis of adjuvant arthritis in rats. J Immunol. 1991;147:4167–4171. [PubMed] [Google Scholar]
- 16.Issekutz AC. Adhesion molecules mediating neutrophil migration to arthritis in vivo and across endothelium and connective tissue barriers in vitro. Inflamm Res. 1998;47:S123–S132. doi: 10.1007/s000110050300. [DOI] [PubMed] [Google Scholar]
- 17.Kakimoto K, Nakamura T, Ishii K, Takashi T, Iigou H, Yagita H, Okumura K, Onoue K. The effect of anti-adhesion molecule antibody on the development of collagen-induced arthritis. Cell Immunol. 1992;142:326–337. doi: 10.1016/0008-8749(92)90294-y. [DOI] [PubMed] [Google Scholar]
- 18.Kavanaugh AF, Davis LS, Nichols LA, Norris SH, Rothlein R, Scharschmidt LA, Lipsky PE. Treatment of refractory rheumatoid arthritis with a monoclonal antibody to intercellular adhesion molecule 1. Arthritis Rheum. 1994;37:992–999. doi: 10.1002/art.1780370703. [DOI] [PubMed] [Google Scholar]
- 19.Machelska H, Stein C. Pain control by immune-derived opioids. Clin Exp Pharm Physiol. 2000;27:533–536. doi: 10.1046/j.1440-1681.2000.03287.x. [DOI] [PubMed] [Google Scholar]
- 20.Machelska H, Cabot PJ, Mousa SA, Zhang Q, Stein C. Pain control in inflammation governed by selectins. Nat Med. 1998;4:1425–1428. doi: 10.1038/4017. [DOI] [PubMed] [Google Scholar]
- 21.Machelska H, Mousa SA, Stein C. Pain and immune function. In: Ader R, Felten D, Cohnen N, editors. Psychoneuroimmunology, Ed 3, Vol 2. Academic; San Diego: 2001. pp. 111–121. [Google Scholar]
- 22.Marvin MR, Southall JC, Trokhan S, DeRosa C, Chabot J. Liver metastases are enhanced in homozygous deletionally mutant ICAM-1 or LFA-1 mice. J Surg Res. 1998;80:143–148. doi: 10.1006/jsre.1998.5322. [DOI] [PubMed] [Google Scholar]
- 23.McCafferty DM, Smith CW, Granger DN, Kubes P. Intestinal inflammation in adhesion molecule-deficient mice: an assessment of P-selectin alone and in combination with ICAM-1 or E-selectin. J Leukoc Biol. 1999;66:67–74. doi: 10.1002/jlb.66.1.67. [DOI] [PubMed] [Google Scholar]
- 24.Mogil JS, Grisel JE. Transgenic studies of pain. Pain. 1998;77:107–128. doi: 10.1016/S0304-3959(98)00093-1. [DOI] [PubMed] [Google Scholar]
- 25.Mousa SA, Machelska H, Schäfer M, Stein C. Co-expression of β-endorphin with adhesion molecules in a rat model of inflammatory pain. J Neuroimmunol. 2000;108:160–170. doi: 10.1016/s0165-5728(00)00284-8. [DOI] [PubMed] [Google Scholar]
- 26.Mousa SA, Zhang Q, Sitte N, Ji R-R, Stein C. β-Endorphin containing memory-cells and μ-opioid receptors undergo site-directed transport into peripheral inflamed tissue. J Neuroimmunol. 2001;115:71–78. doi: 10.1016/s0165-5728(01)00271-5. [DOI] [PubMed] [Google Scholar]
- 27.Mulligan MS, Miyasaka M, Tamatani T, Jones ML, Ward PA. Requirements for L-selectin in neutrophil-mediated lung injury in rats. J Immunol. 1994;152:832–840. [PubMed] [Google Scholar]
- 28.Nwariaku F, Sikes PJ, Lightfoot E, Mileski WJ. Inhibition of selectin- and integrin-mediated inflammatory response after burn injury. J Surg Res. 1996;63:355–358. doi: 10.1006/jsre.1996.0275. [DOI] [PubMed] [Google Scholar]
- 29.Petruzzelli L, Takami M, Humes D. Structure and function of cell adhesion molecules. Am J Med. 1999;106:467–476. doi: 10.1016/s0002-9343(99)00058-3. [DOI] [PubMed] [Google Scholar]
- 30.Przewlocki R, Hassan AHS, Lason W, Epplen C, Herz A, Stein C. Gene expression and localization of opioid peptides in immune cells of inflamed tissue: functional role in antinociception. Neuroscience. 1992;48:491–500. doi: 10.1016/0306-4522(92)90509-z. [DOI] [PubMed] [Google Scholar]
- 31.Qiu C, Sora I, Ren K, Uhl G, Dubner R. Enhanced delta-opioid receptor-mediated antinociception in mu-opioid receptor-deficient mice. Eur J Pharmacol. 2000;387:163–169. doi: 10.1016/s0014-2999(99)00813-4. [DOI] [PubMed] [Google Scholar]
- 32.Rittner HL, Brack A, Machelska H, Mousa SA, Bauer M, Schäfer M, Stein C. Opioid peptide expressing leukocytes: identification, recruitment and simultaneously increasing inhibition of inflammatory pain. Anesthesiology. 2001;95:500–508. doi: 10.1097/00000542-200108000-00036. [DOI] [PubMed] [Google Scholar]
- 33.Roques BP, Noble F, Dauge V, Fournie-Zaluski MC, Beaumont A. Neutral endopeptidase 24.11: structure, inhibition, and experimental and clinical pharmacology. Pharmacol Rev. 1993;45:87–146. [PubMed] [Google Scholar]
- 34.Salmi M, Rajala P, Jalkanen S. Homing of mucosal leukocytes to joints. Distinct endothelial ligands in synovium mediate leukocyte-subtype specific adhesion. J Clin Invest. 1997;99:2165–2172. doi: 10.1172/JCI119389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Samoilova EB, Horton JL, Chen Y. Experimental autoimmune encephalomyelitis in intercellular adhesion molecule-1-deficient mice. Cell Immunol. 1998;190:83–89. doi: 10.1006/cimm.1998.1395. [DOI] [PubMed] [Google Scholar]
- 36.Schäfer M, Carter L, Stein C. Interleukin-1 beta and corticotropin-releasing-factor inhibit pain by releasing opioids from immune cells in inflamed tissue. Proc Natl Acad Sci USA. 1994;91:4219–4223. doi: 10.1073/pnas.91.10.4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schäfer M, Mousa SA, Zhang Q, Carter L, Stein C. Expression of corticotropin-releasing factor in inflamed tissue is required for intrinsic peripheral opioid analgesia. Proc Natl Acad Sci USA. 1996;93:6096–6100. doi: 10.1073/pnas.93.12.6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shu XQ, Mendell LM. Neurotrophins and hyperalgesia. Proc Natl Acad Sci USA. 1999;96:7693–7696. doi: 10.1073/pnas.96.14.7693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sligh JE, Jr, Ballantyne CM, Rich SS, Hawkins HK, Smith CW, Bradley A, Beaudet AL. Inflammatory and immune responses are impaired in mice deficient in intercellular adhesion molecule 1. Proc Natl Acad Sci USA. 1993;90:8529–8533. doi: 10.1073/pnas.90.18.8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Springer TA. Adhesion receptors of the immune system. Nature. 1990;346:425–434. doi: 10.1038/346425a0. [DOI] [PubMed] [Google Scholar]
- 41.Stein C, Gramsch C, Herz A. Intrinsic mechanisms of antinociception in inflammation. Local opioid receptors and β-endorphin. J Neurosci. 1990a;10:1292–1298. doi: 10.1523/JNEUROSCI.10-04-01292.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stein C, Hassan AH, Przewlocki R, Gramsch C, Peter K, Herz A. Opioids from immunocytes interact with receptors on sensory nerves to inhibit nociception in inflammation. Proc Natl Acad Sci USA. 1990b;7:5935–5939. doi: 10.1073/pnas.87.15.5935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stein C, Hassan AH, Lehrberger K, Giefing J, Yassouridis A. Local analgesic effect of endogenous opioid peptides. Lancet. 1993;342:321–324. doi: 10.1016/0140-6736(93)91471-w. [DOI] [PubMed] [Google Scholar]
- 44.Szekanecz Z, Haines GK, Lin TR, Harlow LA, Goerdt S, Rayan G, Koch AE. Differential distribution of intercellular adhesion molecules (ICAM-1, ICAM-2, and ICAM-3) and the MS-1 antigen in normal and diseased human synovia. Their possible pathogenetic and clinical significance in rheumatoid arthritis. Arthritis Rheum. 1994;37:221–231. doi: 10.1002/art.1780370211. [DOI] [PubMed] [Google Scholar]
- 45.Tamatani T, Miyasaka M. Identification of monoclonal antibodies reactive with the rat homolog of ICAM-1, and evidence for a differential involvement of ICAM-1 in the adherence of resting versus activated lymphocytes to high endothelial cells. Int Immunol. 1990;2:165–171. doi: 10.1093/intimm/2.2.165. [DOI] [PubMed] [Google Scholar]
- 46.Taylor BM, Kolbasa KP, Chin JE, Richards IM, Fleming WE, Griffin RL, Fidler SF, Sun FF. Roles of adhesion molecules ICAM-1 and alpha4 integrin in antigen-induced changes in microvascular permeability associated with lung inflammation in sensitized brown Norway rats. Am J Respir Cell Mol Biol. 1997;17:757–766. doi: 10.1165/ajrcmb.17.6.2697. [DOI] [PubMed] [Google Scholar]
- 47.Veihelmann A, Harris AG, Krombach F, Schutze E, Refior HJ, Messmer K. In vivo assessment of synovial microcirculation and leukocyte-endothelial cell interaction in mouse antigen-induced arthritis. Microcirculation. 1999;6:281–290. [PubMed] [Google Scholar]
- 48.Vuorte J, Lindsberg PJ, Kaste M, Meri S, Jansson SE, Rothlein R, Repo H. Anti-ICAM-1 monoclonal antibody R6.5 (Enlimomab) promotes activation of neutrophils in whole blood. J Immunol. 1999;162:2353–2357. [PubMed] [Google Scholar]
- 49.Watkins LR, Maier SF. The pain of being sick: implications of immune-to-brain communication for understanding pain. Annu Rev Psychol. 2000;51:29–57. doi: 10.1146/annurev.psych.51.1.29. [DOI] [PubMed] [Google Scholar]
- 50.Willenborg DO, Simmons RD, Tamatani T, Miyasaka M. ICAM-1-dependent pathway is not critically involved in the inflammatory process of autoimmune encephalomyelitis or in cytokine-induced inflammation of the central nervous system. J Neuroimmunol. 1993;45:147–154. doi: 10.1016/0165-5728(93)90175-x. [DOI] [PubMed] [Google Scholar]