

Abstract
The first mutations of the GABAA receptor channel linked to familial epilepsy in humans were reported recently (Baulac et al., 2001; Wallace et al., 2001). Preliminary functional analysis of α1β2γ2 GABAA receptors expressed inXenopus oocytes suggested that the γ2 subunit R43Q mutation abolished current enhancement by the benzodiazepine, diazepam, and that the γ2 subunit K289M mutation decreased current amplitudes. We used single-channel recording and concentration jump techniques applied to outside out patches to evaluate the impact of these mutations on GABAA receptor channel function of the highly conserved rat ortholog subunits expressed in human embryonic kidney cells. When coexpressed with α1 and β3 subunits, no differences were observed between wild-type and mutant GABAA receptor current activation rates or rates or extent of desensitization during prolonged (400 msec) GABA application (1 mm). Although deactivation after brief (5 msec) or prolonged (400 msec) GABA application was unaltered by the R43Q mutation, deactivation (a correlate of IPSC duration) was accelerated for the K289M mutation. Faster deactivation was likely a consequence of altered gating, because single-channel openings had shorter mean duration. Interestingly, the R43Q mutation did not alter diazepam potentiation. It did, however, substantially decrease current amplitude, which was not caused by decreased single-channel conductance or open time, suggesting reduced surface expression of functional receptors. The two γ2 subunit mutations likely produce disinhibition and familial epilepsy by distinct mechanisms, suggesting that maintenance of neuronal inhibition depends not only on the peak amplitude of IPSCs, but also on their time course.
Keywords: GABAA receptor, mutation, epilepsy, concentration-jump, benzodiazepine, deactivation
GABAAreceptors mediate the majority of fast synaptic inhibition in the brain. The heteropentameric receptor complex is comprised of subunits drawn from at least seven families (α, β, γ, δ, ε, π, θ), although the majority of receptors are thought to be αβγ and αβδ isoforms (McKernan and Whiting, 1996). Subunit composition has been shown to influence the pharmacology, kinetics, and subcellular localization of GABAA receptors (Macdonald and Olsen, 1994; Sieghart, 1995). Pharmacological enhancement of GABAergic inhibition has been used in the treatment of several clinical disorders, including anxiety and epilepsy, and certain anesthetics mediate their CNS actions at least in part through enhancement of GABAA receptor function.
Two recent studies demonstrated for the first time a genetic linkage between familial epilepsy syndromes and mutations in the γ2 subunit of the GABAA receptor. Wallace et al. (2001)reported a missense mutation, R43Q (in the N terminal extracellular domain of the γ2 subunit), in affected individuals of a large family having both childhood absence epilepsy and febrile seizures. Recordings from Xenopus oocytes injected with α1β2γ2(R43Q) GABAA receptor subunits suggested no differences in GABA EC50, current amplitude, or apparent desensitization. However, the receptors were insensitive to functional modulation by the benzodiazepine diazepam. Although this raised the interesting possibility of an endogenous ligand at the benzodiazepine recognition site, that interpretation depends on the exclusion of any other functional consequences of the mutation. Baulac et al. (2001) reported that a family with an epilepsy disorder similar to generalized epilepsy with febrile seizures plus (GEFS+) had a K289M mutation in the γ2 subunit of the GABAA receptor. This residue is located in the short extracellular loop between transmembrane domains TM2 and TM3, a region that has been implicated in the gating of ligand-gated ion channels (Campos-Caro et al., 1996; Lynch et al., 1997). Electrophysiological recordings from oocytes expressing α1β2γ2(K289M) GABAA receptors revealed smaller amplitude currents relative to wild-type receptor current amplitudes.
Although the pathophysiology of epilepsy is complex, and heritable epilepsies represent a small fraction of all patients with epilepsy, mutations such as those discovered by Baulac et al. (2001)and Wallace et al. (2001) may shed light on the importance of specific aspects of GABAA receptor function in the maintenance of central synaptic inhibition. Because γ2 subunit-containing receptors are primarily localized to synapses and inhibitory synaptic transmission occurs in the millisecond time domain, it is necessary to consider the possible impact of these mutations on channel properties relevant to the time scale of synaptic transmission. Thus, we used an ultrafast perfusion technique to ensure resolution of rapid kinetic properties such as activation, desensitization, and deactivation. Finally, we studied both mutations at the single-channel level to determine the basis of our macroscopic observations.
MATERIALS AND METHODS
Expression of recombinant GABAAreceptors. The cDNAs encoding rat α1, β3, and γ2L, GABAA receptor subunit subtypes were individually subcloned into the plasmid expression vector pCMVNeo. Initially we performed experiments with these rat subunits because human subunits were unavailable to us. The rat γ2L subunit mature peptide differs from the reported human γ2L subunit sequence (Pritchett et al., 1989; accession NM 000816) by two residues (of 428): the rat sequence contains a T at position 81 (M in human) and a T at position 142 (S in human). After our observation of robust diazepam enhancement of the γ2L(R43Q) mutation (which contrasted Wallace et al., 2001) (Fig.1), we obtained the cDNAs encoding human α1, β3, and γ2S subunits in the expression vector pCIneo, provided by Dr. Erwin Sigel (University of Bern, Switzerland), to verify that species differences in subunit sequence did not account for our results. Interestingly, the human γ2S subunit contained the rat amino acid sequence at these two sites, resulting in 100% amino acid sequence identity. This γ2 sequence has also been reported by the NCBI Annotation Project (accession XM 003986.2). Nevertheless, we mutated the two residues to match the original published human γ2 sequence to evaluate diazepam sensitivity of the γ2L(R43Q) mutation in both contexts (Fig. 1D). Twenty-four bases of the cytoplasmic loop were introduced into the human γ2S subunit to generate the human γ2L splice form used in these experiments. Point mutants were generated using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA). All subunits and mutations were confirmed by sequencing. Oligonucleotide primers were synthesized by the University of Michigan DNA synthesis core facility (Ann Arbor, MI). Human embryonic kidney cells (HEK293T; a gift from P. Connely, COR Therapeutics, San Francisco, CA) were maintained in DMEM, supplemented with 10% fetal bovine serum, at 37°C in 5% CO2 and 95% air. For expression of both rat and human isoforms, cells were transfected with 2–4 μg of each subunit plasmid along with 1–2 μg of pHOOK (Invitrogen, Carlsbad, CA) (in a ratio of 1:1:1:0.5) for immunomagnetic bead separation (Greenfield et al., 1997), using a modified calcium phosphate coprecipitation technique, as previously described (Angelotti et al., 1993). The next day, cells were replated on collagen-treated culture dishes, and recordings were made at room temperature 18–30 hr later.
Fig. 1.
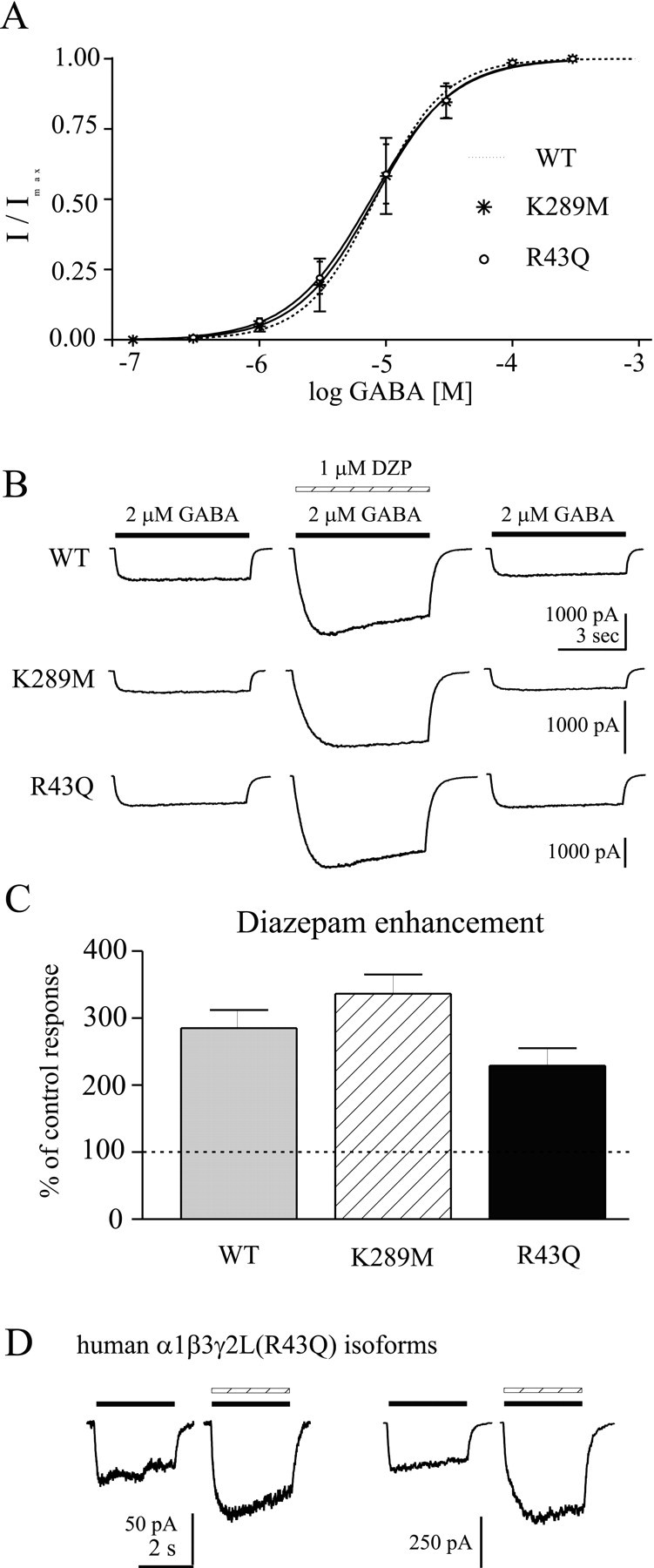
Sensitivity to GABA and diazepam.A, The EC50 for GABA was determined by whole-cell responses to increasing GABA concentration, normalized to the peak current amplitude in each cell. Cells were voltage clamped at –10 to –50 mV, and data were obtained from three cells for each mutation. The dotted line is the concentration–response of wild-type α1β3γ2L GABAAreceptors (Bianchi et al., 2001). B, Diazepam sensitivity was determined by coapplication of 1 μmdiazepam with 2 μm GABA (∼EC20) to whole cells expressing α1β3γ2L (top), α1β3γ2L(K289M), or α1β3γ2L(R43Q) GABAAreceptors. C, Diazepam enhancement is shown as the percentage of control responses (averagepeak current before and after diazepam coapplication). Neither mutation significantly affected modulation of GABA-evoked currents by diazepam. D, Representative currents are shown for two human α1β3γ2L(R43Q) GABAA receptor isoforms. The left pair of traces indicated the response of receptors containing a γ2L subunit with amino acid sequence corresponding to the original published γ2L sequence, whereas theright pair of traces indicated the response of receptors containing a γ2L subunit with amino acid sequence corresponding to that of the rat γ2L subunit (see Materials and Methods). For both pairs of traces, the left current was evoked with 10 μmGABA (solid bar), and the right current was evoked with coapplied 1 μm diazepam (hatched bar) in the same cell. Similar results were obtained in five cells.
Electrophysiology. Patch-clamp recordings were performed on membrane patches excised from transfected fibroblasts bathed in an external solution consisting of (in mm): NaCl 142; KCl 8; MgCl2 6; CaCl21; HEPES 10; glucose 10, pH 7.4, 325 mOsm. Electrodes were pulled from thick-walled borosilicate glass (World Precision Instruments, Pittsburgh, PA) with a Flaming Brown electrode puller (Sutter Instruments, San Rafael, CA), fire-polished to resistances of 4–12 MΩ when filled with an internal solution consisting of (in mm): KCl 153; MgCl2 1; MgATP 2; HEPES 10; EGTA 5, pH 7.3, 300 mOsm. Electrodes used for single-channel recording were coated with polystyrene Q-dope (GC Electronics). This combination of internal and external solutions produced a chloride equilibrium potential of ∼0 mV. All outside-out patches used for macroscopic kinetic experiments were voltage-clamped at −50 mV using an Axon Instruments (Foster City, CA) 200A amplifier. All single-channel recordings were performed at −75 mV. For macroscopic experiments, drugs were applied (via gravity) to whole cells and excised outside-out patches using a rapid perfusion system consisting of pulled multibarrel square glass connected to a Warner Instrument Corp. (Hamden, CT) Perfusion Fast-Step. The glass was pulled to a final size of ∼200 μm. The solution exchange time was determined after each excised patch recording by stepping a dilute external solution across the open electrode tip to measure a liquid junction current. The 10–90% rise times for solution exchange were consistently <400 μsec. For whole-cell experiments, lower resistance thin-walled borosilicate electrodes were used (0.8–1.5 MΩ), and whole cells were generally voltage clamped at −20 mV, although more hyperpolarized potentials were used to increase the chloride ion driving force when small currents were observed. Exchange time around whole cells was probably slower than the open tip rise times. For single-channel recordings, GABA (1 mm) was applied directly to the bath solution and was present for at least 2 min before recordings were started. The sampling intervals of currents used for figure traces were reduced by decimation (averaging of adjacent points) and/or additional filtering for display purposes only.
Analysis. Macroscopic currents were low-pass filtered at 2–5 kHz, digitized at 10 kHz, and analyzed using the pClamp8 software suite (Axon Instruments). The desensitization and deactivation time courses of GABAA receptor currents were fit using the Levenberg–Marquardt least squares method with one or two component exponential functions of the form Σane(-t/τn), where n is the best number of exponential components,a is the relative amplitude of the component, tis time, and τ is the time constant. Three component fits were not considered for the 400 msec duration pulses used in this study. A second component was accepted only if a significant improvement of the fit occurred, as determined by an F-test performed automatically by Clampfit 8.1 analysis software on the sum of squared residuals. In every patch, the second exponential function improved the desensitization fit. Note that faster phases of apparent desensitization could not be resolved if they occurred with time constants below the solution exchange time of our system (∼400 μsec) or if they were faster than the current rise time (< 1 msec). For comparison of deactivation time courses a weighted summation of the fast and slow decay components was used, in which the time constants of the fast and slow phases were weighted by their relative amplitudes. For paired pulse inhibition analysis, the relative amplitude of the second peak was measured for comparison with the first peak of each sweep pair. When the second pulse occurred before the current of the first pulse had relaxed completely (i.e., for brief interpulse intervals), the current remaining from the first pulse was subtracted from the absolute peak of the second amplitude, thus generating a relative amplitude measurement.
Single-channel data were digitized at 20 kHz, filtered at 2 kHz via the internal Axon 200A amplifier filter, and stored on VHS videotape for off-line analysis. Most patches used for single-channel analysis contained more than one channel, based on the presence of overlapped openings. Stretches of single-channel activity that contained few or no overlapped openings were analyzed using the 50% threshold detection method of Fetchan 6.0 (pClamp 8.1). Overlapped openings and bursts were not included in the analysis. Events with durations <150 μsec (∼1.5 times the estimated system dead time) were shown in the histograms but not considered in the fitting routine. Logarithmic binning was used as previously described (Haas and Macdonald, 1999), and fitted with a maximum likelihood routine by the Interval5 software (Dr. Barry Pallotta, University of North Carolina). The number of exponential functions required to fit the distributions was incremented until additional exponentials failed to significantly improve the fit, as determined automatically by the software (analysis of residuals). Mean open times were calculated from the open duration fitted parameters of a histogram containing the combined results from several patches.
Numerical data were expressed as mean ± SEM. Statistical significance, using Student's unpaired t test (with a Welch's correction for unequal variances where appropriate) was taken as p < 0.05.
RESULTS
The biophysical properties of the rat α1β3γ2L GABAA receptor isoform have been extensively studied (Fisher and Macdonald, 1997; Haas and Macdonald, 1999). The well characterized transient and steady-state kinetics offered an appropriate context for evaluation of the K289M and R43Q mutations in the γ2 subunit recently linked to human epilepsy. Initial characterization was performed at the whole-cell level. The EC50 for GABA was determined, using relatively rapid perfusion (see Materials and Methods), by applying increasing concentrations of GABA to whole cells voltage clamped at −10 to −50 mV. As shown in Figure 1A, neither mutation changed the GABA EC50, consistent with Baulac et al. (2001) and Wallace et al. (2001). Modulation of GABAA receptors by the benzodiazepine diazepam depends on the presence of a γ subunit and the appropriate α subunit subtype (Sigel and Buhr, 1997). We tested the effects of both mutations on benzodiazepine sensitivity, as indicated by the diazepam modulation of currents elicited by a low concentration of GABA (∼EC20). Diazepam (1 μm) significantly and reversibly enhanced α1β3γ2L(K289M) and α1β3γ2L(R43Q) GABAA receptor currents, and the extent of enhancement was not different from that observed for wild-type α1β3γ2L GABAA receptor currents (Fig.1B,C). We also examined the diazepam sensitivity of two human isoforms of the γ2L(R43Q) subunit (see Materials and Methods) expressed with human α1 and human β3 subunits. Diazepam significantly potentiated these human isoforms, despite the R43Q mutation (Fig. 1D). Although we used the β3 subunit, and the previous studies used the β2 subunit, this should not affect our results because diazepam sensitivity is known to depend on the α and γ subunits, but not the β subunit subtype. This was consistent with the results of Baulac et al. (2001), who found diazepam enhancement of receptors containing the γ2(K289M) mutation, but contrasted with the results of Wallace et al. (2001), in which α1β2γ2(R43Q) GABAA receptors were shown to be insensitive to diazepam (1 μm) when expressed in Xenopus oocytes. In that study, the possibility of endogenous benzodiazepine ligand or ligands had been raised as a result of that finding.
One major limitation of standard “whole-cell” electrophysiology is the failure to accurately resolve rapid kinetic processes because of the technical difficulty in perfusing whole cells sufficiently fast to avoid “blurring” rapid channel responses to agonist. To overcome this limitation and allow investigation of the kinetic behavior of GABAA receptors containing the reported mutations, we used the concentration jump technique applied to outside out patches excised from transfected human fibroblasts (Fig.2A). This permitted analysis of receptor properties most relevant to the time scale of synaptic currents, including activation, desensitization, and deactivation. We have previously shown that 400 msec pulses of GABA (1 mm) elicit rapidly activating currents from α1β3γ2L receptors that desensitize with two phases and deactivate slowly (Haas and Macdonald, 1999; Bianchi et al., 2001). This duration of application is appropriate for resolving the fast phase of desensitization (τ ∼10 msec) thought to play a critical role in shaping GABAA receptor current deactivation and by extension, IPSC duration (Jones and Westbrook, 1995;Dominguez-Perrot et al., 1997; Haas and Macdonald, 1999). The pattern of current desensitization during the 400 msec GABA application was well described by the sum of two exponential functions in every patch (see Materials and Methods). The rates (Fig.2B1,C1) and relative contributions (Fig.2B2,C2) of the fast and slow phases of macroscopic desensitization were unaffected by the mutations. Neither mutation significantly altered the macroscopic activation rate, as indicated by the 10–90% current rise time (Fig. 2D). The mean peak amplitude of α1β3γ2L(R43Q) currents was significantly smaller (p < 0.01) than either α1β3γ2L or α1β3γ2L(K289M) currents (which were not different from each other) (Fig. 2E). Also, the α1β3γ2L(K289M) currents deactivated significantly faster (p < 0.001) than either α1β3γ2L or α1β3γ2L(R43Q) currents, which were not different from each other (Fig. 2F).
Fig. 2.

Macroscopic kinetic properties. A, Representative current traces obtained from wild-type or mutated receptors during 400 msec jumps into 1 mm GABA. Time scale of top trace applies to all three traces.B1–C2, Neither the fast (B1) nor the slow (C1) time constant of desensitization, nor their relative contributions (B2,C2) were significantly altered by the mutations.D, Current activation rate, as indicated by the 10–90% rise time of the current, was not significantly altered by the mutations. E, Peak current amplitudes were significantly smaller for α1β3γ2L(R43Q) GABAA receptors. *p < 0.01. F, Current deactivation after removal of GABA was significantly faster for α1β3γ2L(K289M) GABAA receptors. *p < 0.001. Data were obtained from 8–13 patches.
To more closely approximate the conditions of synaptic currents, we applied GABA (1 mm) to patches for short durations (2–5 msec) (Fig. 3A1–A3). The traces (dark lines) are shown at the same vertical scale to demonstrate the dramatic effect of the R43Q mutation on peak current. The gray trace in Figure 3A3 is a 10-fold vertical expansion shown for comparison of the deactivation time course. Such applications may reasonably replicate the time course of IPSCs (Jones and Westbrook, 1995; Haas and Macdonald, 1999), and provide information about GABAA receptor current deactivation under nonequilibrium conditions. Deactivation currents relaxed with a biphasic time course for all three isoforms. α1β3γ2L(K289M) currents deactivated significantly faster than α1β3γ2L currents (p < 0.05), whereas the deactivation rate was unaffected by the γ2L(R43Q) mutation (Fig. 3B). For comparison purposes, only the weighted deactivation rates are shown in Figure 3B.
Fig. 3.

Deactivation after brief GABA pulses.A, Representative currents illustrate deactivation rates of α1β3γ2L (A1), α1β3γ2L(K289M) (A2), and α1β3γ2L(R43Q) (A3) GABAA receptors in response to brief (<5 msec) pulses of GABA (1 mm). Scale bars apply to all threesolid traces. The solid trace inA3 is expanded 10-fold (gray trace) for comparison of deactivation current time course.B, Weighted time constants of deactivation (see Materials and Methods) are shown for wild-type and mutated channels. Deactivation was significantly faster for α1β3γ2L(K289M) GABAA receptors (hatched bar). *p < 0.05. α1β3γ2L (R43Q) GABAAreceptor deactivation (solid bar) was not different than that of wild-type receptors (gray bar). Data were obtained from 9–13 patches for each isoform.
One consequence of GABAA receptor entry into relatively long-lasting desensitized states (even after brief exposure to GABA) is that subsequent exposure to GABA will elicit smaller current responses. This phenomenon is known as paired pulse inhibition, in which the smaller amplitude of the second response of paired GABA applications reflects a fraction of receptors that cannot be activated because they remain desensitized. As the interpulse interval is increased, channels recover from desensitization, and progressively less depression is observed in the peak current of the second GABA pulse. We investigated paired pulse inhibition using paired brief applications of 1 mm GABA separated by varying interpulse intervals (30–1000 msec). Prominent paired pulse inhibition was observed for α1β3γ2L and α1β3γ2L(R43Q) GABAA receptors (Fig.4A, top andbottom panels), and the degree of inhibition for each interval was not different between these two isoforms. Although paired pulse inhibition was observed for α1β3γ2L(K289M) GABAA receptor channels (Fig. 4A, middle trace), the degree of inhibition was smaller for each interpulse interval (p < 0.05) (Fig.4B). In other words, more current activation occurred (as indicated by greater current amplitude) in the second pulse of each pair compared with the same interpulse intervals for α1β3γ2L GABAA receptors. Single exponential fitting of the mean relative amplitude data revealed that α1β3γ2L and α1β3γ2L(R43Q) receptors recovered with similar time constants of 130 and 170 msec, respectively, whereas α1β3γ2L(K289M) receptors recovered with a significantly faster time constant of 57 msec (p < 0.05).
Fig. 4.
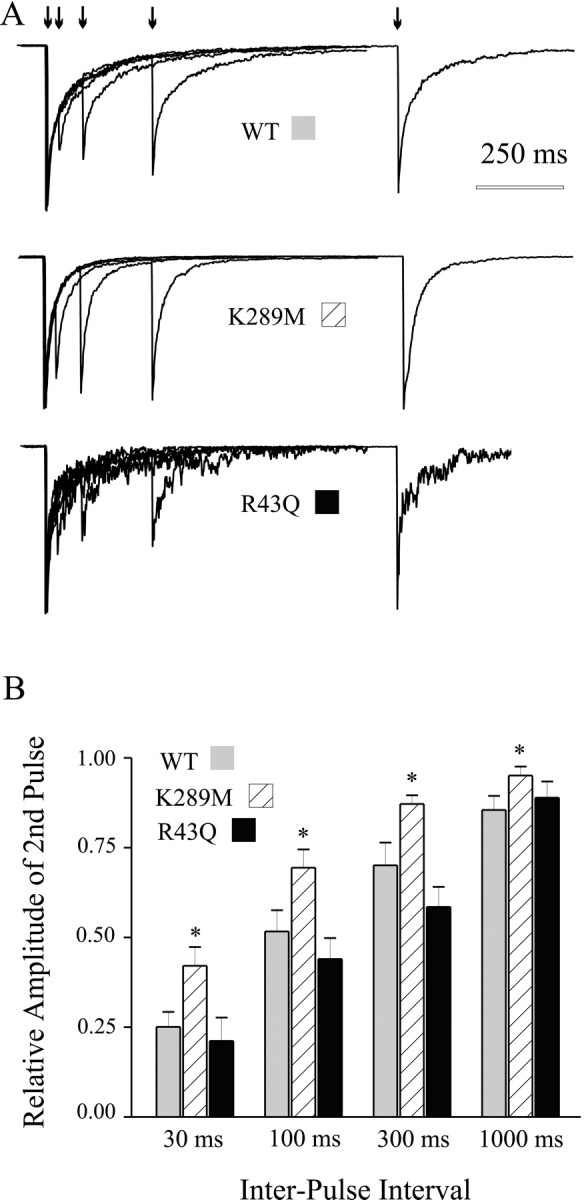
Paired pulse inhibition. Pairs of 5 msec GABA (1 mm) pulses were delivered to outside-out patches at interpulse intervals of 30, 100, 300, and 1000 msec. A, Representative currents from wild-type and mutated GABAAreceptors. B, Summary plot showing the relative amplitude of the second pulse of each pair, for each four interpulse intervals. Less inhibition was observed for α1β3γ2L(K289M) GABAA receptors for each interpulse interval.
Finally, we investigated the single-channel properties of α1β3γ2L(K289M) and α1β3γ2L(R43Q) GABAA receptors to determine whether changes in gating or conductance might explain the faster deactivation and smaller amplitude currents, respectively. Single-channel currents were recorded from outside-out patches during steady-state application of GABA (1 mm). Neither mutation appeared to change the single-channel conductance, because the majority of openings for each isoform was ∼2 pA at −75 mV (Fig. 5A), as previously reported for α1βxγ2 GABAAreceptors (Angelotti et al., 1993; Fisher and Macdonald, 1997; Haas and Macdonald, 1999). The R43Q mutation did not appear to substantially change the pattern of burst-like openings (three fitted open states; data not shown). The mean open duration was slightly but significantly shorter than wild-type (1.3 msec compared with 2.0 msec) (see Materials and Methods). Interestingly, the open durations of α1β3γ2L(K289M) GABAA receptor channels were substantially shorter in duration than both α1β3γ2L and α1β3γ2L(R43Q) mean open times, although they still also tended to occur in bursts. The mean open duration was only 0.5 msec, fourfold shorter than that observed for α1β3γ2L. The open duration histogram is shown in Figure 5D to illustrate the predominance of the first, brief duration open state, and the almost complete absence of the longer, third open state characteristic of α1β3γ2L single-channel currents.
Fig. 5.
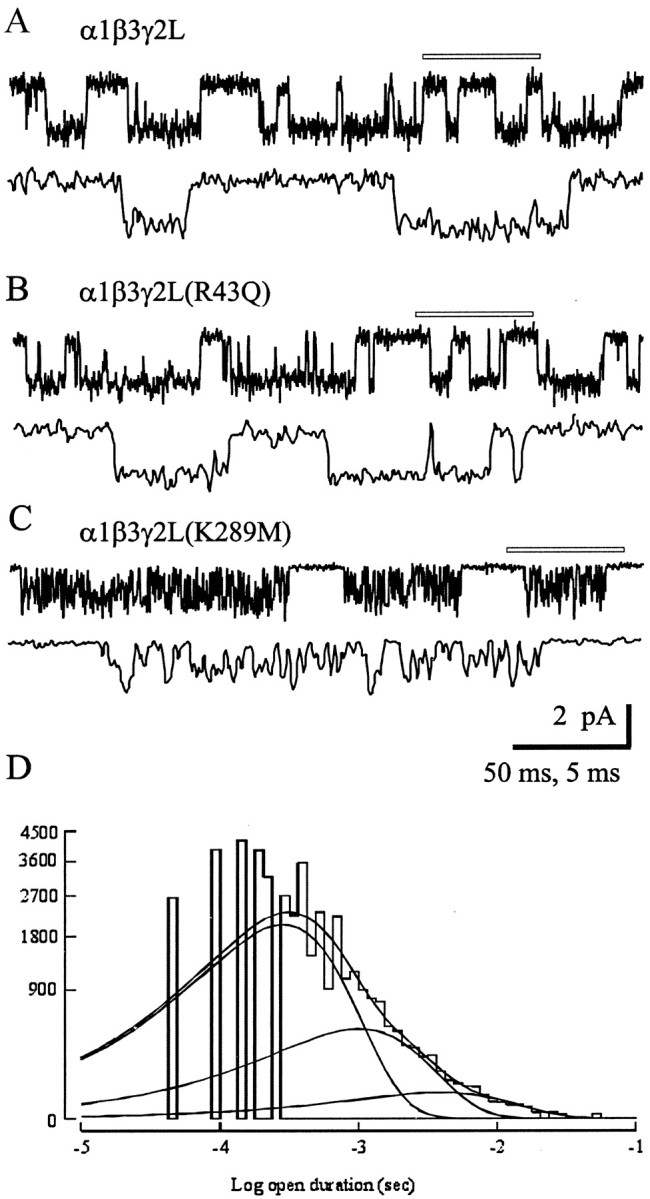
Single-channel analysis. Single-channel records obtained in patches held at −75 mV in the presence of 1 mmGABA from α1β3γ2L (A), α1β3γ2L(R43Q) (B), and α1β3γ2L(K289M) (C) GABAA receptors. A portion of thetop trace in each pair (indicated by the open bar) is expanded below that trace. Calibration bars apply to all three panels. Openings are downward. Similar results were observed from three α1β3γ2L, five α1β3γ2L(R43Q), and seven α1β3γ2L(K289M) patches. D, Open duration histogram for α1β3γ2L(K289M) single channels. The distribution was best described by the sum of three exponential functions, with each exponential fit shown as a smooth curve. Although three functions were required, the relative contribution of the shortest open state (leftmost curve) was highest, indicating that most openings were brief in duration. The time constants were 0.33, 1.17, and 4.54 msec with relative areas 0.81, 0.17, and 0.02, respectively. Data were pooled from seven α1β3γ2L(K289M) patches.
DISCUSSION
The goal of this study was to determine the functional consequences of two recently described mutations of the GABAA receptor γ2 subunit linked to human epilepsy. Specifically, the concentration jump technique was used to explore the rapid kinetic properties thought to be most relevant for the time scale of synaptic transmission. Our results differ from the initial reports of functional deficits associated with these mutations. First, we found unaltered diazepam potentiation for GABAA receptors containing the γ2L(R43Q) mutated subunit. Wallace et al. (2001) suggested the possible existence of endogenous benzodiazepines based on the diazepam insensitivity conferred by this mutation in their in vitro assay. Second, we found no difference in the amplitudes of currents obtained from GABAA receptors containing the γ2L(K289M) mutated subunit, in contrast with the systematically smaller currents observed in oocytes by Baulac et al. (2001). We did, however, observe functional consequences of these mutations that would be consistent with decreased neuronal inhibition: faster deactivation rates for α1β3γ2L(K289M) GABAA receptors (which predicts shorter duration IPSCs) and smaller currents for α1β3γ2L(R43Q) GABAA receptors (which predicts smaller amplitude IPSCs).
The basis for the discrepancies between our results and those ofWallace et al. (2001) and Baulac et al. (2001) remain unclear. The functional differences may be associated with the expression system (oocytes versus a human cell line). For example, GABAA receptor assembly in mammalian cells has been shown to be less promiscuous than in Xenopus oocytes (Sieghart, 1995). Additionally, both previous studies used 10-fold overexpression of the γ2 subunit to drive assembly of ternary receptors; it is not known how such overexpression might alter receptor assembly or function. We achieved consistent expression of ternary GABAA receptor channels with equimolar cDNA ratios, based on various functional criteria such as modulator pharmacology, single-channel conductance, and rapid kinetic properties (Angelotti et al., 1993; Fisher and Macdonald, 1997; Haas and Macdonald, 1999). Also, we tested two human γ2L(R43Q) subunits (see Materials and Methods) expressed with human α1 and human β3 subunits and observed robust diazepam enhancement, indicating that species differences in amino acid sequence could not account for the contrasting benzodiazepine sensitivity (Wallace et al., 2001).
GABAA receptor deactivation is the major determinant of IPSC duration and is influenced by transitions among open, closed, and desensitized states, as well as GABA unbinding steps. Although GABA unbinding is the terminating event for current deactivation, open, pre-open and desensitized states detain GABA on the receptor, thereby prolonging the IPSC time course (Jones and Westbrook, 1995; Chang and Weiss, 1999; Bianchi and Macdonald, 2001a). Thus, there are several mechanisms by which deactivation might be accelerated. Because there was no change in apparent GABA EC50, it is unlikely that decreased microscopic affinity alone could account for the faster deactivation of α1β3γ2L(K289M) currents. Macroscopic desensitization was unaltered by the mutation, arguing against changes in desensitized states that contribute to the prolonged deactivation rate. Single-channel gating efficacy (opening and closing rates) can also affect deactivation (Bianchi and Macdonald, 2001b). We observed significantly decreased mean open times for α1β3γ2L(K289M) GABAA receptor single channels. This finding is consistent with the accelerated deactivation, because channels would spend less time in the open state for any given opening before eventually unbinding GABA. Channels that were open at the end of the GABA pulse would close faster than wild-type channels, and although “late” re-openings (that normally underlie slow deactivation) would still occur, they would contribute less current to the deactivation time course, resulting in an overall accelerated current relaxation.
This phenotype of fast deactivation despite unaltered desensitization time course is similar to that observed for receptors containing mutations in TM1 of the γ2L subunit (Bianchi et al., 2001). In that study, we concluded that desensitization and deactivation had been “uncoupled” by the mutation, and we raised two possible explanations. It might be that TM1 was a critical site for structural interactions between conformations of TM2 (related to the presumed channel gate) and GABA binding sites in the N terminus. An alternative is that a decrease in gating efficacy would accelerate deactivation independent of desensitization. The K-M mutation appears to significantly decrease the mean open time of single-channel events, consistent with the latter possibility. Interestingly, we have found shorter mean open times (our unpublished data) with the TM1 mutation that uncoupled desensitization and deactivation. These findings suggest that gating and desensitization can be modified independently and that gating efficacy is also a critical determinant of deactivation rate and thus IPSC time course. Consistent with these suggestions, we recently presented evidence that a mutation that increased gating efficacy prolonged deactivation without altering desensitized states (Bianchi and Macdonald, 2001b).
Interestingly, although shorter duration of individual synaptic events would be a predicted outcome of the K289M mutation, less paired pulse inhibition would suggest a decrease in the temporal constraints normally imposed on repetitive firing at GABAergic synapses. Whereas the shorter deactivation predicts less inhibition of individual events, the potential for increased inhibition allowed by less effective paired pulse inhibition might partially compensate for the decreased inhibition of individual events predicted by the faster deactivation. How these two predictions may interact in vivo is not yet known, but may provide insight into the mechanism of epileptogenesis. It remains unclear whether the decreased paired pulse inhibition is a result of faster recovery from desensitization or an indirect consequence of the decreased gating efficacy.
Our data are consistent with previous studies that suggested the importance of the TM2–TM3 loop in the gating of ligand-gated ion channels (Campos-Caro et al., 1996; Lynch et al., 1997; Sigel et al., 1999). In some cases, mutations in this domain result in clinical phenotypes. For example, slow channel myasthenic syndrome can be caused by TM2–TM3 loop mutation in nicotinic acetylcholine receptors (Gomez et al., 1997). Also, glycine receptor mutations in this loop region have been shown to result in hereditary hyperekplexia (for review, see Lewis and Schofield, 1999).
In any transfection paradigm, there is inherent variability in protein expression of exogenous cDNAs among individual cells. However, interfering with some aspect of expression or assembly can result in a systematic decrease in current amplitude. Note that current amplitude, even under conditions of rapid agonist application, may be affected by kinetic properties of the channels as well as the level of surface expression (for example, see Haas and Macdonald, 1999). However, α1β3γ2L(R43Q) GABAA receptor currents did not differ significantly in activation, desensitization, or deactivation rates and did not have decreased single-channel conductance or significantly altered gating kinetics. Although the mean open time was shorter compared with wild-type, this difference could not quantitatively account for the smaller currents. Therefore these observations suggest that receptor expression or assembly has been compromised by the mutation. It is known that N-terminal sequences are important for assembly of GABAA receptors (Klausberger et al., 2000). Although the sequence around R43 was not specifically identified as important for intersubunit contacts, the absolute conservation of the R43 residue across subunits and species is more consistent with a role in assembly (which is required of all subunits) than with benzodiazepine modulation (which is highly subunit- and subtype-selective). Indeed, several human diseases have been linked to mutations in conserved arginines (among other residues) that result in altered protein folding and subsequent degradation (Bross et al., 1999). Also, a recent study showed that mutation of a conserved basic arg-lys sequence in the intracellular domain of nicotinic acetylcholine receptors altered intracellular trafficking mechanisms (Keller et al., 2001). Further studies are necessary to determine whether this mutation alters protein folding or processing.
How these mutations actually contribute to complex seizure disorders remains unclear. The decreased inhibitory drive caused by faster current deactivation (K289M) or smaller current amplitudes (R43Q) are both consistent with imbalances of neuronal activity that would favor a hyperexcitable state. It should be noted, however, that native synaptic GABAA receptors likely contain a single γ2 subunit (Chang et al., 1996) resulting in a mixture of wild-type and mutant receptors in patients with either mutation. Although the portion of synaptic receptors containing the K289M mutation would accelerate IPSC time courses, alterations in assembly related to the R43Q mutant subunits might simply result in decreased peak currents. The extent of this effect would depend on whether compensatory increases in expression of the wild-type subunit occurred, which is not known. Further experiments involving expression of a mixture of wild-type and mutated subunits would clarify these potentially intermediate functional consequences. Also, this study did not address possible functional consequences that might depend on interaction of mutated γ2 subunits and other α or β subunits or manifestations of these mutations that depend on a neuronal milieu. Knock-in animal models may further elucidate the molecular mechanisms through which these mutations contribute to seizure disorders in vivo.
Footnotes
This work was supported National Institutes of Health Grant R01-NS33300 (R.L.M.) and National Institute on Drug Abuse training Fellowship T32-DA07281–03 (M.T.B.). We thank Gallia Levy for valuable discussions.
Correspondence should be addressed to Dr. Robert L. Macdonald, Department of Neurology, Vanderbilt University, 2100 Pierce Avenue, Nashville, TN 37212. E-mail: Robert.Macdonald@mcmail.vanderbilt.edu.
REFERENCES
- 1.Angelotti TP, Uhler MD, Macdonald RL. Assembly of GABAA receptor subunits: analysis of transient single-cell expression utilizing a fluorescent substrate/marker gene technique. J Neurosci. 1993;13:1418–1428. doi: 10.1523/JNEUROSCI.13-04-01418.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bianchi MT, Macdonald RL. Agonist trapping by GABAA receptor channels. J Neurosci. 2001a;21:9083–9091. doi: 10.1523/JNEUROSCI.21-23-09083.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bianchi MT, Macdonald RL. Mutation of the 9′ leucine in the GABAA receptor γ2L subunit produces an apparent decrease in desensitization by stabilizing open states without altering desensitized states. Neuropharmacology. 2001b;41:737–744. doi: 10.1016/s0028-3908(01)00132-0. [DOI] [PubMed] [Google Scholar]
- 4.Bianchi MT, Haas KF, Macdonald RL. Structural determinants of fast desensitization and desensitization-deactivation coupling in GABAA receptors. J Neurosci. 2001;21:1127–1136. doi: 10.1523/JNEUROSCI.21-04-01127.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baulac S, Huberfeld G, Gourfinkel-An I, Mitropoulou G, Beranger A, Prud'homee J-F, Baulac M, Brice A, Bruzzone R, LeGuern E. First genetic evidence of GABAA receptor dysfunction in epilepsy: a mutation in the γ2-subunit gene. Nat Genet. 2001;28:46–48. doi: 10.1038/ng0501-46. [DOI] [PubMed] [Google Scholar]
- 6.Bross P, Corydon TJ, Andresen BS, Jorgensen MM, Bolund L, Gregersen N. Protein misfolding and degradation in genetic diseases. Hum Mutat. 1999;14:186–198. doi: 10.1002/(SICI)1098-1004(1999)14:3<186::AID-HUMU2>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 7.Campos-Caro A, Sala S, Ballesta JJ, Vincente-Agullo F, Criado M, Sala F. A single residue in the M2–M3 loop is a major determinant of coupling between binding and gating in neuronal nicotinic receptors. Proc Natl Acad Sci USA. 1996;93:6118–6123. doi: 10.1073/pnas.93.12.6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang Y, Weiss DS. Channel opening locks agonist onto the GABAC receptor. Nat Neurosci. 1999;2:219–225. doi: 10.1038/6313. [DOI] [PubMed] [Google Scholar]
- 9.Chang Y, Wang R, Barot S, Weiss DS. Stoichiometry of a recombinant GABAA receptor. J Neurosci. 1996;16:5415–5424. doi: 10.1523/JNEUROSCI.16-17-05415.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dominguez-Perrot C, Feltz P, Poulter MO. Recombinant GABAA receptor desensitization: the role of the gamma2 subunit and its physiological significance. J Physiol (Lond) 1997;497:145–159. doi: 10.1113/jphysiol.1996.sp021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fisher JL, Macdonald RL. Single channel properties of recombinant GABAA receptors containing gamma 2 or delta subtypes expressed with alpha 1 and beta 3 subtypes in mouse L929 cells. J Physiol (Lond) 1997;505:283–297. doi: 10.1111/j.1469-7793.1997.283bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gomez CM, Maselli R, Gundeck JE, Chao M, Day JW, Tamamizu S, Lasalde JA, McNamee M, Wollman RL. Slow-channel transgenic mice: a model of postsynaptic organellar degeneration at the neuromuscular junction. J Neurosci. 1997;17:4170–4179. doi: 10.1523/JNEUROSCI.17-11-04170.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Greenfield LJ, Jr, Sun F, Neelands TR, Burgard EC, Donnelly JL, Macdonald RL. Expression of functional GABAA receptors in transfected L929 cells isolated by immunomagnetic bead separation. Neuropharmacology. 1997;36:63–73. doi: 10.1016/s0028-3908(96)00150-5. [DOI] [PubMed] [Google Scholar]
- 14.Haas KF, Macdonald RL. GABAA receptor subunit γ2 and δ subtypes confer unique kinetic properties on recombinant GABAA receptor currents in mouse fibroblasts. J Physiol (Lond) 1999;514(1):27–45. doi: 10.1111/j.1469-7793.1999.027af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jones MV, Westbrook GL. Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron. 1995;15:181–191. doi: 10.1016/0896-6273(95)90075-6. [DOI] [PubMed] [Google Scholar]
- 16.Keller SH, Lindstrom J, Ellisman M, Taylor P. Adjacent basic amino acid residues recognized by the COP I complex and ubiquitination govern endoplasmic reticulum to cell surface trafficking of the nicotinic acetylcholine receptor alpha-subunit. J Biol Chem. 2001;276:18384–18391. doi: 10.1074/jbc.M100691200. [DOI] [PubMed] [Google Scholar]
- 17.Klausberger T, Fuchs K, Mayer B, Ehya N, Sieghart W. GABAA receptor assembly: identification and structure of γ2 sequences forming the intersubunit contacts with α1 and β3 subunits. J Biol Chem. 2000;275:8921–8928. doi: 10.1074/jbc.275.12.8921. [DOI] [PubMed] [Google Scholar]
- 18.Lewis TM, Schofield PR. Structure-function relationships of the human glycine receptor: insights from hyperekplexia mutations. Ann NY Acad Sci. 1999;868:681–684. doi: 10.1111/j.1749-6632.1999.tb11345.x. [DOI] [PubMed] [Google Scholar]
- 19.Lynch JW, Rajendra S, Pierce KD, Handford CA, Barry PH, Schofield PR. Identification of intracellular and extracellular domains mediating signal transduction in the inhibitory glycine receptor chloride channel. EMBO J. 1997;16:110–120. doi: 10.1093/emboj/16.1.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Macdonald RL, Olsen RW. GABAA receptor channels. Annu Rev Neurosci. 1994;17:569–602. doi: 10.1146/annurev.ne.17.030194.003033. [DOI] [PubMed] [Google Scholar]
- 21.McKernan RM, Whiting PJ. Which GABAA-receptor subtypes really occur in the brain? Trends Neurosci. 1996;19:139–143. doi: 10.1016/s0166-2236(96)80023-3. [DOI] [PubMed] [Google Scholar]
- 22.Pritchett DB, Sontheimer H, Shivers BD, Ymer S, Kettenmann H, Schofield PR, Seeburg PH. Importance of a novel GABAA receptor subunit for benzodiazepine pharmacology. Nature. 1989;338:582–585. doi: 10.1038/338582a0. [DOI] [PubMed] [Google Scholar]
- 23.Sieghart W. Structure and pharmacology of γ-aminobutyric acidA receptor subtypes. Pharmacol Rev. 1995;47:181–234. [PubMed] [Google Scholar]
- 24.Sigel E, Buhr A. The benzodiazepine binding site of GABAA receptors. Trends Pharmacol Sci. 1997;18:425–429. doi: 10.1016/s0165-6147(97)01118-8. [DOI] [PubMed] [Google Scholar]
- 25.Sigel E, Buhr A, Baur R. Role of the conserved lysine residue in the middle of the predicted extracellular loop between M2 and M3 in the GABA(A) receptor. J Neurochem. 1999;73:1758–1764. doi: 10.1046/j.1471-4159.1999.731758.x. [DOI] [PubMed] [Google Scholar]
- 26.Wallace RH, Marini C, Petrou S, Harkin L, Bowser DN, Panchal RG, Williams DA, Sutherland GR, Mulley JC, Scheffer IE, Berkovic SF. Mutant GABAA receptor γ2 subunit in childhood absence epilepsy and febrile seizures. Nat Genet. 2001;28:49–52. doi: 10.1038/ng0501-49. [DOI] [PubMed] [Google Scholar]