

Abstract
Bcl-xL is a well characterized death-suppressing molecule of the Bcl-2 family. Bcl-xL is expressed in embryonic and adult neurons of the CNS and may play a critical role in preventing neuronal apoptosis that occurs during brain development or results from diverse pathologic stimuli, including cerebral ischemia. In this study, we used a novel approach to study the potential neuroprotective effect of Bcl-xL as a therapeutic agent in the murine model of focal ischemia/reperfusion. We created a Bcl-xL fusion protein, designated as PTD-HA-Bcl-xL, which contains the protein transduction domain (PTD) derived from the human immunodeficiency TAT protein. We demonstrated that this fusion protein is highly efficient in transducing into primary neurons in cultures and potently inhibited staurosporin-induced neuronal apoptosis. Furthermore, intraperitoneal injection of PTD-HA-Bcl-xL into mice resulted in robust protein transduction in neurons in various brain regions within 1–2 hr, and decreased cerebral infarction (up to ∼40%) in a dose-dependent manner, as determined at 3 d after 90 min of focal ischemia. PTD-HA-Bcl-xL was effective even when it was administered after the completion of ischemia (up to 45 min), and the protective effect was independent of the changes in cerebral blood flow or other physiological parameters. Finally, as shown by immunohistochemistry, Western blotting, and substrate–cleavage assays, PTD-HA-Bcl-xL attenuated ischemia-induced caspase-3 activation in ischemic neurons. These results thus confirm the neuroprotective effect of Bcl-xL against ischemic brain injury and provide the first evidence that the PTD can be used to efficiently transduce a biologically active neuroprotectant in experimental cerebral ischemia.
Keywords: cerebral ischemia; stroke; protein transduction; Bcl-2; caspase-3, cytochrome c
Emerging evidence has suggested that a significant portion of neuronal death after cerebral ischemia is attributable to an active type of cell death reminiscent of apoptosis, in which a number of apoptosis-regulatory gene products are activated (Lipton, 1999; Schulz et al., 1999; Sharp et al., 2000; Graham and Chen, 2001). Of the prime gene products involved in ischemic neuronal death, the Bcl-2 family proteins may play a critical role (Graham et al., 2000; Graham and Chen, 2001). Although the pro-apoptotic proteins such as Bax and Bid promote cell death after ischemia (Chen and Yin, 2000; Plesnila et al., 2001), the anti-apoptotic proteins Bcl-2 and Bcl-xL may enhance cell survival. Several studies demonstrated that enhanced expression of Bcl-2 or Bcl-xL in rodent brain was associated with markedly increased resistance to ischemic injury (Martinou et al., 1994; Linnik et al., 1995; Lawrence et al., 1996, 1997; Parsadanian et al., 1998; Wiessner et al., 1999). These observations strongly imply that conditional augmentation of certain Bcl-2-like death-suppressing proteins in the brain could be a clinically relevant strategy to ameliorate ischemic brain injury.
Bcl-xL is a well characterized anti-apoptotic member of the Bcl-2 family, which is expressed in embryonic and adult neurons of the CNS and plays an essential role in preventing neuronal cell death (Gonzalez-Garcia et al., 1995; Motoyama et al., 1995; Blomer et al., 1998; Parsadanian et al., 1998). In a manner similar to that of Bcl-2, overexpression of Bcl-xL potently suppresses apoptotic cell death induced by diverse stimuli in neurons and many other cell types (Frankowski et al., 1995; Gonzalez-Garcia et al., 1995; Kharbanda et al., 1997; Sastry and Rao, 2000; Shinoura et al., 2000) and, to a lesser extent, suppresses hypoxia-induced necrosis (Tsujimoto et al., 1997). Bcl-xL is an integral membrane protein localized primarily in the mitochondrial membrane and the nuclear envelope (Tsujimoto and Shimizu, 2000), and it suppresses cell death presumably by preventing the release of apoptogenic factors from the mitochondria (Kluck et al., 1997; Yang et al., 1997; Susin et al., 1999) and by directly interacting with caspases (Hu et al., 1998; Pan et al., 1998). The protein expression pattern of Bcl-xL has been studied previously in rodent models of cerebral ischemia, and it was found that the expression of Bcl-xL was dramatically decreased in dying ischemic neurons but was retained in surviving neurons (Gillardon et al., 1996;Isenmann et al., 1998; Kitagawa et al., 1998). Thus, Bcl-xL appears to be a robust endogenous neuronal survival factor and an attractive target for molecular therapeutic intervention in cerebral ischemia.
In this study, we generated a biologically active Bcl-xL fusion protein containing the 11 aa protein transduction domain (PTD) derived from the human immunodeficiency virus TAT protein (Nagahara et al., 1998). Taking advantage of the powerful capability of the PTD fusion proteins to transduce across the blood–brain barrier (Schwarze et al., 1999), we studied the neuroprotective effect of Bcl-xL as a potential therapeutic agent in a murine model of transient focal cerebral ischemia.
MATERIALS AND METHODS
Generation of Bcl-xL fusion protein containing the PTD
The cDNA encoding the entire reading frame of rat Bcl-xL was isolated using PCR from a rat brain cDNA library (Chen et al., 1998). PCR was performed using the primers 5′-ATGCGGCCGCGTCACTTCCGACTGAAGAGTGAG-3′ (sense) and 5′-ACGATATCATGTCTCAGAGCAACCGGGAGCTG-3′ (antisense). The products generated by PCR were subcloned into the pSPORT1 vector (Invitrogen, San Diego, CA), amplified, and purified, and the sequence was verified by sequencing analysis (University of Pittsburgh Service Core Facility).
The ET-30 (a+) expression plasmid was reconstructed according to the cloning sites provided by the manufacturer (Novagen, Madison, WI) to allow the generation of hemagglutinin (HA)-tagged Bcl-xL fusion protein that contains the PTD sequence. The final cDNA (which replaced the enterokinase cloning site in the ET-30 plasmid) encodes peptide sequences in the following order: his-6 tag (MHHHHHH), thrombin cleavage site (SGLVPRGS), PTD (YGRKKRRQRRR), HA tag (YPYDVPDVA), and Bcl-xL (233 aa), and the resulting vector is designated as pET-PTD-HA-Bcl-xL. Two control plasmids were also constructed: pET-PTD-HA-green fluorescent protein (GFP), in which α-fetoprotein replaced Bcl-xL, and pET-HA-Bcl-xL, which contained the same sequences as pET-PTD-HA-Bcl-xL but without the PTD sequence.
To produce the fusion proteins, the plasmid was transformed intoEscherichia coli BL21 cells and protein expression was induced using 0.5 mmisopropylthiogalactoside at 37°C for 4 hr. The fusion proteins were purified using an Ni-NTA superflow agarose column (Qiagen, Hilden, Germany) according to the manufacturer's instructions. The resulting fusion proteins were dialyzed twice against PBS using the 10K Dialysis Cassette (Pierce, Rockford, IL) and then digested by thrombin for 16 hr at room temperature to cleave the his-6 tag. This was followed by secondary purification with the Ni-NTA superflow agarose column to remove the his-6 tag and nonspecific binding proteins. The purified proteins were verified by Coomassie blue staining and Western blot analysis and were then stored in 10% glycerol/PBS at −80°C until use.
Primary cortical neuronal culture and induction of apoptosis
Primary cultures of cortical neurons were prepared from 16- to 17-d-old Sprague Dawley rat embryos as described previously (Cao et al., 2001b). Experiments were conducted at 14 d in vitro, when cultures consisted primarily of neurons (∼95%) as determined using cell phenotype-specific immunocytochemistry (Cao et al., 2001b).
Apoptosis was induced in cultured neurons by incubating the cells at the indicated concentrations with staurosporin (STS) (Biomol, Plymouth Meeting, PA). Apoptosis was evaluated 24 hr after STS incubation using propidium iodide (PI) nuclear staining (Chen et al., 2000). The percentage of cells showing apoptotic changes (chromatin condensation and/or fragmentation) was quantified by counting ≥3000 cells under each experimental condition (three randomly selected fields per well, four to six wells per condition per experiment, and three independent experiments). Cell viability was also measured in selective experiments using the LIVE/DEAD viability/cytotoxicity kit (Molecular Probes, Eugene, OR) according to the manufacturer's instructions.
Murine model of transient focal ischemia
Animal surgery. Focal cerebral ischemia was produced by intraluminal occlusion of the left middle cerebral artery (MCA) with a nylon monofilament suture (Yang et al., 1994; Kondo et al., 1997). Male 2- to 3-month-old C57BL/6 mice (25–30 gm each; The Jackson Laboratory, Bar Harbor, ME) were anesthetized with 1.5% isoflurane in a 30% O2/70% N2O mixture under spontaneous breathing. The rectal temperature was controlled at 37.0 ± 0.5°C during surgery and MCA occlusion via a temperature-regulated heating pad. Mean arterial blood pressure was monitored during MCA occlusion through a tail cuff, and arterial blood gas was analyzed at 15 min after the onset of ischemia. The animals underwent MCA occlusion for 90 min and then reperfusion for ≤72 hr. After recovering from anesthesia, the animals were maintained in an air-conditioned room at 20°C.
Measurement of cerebral blood flow. Changes in regional cerebral blood flow before, during, and after MCA occlusion were evaluated in animals using laser–Doppler flowmetry (Yang et al., 1994).
Neurological deficits. At 24 hr after ischemia, neurological examination was performed in surviving animals by an observer who was blinded to the experimental conditions. Neurological deficits were scored on the 0–5 scale (Murakami et al., 1998): 0, no neurological deficit; 1, failure to fully extend the right forepaw; 2, circling to the right; 3, falling to the right; 4, unable to walk spontaneously; and 5, dead.
Determination of infarct volume. At 72 hr after MCA occlusion, brains were removed and the forebrain was sliced into eight coronal sections (1 mm thick). Sections were stained with 2,3,5-triphenyltetrazolium (3%). Infarct volume was determined using the microcomputer imaging device image analysis system (Imaging Research Inc., St. Catharine's, Ontario, Canada) as described previously (Chen et al., 1996). Animals that had a massive hematoma in the brain or no infarction in the brain were omitted from additional neurological and histological analysis.
In vivo administration of Bcl-x fusion proteins. To test the therapeutic effect of PTD-HA-Bcl-xL against ischemic injury, the purified fusion proteins at the indicated amounts were injected intraperitoneally at the indicated times into mice in 300 μl of PBS and 10% glycerol. The animals received injection of PTD-HA-Bcl-xL or control proteins (HA-Bcl-xL or PTD-HA-GFP) and were assigned randomly to the experimental groups consisting of 8–12 animals each.
Western blot analysis and immunohistochemistry
Western blot analysis was performed using standard methods (Chen et al., 1998). For immunohistochemistry, animals were anesthetized with 8% chloral hydrate and were then perfused with heparinized 0.9% saline followed by 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4. The brains were removed and postfixed in 30% sucrosis/4% paraformaldehyde. Coronal sections 40 μmthick were cut throughout the forebrain. For immunohistochemical staining of HA (to detect the Bcl-xL fusion protein) or active caspase-3, rabbit polyclonal antibodies were used at the dilution of 1:500 (anti-HA; Sigma, St. Louis, MO) and 1:100 (anti-active caspase-3; Cell Signaling Technology, Inc., Beverly, MA), respectively. For double-label immunofluorescence staining, sections were first incubated with the rabbit anti-HA antibody at 4°C for 48 hr followed by incubation for 2 hr at room temperature with goat anti-rabbit Cy3.18 immunoconjugate (Jackson ImmunoResearch, West Grove, PA) at 1:2500 dilutions. Sections were then subjected to incubation for 48 hr in mouse anti-neuron-specific enolase (NSE) antibody (dilution at 1:200; Capricorn Products, Inc., Scarborough, ME), mouse anti-neuronal-specific nuclear protein (NeuN) antibody (dilution at 1:500; Chemicon, Temecula, CA), or mouse anti-glial fibrillary acidic protein antibody (dilution at 1:200; Chemicon). This was followed by incubation with biotin-conjugated anti-mouse antibody (dilution at 1:3000) and then fluorescein–avidin D (Vector Laboratories, Burlingame, CA) at 8 μg/ml. The sections were washed four times in PBS, mounted in gelvatol, and coverslipped. For the assessment of nonspecific staining, alternating sections from each experimental condition were incubated without the primary antibody or with the antibody that had been reabsorbed by excessive amounts of antigen.
Measurement of caspase-3-like protease activity
Caspase-3-like protease activity was measured in cell extracts using the fluorogenic substrateAc-Acetyl-Asp-Glu-Val-Asp (DEVD) 7-amino-4-trifluoromethyl coumarin (AFC) as described previously (Cao et al., 2001a). One unit of the caspase-3-like activity corresponded to the caspase-like activity that cleaves 1 pmol of AFC per minute at 37°C at saturating substrate concentrations. To detect nonspecific protease activity, in parallel experiments, the protein extracts were incubated in the reaction buffer with a 5 μmconcentration of the caspase-3 inhibitor DEVD-aldehyde at room temperature for 30 min before the addition of assay substrates, and the values were subtracted from those obtained without the inhibitor.
Statistical analysis
Results are reported as mean values ± SEM. The significance of differences between means was assessed by Student'st test (single comparisons) or by ANOVA and post hoc Scheffe's tests, with p < 0.05 considered statistically significant.
RESULTS
Creation of a biologically active Bcl-xL fusion protein
To generate a Bcl-xL fusion protein containing the PTD (Fig.1a), the full-length rat Bcl-xL cDNA was isolated and subsequently subcloned into the ET-30 (a+) plasmid that had been reconstructed to contain the sequences for two tag peptides (his-6 and HA) and the PTD. Additional constructs lacking either PTD (pET-HA-Bcl-xL) or Bcl-xL (PTD-HA-GFP) were also made. The nucleotide sequences of all constructs were verified by sequencing analysis before protein production. To prepare the Bcl-xL fusion proteins for in vivo delivery, the proteins were expressed in E. coli and subsequently purified to near homogeneity, as shown by Coomassie blue staining (Fig. 1b). The protein composition of these fusion proteins was further confirmed using Western blot analysis with anti-HA or anti-Bcl-xL antibody (Fig.1b).
Fig. 1.

The death-suppressing effect of PTD-HA-Bcl-xL in primary cultures of cortical neurons. a, Structures of the Bcl-xL fusion proteins. PTD-HA-Bcl-xL contains both HA tag and the PTD, whereas HA-Bcl-xL does not contain the PTD; the latter served as the control protein in subsequent studies. b, Verification of the Bcl-xL fusion proteins. Coomassie blue (CB) staining shows that the proteins have been purified to near homogeneity. Western blots show that the proteins could be detected using either anti-HA or anti-Bcl-x antibody.c, Representative immunofluorescent images show the transduction and death-suppressing effect in cortical neurons. PTD-HA-Bcl-xL (B, low power; C, high power) but not HA-Bcl-xL (A) transduces into neurons within 15 min of exposure, as determined using HA immunostaining. Immunofluorescence for HA (D) and CCOX IV (E) are partially colocalized (F, overlay), suggesting that a portion of the transduced PTD-HA-Bcl-xL is associated with the mitochondria. The addition of PTD-HA-Bcl-xL (H) but not HA-Bcl-xL (I) reduces the amounts of nuclei showing apoptotic changes (arrows), compared with the vehicle control (G). Immunofluorescence for cytochrome c is preserved in PTD-HA-Bcl-xL-treated neurons (K) but not in HA-Bcl-xL-treated neurons (L) or vehicle-treated neurons (J), consistent with the speculation that PTD-HA-Bcl-xL inhibits STS-induced cytochrome c release.G–L were obtained 24 hr after STS. Scale bar, 20 μm. d, e, PTD-HA-Bcl-xL but not HA-Bcl-xL or PTD-GFP inhibits STS-induced apoptosis in a dose-dependent manner (d). PTD-HA-Bcl-xL was equally effective when it was added to the cultures before or ≤1 hr after STS (e). Apoptosis was quantified after PI staining at 24 hr after STS exposure (0.3 μm). Data are mean ± SE from three independent experiments. **p < 0.01 versus vehicle controls (ANOVA and post hocScheffe's tests). f, Western blots demonstrate that PTD-HA-Bcl-xL treatment attenuated STS-induced cytochromec release to the cytoplasm in cultured neurons. Eachlane contains 20 μg of cytosolic protein, prepared from noninduced (control) or STS-induced (0.3 μm for 6 hr) neurons, respectively. The blots are representative of three independent experiments with similar results.
The cellular-transduction and death-suppressing capabilities of Bcl-xL fusion proteins were evaluated in the primary cultures of cortical neurons. The addition of PTD-HA-Bcl-xL (1–10 μg/ml) but not HA-Bcl-xL to the cultures resulted in rapid protein transduction into nearly 100% of cells within 15 min of incubation, as shown by anti-HA immunofluorescence (Fig. 1c, A–C). Transduction of PTD-HA-Bcl-xL at the above concentrations did not show detectable cytotoxicity within 30 hr (measurement of lactate dehydrogenase release and Hoechst 33258 staining). Double-label immunofluorescence of HA and cytochrome c oxidase (CCOX) IV showed overlapping between these two antigens (Fig. 1c,D–F). However, the PTD-HA-Bcl-xL immunofluorescence showed a more diffusive intracellular pattern than cytochromec oxidase IV, suggesting that PTD-HA-Bcl-xL was distributed in both mitochondria components and nonmitochondria components.
Cellular transduction of PTD-HA-Bcl-xL attenuated STS-induced apoptosis in cortical neurons in a dose-dependent manner (Fig. 1d). STS treatment at the concentration of 0.3 μmtriggered apoptosis in ∼65% of neurons within 24 hr. When PTD-HA-Bcl-xL was added 15 min before STS treatment, cell death was markedly decreased, as determined using nuclear staining (Fig.1d) or the LIVE/DEAD viability/cytotoxicity assay kit (data not shown). In contrast, neither HA-Bcl-xL nor PTD-HA-GFP had a protective effect at compatible concentrations. The concentrations of PTD-HA-Bcl-xL that reached the half-maximal and maximal effect against STS-induced apoptosis were ∼1 μg/ml (33 nm) and ∼3 μg/ml (100 nm), respectively, whereas for the broad-spectrum caspase inhibitorz-VAD-fmk to reach a similar protective effect, concentrations of 30–100 μm were required (data not shown). These results indicated that PTD-HA-Bcl-xL is an extremely potent anti-apoptotic agent. To determine the time window of efficacy against STS neurotoxicity, PTD-HA-Bcl-xL was added to the culture media at various time points before and after STS exposure. The post-treatment of PTD-HA-Bcl-xL ≤1 hr after STS exposure was as effective as most pretreatment regimens (Fig. 1e).
Because Bcl-xL is known to inhibit apoptosis by blocking cytochromec release, immunofluorescent staining for cytochromec was performed in cultures 24 hr after STS (0.3 μm) treatment without or with PTD-HA-Bcl-xL (3 μg/ml) treatment (Fig. 1c, J–L). PTD-HA-Bcl-xL significantly increased the percentages of neurons that retained cytochrome c immunofluorescence from 46.6 ± 3.8 to 84.1 ± 2.7% (p < 0.001). The effect of PTD-HA-Bcl-xL on cytochrome c release was also confirmed by Western blot analysis (Fig. 1f).
In vivo transduction of Bcl-xL fusion protein in murine brain
The in vivo transduction capability of PTD-HA-Bcl-xL to the brain was evaluated in adult male mice. Our preliminary studies suggested that PTD-HA-GFP could achieve robust transduction in the brain 2–4 hr after systemic injection. Therefore, we initially performed Western blotting to detect PTD-HA-Bcl-xL in brain tissues at 4 hr after intraperitoneal injection of the protein. Marked dose-dependent increases in the amounts of PTD-HA-Bcl-xL protein were detectable by either anti-HA or anti-Bcl-x antibody in various brain regions, including the cortex (Fig.2a), caudate–putamen, hippocampus, cerebellum, and spinal cord. In contrast, injection of HA-Bcl-xL did not achieve detectable protein transduction. The quantitative ELISA confirmed that the levels of Bcl-xL were increased approximately twofold and sevenfold in the cortex after single injections of PTD-HA-Bcl-xL at the doses of 3 and 9 mg/kg, respectively (Fig. 2b). The time course for protein transduction of PTD-HA-Bcl-xL in brain was also determined using ELISA after single injections of the protein (9 mg/kg). Increases in PTD-HA-Bcl-xL were clearly detectable at 1–2 hr after injection, peaked at 4–8 hr, and were primarily retained at 24 hr (Fig. 2b).
Fig. 2.

In vivo protein transduction of PTD-HA-Bcl-xL in murine brain. a, Western blotting with either the anti-Bcl-x or anti-HA antibody detects the transduction of PTD-HA-Bcl-xL into the cortex 4 hr after intraperitoneal injection of the protein at various doses (left); the HA-Bcl-xL protein lacking PTD failed to transduce across the blood–brain barrier (right). In all blots, the fusion proteins serve as the positive controls (PC). Immunoblotting of α-tubulin serves as a control for sample loadings (30 μg perlane). Note that the endogenous Bcl-x proteins are detectable in all brains. b, c, Quantitation of PTD-HA-Bcl-xL transduction in the brain by ELISA. Intraperitoneal injection of PTD-HA-Bcl-xL results in dose-dependent (b, 4 hr after injection) and time-dependent (c, injection at the dose of 9 mg/kg) protein transduction into the murine cerebral cortex. Data are mean ± SE (three animals per group). *p < 0.05 versus controls; **p < 0.01 versus controls. d, Immunofluorescent detection (using anti-HA antibody) of PTD-HA-Bcl-xL transduction in the cortex/caudate (A) and hippocampal (Hipp) formation (B) 4 hr after intraperitoneal injection; the immunofluorescence is lost after the primary antibody was preabsorbed with the fusion protein (C). Injection of HA-Bcl-xL serves as the negative control (Ctr) (D). In the cerebral parenchyma, most of the cells transduced with PTD-HA-Bcl-xL (HA immunoreactive; E, H, red) are neurons, being immunoreactive for the neuronal markers NSE (F, arrows in G; cortex) or NeuN (I, arrows in J; hippocampal dentate). Some astrocytes (GFAP-immunoreactive;L, green) in the caudate are also transduced (arrows in K–M).Str, Striatum.
Transduction of PTD-HA-Bcl-xL in the brain was evaluated at cellular levels using immunohistochemistry at 4 hr after protein injection (9 mg/kg). As determined in the cortex, caudate–putamen, and hippocampus, robust HA immunoreactivity was detected in most neurons throughout the forebrain after injection of PTD-HA-Bcl-xL but not HA-Bcl-xL (Fig.2d, A–C). Sections that were incubated in the anti-HA antibody preabsorbed with PTD-HA-Bcl-xL resulted in loss of HA immunoreactivity (Fig. 2d, D), thus confirming the specificity of the detected immunoreactivity. Double-label immunofluorescent staining revealed that in cerebral parenchyma, most of the transduced cells were neurons (Fig. 2d,E–J; NSE or NeuN positive); however, transduction also occurred in some astrocytes in the caudate–putamen (Fig.2d, K–M, GFAP positive). Astroglial transduction of PTD-HA-Bcl-xL primarily occurred in astrocyte-enriched regions, including vessel walls and white matter such as the corpus callosum (data not shown).
In vivo systemic delivery of Bcl-xL fusion protein protects against focal ischemic injury
MCA occlusion produced ipsilateral cerebral infarction averaging ∼65 mm3 in volume, as determined at 72 hr based on the loss of triphenyltetrazolium chloride (TTC) staining (Fig. 3a). Administration with PTD-HA-Bcl-xL but not HA-Bcl-xL or PTD-HA-GFP 2 hr before the onset of ischemia reduced the infarct volume in a dose-dependent manner (Fig. 3d). A significant reduction averaging ∼40% was reached at 9 mg/kg (p < 0.0001 vs vehicle controls), but an additional increase in the dose to 18 mg/kg did not result in an additional reduction in the infarct volume. Cerebral salvage occurred primarily in the periphery of the ischemic tissue, leading to enhanced survival of penumbral cortical tissue. As determined at 24 hr after ischemia, PTD-HA-Bcl-xL (9 mg/kg)-treated animals showed improved neurological scores (Fig.3c). Reduction of infarct size by PTD-HA-Bcl-xL (9 mg/kg) was not accompanied by significant alterations in arterial blood pressure and blood gases compared with control animals that received vehicle alone (data not shown). As determined using laser–Doppler flowmetry, animals that received PTD-HA-Bcl-xL (9 mg/kg) or vehicle showed similar cortical blood flow changes during and after MCA occlusion (Fig. 3b). Delayed treatment with PTD-HA-Bcl-xL (9 mg/kg) continued to significantly reduce infarct volume when administered at 5 or 45 min after the completion of ischemia, but the effect was lost when protein injection was delayed by 120 min (Fig.3d).
Fig. 3.
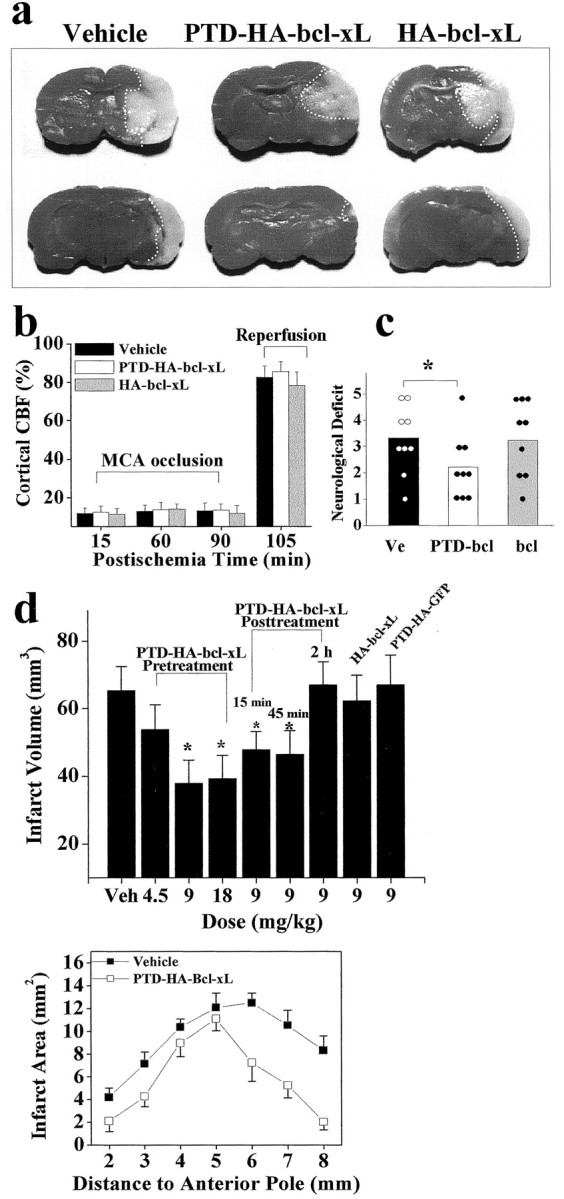
Systemic delivery of PTD-HA-Bcl-xL protects against focal ischemic infarction. a, Representative photographs of TTC-stained coronal sections of mouse brains recovered from 90 min of focal ischemia and 72 hr of reperfusion. The sections were chosen at the levels of 4 mm (top) and 6 mm (bottom) from the anterior pole. The dotted white lines illustrate the infarct border for each section.b, Changes in cortical blood flow, as determined using laser–Doppler flowmetry, are not different between fusion protein-treated and vehicle-treated brains during or after ischemia (n = 6 per group). CBF, Cerebral blood flow. c, Neurological deficit scores in vehicle-treated (Ve), PTD-HA-Bcl-xL-treated (PTD-bcl), and HA-Bcl-xL-treated (bcl) mice at 24 hr after ischemia.Dots and columns represent the scores of each animal and the mean scores, respectively. The neurological scores were graded according to the scale described previously (Murakami et al., 1998). d, Effects of intraperitoneal injection ofPTD-HA-Bcl-xL on infarct volumes in the brain after 90 min of focal ischemia. Vehicle (Veh), PTD-HA-Bcl-xL, or the control proteins were injected at the indicated doses either 1 hr before or at the indicated time after ischemia, and the infarct volume was measured at 72 hr after ischemia. Data are mean ± SE;n = 6–8 per group. *p < 0.05 versus vehicle treatment (ANOVA and post hoc Scheffe's tests). The graph at the bottomillustrates the comparison of infarct sizes in each brain section between vehicle-treated and PTD-HA-Bcl-xL-treated mice.
Bcl-xL fusion protein inhibits an apoptotic component of ischemic neuronal death
To determine whether the protective effect of PTD-HA-Bcl-xL against ischemic injury involves an anti-apoptotic mechanism, animals treated with PTD-HA-Bcl-xL (9 mg/kg) or vehicle were subjected to MCA occlusion, and the cerebral cortical tissues were assayed using the DEVD-AFC cleavage assay for caspase-3-like activity at 3 and 24 hr after ischemia. In vehicle-treated animals, there were significant increases in caspase-3-like activity at 3 hr and, to a lesser extent, 24 hr after ischemia (Fig.4a). However, the increased caspase-3-like activity was almost completely attenuated in PTD-HA-Bcl-xL-treated animals (p < 0.001). This effect by PTD-HA-Bcl-xL was also confirmed by Western blotting. As shown (Fig. 4b), the immunoreactivity for the active caspase-3 (p17) was increased in the vehicle-treated but not the PTD-HA-Bcl-xL-treated animals.
Fig. 4.

PTD-HA-Bcl-xL inhibits an apoptotic component of ischemic neuronal death. a, Effects of PTD-HA-Bcl-xL on caspase-3-like protease activity in the brain at 3 and 24 hr after ischemia, as determined in the cortical cell extracts using the fluorogenic substrate DEVD-AFC. Data are mean ± SE; n = 5 per group. **p < 0.01 versus vehicle treatment. b, Attenuation of caspase-3 activation after ischemia by PTD-HA-Bcl-xL, as demonstrated by Western blotting. The active (p17) caspase-3 fragment is increased in the ischemic hemisphere (L) compared with the nonischemic side (R) in vehicle-treated but not in PTD-HA-Bcl-xL-treated brains. The blots are representative of three independent experiments with similar results. c, Coronal sections obtained from vehicle-treated and PTD-HA-Bcl-xL-treated brains, respectively, 24 hr after ischemia. The red boxes mark the approximate locations from which the immunofluorescent images in c were obtained.d, Representative immunofluorescent images show that PTD-HA-Bcl-xL attenuates caspase-3 activation after ischemia. Immunostaining was accomplished using the antibody recognizing the active caspase-3. A–E, Low power (25×):A, Nonischemic cortex. B, At 3 hr after ischemia, caspase-3-immunoreactive cells are present primarily in layers I-II and V of the cortex. C, The mounts of caspase-3-immunoreactive cells are decreased in PTD-HA-Bcl-xL-treated brain. D, 24 hr after ischemia, caspase-3-immunoreactive cells are localized in the border zone of the infarction (*). Thedotted lines in D and E mark the estimated infarct border. E, Few caspase-3-immunoreactive cells are present in PTD-HA-Bcl-xL-treated brain. F–I, high power (400×). Scale bar, 20 μm. F, At 3 hr after ischemia (vehicle treated), caspase-3 immunoreactivity primarily shows a cytosolic location. G, As indicated by the arrows, the cells in F counterstained with the DNA dye PI.H, At 24 hr after ischemia, caspase-3 immunoreactivity is localized in the nucleus. I, As indicated by thearrows, the cells in H counterstained with PI. J–L, Double-label immunofluorescence of PTD-HA-Bcl-xL (J) and caspase-3 (K) in the border zone of the cortical infarction at 24 hr after ischemia (PTD-HA-Bcl-xL-treated brain); Lis the overlay of J and K. Note that there is no colocalization of caspase-3 and PTD-HA-Bcl-xL in neurons (L, arrows indicate protein-transduced neurons). e, Apoptotic DNA fragmentation is attenuated by PTD-HA-Bcl-xL after ischemia, as demonstrated by DNA electrophoresis. The DNA was prepared from the cortical tissues 72 hr after ischemia, using the brains pretreated with vehicle, PTD-HA-Bcl-xL, or HA-Bcl-xL. The gel is representative of two independent experiments with similar results.
The effect of PTD-HA-Bcl-xL on caspase-3 activation was also demonstrated at the cellular level (Fig. 4d). Using the same antibody as that used in Western blots to detect active caspase-3, we detected markedly increased immunofluorescence in neurons widely distributed in the frontoparietal cortex 3 hr after ischemia in vehicle-treated animals (Fig. 4d, B,D), and the signals were localized primarily in the cytosol of neurons (Fig.4d, F,G). At 24 hr after ischemia, caspase-3 immunofluorescent cells were distributed primarily in the border zone of cortical infarction, and the immunofluorescence was present primarily in the nucleus of the neurons (Fig. 4d,H,I). These results confirm previous findings by others in a similar animal model (Namura et al., 1998). In contrast, the amounts of caspase-3 immunofluorescent neurons were greatly decreased in the cerebral cortex in PTD-HA-Bcl-xL-treated animals at both postischemic time points (Fig. 4d, C,E). Double-label immunofluorescent staining revealed that there was no colocalization of caspase-3 and PTD-HA-Bcl-xL immunofluorescence within the protected cortex after ischemia (Fig. 4d,J–L), further confirming the anti-apoptotic effect of PTD-HA-Bcl-xL protein transduction in the ischemic brain.
DISCUSSION
Using recombinant technology, we produced a biologically active Bcl-xL fusion protein containing the PTD that allows the in vivo delivery of the protein across the blood–brain barrier and robust protein transduction in the brain. The death-suppressing effect of this fusion protein was initially confirmed in primary cultures of cortical neurons, in which PTD-HA-Bcl-xL but not HA-Bcl-xL or PTD-HA-GFP inhibited STS-induced apoptosis in both dose-dependent and time-dependent manners. When PTD-HA-Bcl-xL was injected intraperitoneally, the protein was efficiently transduced into brain cells within 2–4 hr. The PTD-HA-Bcl-xL-transduced brains but not control protein-treated brains showed significantly decreased infarct sizes after 90 min of focal ischemia, and this protective effect was detectable when the protein was administered before or ≤45 min after ischemia. Furthermore, ischemia-induced activation of caspase-3 was markedly attenuated in PTD-HA-Bcl-xL-transduced brains. These results provide the first evidence that brain transduction of a nonsecretory protein engineered to contain the PTD shows biological activity and protects the brain against ischemic/reperfusion injury.
The data presented herein demonstrate that the PTD-HA-Bcl-xL protein was transduced efficiently into neurons in vitro andin vivo. The fusion protein contains an HA tag, which allows immunodetection of the transduced protein to be distinguished from endogenous Bcl-xL. When directly applied to the culture media, PTD-HA-Bcl-xL was detected in ∼100% of the neurons within 15 min (Fig. 1). This observation confirms the reported capability of PTD to help achieve rapid protein transduction in mammalian cells (Nagahara et al., 1998; Schwarze et al., 1999). Notably, the death-suppressing effect of PTD-HA-Bcl-xL in neurons was equally potent when the protein was added either 4 hr before or 1 hr after STS exposure. This suggests that the transduced protein can rapidly achieve its biological activity in neurons, presumably through chaperone-mediated conformational changes, and that its death-suppressing efficacy is stable. The recent study by Liu et al. (2001) showed a similar death-suppressing effect by a Bcl-xL fusion protein, LFn-Bcl-xL, against STS-induced apoptosis in rat cerebellar granule cells and macrophages, in which Bcl-xL was fused into a 254 aa nontoxic derivative (LFn) of anthrax toxin. The transduction of LFn-Bcl-xL requires the action of another anthrax toxin component, protective antigen (Liu et al., 2001), which binds to an unidentified cell surface receptor and subsequently transports LFn into cells (Friedlander, 1986). Compared with the anthrax toxin delivery system, the PTD transduction system described in this study may have several advantages. First, the PTD consists of only 11 aa, which does not substantially increase the particle mass of the fusion protein. Second, the transduction of PTD-containing fusion protein does not require a helper protein. Third, the PTD mediates protein transduction by targeting the lipid bilayer component of the cell membrane; thus, theoretically, a protein containing PTD may be transduced into all mammalian cell types (Schwarze et al., 1999).
The cellular transduction of PTD-HA-Bcl-xL in murine brain and its protective effect against focal stroke are robust. Using quantitative ELISA, we detected ∼4 ng of PTD-HA-Bcl-xL (per milligram of tissue protein) in the cortex within 2 hr after systemic injection (9 mg/kg), which was within the range of the effective concentrations of PTD-HA-Bcl-xL in cultures against STS-induced apoptosis. At the dosage of 9 mg/kg, we demonstrated that administration of PTD-HA-Bcl-xL resulted in an up to ∼40% reduction in infarct sizes 72 hr after focal ischemia. This efficacy likely resulted from the direct protection by PTD-HA-Bcl-xL, because the protection occurred without altering cortical blood flow, body temperature, or other physiological parameters during or after ischemia.
The protective effect by PTD-HA-Bcl-xL against ischemic injury may be mediated by several mechanisms. There is clear evidence that PTD-HA-Bcl-xL prevented caspase-3 activation in the ischemic brain (Fig. 4). This effect by PTD-HA-Bcl-xL could be the consequence of its inhibitory effect on cytochrome c release from mitochondria and/or its direct interaction with the Apaf-1/caspase-9 apoptosome, because both mechanisms are upstream of caspase-3 activation and appear to be major targets for Bcl-xL (Kluck et al., 1997; Hu et al., 1998;Pan et al., 1998). Furthermore, there is strong evidence that these mechanisms may participate in the molecular cascade leading to neuronal apoptosis after cerebral ischemia (Fujimura et al., 1999;Sugawara et al., 1999; Noshita et al., 2001; Cao et al., 2002). Moreover, PTD-HA-Bcl-xL could block mitochondrial release of other apoptogenic factors such as apoptosis-inducing factor; the latter may mediate ischemic cell death via caspase-independent mechanisms (Graham and Chen, 2001). Finally, the speculative role for Bcl-xL in protecting against hypoxia-induced necrosis (Tsujimoto et al., 1997) may have particular relevance in explaining its potent death-suppressing effect demonstrated in this study, because it is very likely that ischemic neuronal death involves both apoptotic and necrotic mechanisms.
The significance of the results documented in this report is twofold. First, the powerful capability of the PTD in accelerating in vivo protein transduction in the brain and in cultured neurons provides an excellent tool for mechanistic studies, through which many important biological issues such as cell death pathways or signal transduction mechanisms in the CNS can be addressed. Second, the PTD recombinant technology will allow us to screen a large number of potentially potent peptide neuroprotectants in animal models of stroke or other neurological disorders, which may have clinical implications as a novel molecular therapeutic intervention.
Footnotes
This work was supported by National Institutes of Health Grants NS38560, NS36736, and NS35965 to J.C., from the National Institute of Neurological Disorders and Stroke. J.C. is a recipient of the Established Investigator Award from the American Heart Association. J.C. and S.H.G. were also supported in part by the Geriatric Research, Education and Clinical Center, Veterans Affairs Pittsburgh Health Care System (Pittsburgh, PA). We thank Cristine O'Horo for technical assistance, Carol Culver for editorial assistance, and Pat Strickler for secretarial support.
Correspondence should be addressed to Dr. Jun Chen, Department of Neurology, S-507, Biomedical Science Tower, University of Pittsburgh School of Medicine, Pittsburgh, PA 15213. E-mail: jun@med.pitt.edu.
REFERENCES
- 1.Blomer U, Kafri T, Randolph-Moore L, Verma IM, Gage FH. Bcl-xL protects adult septal cholinergic neurons from axotomized cell death. Proc Natl Acad Sci USA. 1998;95:2603–2608. doi: 10.1073/pnas.95.5.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cao G, Minami M, Pei W, Yan C, Chen D, O'Horo C, Graham SH, Chen J. Intracellular Bax translocation after transient cerebral ischemia: implications for a role of the mitochondrial apoptotic signaling pathway in ischemic neuronal death. J Cereb Blood Flow Metab. 2001a;21:321–333. doi: 10.1097/00004647-200104000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Cao G, Pei W, Lan J, Stetler RA, Luo Y, Nagayama T, Graham SH, Yin XM, Simon RP, Chen J. Caspase-activated DNase/DNA fragmentation factor 40 mediates apoptotic DNA fragmentation in transient cerebral ischemia and in neuronal cultures. J Neurosci. 2001b;21:4678–4690. doi: 10.1523/JNEUROSCI.21-13-04678.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cao G, Luo Y, Nagayama T, Pei W, Stetler RA, Graham SH, Chen J. Cloning and characterization of rat caspase-9: implications for a role in mediating caspase-3 activation and hippocampal cell death after transient cerebral ischemia. J Cereb Blood Flow Metab. 2002;22:534–546. doi: 10.1097/00004647-200205000-00005. [DOI] [PubMed] [Google Scholar]
- 5.Chen D, Stetler RA, Cao G, Pei W, O'Horo C, Yin XM, Chen J. Characterization of the rat DNA fragmentation factor 35/inhibitor of caspase-activated DNase (short form): the endogenous inhibitor of caspase-dependent DNA fragmentation in neuronal apoptosis. J Biol Chem. 2000;275:38508–38517. doi: 10.1074/jbc.M003906200. [DOI] [PubMed] [Google Scholar]
- 6.Chen J, Yin XM. Regulation of active cell death in ischemic brain injury: the role of Bax and the mitochondrial pathway. In: Krieglstein J, Klumpp S, editors. Pharmacology of cerebral ischemia. Scientific Publishers; Stuttgart: 2000. pp. 189–198. [Google Scholar]
- 7.Chen J, Graham SH, Zhu RL, Simon RP. Stress proteins and tolerance to focal cerebral ischemia. J Cereb Blood Flow Metab. 1996;16:566–577. doi: 10.1097/00004647-199607000-00006. [DOI] [PubMed] [Google Scholar]
- 8.Chen J, Nagayama T, Jin K, Stetler RA, Zhu RL, Graham SH, Simon RP. Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci. 1998;18:4914–4928. doi: 10.1523/JNEUROSCI.18-13-04914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Frankowski H, Missotten M, Fernandez PA, Martinou I, Michel P, Sadoul R, Martinou JC. Function and expression of the Bcl-x gene in the developing and adult nervous system. NeuroReport. 1995;6:1917–1921. doi: 10.1097/00001756-199510020-00023. [DOI] [PubMed] [Google Scholar]
- 10.Friedlander AM. Macrophages are sensitive to anthrax lethal toxin through an acid-dependent process. J Biol Chem. 1986;261:7123–7126. [PubMed] [Google Scholar]
- 11.Fujimura M, Morita-Fujimura Y, Kawase M, Copin JC, Calagui B, Epstein CJ, Chan PH. Manganese superoxide dismutase mediates the early release of mitochondrial cytochrome C and subsequent DNA fragmentation after permanent focal cerebral ischemia in mice. J Neurosci. 1999;19:3414–3422. doi: 10.1523/JNEUROSCI.19-09-03414.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gillardon F, Lenz C, Waschke KF, Krajewski S, Reed JC, Zimmermann M, Kuschinsky W. Altered expression of Bcl-2, Bcl-X, Bax, and c-Fos colocalizes with DNA fragmentation and ischemic cell damage following middle cerebral artery occlusion in rats. Brain Res Mol Brain Res. 1996;40:254–260. doi: 10.1016/0169-328x(96)00059-9. [DOI] [PubMed] [Google Scholar]
- 13.Gonzalez-Garcia M, Garcia I, Ding L, O'Shea S, Boise LH, Thompson CB, Nunez G. bcl-x is expressed in embryonic and postnatal neural tissues and functions to prevent neuronal cell death. Proc Natl Acad Sci USA. 1995;92:4304–4308. doi: 10.1073/pnas.92.10.4304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graham SH, Chen J. Programmed cell death in cerebral ischemia. J Cereb Blood Flow Metab. 2001;21:99–109. doi: 10.1097/00004647-200102000-00001. [DOI] [PubMed] [Google Scholar]
- 15.Graham SH, Chen J, Clark RS. Bcl-2 family gene products in cerebral ischemia and traumatic brain injury. J Neurotrauma. 2000;17:831–841. doi: 10.1089/neu.2000.17.831. [DOI] [PubMed] [Google Scholar]
- 16.Hu Y, Benedict MA, Wu D, Inohara N, Nunez G. Bcl-XL interacts with Apaf-1 and inhibits Apaf-1-dependent caspase-9 activation. Proc Natl Acad Sci USA. 1998;95:4386–4391. doi: 10.1073/pnas.95.8.4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Isenmann S, Stoll G, Schroeter M, Krajewski S, Reed JC, Bahr M. Differential regulation of Bax, Bcl-2, and Bcl-X proteins in focal cortical ischemia in the rat. Brain Pathol. 1998;8:49–63. doi: 10.1111/j.1750-3639.1998.tb00134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kharbanda S, Pandey P, Schofield L, Israels S, Roncinske R, Yoshida K, Bharti A, Yuan ZM, Saxena S, Weichselbaum R, Nalin C, Kufe D. Role for Bcl-xL as an inhibitor of cytosolic cytochrome C accumulation in DNA damage-induced apoptosis. Proc Natl Acad Sci USA. 1997;94:6939–6942. doi: 10.1073/pnas.94.13.6939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kitagawa K, Matsumoto M, Tsujimoto Y, Ohtsuki T, Kuwabara K, Matsushita K, Yang G, Tanabe H, Martinou JC, Hori M, Yanagihara T. Amelioration of hippocampal neuronal damage after global ischemia by neuronal overexpression of BCL-2 in transgenic mice. Stroke. 1998;29:2616–2621. doi: 10.1161/01.str.29.12.2616. [DOI] [PubMed] [Google Scholar]
- 20.Kluck RM, Bossy-Wetzel E, Green DR, Newmeyer DD. The release of cytochrome c from mitochondria: a primary site for Bcl-2 regulation of apoptosis. Science. 1997;275:1132–1136. doi: 10.1126/science.275.5303.1132. [DOI] [PubMed] [Google Scholar]
- 21.Kondo T, Reaume AG, Huang TT, Carlson E, Murakami K, Chen SF, Hoffman EK, Scott RW, Epstein CJ, Chan PH. Reduction of CuZn-superoxide dismutase activity exacerbates neuronal cell injury and edema formation after transient focal cerebral ischemia. J Neurosci. 1997;17:4180–4189. doi: 10.1523/JNEUROSCI.17-11-04180.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lawrence MS, Ho DY, Sun GH, Steinberg GK, Sapolsky RM. Overexpression of Bcl-2 with herpes simplex virus vectors protects CNS neurons against neurological insults in vitro and in vivo. J Neurosci. 1996;16:486–496. doi: 10.1523/JNEUROSCI.16-02-00486.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lawrence MS, McLaughlin JR, Sun GH, Ho DY, McIntosh L, Kunis DM, Sapolsky RM, Steinberg GK. Herpes simplex viral vectors expressing Bcl-2 are neuroprotective when delivered after a stroke. J Cereb Blood Flow Metab. 1997;17:740–744. doi: 10.1097/00004647-199707000-00003. [DOI] [PubMed] [Google Scholar]
- 24.Linnik MD, Zahos P, Geschwind MD, Federoff HJ. Expression of bcl-2 from a defective herpes simplex virus-1 vector limits neuronal death in focal cerebral ischemia. Stroke. 1995;26:1670–1675. doi: 10.1161/01.str.26.9.1670. [DOI] [PubMed] [Google Scholar]
- 25.Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- 26.Liu XH, Collier RJ, Youle RJ. Inhibition of axotomy-induced neuronal apoptosis by extracellular delivery of a Bcl-XL fusion protein. J Biol Chem. 2001;276:46326–46332. doi: 10.1074/jbc.M108930200. [DOI] [PubMed] [Google Scholar]
- 27.Martinou JC, Dubois-Dauphin M, Staple JK, Rodriguez I, Frankowski H, Missotten M, Albertini P, Talabot D, Catsicas S, Pietra C. Overexpression of BCL-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron. 1994;13:1017–1030. doi: 10.1016/0896-6273(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 28.Motoyama N, Wang F, Roth KA, Sawa H, Nakayama K, Negishi I, Senju S, Zhang Q, Fujii S, Loh DY. Massive cell death of immature hematopoietic cells and neurons in Bcl-x-deficient mice. Science. 1995;267:1506–1510. doi: 10.1126/science.7878471. [DOI] [PubMed] [Google Scholar]
- 29.Murakami K, Kondo T, Kawase M, Li Y, Sato S, Chen SF, Chan PH. Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J Neurosci. 1998;18:205–213. doi: 10.1523/JNEUROSCI.18-01-00205.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagahara H, Vocero-Akbani AM, Snyder EL, Ho A, Latham DG, Lissy NA, Becker-Hapak M, Ezhevsky SA, Dowdy SF. Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration. Nat Med. 1998;4:1449–1452. doi: 10.1038/4042. [DOI] [PubMed] [Google Scholar]
- 31.Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J, Moskowitz MA. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci. 1998;18:3659–3668. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Noshita N, Sugawara T, Fujimura M, Morita-Fujimura Y, Chan PH. Manganese superoxide dismutase affects cytochrome c release and caspase-9 activation after transient focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2001;21:557–567. doi: 10.1097/00004647-200105000-00010. [DOI] [PubMed] [Google Scholar]
- 33.Pan G, O'Rourke K, Dixit VM. Caspase-9, Bcl-XL, and Apaf-1 form a ternary complex. J Biol Chem. 1998;273:5841–5845. doi: 10.1074/jbc.273.10.5841. [DOI] [PubMed] [Google Scholar]
- 34.Parsadanian AS, Cheng Y, Keller-Peck CR, Holtzman DM, Snider WD. Bcl-xL is an antiapoptotic regulator for postnatal CNS neurons. J Neurosci. 1998;18:1009–1019. doi: 10.1523/JNEUROSCI.18-03-01009.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Plesnila N, Zinkel S, Le DA, Amin-Hanjani S, Wu Y, Qiu J, Chiarugi A, Thomas SS, Kohane DS, Korsmeyer SJ, Moskowitz MA. BID mediates neuronal cell death after oxygen/glucose deprivation and focal cerebral ischemia. Proc Natl Acad Sci USA. 2001;98:15318–15323. doi: 10.1073/pnas.261323298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sastry PS, Rao KS. Apoptosis and the nervous system. J Neurochem. 2000;74:1–20. doi: 10.1046/j.1471-4159.2000.0740001.x. [DOI] [PubMed] [Google Scholar]
- 37.Schulz JB, Weller M, Moskowitz MA. Caspases as treatment targets in stroke and neurodegenerative diseases. Ann Neurol. 1999;45:421–429. doi: 10.1002/1531-8249(199904)45:4<421::aid-ana2>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 38.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 39.Sharp FR, Lu A, Tang Y, Millhorn DE. Multiple molecular penumbras after focal cerebral ischemia. J Cereb Blood Flow Metab. 2000;20:1011–1032. doi: 10.1097/00004647-200007000-00001. [DOI] [PubMed] [Google Scholar]
- 40.Shinoura N, Satou R, Yoshida Y, Asai A, Kirino T, Hamada H. Adenovirus-mediated transfer of Bcl-X(L) protects neuronal cells from Bax-induced apoptosis. Exp Cell Res. 2000;254:221–231. doi: 10.1006/excr.1999.4751. [DOI] [PubMed] [Google Scholar]
- 41. Sugawara T, Fujimura M, Morita-Fujimura Y, Kawase M, Chan PH. Mitochondrial release of cytochrome c corresponds to the selective vulnerability of hippocampal CA1 neurons in rats after transient global cerebral ischemia. J Neurosci 19 1999. RC39:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 43.Tsujimoto Y, Shimizu S. Bcl-2 family: life-or-death switch. FEBS Lett. 2000;466:6–10. doi: 10.1016/s0014-5793(99)01761-5. [DOI] [PubMed] [Google Scholar]
- 44.Tsujimoto Y, Shimizu S, Eguchi Y, Kamiike W, Matsuda H. Bcl-2 and Bcl-xL block apoptosis as well as necrosis: possible involvement of common mediators in apoptotic and necrotic signal transduction pathways. Leukemia. 1997;11 [Suppl 3]:380–382. [PubMed] [Google Scholar]
- 45.Wiessner C, Allegrini PR, Rupalla K, Sauer D, Oltersdorf T, McGregor AL, Bischoff S, Bottiger BW, van der Putten H. Neuron-specific transgene expression of Bcl-XL but not Bcl-2 genes reduced lesion size after permanent middle cerebral artery occlusion in mice. Neurosci Lett. 1999;268:119–122. doi: 10.1016/s0304-3940(99)00392-4. [DOI] [PubMed] [Google Scholar]
- 46.Yang G, Chan PH, Chen J, Carlson E, Chen SF, Weinstein P, Epstein CJ, Kamii H. Human copper-zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke. 1994;25:165–170. doi: 10.1161/01.str.25.1.165. [DOI] [PubMed] [Google Scholar]
- 47.Yang J, Liu X, Bhalla K, Kim CN, Ibrado AM, Cai J, Peng TI, Jones DP, Wang X. Prevention of apoptosis by Bcl-2: release of cytochrome c from mitochondria blocked. Science. 1997;275:1129–1132. doi: 10.1126/science.275.5303.1129. [DOI] [PubMed] [Google Scholar]