

Abstract
The mesolimbic dopamine (DA) system originating in the ventral tegmental area (VTA) is involved in many drug-related behaviors, including ethanol self-administration. In particular, VTA activity regulating ethanol consummatory behavior appears to be modulated through GABAA receptors. Previous exposure to ethanol enhances ethanol self-administration, but the mechanisms underlying this phenomenon are not well understood. In this study, we examined changes occurring at GABA synapses onto VTA DA neurons after a singlein vivo exposure to ethanol. We observed that evoked GABAA IPSCs in DA neurons of ethanol-treated animals exhibited paired-pulse depression (PPD) compared with saline-treated animals, which exhibited paired-pulse facilitation (PPF). Furthermore, PPD was still present 1 week after the single exposure to ethanol. An increase in frequency of spontaneous miniature GABAA IPSCs (mIPSCs) was also observed in the ethanol-treated animals. Additionally, the GABAB receptor antagonist (3-aminopropyl)(diethoxymethyl) phosphinic acid shifted PPD to PPF, indicating that presynaptic GABAB receptor activation, likely attributable to GABA spillover, might play a role in mediating PPD in the ethanol-treated mice. The activation of adenylyl cyclase by forskolin increased the amplitude of GABAA IPSCs and the frequency of mIPSCs in the saline- but not in the ethanol-treated animals. Conversely, the protein kinase A (PKA) inhibitorN-[z-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide significantly decreased both the frequency of spontaneous mIPSCs and the amplitude of GABAA IPSCs in the ethanol-treated mice but not in the saline controls. The present results indicate that potentiation of GABAergic synapses, via a PKA-dependent mechanism, occurs in the VTA after a single in vivo exposure to ethanol, and such potentiation might be a key synaptic modification underlying increased ethanol intake.
Keywords: ventral tegmental area, ethanol, probability of GABA release, presynaptic plasticity, cAMP, PKA
The mesolimbic dopamine (DA) system originates in the ventral tegmental area (VTA), and it projects to structures associated with the limbic system, primarily the nucleus accumbens (NAcc), the amygdala, the hippocampus, and the prefrontal cortex (Fuxe et al., 1974; Oades and Halliday, 1987). A growing body of evidence implicates the mesolimbic DA system in the regulation of ethanol self-administration (Rassnick et al., 1993a; Samson et al., 1993; Ng and George, 1994; Koob et al., 1998; McBride et al., 1999;Kaczmarek and Kiefer, 2000; Nowak et al., 2000). An important role of DA in ethanol reinforcement has been suggested by studies showing that DA receptor antagonists, injected systemically or directly into the terminal regions of the mesolimbic DA system, decrease lever pressing for ethanol (Samson et al., 1993; Ng and George, 1994). Furthermore, a variety of pharmacological manipulations within this pathway, affecting the activity of DA neurons, produced changes in ethanol consumption, suggesting that DA neuronal activity within the VTA may be important for maintaining ethanol consummatory behavior (Rassnick et al., 1993a;Ng and George, 1994; Kaczmarek and Kiefer, 2000; Nowak et al., 2000). In fact, a marked reduction of the spontaneous activity of mesolimbic DA neurons (Diana et al., 1993; Bailey et al., 1998), resulting in decreased extracellular DA levels in NAcc (Diana et al., 1993), has been observed during acute withdrawal from chronic ethanol. Moreover, the fact that ethanol intake in dependent rats greatly exceeds that of nondependent rats during acute withdrawal, and that increased self-administration restores DA levels to normal in NAcc, suggests that decreased DA levels may trigger ethanol-seeking behavior (Weiss et al., 1996). The above-mentioned studies indicate that changes in activity of VTA DA cells, correlated with extracellular DA levels in the NAcc, might regulate ethanol consumption (McBride et al., 1995; Weiss et al., 1996; Hodge et al., 1997; Ikemoto et al., 1997), and accordingly, DA antagonists impair alcohol self-administration. In the midbrain dopamine systems, GABAergic neurons exert an inhibitory control on DA neurons (Johnson and North, 1992b; Hausser and Yung, 1994; Paladini et al., 1999). Therefore, hyperactivity of VTA GABA cells observed during acute withdrawal from chronic ethanol (Gallegos et al., 1999) could account, at least in part, for the reduced DAergic activity. Because the hypofunction of the DAergic system outlasts the somatic signs of acute withdrawal (Diana et al., 1996), such an increase of GABAergic synaptic transmission might not only represent a functional correlate of acute withdrawal from ethanol but also play a role in both short- and long-term consequences produced by ethanol exposure.
Interestingly, the influence of the initial exposure to ethanol and the patterns of its subsequent consumption have been observed in both humans and laboratory animals (Haertzen et al., 1983; Camarini et al., 2000; Files et al., 2000). Unfortunately, the relationship between the reinforcing quality of the first experience and subsequent habits of ethanol consumption is still unclear. Whether this change in behavior is attributable to a reduced sensitivity to the stimulant effects of ethanol (Phillips et al., 1995) or a blockade of the development of ethanol-induced conditioned taste aversion (Risinger and Cunningham, 1995) remains to be elucidated. Because both systemic and intra-VTA administration of GABAA receptor agonists facilitate, whereas antagonists decrease, the acquisition of voluntary ethanol drinking in rats (Smith et al., 1992; Nowak et al., 1998), the GABAergic transmission within the VTA might play an important role.
Although the acute effects of ethanol in the mesolimbic system have been studied extensively (Brodie et al., 1990, 1999; Nie et al., 1993,1994; Brodie and Appel, 1998, 2000; Steffensen et al., 2000), there are no studies directly examining whether synaptic changes occur in the VTA after exposure to ethanol.
To address this issue, we studied GABAA-mediated IPSCs in VTA DA neurons 24 hr after a single injection of either ethanol (2 gm/kg, i.p.) or saline.
MATERIALS AND METHODS
Slice preparation. The preparation of VTA slices was as described previously (Thomas et al., 2000). Briefly, C57BL/6J mice (21–35 d; Charles River, Hollister, CA) were anesthetized with halothane and killed. A block of tissue containing the midbrain was sliced in the horizontal plane (230 μm) with a vibratome (Leica, Nussloch, Germany) in ice-cold low-Ca2+solution containing (in mm): 126 NaCl, 1.6 KCl, 1.2 NaH2PO4, 1.2 MgCl, 0.625 CaCl2, 18 NaHCO3, and 11 glucose. Slices (two per animal) were transferred in a holding chamber with a bicarbonate-buffered solution (32–34°C) saturated with 95%O2 and 5%CO2containing (in mm): 126 NaCl, 1.6 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 18 NaHCO3, and 11 glucose. Slices were allowed to recover for at least 1 hr before being placed in the recording chamber and superfused with the bicarbonate-buffered solution (32–34°C) saturated with 95%O2 and 5%CO2.
Whole-cell recording. Only one cell for each experimental procedure was recorded per mouse. Cells were visualized with an upright microscope with infrared illumination, and whole-cell voltage-clamp recordings were made by using an Axopatch 1D amplifier (Axon Instruments, Foster City, CA). All GABAA IPSC recordings were made with electrodes filled with an internal solution containing the following (in mm): 144 KCl, 1 CaCl2, 3.45 K4BAPTA, 10 HEPES, 2 Mg2ATP, and 0.25 Mg2GTP, pH 7.2–7.4. Experiments were begun only after series resistance had stabilized (typically 15–40 MΩ). Series and input resistance were monitored continuously on-line with a 4 mV depolarizing step (25 msec). Data were filtered at 2 kHz, digitized at 10 kHz, and collected on-line with acquisition software (Igor Pro, Lake Oswego, OR). Because of the composition of the internal solution, the GABAA IPSCs were inward at a membrane potential of −70 mV and were completely blocked by picrotoxin (100 μm). DA cells were identified by the presence of a largeIh current (Johnson and North, 1992a) that was assayed immediately after break-in, using a series of incremental 10 mV hyperpolarizing steps from a holding potential of −70 mV. A bipolar stainless steel stimulating electrode was placed 100 μm rostral to the recording electrode and was used to stimulate at a frequency of 0.1 Hz. Neurons were voltage-clamped at a membrane potential of −70 mV. All GABAA IPSCs were recorded in the presence of 2-amino-5-phosphonopentanoic acid (AP5; 100 μm), 6-cyano-2,3-dihydroxy-7-nitro-quinoxaline (10 μm), strychnine (1 μm), and eticlopride (100 nm) to block NMDA, AMPA, glycine, and dopamine D2-mediated synaptic currents, respectively. As described previously (Bonci and Williams, 1997), this solution had no effect on the holding current of the dopamine cells. The amplitudes of IPSCs were calculated by taking a 1 msec window around the peak of the IPSC and comparing this with the 5 msec window immediately before the stimulation artifact. Paired stimuli were given with an interstimulus interval of 50 msec, and the ratio between the second and the first IPSCs was calculated and averaged for a 10 min baseline. Drugs were applied in known concentrations to the superfusion medium. The spontaneous miniature IPSCs (mIPSC) were collected in the presence of lidocaine (500 μm) and analyzed (120 sweeps for each condition, 1 sec/sweep) using Mini Analysis program (Synaptosoft). To accurately determine the mIPSC amplitude, only mIPSCs that were >8 pA were accepted for analysis. The choice of this cutoff amplitude for acceptance of mIPSCs was made to obtain a high signal-to-noise ratio.
Alcohol self-administration. Sixteen C57BL/6J mice (Charles River Laboratories, Wilmington, MA) were housed individually in polycarbonate cages, with food and water available ad libitum, and habituated to their home cage for 1 week before the experiment. The colony room was maintained on a 12 hr light/dark cycle with lights on at 6 A.M. All experimental procedures were conducted under institutional and National Institutes of Health guidelines. Oral ethanol self-administration was examined using a two-bottle choice protocol (Phillips et al., 1998; Hodge et al., 1999). Mice were offered the choice between 2% (v/v) ethanol versus water for 5 d. The ethanol concentration was increased to 5%, and the ethanol consumption was measured for 5 more days. Fluid volumes consumed were recorded every day, and the bottle positions were alternated daily. Each day, the mice were weighed, and then the ethanol consumption was calculated as grams of ethanol per kilogram.
Blood alcohol determination. Blood tail collection of 3- to 4-week-old mice did not provide enough volume for measurement; therefore, blood ethanol concentration was measured by drawing a 40 μl blood sample from the trunk. Twenty-four hours after either ethanol (2 gm/kg, i.p.) or saline exposure, blood samples were collected at 10, 30, 60, and 90 min after an intraperitoneal 4 gm/kg injection of ethanol (four or five mice were used for each group and for every time point). Blood plasma was extracted with trichloroacetic acid, and plasma ethanol content was measured using a 332 alcohol diagnostic kit (Sigma, St. Louis, MO).
Results in the text and figures are presented as the mean ± SEM. Results between groups were compared using a t test, either paired or unpaired where appropriate; p < 0.05 was taken as indicating statistical significance.
RESULTS
In the present study, we investigated the properties of GABAA IPSCs recorded in VTA DA cells from mice that received a single in vivo injection of ethanol (2 gm/kg, i.p.) or saline the day before the recordings.
First, to determine whether changes in the probability of GABA release occurred at these synapses in ethanol-treated mice, we use the paired-pulse stimulation protocol to test for changes in synaptic strength elicited by paired stimuli given at an interval of 50 msec. It has been shown that changes in transmitter release would generally affect the paired pulse ratio (Khazipov et al., 1995, Mennerick and Zorumski, 1995; Debanne et al., 1996; Salin et al., 1996; Stoop and Poo, 1996; Murthy et al., 1997; Gottschalk et al., 1998;Hernandez-Echeagaray et al., 1998; Lessmann and Heumann, 1998; Emmerson and Miller, 1999; Niittykoski et al., 1999; Stanford and Cooper, 1999;Steffensen et al., 1999; Sullivan, 1999; Jiang et al., 2000; Poncer et al., 2000; Yun et al., 2000; Cooper and Stanford, 2001; Rozov et al., 2001). Although some inconsistencies have recently been reported for some brain areas (Brody and Yue, 2000; Kraushaar and Jonas, 2000;Waldeck et al., 2000), it is well established that changes in transmitter release affect the paired-pulse ratio (PPR) in the VTA (Bonci and Williams, 1997; Manzoni and Williams, 1999). In slices from saline-treated mice, we observed paired-pulse facilitation (PPF), with the second pulse evoking a GABAA IPSC (IPSC2) that was significantly larger than the first (Fig.1a,e; IPSC2/IPSC1 = 1.3 ± 0.06; n = 16). Conversely, ethanol-treated mice exhibited paired pulse depression (PPD; Fig. 1b,e; IPSC2/IPSC1 = 0.8 ± 0.02; n = 17;p < 0.05). Thus, after a single in vivoexposure to ethanol, the paired-pulse protocol resulted in PPD (Fig.1c–e). PPF or PPD did not depend on the size of the first GABAA IPSC (IPSC1; Fig. 1c) and was not affected by changes in stimulus intensity (Fig. 1d). In addition, we analyzed the PPR by dividing the mean of IPSC2 by the mean of IPSC1, and we find this method yields similar results (p < 0.05; ethanol, IPSC2/IPSC1 = 0.7 ± 0.03; n = 17; saline, IPSC2/IPSC1 = 1.1 ± 0.06; n = 16).
Fig. 1.

Increased probability of GABA release 24 hr after a single in vivo exposure to ethanol. GABAAIPSCs from ethanol-treated mice show PPD compared with saline controls, which show PPF. a, b, Examples of recordings from saline-treated (a) and ethanol-treated (b) animals. c, No correlation was found between the amplitude of IPSC1 and the IPSC2/IPSC1 ratio in both saline-treated mice (n = 16) and ethanol-treated mice (n = 17). d, The IPSC2/IPSC1 ratio is independent of the stimulus strength. Results are the average from four cells in each group of animals. e, PPD in ethanol-treated mice is a long-lasting phenomenon. The bar graph shows the average IPSC2/IPSC1 ratio (mean ± SEM) of saline- and ethanol-treated mice after 1 d (n= 16 and 17 for saline and ethanol, respectively; *p < 0.05), 1 week (n = 9 per group; *p < 0.05), and 2 weeks (n = 8 per group; p > 0.05) after ethanol pre-exposure. f, Ethanol pre-exposure does not change either number or function of postsynaptic GABAAreceptors. Bath application of GABA (100 μm, 3 min) in the presence of GABAB receptor antagonist CGP 35348 (100 μm) elicited a similar current (30 sec bins) in both groups of animals (n = 4; p > 0.05) when neurons were voltage-clamped at −70 mV.
A change in PPR toward PPD might reflect changes in probability of release, function of postsynaptic GABAAreceptors, or a combination of these. To determine whether changes in GABAA receptor function, number, or both occurred after in vivo ethanol administration, we bath applied GABA (100 μm, 3 min) in presence of the GABAB receptor antagonist (3-aminopropyl)(diethoxymethyl) phosphinic acid (CGP35348) (100 μm). The amplitude of the inward current elicited by GABA did not differ between ethanol- and saline-treated animals (n = 4 for each group;p > 0.05; Fig. 1f). These results suggest that modifications in transmitter release, rather than in postsynaptic receptors, occur after an in vivo exposure to ethanol.
To further test this possibility, we examined spontaneous GABAA mIPSCs. Figure2, a–c, shows that the frequency of mIPSCs was significantly higher in ethanol- than in saline-treated animals (ethanol, 2.8 ± 0.4 Hz; n= 9; saline, 0.7 ± 0.1 Hz; n = 7;p < 0.05). Furthermore, there was no significant difference in the amplitude of mIPSCs in the two groups, with mean amplitudes of 30.4 ± 4.2 and 29.2 ± 3.4 pA in saline- and ethanol-treated mice, respectively (Fig. 2c,d;p > 0.05). Because an increase in frequency but not amplitude of mIPSCs is generally thought to reflect a presynaptic increase in probability of transmitter release (Malenka and Nicoll, 1999), both the paired pulse protocol and the increased frequency of spontaneous events indicate that the probability of GABA release in the VTA was increased 1 d after a single exposure to ethanol.
Fig. 2.

Ethanol pre-exposure increased the frequency, but not amplitude, of spontaneous mIPSCs. a, Samples of mIPSCs from saline-treated mice (top traces) and ethanol-treated mice (bottom traces). b,Bar graph showing the average (mean ± SEM) frequency for saline-treated animals (n = 7) and ethanol-treated animals (n = 9; *p < 0.05). c, Bar graph (10 pA bins) showing an amplitude histogram of mIPSCs for ethanol-treated (n = 9) versus saline-treated (n = 7) mice. d, Bar graph showing the average (mean ± SEM) amplitude for saline-treated animals (n = 7) and ethanol-treated animals (n = 9).
Previous in vivo studies have shown that reduced DAergic activity persists long after somatic signs of withdrawal have subsided (Diana et al., 1996). Therefore, we collected evoked GABAA IPSCs by using the paired-pulse protocol both 1 and 2 weeks after the single in vivo exposure to ethanol to determine how long the PPD lasted. Figure 1eshows that PPD was still present after 1 week (ethanol, IPSC2/IPSC1 = 0.9 ± 0.08; n = 9;p < 0.05; saline, IPSC2/IPSC1 = 1.2 ± 0.08;n = 7), but that within 2 weeks, the phenomenon had subsided (ethanol, IPSC2/IPSC1 = 1.1 ± 0.06;n = 8; p > 0.05; saline, IPSC2/IPSC1 = 1.2 ± 0.05; n = 7). These data are consistent with the idea that increased probability of GABA release might contribute to the long-lasting hypoactivity of DA neurons that has been observed in vivo after ethanol exposure (Diana et al., 1996).
Behavioral studies reported that a single in vivo exposure to ethanol produced an increase in subsequent self-administration of ethanol in rodents (Camarini et al., 2000; Files et al., 2000). In addition, activation of GABAA receptors plays a role in ethanol self-administration, because GABAA agonists facilitate acquisition of voluntary ethanol drinking in rats (Smith et al., 1992; Nowak et al., 1998). We therefore decided to measure the ethanol consumption in mice that underwent the same experimental protocol. To perform these experiments, 24 hr after a single exposure to ethanol (2 gm/kg, i.p.), mice were given a two-bottle choice test with two concentrations of ethanol versus water for 5 d at each concentration (2 and 5% v/v, respectively). We began with the lower concentration for 5 d to allow the animals to acclimate to the taste of ethanol. As reported previously (Camarini et al., 2000), C57BL/6J mice pretreated with ethanol consumed more ethanol than saline controls after 10 d of housing in the continuous access situation (Fig.3a). We found that 10 d after the acute exposure, the mean ethanol intake was 9.3 ± 0.6 and 7.4 ± 0.4 gm/kg for ethanol-treated (n = 8) and saline-treated (n = 8) mice, respectively (p < 0.05). Although both groups preferred water to ethanol, ethanol-treated mice showed an increased preference for ethanol (46.9 ± 0.5%; n = 8;p < 0.05; Fig. 3b) when compared with saline controls (41.1 ± 1.5%; n = 8). However, because differential absorption, distribution, or clearance of ethanol may contribute to the increased ethanol intake observed in ethanol-treated mice, we measured blood ethanol concentrations 10–90 min after administration of ethanol (4 gm/kg, i.p.). Figure3c shows that ethanol clearance did not differ between the two groups of animals, although it showed lower blood ethanol levels than commonly reported (Hodge et al., 1999; Thiele et al., 2000; Wand et al., 2001). One possible explanation for such low levels might be the faster metabolism of younger mice used for the present study compared with the 2- to 4-month-old mice tested in other studies (Hodge et al., 1999; Thiele et al., 2000; Wand et al., 2001). Nevertheless, taken together, these results support the hypothesis that an increased GABAergic transmission in the VTA may be involved in facilitating or maintaining ethanol consumption (Smith et al., 1992; Nowak et al., 1998).
Fig. 3.
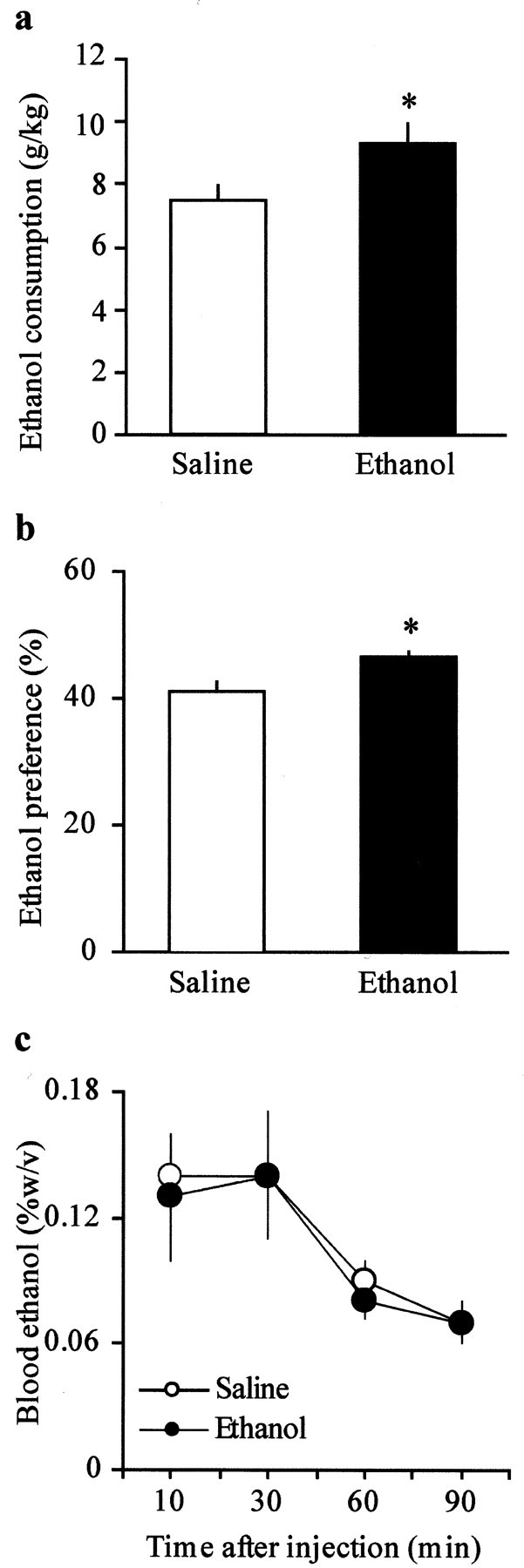
One week after the pre-exposure, ethanol-treated mice show increased ethanol intake and preference compared with saline controls. a, Voluntary 24 hr ethanol consumption (grams per kilogram) in C57BL/6J mice pretreated with ethanol (n = 8; *p < 0.05) and saline (n = 8). b, Ethanol preference, calculated as 100 × milliliters of ethanol per total milliliters consumed. C57BL/6J mice pretreated with ethanol demonstrated a significant increase in ethanol preference (n = 8; *p < 0.05) when compared with saline controls (n = 8). c, Blood ethanol clearance after acute administration of ethanol (4 gm/kg, i.p.) did not differ between ethanol- and saline-treated mice. Data (mean ± SEM) represent four animals per each group at every time point.
The observed difference in PPR between the two groups of animals might result indirectly from activation of presynaptic receptors. In the midbrain, GABAB receptors are present presynaptically and postsynaptically, and it has been shown that the activation of presynaptic GABAB receptors causes inhibition of GABAA IPSCs (Johnson and North, 1992b; Hausser and Yung, 1994). Therefore, to test the possibility that increased probability of GABA release might raise GABA levels and thus activate presynaptic GABAB receptors, we measured the PPR in the presence of the GABAB receptor antagonist CGP35348 (100 μm, 5 min). Figure4, a and b, shows that CGP35348 significantly shifted the PPD to PPF in ethanol-treated animals (IPSC2/IPSC1 = 0.7 ± 0.04–1.2 ± 0.08;n = 10; p < 0.05) by increasing the amplitude of the second evoked GABAA IPSC (IPSC2, 140 ± 9%; data not shown) without affecting either GABAA IPSC in the saline-treated animals (IPSC2/IPSC1 = 1.5 ± 0.1–1.4 ± 0.2; n= 5; p > 0.05). In addition, both the frequency and the amplitude of mIPSCs were unaffected by CGP35348 (100 μm, 5 min) in both groups of mice (frequency: ethanol, 2.9 ± 0.2–2.6 ± 0.3 Hz; n = 7; saline, 1.1 ± 0.1–1.1 ± 0.1 Hz; n = 7; amplitude: ethanol, 33.6 ± 1.8–33 ± 2.9 pA;n = 5; saline, 33.2 ± 2.1–26.9 ± 2.6 pA;n = 7; Fig. 4c,d). Thus, the PPD observed in the ethanol-treated mice could result from an increased probability of GABA release, which might in turn lead to activation of presynaptic GABAB receptors and decrease the IPSC2.
Fig. 4.

Effect of the GABAB receptor antagonist CGP35348 (100 μm) on IPSC2/IPSC1 ratio and spontaneous mIPSCs. a, CGP35348 (100 μm, 5 min) shifts the PPD to PPF in ethanol-treated mice (n = 10; *p < 0.05), without affecting the PPF in saline-treated mice (n = 5).b, The IPSC2/IPSC1 ratio is plotted as a function of time in cells recorded from saline- and ethanol-treated mice and normalized against the mean of the first 10 min for each cell.c, CGP35348 does not change the frequency of mIPSCs in either group (n = 7 per each group).d, No changes in amplitude of spontaneous mIPSCs were found in either group (n = 7 per each group).
An alternative interpretation of the present results is that the sensitivity of presynaptic GABAB receptors might be enhanced in the ethanol-treated animals. Therefore, we tested differences in sensitivity of presynaptic GABABreceptors by comparing the inhibition caused by the GABAB receptor agonist baclofen in slices from both saline- and ethanol-treated animals. Figure5a shows that the concentration–response curves to baclofen were similar in the saline- and ethanol-treated mice (baclofen 0.1 μm: ethanol, 22.3 ± 8.9%; n = 5; saline, 25.4 ± 3.1%; n = 5; baclofen 1 μm: ethanol, 42.8 ± 12.3%; n = 5; saline, 44.5 ± 2.3%; n = 5; baclofen 10 μm: ethanol, 70.6 ± 11.8%;n = 5; saline, 72.7 ± 7.2%; n = 5). Because the amplitude of the IPSC1 was decreased in both groups of animals to the same extent at all doses tested (Fig. 5a,b), we concluded that the increase in the IPSC2 observed in the presence of CGP35348 in ethanol-treated animals did not result from altered sensitivity of GABAB receptors to endogenous GABA. Additionally, a high dose of baclofen (10 μm; Fig. 5c,d) reverted the PPD to PPF in ethanol-treated mice (IPSC2/IPSC1 = 0.7 ± 0.02–1.2 ± 0.1; n = 5; p < 0.05), but it produced a nonsignificant increase in PPF in saline-treated animals (IPSC2/IPSC1 = 1.2 ± 0.1–1.3 ± 0.1; n = 5). These results further support the hypothesis that GABA levels are increased after ethanol exposure, leading to spillover onto presynaptic GABABreceptors, whose activation leads to inhibition of release (Hausser and Yung, 1994). Because changes in the paired-pulse ratio generally reflect changes in the probability of release, we expected baclofen to increase PPF in the saline group. However, we did detect a nonsignificant increase in the PPF during the application of baclofen at this concentration (IPSC2/IPSC1 = 1.2 ± 0.09–1.3 ± 0.02; n = 5; p > 0.05) and at 1 μm (IPSC2/IPSC1 = 1.1 ± 0.07–1.2 ± 0.08; n = 5; p > 0.05) in the saline-treated animals. This indicates that the PPR measure is not sensitive enough in this range to detect a decrease in GABA release, possibly as a result of a ceiling effect.
Fig. 5.

Effect of the GABAB receptor agonist baclofen on evoked IPSCs. a, Concentration–response curve for baclofen measuring the amplitude of IPSC1 from saline- and ethanol-treated animals (n = 5 per each group at all doses tested). b, Baclofen (1 μm, 10 min) decreases the amplitude of IPSC1 in both saline- and ethanol-treated mice to the same extent (n = 5 per each group). c, Baclofen (10 μm, 10 min) shifts the PPD to PPF in ethanol-treated mice (n = 5; *p < 0.05) without affecting the PPF in saline-treated mice (n = 5). d, IPSC2/IPSC1 ratio plotted as a function of time in cells recorded from saline- and ethanol-treated mice (n = 5 per group; *p < 0.05) and normalized against the mean of the first 10 min for each cell.
An increase in probability of GABA release has been described previously during acute withdrawal from chronic morphine in several brain regions, including the VTA (Bonci and Williams, 1997). In particular, this phenomenon has been characterized as being cAMP-dependent in the VTA (Bonci and Williams, 1997), the periaqueductal gray (Ingram et al., 1998), the NAcc (Chieng and Williams, 1998), and the dorsal raphe nucleus (Jolas et al., 2000). Ethanol and other drugs of abuse are known to modulate the cAMP–protein kinase A (PKA) cascade within the mesolimbic system (Hoffman and Tabakoff, 1990; Self et al., 1998; Spanagel and Weiss, 1999). Therefore, to examine the possibility that the cAMP-dependent pathway was modified in the ethanol-treated animals, we directly activated adenylyl cyclase (AC) by bath applying forskolin. Forskolin (10 μm, 10 min) augmented the IPSC1 in saline-treated animals (Fig. 6a; 111.6 ± 17.4%; n = 5; p < 0.05) but had no effect on the amplitude of the IPSC1 in ethanol-treated mice (23.1 ± 13.3%; n = 5). Thus, application of forskolin decreased the paired-pulse ratio toward depression in slices from saline-treated animals (Fig. 6b; IPSC2/IPSC1 = 1.4 ± 0.1–0.9 ± 0.1; n = 5; p < 0.05) but was without effect in slices from ethanol-treated animals (IPSC2/IPSC1 = 0.8 ± 0.05–0.7 ± 0.1;n = 5). This supports the idea that activation of AC increased the probability of GABA release. In addition, the frequency of spontaneous mIPSCs was also significantly increased by forskolin in saline-treated mice (0.7 ± 0.1–1.7 ± 0.3 Hz;n = 7; p < 0.05; Fig. 6c) but not in the ethanol-treated animals (2.6 ± 0.7–2.9 ± 1.0 Hz; n = 8; Fig. 6c). There was no significant difference in the amplitude of the mIPSCs in the absence or presence of forskolin in slices from either group of animals (saline, 29.4 ± 2.5–31 ± 2.9 pA; n = 7; ethanol, 34 ± 4.2–26.5 ± 1.7 pA; n = 8; Fig.6d). To rule out the possibility of nonspecific effects of forskolin, we tested 1,9-dideoxyforskolin, an inactive analog of forskolin (Seamon and Daly, 1985). Superfusion of 1,9-dideoxyforskolin (10 μm, 10 min) had no effect on IPSCs in either group of animals (7.7 ± 3.3 and 1.1 ± 2.6% in ethanol- and saline-treated mice, respectively; n = 4 for each group; data not shown). Taken together, these results suggest that a saturation of AC might occur after a single in vivoexposure to ethanol.
Fig. 6.

Effect of forskolin on evoked and spontaneous IPSCs. a, Forskolin (10 μm, 10 min) increases the amplitude of evoked IPSC1 in saline-treated mice (n = 5; *p < 0.05) but not ethanol-treated mice (n = 5). b, Forskolin (10 μm, 10 min) shifts the PPF to PPD in saline-treated mice (n = 5; *p< 0.05) without affecting the PPD in ethanol-treated mice (n = 5). The IPSC2/IPSC1 ratio is plotted as function of time in cells recorded from saline- and ethanol-treated mice and normalized against the mean of the first 10 min for each cell.c, Forskolin (10 μm, 10 min) induces a significant increase in the frequency of mIPSCs in saline-treated mice (n = 7; *p < 0.05) but not in ethanol-treated mice (n = 9). d, No changes in amplitude were found in either group.
Because changes of intracellular cAMP levels subsequently alter PKA activity, we tested the effect ofN-[2(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H89), which inhibits PKA in a competitive manner against ATP (Chijiwa et al., 1990). Superfusion of H89 (10 μm, 20 min) significantly reduced the amplitude of IPSC1 in ethanol-treated mice (38.6 ± 5.4%; n = 5; p < 0.05; Fig. 7a) but had no effect in slices from control animals (3.4 ± 8.1%; n = 5). The subsequent application of forskolin (10 μm, 10 min) in the presence of H89, used to test the activity of H89 in control animals, did not change the amplitude of the IPSC1 in either group (Fig. 7a). Furthermore, H89, by reducing the size of IPSC1, shifted the PPD to PPF in slices from ethanol-treated animals (IPSC2/IPSC1 = 0.8 ± 0.02–1.1 ± 0.01;n = 5; p < 0.05; Fig. 7b). Consistent with these results, H89 reduced the frequency of spontaneous mIPSC in the ethanol-treated animals (2.6 ± 0.1–1.6 ± 0.1 Hz; n = 6; p < 0.05; Fig.7c) but not in the saline-treated animals (0.9 ± 0.1–0.8 ± 0.1 Hz; n = 5); in the presence of H89, the amplitude of spontaneous mIPSCs was not changed in either saline-treated mice (31.7 ± 5.2–31.3 ± 2.6 pA;n = 5) or ethanol-treated mice (28.7 ± 3.1–24.8 ± 2.9 pA; n = 6). In conclusion, these experiments indicate that PKA activity is significantly enhanced by a single exposure to ethanol, and that such a phenomenon increases the probability of GABA release in the VTA.
Fig. 7.

Effect of H89 on evoked and spontaneous IPSCs.a, H89 (10 μm, 20 min) decreases the amplitude of evoked IPSCs in ethanol-treated mice (n = 5; *p < 0.05), but not saline-treated mice (n = 5). b, H89 (10 μm, 20 min) shifts the PPD to PPF in ethanol-treated mice (n = 5; *p < 0.05) without affecting the PPF in saline-treated mice. c, H89 (10 μm, 20 min) induces a significant decrease in the frequency of mIPSCs in ethanol-treated mice (n = 6; *p < 0.05) but not saline-treated mice (n = 5). d, No changes in amplitude were found in either group.
DISCUSSION
In the present study, we observe that a single in vivoexposure to ethanol produces a long-lasting increase in the probability of GABA release in the VTA, and that such an increase is dependent on the activation of the cAMP–PKA signaling cascade. We hypothesize that this type of plasticity may play an important role in determining the increased alcohol consumption observed after a single exposure to ethanol (Spanagel and Weiss, 1999; Camarini et al., 2000). Our data, together with the fact that we and others have observed increased ethanol consumption when mice were pre-exposed to ethanol, indicate that increased probability of GABA release and increased ethanol self-administration, both produced by the single ethanol injection, might be strictly associated. Indeed, activation of GABAA receptors plays a role in ethanol self-administration, because GABAA agonists facilitate acquisition of voluntary ethanol drinking in rats (Smith et al., 1992; Nowak et al., 1998). Accordingly, a role of GABAA receptors within the VTA in mediating ethanol intake has been suggested (Samson et al., 1987; Boyle et al., 1993; Rassnick et al., 1993b). Indeed, systemic administration of GABAA receptor antagonists reduces intake (Boyle et al., 1993) and operant responding for ethanol in rats (Samson et al., 1987; Rassnick et al., 1993b). Consistent with these and previous findings, intra-VTA infusions of GABAA receptor antagonists decreased ethanol consumption in rats of the alcohol-preferring P line (Nowak et al., 1998). To further support the hypothesis that increased GABAergic activity produced by a singlein vivo exposure to ethanol plays a role in ethanol-related behaviors, it has recently been shown that there is a direct relationship between pretreatment with ethanol and enhanced self-administration of ethanol in mice (Camarini et al., 2000). Specifically, C57BL/6J mice pre-exposed to ethanol exhibited a significant increase of ethanol intake, and DBA/2J mice, which normally avoid oral ingestion of ethanol, did start to self-administer ethanol in a two-bottle choice test.
In our first set of experiments, we show that a single injection of ethanol shifted the paired-pulse modulation of GABAA IPSCs from PPF to PPD. The paired pulse stimulation is typically used as an electrophysiological protocol to test for changes in probability of transmitter release (Zucker, 1989;Stuart and Redman, 1991; Manabe et al., 1993; Mennerick and Zorumski, 1995; Debanne at al., 1996). Although this phenomenon is not always use-dependent (Brody and Yue, 2000; Kraushaar and Jonas, 2000; Waldeck et al., 2000), a variety of manipulations that increase transmitter release, including exposure to drugs of abuse, have been found to shift the paired-pulse ratio from facilitation toward depression in the hippocampus (Mennerick and Zorumski, 1995; Salin et al., 1996) and the VTA (Bonci and Williams, 1997). Furthermore, the persistence of PPD 1 week after the ethanol injection suggests that increased GABAA-mediated inhibition may be considered a measure of changes occurring at these synapses, eventually contributing to the expression of ethanol-seeking behavior.
However, an increase in the probability of GABA release might simply be one of many factors determining the shift from PPF to PPD in the ethanol-treated animals. Although a desensitization of postsynaptic GABAA receptors could account for PPD, we tend to rule out that possibility, because the ethanol-treated animals show an increase in mISPCs frequency but not in amplitude when compared with the saline-treated animals. Furthermore, bath application of GABA, in the presence of a GABAB receptor antagonist, produces similar responses in saline- and ethanol-treated animals.
An alternative explanation for the observed PPD in the ethanol-treated animals, is that activation of presynaptic GABABreceptors might occur as a consequence of increased GABA levels produced by the first evoked stimulus, thus reducing the amplitude of IPSC2. Indeed, it has been shown that activation of GABAB receptors inhibits GABAA IPSCs in the midbrain via a presynaptic mechanism and therefore are considered to serve also as autoreceptors (Hausser and Yung, 1994). Our results showing that the GABAB antagonist CGP35348 shifted PPD to PPF in animals injected with ethanol, but not in the saline controls, indicate that presynaptic GABAB receptors might play a minor role when probability of GABA release is relatively low, as in the saline-injected animals. However, they might act as a negative feedback mechanism to regulate GABAergic transmission within the VTA when probability of GABA release is increased, such as after a singlein vivo exposure to ethanol. Thus, our data indicate that increased GABA levels, by changing the spatial range of synaptically released GABA, allow the activation of presynaptic GABAB receptors located on the GABAergic interneurons, which in turn would prevent excessive GABAA-mediated synaptic transmission (McCarren and Alger, 1985; Deisz and Prince, 1989; Davies et al., 1990; Isaacson et al., 1993).
Our results also suggest that the ethanol-induced increase in the probability of GABA release was a result of saturation of the AC cascade within GABAergic terminals. Forskolin, which enhanced the amplitude of evoked IPSCs and the frequency of mIPSCs in saline-treated mice, had no effect in the ethanol-treated mice. In addition, the PKA inhibitor H89 reduced the amplitude of evoked IPSCs and the frequency of mIPSCs only in ethanol-treated animals, whereas it had no effect in saline-treated animals. These findings suggest that a single in vivo exposure to ethanol results in persistent enhancement of PKA-dependent processes in GABAergic terminals in the VTA. It is possible that because of high cAMP levels after the exposure to ethanol, the catalytic subunits of the PKA complex become unbound and freely diffuse within the terminals. In fact, in slices from ethanol-treated animals, H89 revealed an increased basal activity of PKA, and the activation of AC by forskolin was blunted. Consistent with our findings, reduced signaling through the cAMP–PKA system, whether because of decreased expression of the α subunit of the stimulatory G-protein (Gsα) or inhibition of PKA, changed C57BL/6J mice, considered to be an ethanol-preferring line of mice, into ethanol nonpreferring mice (Wand et al., 2001). In addition, alcohol-preferring rats show increased AC activity and expression of Gsα in mesolimbic regions when compared with alcohol-nonpreferring rats (Froehlich and Wand, 1997). More generally, our results are in agreement with previous studies reporting that genetic manipulations of the cAMP–PKA pathway modulate ethanol intake and sensitivity to its sedative effects (Thiele et al., 2000; Wand et al., 2001). Although the relationships between the sedative and rewarding effects of ethanol are complex, it is also important to mention that the cAMP–PKA system has been implicated in neural plasticity associated with drug tolerance and dependence (Self and Nestler, 1995; Moore et al., 1998; Andretic et al., 1999; Yoshimura and Tabakoff, 1999). Finally, it has been shown that chronic exposure to many drugs of abuse, including ethanol, also leads to increased activity of cAMP-dependent processes (Terwilliger et al., 1991; Dohrman et al., 1996; Bonci and Williams, 1997).
In conclusion, our results provide evidence that a single in vivo exposure to ethanol produces a long-lasting potentiation of GABAergic synapses in the VTA. This cAMP–PKA-dependent plasticity occurring at these synapses might represent an important cellular signaling event underlying increased ethanol consumption. Whether these changes are the result of a compensation for acute effects of ethanol or manifestation of a long-lasting effect of acute ethanol remains to be elucidated. Although acute effects of ethanol on the GABAergic systems in the CNS are still a matter of debate, we tend to support the latter possibility, because ethanol has been found to enhance GABAergic transmission in several brain regions (Celentano et al., 1988; Deitrich et al., 1989; Aguayo and Pancetti, 1994; Mehta and Ticku, 1994; Wan et al., 1996; Nie et al., 2000), including the VTA (M. Melis and A. Bonci, unpublished observations). In conclusion, we observed that a singlein vivo ethanol exposure induces a long-lasting potentiation of GABA synaptic transmission within the VTA via a cAMP–PKA mechanism, and that it appears to be directly related to increased ethanol consumption and may therefore be involved in the development of alcoholism.
Footnotes
This work was supported by funds provided by the State of California for medical research on alcohol and substance abuse through the University of California, San Francisco, and by the Department of the Army, award number DAMD17-01-10736, United States Army Medical Research Acquisition Activity (Fort Detrick, MD). The content of the information does not necessarily reflect the position or the policy of the Government, and no official endorsement should be inferred. We thank all members of the Bonci Lab for many helpful discussions and inputs. We also deeply thank the late Dr. Thomas Dunwiddie for his many helpful insights. He will be sorely missed in the future.
Correspondence should be addressed to Antonello Bonci at the above address. E-mail: bonci@itsa.ucsf.edu.
REFERENCES
- 1.Aguayo LG, Pancetti FC. Ethanol modulation of the gamma-aminobutyric acidA- and glycine-activated Cl− current in cultured mouse neurons. J Pharmacol Exp Ther. 1994;270:61–69. [PubMed] [Google Scholar]
- 2.Andretic R, Chaney S, Hirsh J. Requirement of circadian genes for cocaine sensitization in Drosophila. Science. 1999;285:1066–1068. doi: 10.1126/science.285.5430.1066. [DOI] [PubMed] [Google Scholar]
- 3.Bailey CP, Manley SJ, Watson WP, Wonnacott S, Molleman A, Little HJ. Chronic ethanol administration alters activity in ventral tegmental area neurons after cessation of withdrawal hyperexcitability. Brain Res. 1998;803:144–152. doi: 10.1016/s0006-8993(98)00654-4. [DOI] [PubMed] [Google Scholar]
- 4.Bonci A, Williams JT. Increased probability of GABA release during withdrawal from morphine. J Neurosci. 1997;17:796–803. doi: 10.1523/JNEUROSCI.17-02-00796.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boyle AE, Segal R, Smith BR, Amit Z. Bidirectional effects of GABAergic agonists and antagonists on maintenance of voluntary ethanol intake in laboratory rats. Pharmacol Biochem Behav. 1993;46:179–182. doi: 10.1016/0091-3057(93)90338-t. [DOI] [PubMed] [Google Scholar]
- 6.Brodie MS, Appel SB. The effects of ethanol on dopaminergic neurons of the ventral tegmental area studied with intracellular recording in brain slices. Alcohol Clin Exp Res. 1998;22:236–244. [PubMed] [Google Scholar]
- 7.Brodie MS, Appel SB. Dopaminergic neurons in the ventral tegmental area of C57BL/6J and DBA/2J mice differ in sensitivity to ethanol excitation. Alcohol Clin Exp Res. 2000;24:1120–1124. [PubMed] [Google Scholar]
- 8.Brody DL, Yue DT. Release-independent short-term synaptic depression in cultured hippocampal neurons. J Neurosci. 2000;20:2480–2494. doi: 10.1523/JNEUROSCI.20-07-02480.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brodie MS, Shefner SA, Dunwiddie TV. Ethanol increases the firing rate of dopamine neurons of the rat ventral tegmental area in vitro. Brain Res. 1990;508:65–69. doi: 10.1016/0006-8993(90)91118-z. [DOI] [PubMed] [Google Scholar]
- 10.Brodie MS, Pesold C, Appel SB. Ethanol directly excites dopaminergic ventral tegmental area reward neurons. Alcohol Clin Exp Res. 1999;23:1848–1852. [PubMed] [Google Scholar]
- 11.Brundege JM, Dunwiddie TV. Modulation of excitatory synaptic transmission by adenosine released from single hippocampal pyramidal neurons. J Neurosci. 1996;16:5603–5612. doi: 10.1523/JNEUROSCI.16-18-05603.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Camarini R, Mehmert KK, Hodge CW. Effects of ethanol pre-exposure on ethanol self-administration in DBA/2J and C57BL/6 mice. Alcohol Clin Exp Res [Suppl] 2000;24:16A. [Google Scholar]
- 13.Celentano JJ, Gibbs TT, Farb DH. Ethanol potentiates GABA- and glycine-induced chloride currents in chick spinal cord neurons. Brain Res. 1988;455:377–380. doi: 10.1016/0006-8993(88)90098-4. [DOI] [PubMed] [Google Scholar]
- 14.Chieng B, Williams JT. Increased opioid inhibition of GABA release in nucleus accumbens during morphine withdrawal. J Neurosci. 1998;18:7033–7039. doi: 10.1523/JNEUROSCI.18-17-07033.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chijiwa T, Mishima A, Hagiwara M, Sano M, Hayashi K, Inoue T, Naito K, Toshioka T, Hidaka H. Inhibition of forskolin-induced neurite outgrowth and protein phosphorylation by a newly synthesized selective inhibitor of cyclic AMP-dependent protein kinase, N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulfonamide (H-89), of PC12D pheochromocytoma cells. J Biol Chem. 1990;265:5267–5272. [PubMed] [Google Scholar]
- 16.Cooper AJ, Stanford IM. Dopamine D2 receptor mediated presynaptic inhibition of striatopallidal GABA(A) IPSCs in vitro. Neuropharmacology. 2001;41:62–71. doi: 10.1016/s0028-3908(01)00038-7. [DOI] [PubMed] [Google Scholar]
- 17.Davies CH, Davies SN, Collingridge GL. Paired-pulse depression of monosynaptic GABA-mediated inhibitory postsynaptic responses in rat hippocampus. J Physiol (Lond) 1990;424:513–531. doi: 10.1113/jphysiol.1990.sp018080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Debanne D, Guerineau NC, Gahwiler BH, Thompson SM. Paired-pulse facilitation and depression at unitary synapses in rat hippocampus: quantal fluctuation affects subsequent release. J Physiol (Lond) 1996;491:163–176. doi: 10.1113/jphysiol.1996.sp021204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Deisz RA, Prince DA. Frequency-dependent depression of inhibition in guinea-pig neocortex in vitro by GABAB receptor feed-back on GABA release. J Physiol (Lond) 1989;412:513–541. doi: 10.1113/jphysiol.1989.sp017629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deitrich RA, Dunwiddie TV, Harris RA, Erwin VG. Mechanism of action of ethanol: initial central nervous system actions. Pharmacol Rev. 1989;41:489–537. [PubMed] [Google Scholar]
- 21.Diana M, Pistis M, Carboni S, Gessa GL, Rossetti ZL. Profound decrement of mesolimbic dopaminergic neuronal activity during ethanol withdrawal syndrome in rats: electrophysiological and biochemical evidence. Proc Natl Acad Sci USA. 1993;90:7966–7969. doi: 10.1073/pnas.90.17.7966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Diana M, Pistis M, Muntoni AL, Gessa GL. Mesolimbic dopaminergic reduction outlasts ethanol withdrawal syndrome: evidence of protracted abstinence. Neuroscience. 1996;71:411–415. doi: 10.1016/0306-4522(95)00482-3. [DOI] [PubMed] [Google Scholar]
- 23.Dohrman DP, Diamond I, Gordon AS. Ethanol causes translocation of cAMP-dependent protein kinase catalytic subunit to the nucleus. Proc Natl Acad Sci USA. 1996;93:10217–10221. doi: 10.1073/pnas.93.19.10217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Emmerson PJ, Miller RJ. Pre- and postsynaptic actions of opioid and orphan opioid agonists in the rat arcuate nucleus and ventromedial hypothalamus in vitro. J Physiol (Lond) 1999;517:431–445. doi: 10.1111/j.1469-7793.1999.0431t.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Files FJ, Samson HH, Denning CE. Effects of prior ethanol exposure on ethanol self-administration in a continuous access situation using retractable drinking tubes. Alcohol. 2000;21:97–102. doi: 10.1016/s0741-8329(99)00102-0. [DOI] [PubMed] [Google Scholar]
- 26.Froehlich JC, Wand GS. Adenylyl cyclase signal transduction and alcohol-induced sedation. Pharmacol Biochem Behav. 1997;58:1021–1030. doi: 10.1016/s0091-3057(97)00305-5. [DOI] [PubMed] [Google Scholar]
- 27.Fuxe K, Nystrom M, Tovi M, Smith R, Ogren SO. Central catecholamine neurons, behavior and neuroleptic drugs: an analysis to understand the involvement of catecholamines in schizophrenia. J Psychiatr Res. 1974;11:151–161. doi: 10.1016/0022-3956(74)90087-9. [DOI] [PubMed] [Google Scholar]
- 28.Gallegos RA, Lee RS, Criado JR, Henriksen SJ, Steffensen SC. Adaptive responses of gamma-aminobutyric acid neurons in the ventral tegmental area to chronic ethanol. J Pharmacol Exp Ther. 1999;291:1045–1053. [PubMed] [Google Scholar]
- 29.Gottschalk W, Pozzo-Miller LD, Figurov A, Lu B. Presynaptic modulation of synaptic transmission and plasticity by brain-derived neurotrophic factor in the developing hippocampus. J Neurosci. 1998;18:6830–6839. doi: 10.1523/JNEUROSCI.18-17-06830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haertzen CA, Kocher TR, Miyasato K. Reinforcements from the first drug experience can predict later drug habits and/or addiction: results with coffee, cigarettes, alcohol, barbiturates, minor and major tranquilizers, stimulants, marijuana, hallucinogens, heroin, opiates and cocaine. Drug Alcohol Depend. 1983;11:147–165. doi: 10.1016/0376-8716(83)90076-5. [DOI] [PubMed] [Google Scholar]
- 31.Hausser MA, Yung WH. Inhibitory synaptic potentials in guinea-pig substantia nigra dopamine neurons in vitro. J Physiol (Lond) 1994;479:401–422. doi: 10.1113/jphysiol.1994.sp020305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hernandez-Echeagaray E, Galarraga E, Bargas J. 3-Alpha-chloro-imperialine, a potent blocker of cholinergic presynaptic modulation of glutamatergic afferents in the rat neostriatum. Neuropharmacology. 1998;37:1493–1502. doi: 10.1016/s0028-3908(98)00131-2. [DOI] [PubMed] [Google Scholar]
- 33.Hodge CW, Samson HH, Chappelle AM. Alcohol self-administration: further examination of the role of dopamine receptors in the nucleus accumbens. Alcohol Clin Exp Res. 1997;21:1083–1091. doi: 10.1111/j.1530-0277.1997.tb04257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hodge CW, Mehmert KK, Kelley SP, McMahon T, Haywood A, Olive MF, Wang D, Sanchez-Perez AM, Messing RO. Supersensitivity to allosteric GABAA receptor modulators and alcohol in mice lacking PKCε. Nat Neurosci. 1999;2:997–1002. doi: 10.1038/14795. [DOI] [PubMed] [Google Scholar]
- 35.Hoffman PL, Tabakoff B. Ethanol and guanine nucleotide binding proteins: a selective interaction. FASEB J. 1990;4:2612–2622. doi: 10.1096/fasebj.4.9.2161371. [DOI] [PubMed] [Google Scholar]
- 36.Ikemoto S, McBride WJ, Murphy JM, Lumeng L, Li TK. 6-OHDA-lesions of the nucleus accumbens disrupt the acquisition but not the maintenance of ethanol consumption in the alcohol-preferring P line of rats. Alcohol Clin Exp Res. 1997;21:1042–1046. [PubMed] [Google Scholar]
- 37.Ingram SL, Vaughan CW, Bagley EE, Connor M, Christie MJ. Enhanced opioid efficacy in opioid dependence is caused by an altered signal transduction pathway. J Neurosci. 1998;18:10269–10276. doi: 10.1523/JNEUROSCI.18-24-10269.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Isaacson JS, Solis JM, Nicoll RA. Local and diffuse synaptic actions of GABA in the hippocampus. Neuron. 1993;10:165–175. doi: 10.1016/0896-6273(93)90308-e. [DOI] [PubMed] [Google Scholar]
- 39.Jiang L, Sun S, Nedergaard M, Kang J. Paired-pulse modulation at individual GABAergic synapses in rat hippocampus. J Physiol (Lond) 2000;523:425–439. doi: 10.1111/j.1469-7793.2000.t01-1-00425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Johnson SW, North RA. Two types of neurone in the rat ventral tegmental area and their synaptic inputs. J Physiol (Lond) 1992a;450:455–468. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. 1992b;12:483–488. doi: 10.1523/JNEUROSCI.12-02-00483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jolas T, Nestler EJ, Aghajanian GK. Chronic morphine increases GABA tone on serotonergic neurons of the dorsal raphe nucleus: association with an up-regulation of the cyclic AMP pathway. Neuroscience. 2000;95:433–443. doi: 10.1016/s0306-4522(99)00436-4. [DOI] [PubMed] [Google Scholar]
- 43.Kaczmarek HJ, Kiefer SW. Microinjections of dopaminergic agents in the nucleus accumbens affect ethanol consumption but not palatability. Pharmacol Biochem Behav. 2000;66:307–312. doi: 10.1016/s0091-3057(00)00182-9. [DOI] [PubMed] [Google Scholar]
- 44.Khazipov R, Congar P, Ben-Ari Y. Hippocampal CA1 lacunosum-moleculare interneurons: modulation of monosynaptic GABAergic IPSCs by presynaptic GABAB receptors. J Neurophysiol. 1995;74:2126–2137. doi: 10.1152/jn.1995.74.5.2126. [DOI] [PubMed] [Google Scholar]
- 45.Koob GF, Roberts AJ, Schulteis G, Parsons LH, Heyser CJ, Hyytia P, Merlo-Pich E, Weiss F. Neurocircuitry targets in ethanol reward and dependence. Alcohol Clin Exp Res. 1998;22:3–9. [PubMed] [Google Scholar]
- 46.Kraushaar U, Jonas P. Efficacy and stability of quantal GABA release at a hippocampal interneuron-principal neuron synapse. J Neurosci. 2000;20:5594–5607. doi: 10.1523/JNEUROSCI.20-15-05594.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lessmann V, Heumann R. Modulation of unitary glutamatergic synapses by neurotrophin-4/5 or brain-derived neurotrophic factor in hippocampal microcultures: presynaptic enhancement depends on pre-established paired-pulse facilitation. Neuroscience. 1998;86:399–413. doi: 10.1016/s0306-4522(98)00035-9. [DOI] [PubMed] [Google Scholar]
- 48.Malenka RC, Nicoll RA. Long-term potentiation—a decade of progress? Science. 1999;285:1870–1874. doi: 10.1126/science.285.5435.1870. [DOI] [PubMed] [Google Scholar]
- 49.Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA. Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol. 1993;70:1451–1459. doi: 10.1152/jn.1993.70.4.1451. [DOI] [PubMed] [Google Scholar]
- 50.Manzoni OJ, Williams JT. Presynaptic regulation of glutamate release in the ventral tegmental area during morphine withdrawal. J Neurosci. 1999;19:6629–6636. doi: 10.1523/JNEUROSCI.19-15-06629.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McBride WJ, Bodart B, Lumeng L, Li TK. Association between low contents of dopamine and serotonin in the nucleus accumbens and high alcohol preference. Alcohol Clin Exp Res. 1995;19:1420–1422. doi: 10.1111/j.1530-0277.1995.tb01001.x. [DOI] [PubMed] [Google Scholar]
- 52.McBride WJ, Murphy JM, Ikemoto S. Localization of brain reinforcement mechanisms: intracranial self-administration and intracranial place-conditioning studies. Behav Brain Res. 1999;101:129–152. doi: 10.1016/s0166-4328(99)00022-4. [DOI] [PubMed] [Google Scholar]
- 53.McCarren M, Alger BE. Use-dependent depression of IPSPs in rat hippocampal pyramidal cells in vitro. J Neurophysiol. 1985;53:557–571. doi: 10.1152/jn.1985.53.2.557. [DOI] [PubMed] [Google Scholar]
- 54.Mehta AK, Ticku MK. Ethanol enhancement of GABA-induced 36Cl− influx does not involve changes in Ca2+. Pharmacol Biochem Behav. 1994;47:355–357. doi: 10.1016/0091-3057(94)90022-1. [DOI] [PubMed] [Google Scholar]
- 55.Mennerick S, Zorumski CF. Paired-pulse modulation of fast excitatory synaptic currents in microcultures of rat hippocampal neurons. J Physiol (Lond) 1995;488:85–101. doi: 10.1113/jphysiol.1995.sp020948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Moore MS, DeZazzo J, Luk AY, Tully T, Singh CM, Heberlein U. Ethanol intoxication in Drosophila: genetic and pharmacological evidence for regulation by the cAMP signaling pathway. Cell. 1998;93:997–1007. doi: 10.1016/s0092-8674(00)81205-2. [DOI] [PubMed] [Google Scholar]
- 57.Murthy VN, Sejnowski TJ, Stevens CF. Heterogeneous release properties of visualized individual hippocampal synapses. Neuron. 1997;18:599–612. doi: 10.1016/s0896-6273(00)80301-3. [DOI] [PubMed] [Google Scholar]
- 58.Ng GY, George SR. Dopamine receptor agonist reduces ethanol self-administration in the ethanol-preferring C57BL/6J inbred mouse. Eur J Pharmacol. 1994;269:365–374. doi: 10.1016/0922-4106(94)90044-2. [DOI] [PubMed] [Google Scholar]
- 59.Nie Z, Yuan X, Madamba SG, Siggins GR. Ethanol decreases glutamatergic synaptic transmission in rat nucleus accumbens in vitro: naloxone reversal. J Pharmacol Exp Ther. 1993;266:1705–1712. [PubMed] [Google Scholar]
- 60.Nie Z, Madamba SG, Siggins GR. Ethanol inhibits glutamatergic neurotransmission in nucleus accumbens neurons by multiple mechanisms. J Pharmacol Exp Ther. 1994;271:1566–1573. [PubMed] [Google Scholar]
- 61.Nie Z, Madamba SG, Siggins GR. Ethanol enhances gamma-aminobutyric acid responses in a subpopulation of nucleus accumbens neurons: role of metabotropic glutamate receptors. J Pharmacol Exp Ther. 2000;293:654–661. [PubMed] [Google Scholar]
- 62.Niittykoski M, Ruotsalainen S, Haapalinna A, Larson J, Sirvio J. Activation of muscarinic M3-like receptors and beta-adrenoceptors, but not M2-like muscarinic receptors or alpha-adrenoceptors, directly modulates corticostriatal neurotransmission in vitro. Neuroscience. 1999;90:95–105. doi: 10.1016/s0306-4522(98)00447-3. [DOI] [PubMed] [Google Scholar]
- 63.Nowak KL, McBride WJ, Lumeng L, Li TK, Murphy JM. Blocking GABAA receptors in the anterior ventral tegmental area attenuates ethanol intake of the alcohol-preferring P rats. Psychopharmacology. 1998;139:108–116. doi: 10.1007/s002130050695. [DOI] [PubMed] [Google Scholar]
- 64.Nowak KL, McBride WJ, Lumeng L, Li TK, Murphy JM. Involvement of dopamine D2 autoreceptors in the ventral tegmental area on alcohol and saccharin intake of the alcohol-preferring P rat. Alcohol Clin Exp Res. 2000;24:476–483. [PubMed] [Google Scholar]
- 65.Oades RD, Halliday GM. Ventral tegmental (A10) system: neurobiology. 1. Anatomy and connectivity. Brain Res. 1987;434:117–165. doi: 10.1016/0165-0173(87)90011-7. [DOI] [PubMed] [Google Scholar]
- 66.Paladini CA, Celada P, Tepper JM. Striatal, pallidal, and pars reticulata evoked inhibition of nigrostriatal dopaminergic neurons is mediated by GABA(A) receptors in vivo. Neuroscience. 1999;89:799–812. doi: 10.1016/s0306-4522(98)00355-8. [DOI] [PubMed] [Google Scholar]
- 67.Phillips TJ, Huson M, Gwiazdon C, Burkhart-Kasch S, Shen EH. Effects of acute and repeated ethanol exposures on the locomotor activity of BXD recombinant inbred mice. Alcohol Clin Exp Res. 1995;19:269–278. doi: 10.1111/j.1530-0277.1995.tb01502.x. [DOI] [PubMed] [Google Scholar]
- 68.Phillips TJ, Brown KJ, Burkhart-Kasch S, Wenger CD, Kelly MA, Rubinstein M, Grandy DK, Low MJ. Alcohol preference and sensitivity are markedly reduced in mice lacking dopamine D2 receptors. Nat Neurosci. 1998;1:610–615. doi: 10.1038/2843. [DOI] [PubMed] [Google Scholar]
- 69.Poncer JC, McKinney RA, Gahwiler BH, Thompson SM. Differential control of GABA release at synapses from distinct interneurons in rat hippocampus. J Physiol (Lond) 2000;528:123–130. doi: 10.1111/j.1469-7793.2000.00123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rassnick S, Stinus L, Koob GF. The effects of 6-hydroxydopamine lesions of the nucleus accumbens and the mesolimbic dopamine system on oral self-administration of ethanol in the rat. Brain Res. 1993a;623:16–24. doi: 10.1016/0006-8993(93)90004-7. [DOI] [PubMed] [Google Scholar]
- 71.Rassnick S, D'Amico E, Riley E, Koob GF. GABA antagonists and benzodiazepine partial inverse agonists reduce motivated responding for ethanol. Alcohol Clin Exp Res. 1993b;7:124–130. doi: 10.1111/j.1530-0277.1993.tb00736.x. [DOI] [PubMed] [Google Scholar]
- 72.Risinger FO, Cunningham CL. Genetic differences in ethanol-induced conditioned taste aversion after ethanol pre-exposure. Alcohol. 1995;12:535–539. doi: 10.1016/0741-8329(95)00040-2. [DOI] [PubMed] [Google Scholar]
- 73.Rozov A, Burnashev N, Sakmann B, Neher E. Transmitter release modulation by intracellular Ca2+ buffers in facilitating and depressing nerve terminals of pyramidal cells in layer 2/3 of the rat neocortex indicates a target cell-specific difference in presynaptic calcium dynamics. J Physiol (Lond) 2001;531:807–826. doi: 10.1111/j.1469-7793.2001.0807h.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Salin PA, Malenka RC, Nicoll RA. Cyclic AMP mediates a presynaptic form of LTP at cerebellar parallel fiber synapses. Neuron. 1996;16:797–803. doi: 10.1016/s0896-6273(00)80099-9. [DOI] [PubMed] [Google Scholar]
- 75.Samson HH, Tolliver GA, Pfeffer AO, Sadeghi KC. Oral ethanol reinforcement and the effect of the partial inverse benzodiazepine agonist RO 15-4513. Pharmacol Biochem Behav. 1987;33:601–608. doi: 10.1016/0091-3057(87)90357-1. [DOI] [PubMed] [Google Scholar]
- 76.Samson HH, Hodge CW, Tolliver GA, Haraguchi M. Effect of dopamine agonists and antagonists on ethanol-reinforced behavior: the involvement of the nucleus accumbens. Brain Res Bull. 1993;30:133–141. doi: 10.1016/0361-9230(93)90049-h. [DOI] [PubMed] [Google Scholar]
- 77.Seamon KB, Daly JW. High-affinity binding of forskolin to rat brain membranes. Adv Cyclic Nucleotide Protein Phosphor Res. 1985;19:125–135. [PubMed] [Google Scholar]
- 78.Self DW, Nestler EJ. Molecular mechanisms of drug reinforcement and addiction. Annu Rev Neurosci. 1995;18:463–495. doi: 10.1146/annurev.ne.18.030195.002335. [DOI] [PubMed] [Google Scholar]
- 79.Self DW, Genova LM, Hope BT, Barnhart WJ, Spencer JJ, Nestler EJ. Involvement of cAMP-dependent protein kinase in the nucleus accumbens of cocaine self-administration and relapse of cocaine-seeking behavior. J Neurosci. 1998;18:1848–1859. doi: 10.1523/JNEUROSCI.18-05-01848.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Smith BR, Robidoux J, Amit Z. GABAergic involvement in the acquisition of voluntary ethanol intake in laboratory rats. Alcohol Alcohol. 1992;27:227–231. [PubMed] [Google Scholar]
- 81.Spanagel R, Weiss F. The dopamine hypothesis of reward: past and current status. Trends Neurosci. 1999;22:521–527. doi: 10.1016/s0166-2236(99)01447-2. [DOI] [PubMed] [Google Scholar]
- 82.Stanford IM, Cooper AJ. Presynaptic μ and δ opioid receptor modulation of GABAA IPSCs in the rat globus pallidus in vitro. J Neurosci. 1999;19:4796–4803. doi: 10.1523/JNEUROSCI.19-12-04796.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Steffensen SC, Henriksen SJ, Wilson MC. Transgenic rescue of SNAP-25 restores dopamine-modulated synaptic transmission in the coloboma mutant. Brain Res. 1999;847:186–195. doi: 10.1016/s0006-8993(99)02023-5. [DOI] [PubMed] [Google Scholar]
- 84.Steffensen SC, Nie Z, Criado JR, Siggins GR. Ethanol inhibition of N-methyl-d-aspartate responses involves presynaptic gamma-aminobutyric acid (B) receptors. J Pharmacol Exp Ther. 2000;294:637–647. [PubMed] [Google Scholar]
- 85.Stoop R, Poo MM. Synaptic modulation by neurotrophic factors: differential and synergistic effects of brain-derived neurotrophic factor and ciliary neurotrophic factor. J Neurosci. 1996;16:3256–3264. doi: 10.1523/JNEUROSCI.16-10-03256.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Stuart GI, Redman SJ. Mechanisms of presynaptic inhibition studied using paired-pulse facilitation. Neurosci Lett. 1991;126:179–183. doi: 10.1016/0304-3940(91)90548-8. [DOI] [PubMed] [Google Scholar]
- 87.Sullivan JM. Mechanisms of cannabinoid-receptor-mediated inhibition of synaptic transmission in cultured hippocampal pyramidal neurons. J Neurophysiol. 1999;82:1286–1294. doi: 10.1152/jn.1999.82.3.1286. [DOI] [PubMed] [Google Scholar]
- 88.Terwilliger RZ, Beitner-Johnson D, Sevarino KA, Crain SM, Nestler EJ. A general role for adaptations in G-proteins and the cyclic AMP system in mediating the chronic actions of morphine and cocaine on neuronal function. Brain Res. 1991;548:100–110. doi: 10.1016/0006-8993(91)91111-d. [DOI] [PubMed] [Google Scholar]
- 89. Thiele TE, Willias B, Stadler J, Reynolds JG, Bernestein IL, McKnight GS. High ethanol consumption and low sensitivity to ethanol-induced sedation in protein kinase A-mutant mice. J Neurosci 20 2000. RC75:1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thomas MJ, Malenka RC, Bonci A. Modulation of long-term depression by dopamine in the mesolimbic system. J Neurosci. 2000;20:5581–5586. doi: 10.1523/JNEUROSCI.20-15-05581.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Waldeck RF, Pereda A, Faber DS. Properties and plasticity of paired-pulse depression at a central synapse. J Neurosci. 2000;20:5312–5320. doi: 10.1523/JNEUROSCI.20-14-05312.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wan FJ, Berton F, Madamba SG, Francesconi W, Siggins GR. Low ethanol concentrations enhance GABAergic inhibitory postsynaptic potentials in hippocampal pyramidal neurons only after block of GABAB receptors. Proc Natl Acad Sci USA. 1996;93:5049–5054. doi: 10.1073/pnas.93.10.5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wand G, Levine M, Zweifel L, Schwindinger W, Abel T. The cAMP-protein kinase A signal transduction pathway modulates ethanol consumption and sedative effects of ethanol. J Neurosci. 2001;21:5297–5303. doi: 10.1523/JNEUROSCI.21-14-05297.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Weiss F, Parson LH, Schulteis G, Hyytia P, Lorang MT, Bloom FE, Koob GF. Ethanol self-administration restores withdrawal-associated deficiencies in accumbal dopamine and 5-hydroxytryptamine release in dependent rats. J Neurosci. 1996;16:3474–3485. doi: 10.1523/JNEUROSCI.16-10-03474.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yoshimura M, Tabakoff B. Ethanol's actions on cAMP-mediated signaling in cells transfected with type VII adenylyl cyclase. Alcohol Clin Exp Res. 1999;23:1457–1461. [PubMed] [Google Scholar]
- 96.Yun SH, Cheong MY, Mook-Jung I, Huh K, Lee C, Jung MW. Cholinergic modulation of synaptic transmission and plasticity in entorhinal cortex and hippocampus of the rat. Neuroscience. 2000;97:671–676. doi: 10.1016/s0306-4522(00)00108-1. [DOI] [PubMed] [Google Scholar]
- 97.Zucker RS. Short-term synaptic plasticity. Annu Rev Neurosci. 1989;12:13–31. doi: 10.1146/annurev.ne.12.030189.000305. [DOI] [PubMed] [Google Scholar]