

Abstract
Impairments in mitochondrial energy metabolism are thought to be involved in most neurodegenerative diseases, including Huntington's disease (HD). Chronic administration of 3-nitropropionic acid (3-NP), a suicide inhibitor of succinate dehydrogenase, causes prolonged energy impairments and replicates most of the pathophysiological features of HD, including preferential striatal degeneration. In this study, we analyzed one of the mechanisms that could account for this selective 3-NP-induced striatal degeneration. In chronically 3-NP-infused rats, the time course of motor behavioral impairments and histological abnormalities was determined. Progressive alterations of motor performance occurred after 3 d. By histological analysis and terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end-labeling staining, we found a selective neurodegenerescence in the striatum, occurring first in its dorsolateral (DL) part. Activation of c-Jun N-terminal kinase (JNK) was analyzed from brain sections of these rats, using immunocytochemical detection of its phosphorylated form. Activation of JNK occurred progressively and selectively in the DL of the striatum and was followed by c-Jun activation and expression in the same striatal region. To elucidate the role of the JNK/c-Jun module in 3-NP-induced striatal degeneration, we then used primary striatal neurons in culture, in which we replicated neuronal death by application of 3-NP. We found strong nuclear translocation of activated JNK that was rapidly followed by phosphorylation of the transcription factor c-Jun. Overexpression of a dominant negative version of c-Jun, lacking its transactivation domain and phosphorylation sites for activated JNK, completely abolished 3-NP-induced striatal neurodegeneration. We thus conclude that a genetic program controlled by the JNK/c-Jun module is an important molecular event in 3-NP-induced striatal degeneration.
Keywords: chronic infusion of 3-NP, apoptosis, Huntington's disease, striatal neurons, JNK activation, gene regulation, in vivo analysis
Mitochondrial dysfunctions are critically involved in cellular processes underlying both necrotic and apoptotic cell death, which are thought to have a major role in the pathogenesis of neurodegenerative diseases (for review, see Beal, 2000). One example is Huntington's disease (HD), in which defects in mitochondrial energy metabolism are now documented from several criteria. Individuals with HD show increases in lactate concentration in occipital cortex and striatum (Jenkins et al., 1993, 1998) and decreased mitochondrial enzyme activities (complexes II–III and IV) in the striatum (Gu et al., 1996; Browne et al., 1997). Recently, mitochondria from HD patients were shown to be more sensitive to apoptosis (Sawa et al., 1999). The gene mutation in HD involves expansion of a trinucleotide (CAG) repeat in the N-terminal region of the gene IT15, which encodes the protein called huntingtin (Huntington's Disease Collaborative Research Group, 1993), the function of which is unknown. Although the pathological feature of this disease is the preferential degeneration of striatal neurons, the means by which this neuronal degeneration occurs remain unknown.
Chronic 3-nitropropionic acid (3-NP) administration in rodents and nonhuman primates has provided a useful experimental model for HD (for review, see Brouillet et al., 1999). 3-NP is a suicide inhibitor of succinate dehydrogenase (SDH; complex ΙΙ), which causes prolonged energy impairment and replicates most of the clinical and pathophysiological hallmarks of HD, including spontaneous choreiform and dystonic movements, as well as selective degeneration of striatum. One generally admitted hypothesis to account for 3-NP-induced striatal degeneration is that depletion in ATP levels, produced by a deficit in energy metabolism, can lead to membrane depolarization and can produce NMDA toxicity via the relief of a voltage-dependent Mg2+ block (Beal et al., 1993; Zeevalk et al., 1995; Greene et al., 1998). On the other hand, impaired energy metabolism can produce an oxidative stress, formation of reactive oxygen and nitrogen species (Lipton and Rosenberg, 1994; Schulz et al., 1995), which are suspected to be critically involved in neuronal cell death.
Increasing evidence now implicates the c-Jun N-terminal kinase (JNK) pathway as an important mediator of apoptosis in different model systems. This pathway is activated in response to free radicals generated by ultraviolet radiation, inflammatory cytokines, and direct application of H2O2 (for review, see Davis, 2000) as well as, in striatal neurons, high doses of dopamine (Luo et al., 1998) or glutamate (Schwarzschild et al., 1997). Overexpression of dominant negative components of the JNK pathway reverses apoptosis induced by trophic factor withdrawal in PC12 cells (Xia et al., 1995). Furthermore, knock-out mice for JNK3 show reduced kainate-induced neuronal apoptosis in the hippocampus (Yang et al., 1997). One major target of this pathway is the transcription factor c-Jun, which is regulated at both transcriptional and post-transcriptional levels by activated JNK (Davis, 2000). Although c-Jun can be involved in multiple functions, including survival, differentiation, and neuronal regeneration (Herdegen et al., 1997) recent evidence critically involves this transcription factor in neuronal apoptosis. In this way, blocking c-Jun functions protects neurons from apoptosis in vitro (Estus et al., 1994; Ham et al., 1995) as well as in vivo (Behrens et al., 1999).
We designed the present study to investigate a possible role of a JNK/c-Jun module in striatal degeneration induced by 3-NP. For this aim, we used both in vivo and in vitro model systems, i.e., chronic infusion of 3-NP in rats and continuous 3-NP application in primary striatal neurons in culture. These complementary approaches provide strong arguments for a key role of the JNK/c-Jun module in 3-NP-induced striatal death.
MATERIALS AND METHODS
Animals. Male Lewis rats (body weight, 340–370 gm) were obtained from Iffa-Credo (l'Arbresle, France). Because vulnerability of animals toward 3-NP toxicity increases with age (Brouillet et al., 1993), all animals were the same age (12weeks) at the beginning of the intoxication. All experimental procedures were in straight accordance with standard ethical guidelines (National Institutes of Health publication 85-23, revised 1985; European Community Guidelines on the Care and Use of Laboratory Animals) and approved by the local ethics committee.
3-Nitropropionic acid treatment. 3-NP was prepared and administered as described previously (Ouary et al., 2000). 3-NP (Fluka, Buchs, Switzerland) was dissolved in water, and the solution was brought to pH 7.4 with 5 m sodium hydroxide. The animals were anesthetized (ketamine and xylasine) and implanted with subcutaneous osmotic minipumps (2ML4; Alzet Inc., Palo Alto, CA) loaded with 3-NP. The concentration was calculated for each individual rat so that all animals received 3-NP at a dosage of 38 mg · kg−1 · d−1. Control animals were implanted with empty osmotic pumps.
Behavioral observation. All 3-NP-treated animals and corresponding controls were evaluated for motor deficits using a neurological scale as described previously (Ouary et al., 2000). Briefly, motor abnormalities were determined on the basis of the presence and severity of motor symptoms, consisting of dystonia, gait abnormalities, and recumbency. Furthermore, the capability of the animals to grasp a cage grid with their forepaws and their capability to remain on a small platform for >10 sec was assessed. The presence of these motor abnormalities was studied on a daily basis, and a neurological score was calculated for each animal as the sum of all indices [minimum, 0 (normal score); maximum, 8 (animal showing near-death recumbency)].
Succinate dehydrogenase activity. The brains of controls (10 rats) and 3-NP-treated animals (9 rats) were cut in two, one-half for the analysis by Western blot and the other half for the SDH activity. SDH activity was determined by in situ semiquantitative histochemistry. The brains were rapidly frozen in isopentane (−25°C) and kept at −80°C. The brains were cut in sections of 20 μm and mounted on slides. Sections were incubated for 15 min in 0.1m PBS at 37°C followed by incubation in 0.3 mm nitroblue tetrazolium, 0.05m phosphate buffer, and 0.05m sodium succinate for 30 min at 37°C. For determination of nonspecific staining unrelated to SDH activity, adjacent sections were incubated in the same medium in which succinate was omitted. Finally sections were rinsed in cold PBS for 5 min, fixed in 4% paraformaldehyde (PFA), and rinsed in water. The image of each section was acquired with an IMSTAR image analyzer and quantified as reported previously (Brouillet et al., 1998).
Tissue preparation for immunohistochemistry. Animals received a sublethal dose of pentobarbital (120 mg/kg) and were transcardially perfused with 4% PFA in 0.1 mNa2HPO4/NaH2PO4buffer, pH 7.5, delivered with a peristaltic pump at 50 ml/min for 10 min. Brains were removed, post-fixed in the same fixative solution overnight, cryoprotected in 0.1 m phosphate buffer containing 15% sucrose for 24 hr, and then frozen in isopentane. Thin sections (20 μm) were cut on a microtome and then kept in a solution containing 30% ethylene glycol, 30% glycerol, 0.1 mphosphate buffer, and 0.1% diethyl pyrocarbonate (Sigma, St. Louis, MO) at −20°C until they were processed for immunohistochemistry.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling. The detection of DNA strand breaks was performed using terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end-labeling (TUNEL) according to the procedure of the manufacturers (Roche Molecular Biochemicals, Bagnolet, France) with minor modifications. Briefly, sections were mounted on slides and rehydrated. They were then treated with 0.1% sodium citrate and 0.1% Triton X-100 for 30 min at room temperature and rinsed three times in PBS. They were then incubated in proteinase K (1 mg/ml in PBS, pH 7.4) for 5 min, reimmersed in 4% PFA for 15 min, and rinsed three times in PBS before TUNEL reactions. Sections were then covered with 50 μl of TUNEL mixture for 30 min at 37°C in a humidified chamber. After three washes in PBS, the slides were mounted with Vectashield (Vector Laboratories).
Immunohistochemistry. Free-floating sections were processed for immunohistochemistry as described previously for detection of phosphorylated extracellular signal-regulated kinase (Sgambato et al., 1998) with minor modifications. Sections were incubated overnight at 4°C with the following primary polyclonal antibodies: phospho Thr183-Tyr185JNK (P-JNK, 1:500; Promega), phospho Ser63-c-Jun (P-c-Jun, 1:100; New England Biolabs, Ozyme, France), and c-jun (1:100, New England Biolabs). On the second day, after three rinses in Tris-buffered saline (TBS), they were incubated in biotinylated horse anti-rabbit antibody (1:500; Vector Laboratories) in TBS 2 hr at room temperature. After washing, the sections were incubated for 90 min in an ABC kit (Vector Laboratories). Sections were then rinsed twice in TBS and then in Tris buffer (TB). The reactions were developed in a solution of TB containing 0.1% 3–3′ diaminobenzidine and H2O2(0.02%) for 60 min. After processing, tissue sections were mounted onto gelatin-coated slides and dehydrated through alcohol to xylene for light microscopic examination.
For double labeling of TUNEL and P-c-Jun, the TUNEL reaction was first processed as described above, and then P-c-Jun immunohistochemistry was performed and revealed with an anti-rabbit Cy3-conjugated antibody (1:1000; Amersham Biosciences, Les Ulis, France). After washing, sections were counterstained with Hoechst and mounted onto gelatin-coated slides under coverslips using Vectashield (Vector Laboratories).
Primary striatal cultures. Striata of fetal rat (embryonic day 17) from pregnant Sprague Dawley rats were dissected, and tissues were dissociated by repeated trituration with a pipette in PBS and 0.6% glucose. After decantation for 5 min, cells were collected by centrifugation at 1000 × g for 5 min. Cell pellets were resuspended in Neurobasal media supplemented with B27, glutamine, penicillin-streptomycin (Invitrogen, Gaithersburg, MD), and β-mercaptoethanol (Sigma). Cells were seeded at 960 cells/mm2 into poly-d-lysine (Sigma)-coated 24-well plates. The cultures were maintained at 37°C in a humidified incubator with 5% CO2 and 95% air. On the seventh day in vitro, the medium was removed and replaced by fresh medium containing 3-NP (Fluka) at 1 mm. The cells were then replaced at 37°C for the appropriate time.
Immunocytochemistry. After treatment, cells were fixed with 4% PFA diluted in PBS for 40 min at room temperature and then incubated with a 1:1 ratio of methanol/acetone for 10 min at 4°C. After washing with PBS, plates were preincubated with blocking buffer (10% fetal bovine serum and 1% BSA in PBS) for 2 hr at room temperature and then incubated with the following primary antibodies in PBS containing 1% BSA overnight at 4°C: P-JNK (1:500) and P-c-Jun (1:500). Plates were rinsed and incubated with an anti-rabbit Cy3-conjugated antibody (1:1000; Amersham Biosciences) for 2 hr at room temperature. After washing, plates were counterstained with Hoechst and mounted under coverslips using Vectashield. For each experiment, cells were analyzed under a fluorescent inverted microscope (Nikon) directly into the wells. Images from immunofluorescence were digitized (magnification 400×) in parallel with Hoechst from five independent fields for each experiment (n = 3 for each treatment). The percentage of P-JNK or P-c-Jun neurons was calculated for each experiment.
Western blot. Immunoblot analysis was performed as described previously (Vanhoutte et al., 1999). Neurons were cultured in six-well plates, placed on ice, and lysed in a solubilization buffer containing a mix of protease and phosphatase inhibitors (10 mmTris-Cl, 50 mm NaCl, 1% Triton X-100, 30 mmsodium pyrophosphate, 50 mm NaF, 5 μmZnCl2, 100 μmNa3VO4, 1 mmDTT, 5 nm okadaic acid, 2.5 μg of aprotinin, 2.5 μg of pepstatin, 0.5 μm PMSF, 0.5 mm benzamidine, and 2.5 μg of leupeptin). Insoluble material was removed by centrifugation (13,000 rpm for 20 min at 4°C), and samples were then kept at −80°C. Protein extracts (10 μg) were separated by 10% SDS-PAGE before electrotransfer. Blots were blocked with 5% nonfat milk and incubated with rabbit polyclonal antisera raised against P-JNK (1:1000) overnight at 4°C. After rinsing, the blots were incubated with goat horseradish peroxidase-conjugated antibody (1:2500; Amersham Biosciences) for 2 hr at room temperature before exposure to an ECL kit (Amersham Biosciences).
Transfection. Transient transfection of primary striatal cultures was performed with LipofectAMINE 2000 (Invitrogen) as recommended by the manufacturer's protocol. Cells (1.8 × 105) were transfected with 1 μg of enhanced green fluorescent protein (pEGFP-N3; Clontech, Cambridge, UK) alone or in the presence of 5 μg of the dominant negative c-Jun (FLAGΔ169; Δc-Jun). After 6 hr, the cultures were rinsed with fresh medium. After the treatment with 3-NP, the cells were fixed as described above. Immunocytochemical analysis of Δc-Jun expression was performed using a mouse monoclonal anti-flag M2 antibody (Sigma) with the protocol described above for P-JNK and P-c-Jun immunodetection.
Statistical analysis. Results are expressed as the mean ± SEM. Statistical analysis was performed using one-way ANOVA followed by a post hoc Scheffé test for comparison of multiple experimental conditions. In all cases, significance was set up atp < 0.05.
RESULTS
Systemic administration of 3-NP induces selective and progressive striatal degeneration
We first wished to analyze JNK activation in a model system of chronic striatal degeneration induced, in vivo, by continuous infusion of 3-NP (Brouillet et al., 1993, 1995). We took advantage of the improvement of this model in the Lewis rat strain in which the onset of 3-NP-induced neurodegeneration is highly reproducible (Dautry et al., 2000; Ouary et al., 2000). To assess for the efficacy of 3-NP infusion, during the treatment we measured neurological impairments, which primarily included dystonia, bradykinesia, and gait abnormalities (Ouary et al., 2000). We established neurological scores from 1 (the less severe) to 8 (the most severe) (Fig. 1A). On day 3, 3-NP-treated rats showed mild and intermittent dystonia of the hindlimbs. On day 4, the animals showed more severe permanent dystonia of the hindlimbs associated with wobbling gaits. At later stages, most animals showed severe dystonic posture (score 5–6), and some were recumbent (score 8). As revealed by one-way ANOVA (F(4,64) = 127.882; p< 0.0001), chronic systemic administration of 3-NP induced progressive motor alterations.
Fig. 1.
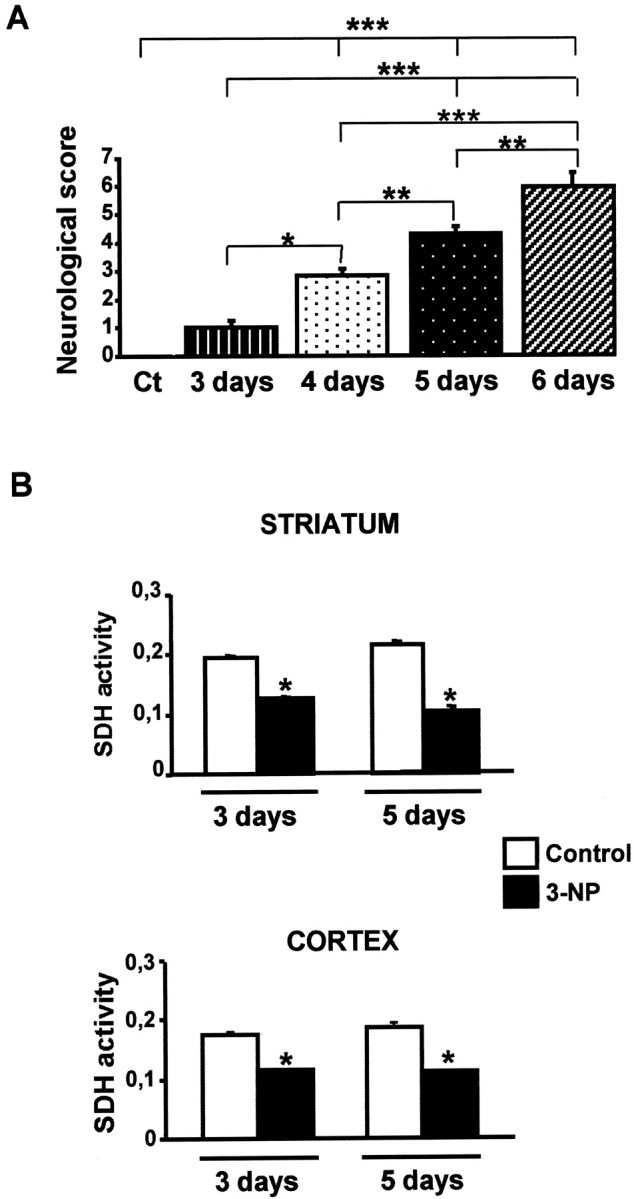
Systemic administration of 3-NP leads to neurological impairment and inhibition of SDH. A, Control (Ct) and 3-NP-treated rats were tested every day for behavioral performance (from day 3). Score 1 corresponded to mild and intermittent dystonia of one hindlimb, scores 2–3, intermittent or permanent appearance of dystonia for two hindlimbs; scores 4–5, wobbling gait and lack of grasping; and scores 6–8, severe dystonic posturing to recumbency. Data are representative of at least six rats per group. Statistical analysis: *p < 0.05; **p < 0.005; ***p < 0.001 when comparing control and 3-NP-treated rats (Scheffé test).B, Striatal and cortical SDH activity was measured by semiquantitative immunohistochemistry. Note the strong decrease in both cortical and striatal slices. *p < 0.001 when comparing 3-NP-treated rats with their corresponding control (Scheffé test).
To assess biochemically the efficacy of 3-NP treatment, brain SDH activity was measured by semiquantitative histochemistry (Brouillet et al., 1998; Fig. 1B). Statistical comparisons revealed a significant reduction (35%) of this activity after 3 d of 3-NP treatment in the striatum and cerebral cortex (F(3,15) = 75.488; p< 0.0001; F(3,15) = 55.994;p < 0.0001, respectively). On day 5 of 3-NP treatment, SDH activity remained in the same range (40%) in the cerebral cortex, whereas it was further reduced (−50%) in the striatum, partly as a direct consequence of degeneration (see below).
Histology was then performed on brain sections from rats that were used for behavioral analysis described above. Of interest, although 3-NP diffuses throughout the brain, as indicated by SDH measurements, the neurotoxin produced a selective and progressive striatal degeneration that was clearly visible in the dorsolateral (DL) part of the structure after 5 d of treatment (Fig.2A). From day 6, this degenerative process extended to the ventral region of the striatum in most animals (Fig. 2A), thus indicating that this limbic-associated area can also be altered by 3-NP treatment, albeit later on. At higher magnification, histological examination showed shrinkage of the cell body (retraction) of most neurons in the DL when compared with the dorsomedial (DM) striatum on days 5 and 6 and control striatum (Fig. 2B). In some cells, cresyl violet staining had apoptotic features, with nuclear fragmentation (see Fig.2B, inset) as reported previously (Ouary et al., 2000).
Fig. 2.
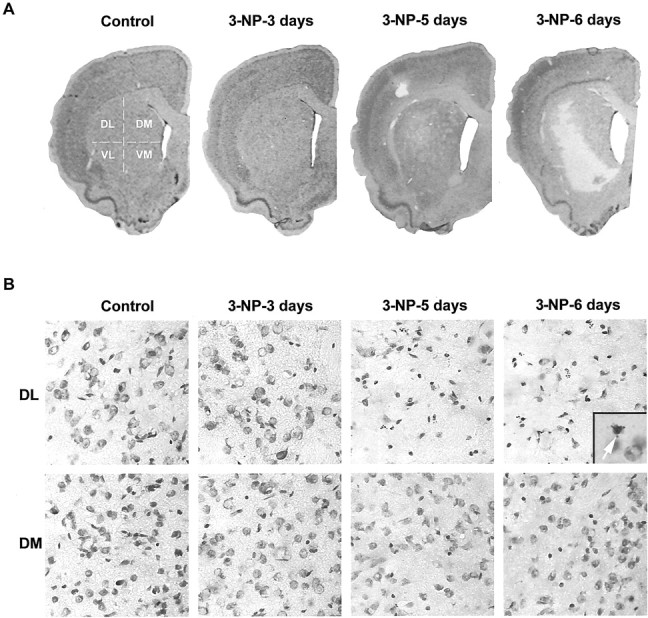
Systemic administration of 3-NP leads to a progressive and selective degeneration in the striatum. Cresyl violet staining was performed on brain sections from control and 3-NP-treated rats. A, Low magnification showing the selective loss of cells in the striatum at day 5. B, High magnification of cresyl violet staining in the DL and DM parts of the striatum. Note the progressive cell loss in the DL striatum on 3-NP treatment when compared with the DM striatum.Inset, Fragmented nucleus displaying apoptotic bodies.VL, Ventrolateral; VM, ventromedial.
We thus investigated, using TUNEL staining, whether striatal degeneration was accompanied by DNA fragmentation, which is classically used as an index of apoptosis. We found strong TUNEL labeling that occurred in the DL striatum, specifically at day 5 of 3-NP treatment (Fig. 3A). This staining then spread along the ventral part of the DL and thereby followed the evolution of striatal degeneration. At higher magnification, TUNEL staining was found in a subpopulation of striatal cells and also showed apoptotic bodies (Fig. 3B). The number of TUNEL-positive cells was quantified in 3-NP-treated animals and compared with the neurological scores, which were considered good indices of striatal degeneration. Although no TUNEL-positive cells were observed for scores of <5, we found a strong and significant increase in TUNEL-positive cells from scores 5–6 to scores 7–8 (Fig. 3C;F(2,17) = 12.415; p < 0.0005). Corresponding time points were 3–4 d for scores of <5, 5 d for scores of 5–6, and 6 d for scores of 7–8.
Fig. 3.
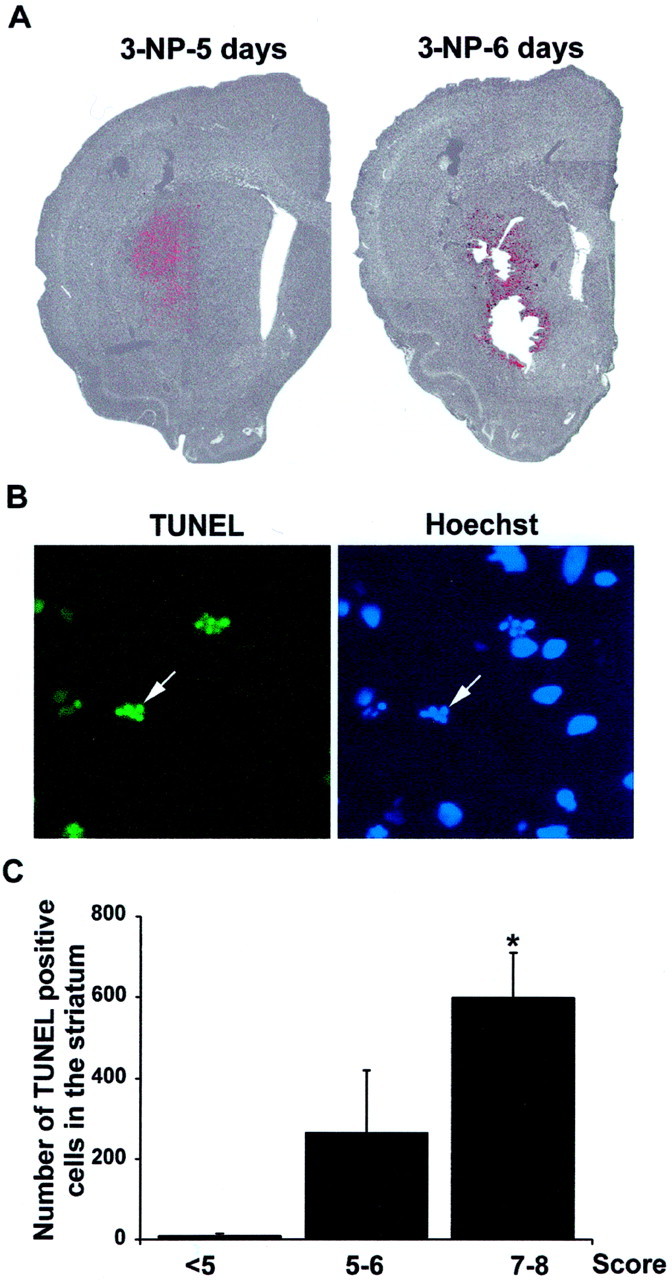
TUNEL immunoreactivity correlates with neurological scores. A, Low magnification showing TUNEL-reactive cells (revealed by FITC and depicted here inred) superimposed with Hoechst staining (showed here ingray) in the striatum. Note the strong TUNEL labeling that appears at 3-NP-5 days in the DL striatum.B, Higher magnification of TUNEL-positive cells showing apoptotic bodies (arrow) at day 5 of the 3-NP treatment showing fragmented nuclei (arrow). Right panel, Corresponding Hoechst staining. C, TUNEL-positive cells were counted in the striatum. The mean value was determined in group of 3-NP-treated rats presenting the same neurological score. <5, Three to 4 d of 3-NP treatment; 5–6, 5 d of treatment;7–8, 6 d of treatment. Statistical analysis: *p < 0.05 when comparing scores7–8 and <5 (Scheffé test).
In conclusion, these data indicate that systemic administration of 3-NP produces selective striatal degeneration. This degenerative process begins in the dorsolateral part and then diffuses to the ventral region of the striatum. This degeneration has some apoptotic characteristics and occurs progressively from day 5 of treatment and is strictly correlated with neurological deficits.
3-NP treatment activates JNK and c-Jun in the striatum: in vivo analysis
We next determined JNK activation from brain sections or 3-NP-treated rats that were studied for motor behavioral impairments and histological abnormalities (see above). For this purpose, immunocytochemical detection of the phosphorylated and thereby active form of JNK (P-JNK) was used. In control rats, no P-JNK immunolabeling was found in the striatum or any other brain regions (Fig.4A). Similarly, no P-JNK immunoreactivity was found before 5 d of 3-NP infusion (data not shown). P-JNK-positive cells then progressively appeared in the DL region of the striatum, specifically after 5 d of treatment, and further increased after 6 d (Fig. 4B). When compared with the neurological score, the number of P-JNK-immunoreactive cells showed a slight but not significant increase at scores of 5–6 and a significant increase from animals scored 7–8 (F(2,22) = 4.123; p < 0.05; Fig. 4C).
Fig. 4.
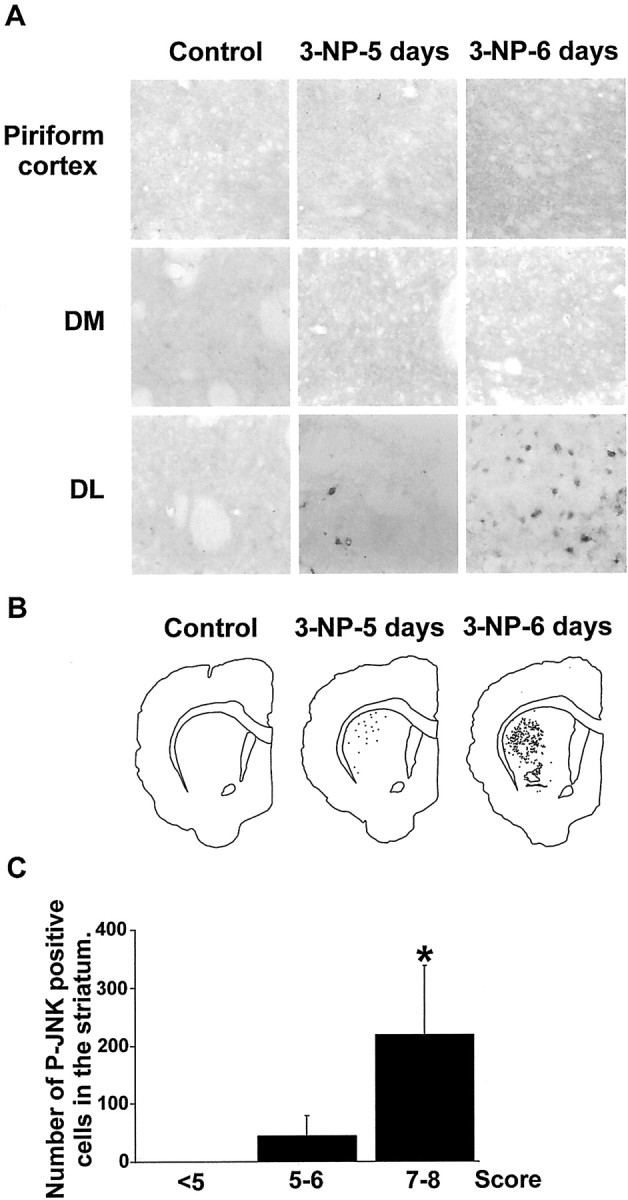
Systemic administration of 3-NP leads to a progressive activation of JNK in the striatum selectively. JNK activation was analyzed by immunocytochemical detection of its active, phosphorylated form (P-JNK) from sections corresponding to the rats used for behavioral, histological, and TUNEL analysis (see previous figures). A, High magnification of P-JNK immunoreactivity in the piriform cortex and DM and DL regions of the striatum. Note the lack of labeling in the Piriform cortex and the DM striatum inControl and 3-NP-treated rats. P-JNK immunoreactivity appears at 3-NP-5 days in theDL striatum specifically. B, P-JNK-immunoreactive cells were depicted in the striatum using an image analyzer. Note their appearance in the DL striatum specifically at 3-NP-5 days treatment. C, Quantification of P-JNK-immunoreactive cells was performed from at least five different rats per neurological score. For each rat, one equivalent striatal section (30 μm) was counted. Shown is the mean value determined in 3-NP-treated rats presenting the same neurological score. <5, Three to 4 d of 3-NP treatment;5–6, 5 d of treatment; 7–8, 6 d of treatment. Statistical analysis: *p < 0.005 when comparing scores 7–8 and <5(Scheffé test).
Within the nucleus, activated JNK is known to control the phosphorylation state of c-Jun as well as its transcriptional control (Angel et al., 1988). Using an antibody specific for the phosphorylated form of c-Jun (P-c-Jun), we found activation of c-Jun at day 5 of 3-NP treatment that was further increased at day 6 (Fig.5). This activation was found in the degenerative area, as analyzed by double staining with TUNEL (Fig. 5). Interestingly, in some cases, P-c-Jun immunoreactivity occurred in the same striatal neurons.
Fig. 5.
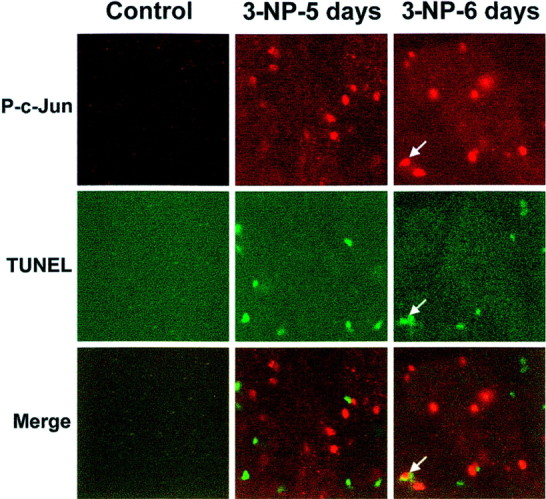
Activation of c-Jun phosphorylation occurs in the degenerative striatal region. P-c-Jun immunoreactivity was performed using an anti-P-c-Jun antibody. Note a strong induction at3-NP-5 days and 3-NP-6 days treatment. Coimmunostaining with TUNEL indicates that P-c-Jun immunoreactivity occurs in the degenerative region. Note the double labeling between P-c-Jun and TUNEL staining in one striatal neuron (bottom right panel, arrow).
Next, we examined c-Jun expression after 3-NP treatment. We failed to detect any c-Jun-immunoreactive cells in the striatum of control rats, although strong basal levels were found in the piriform cortex (Fig.6A,B). c-Jun-immunoreactive cells appeared after 6 d of 3-NP treatment in the DL of the striatum specifically (Fig. 6A,B). c-Jun-positive neurons clearly appeared at high neurological scores (Fig. 6C; F(2,22) = 21.673;p < 0.0001). Thus, c-Jun expression is delayed when compared with JNK activation, a result consistent with the necessary delay for transcription and translation of the protein.
Fig. 6.
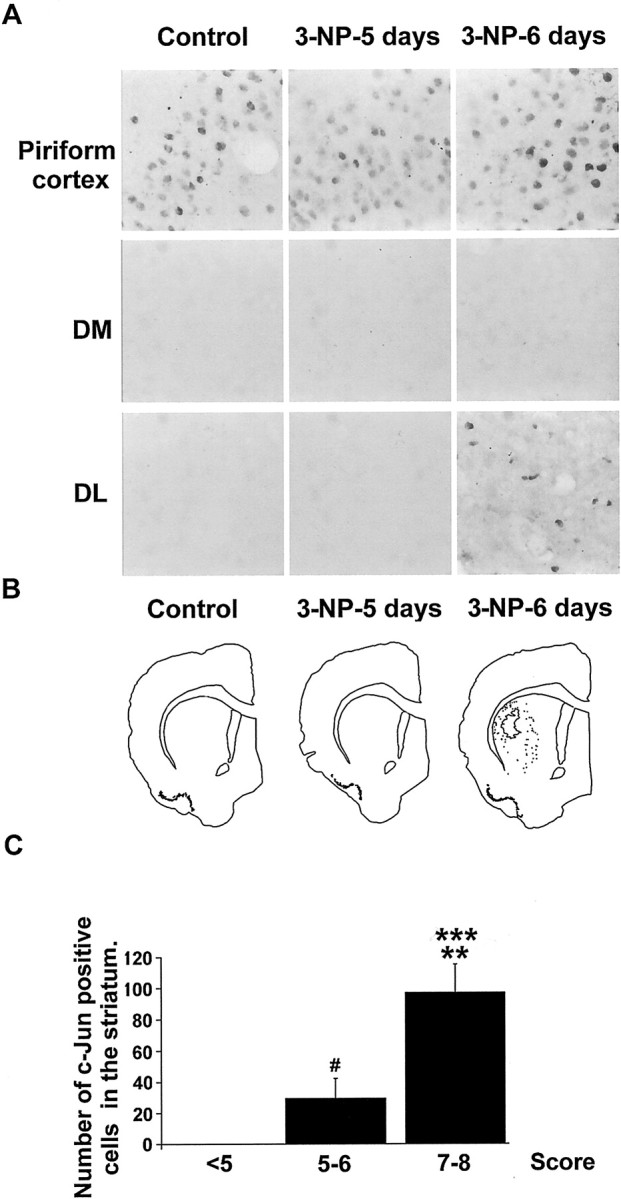
Systemic administration of 3-NP leads to a progressive expression of c-Jun in the striatum, selectively. c-Jun expression was analyzed by immunocytochemistry using a selective antibody. A, High magnification of c-Jun immunoreactivity in the Piriform cortex shows basal expression in this region in both Control and3-NP-treated rats. Note that c-Jun expression occurs in the DL part of the striatum at 3-NP-6 days treatment. B, c-Jun-immunoreactive cells were depicted in the striatum using an image analyzer. Note their appearance in the DL striatum specifically at 3-NP-6 days treatment. C, Quantification of c-Jun-immunoreactive cells was performed as in Figure 4. Statistical analysis: **p < 0.01 when comparing scores5–6 and 7–8; ***p< 0.0001 when comparing scores 7–8 and <5; #p < 0.05 when comparing scores 5–6 and <5(Scheffé test).
3-NP treatment activates JNK and c-Jun in primary striatal neurons in culture
We then used a simplified model system, primary striatal neurons in culture, to evaluate the function of the JNK/c-Jun module in 3-NP-induced striatal degeneration. Time course analysis of neurodegeneration after initial treatment of cultured cells with 3-NP (1 mm) was performed to find out apoptotic features previously described with this dose in hippocampal primary cultures (Pang and Geddes, 1997). In agreement with these experiments, we found ∼50% of neuronal cell loss after 48 hr of 3-NP treatment (Fig.7A,B).
Fig. 7.
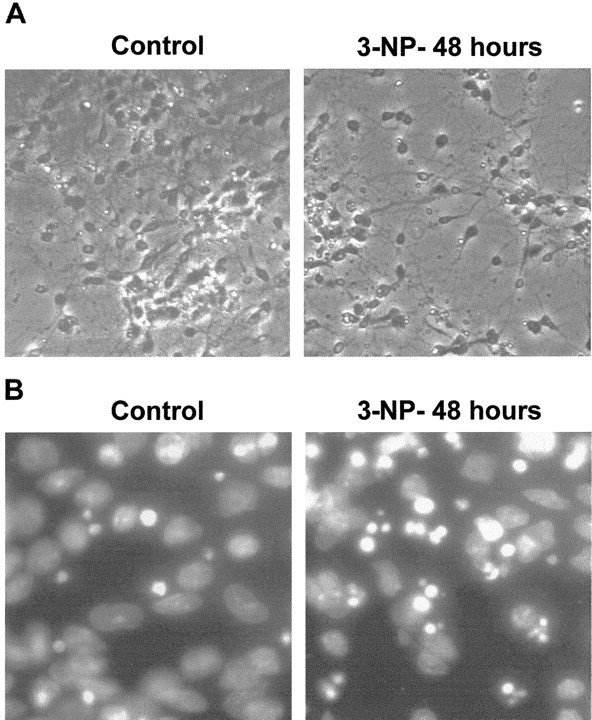
3-NP leads to apoptosis in primary striatal cultures. High magnification is shown of phase contrast (A) and Hoechst staining (B) from primary striatal cultures treated with 3-NP (1 mm) for 48 hr (3-NP-48 hours).Control, Untreated striatal neurons.
Kinetics of JNK activation were analyzed on this model system by immunofluorescence (Fig.8A). A low basal level of P-JNK immunoreactivity was found in control cells that corresponded, nevertheless, to an exclusive neuritic localization. After 2 hr of 3-NP treatment and later on, P-JNK immunoreactivity showed a clear-cut nuclear localization, thus suggesting activation of the kinases. Accordingly, Western blot analysis showed increased levels of P-JNK immunoreactivity after 2 hr of 3-NP treatment (Fig. 8, compare A, B). Counting cells with a nuclear expression of P-JNK showed a peak at 2–3 hr and then a progressive decline (Fig. 8C;F(3,56) = 67.173; p < 0.0001). At 24 and 48 hr of 3-NP treatment, P-JNK immunoreactivity was neuritic again (data not shown). Thus altogether, these data strongly support the idea that JNKs are activated and translocated to the nucleus on 3-NP treatment.
Fig. 8.
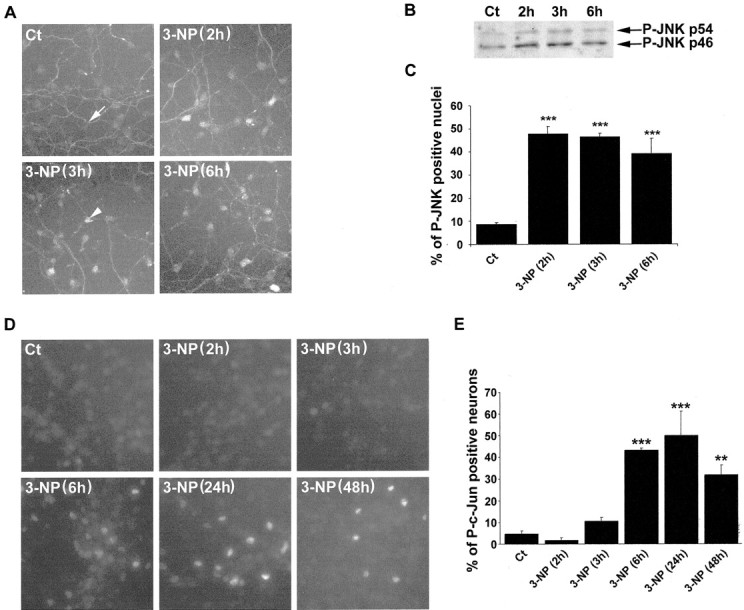
3-NP leads to activation of the JNK/c-Jun module in cultures of primary striatal neurons. A, P-JNK immunoreactivity in control (Ct) and 3-NP-treated cells. Note the low P-JNK immunoreactivity in control cells restricted to the neuritic extension (white arrow). Note also the strong nuclear translocation of P-JNK immunoreactivity after3-NP(3h) treatment (arrowhead).B, Western blot analysis of P-JNK immunoreactivity.C, Quantification of P-JNK-immunoreactive nuclei was performed and compared with the total number of striatal neurons (analyzed by Hoechst staining). Data are representative of three independent experiments (for each experiment, the mean value was calculated from 5 randomly chosen fields, representing 50 neurons each). Statistical analysis: ***p < 0.001 when comparing P-JNK-immunoreactive nuclei between3-NP-treated and control (Ct) neurons (Scheffé test). D, Phosphorylation of c-Jun was determined using an antibody specific for its phosphorylated form (P-c-Jun). Note that P-c-Jun-immunoreactive neurons appear at3-NP(3h) treatment. E, Quantification of P-c-Jun-positive nuclei was performed as detailed for P-JNK. Statistical analysis: **p < 0.001 and ***p < 0.0001 when comparing control (Ct) and 3-NP-treated neurons (Scheffé test).
Within the nucleus, P-c-Jun immunoreactivity occurred in response to 3-NP treatment. In control conditions, no labeling for P-c-Jun was observed. P-c-Jun immunoreactivity occurred after 3 hr of the neurotoxic treatment, with a peak at 6 hr and a slight decrease at 48 hr (Fig. 8D,E;F(5,83) = 46.331; p < 0.0001).
Overexpression of dominant negative c-Jun blocks 3-NP-induced apoptosis in primary striatal neurons
To address the role of c-Jun activation in striatal neurodegeneration induced by 3-NP, we overexpressed a dominant negative form (Δc-Jun) in primary striatal neurons. We used a construct corresponding to a deletion of the first 169 amino acids, the region containing the transactivation domain (i.e., phosphorylation sites for activated JNK) (Ham et al., 1995). Neurons were transfected with GFP alone or in combination with Δc-Jun. GFP expression allowed us to visualize the cytoarchitecture, including neuritic extension of transfected neurons and cell body characteristics (analyzed after 48 hr of 3-NP treatment) of transfected neurons. Immunocytochemical detection of Δc-Jun showed its nuclear localization, as well as coexpression with GFP in double-transfected neurons (Fig.9A).
Fig. 9.
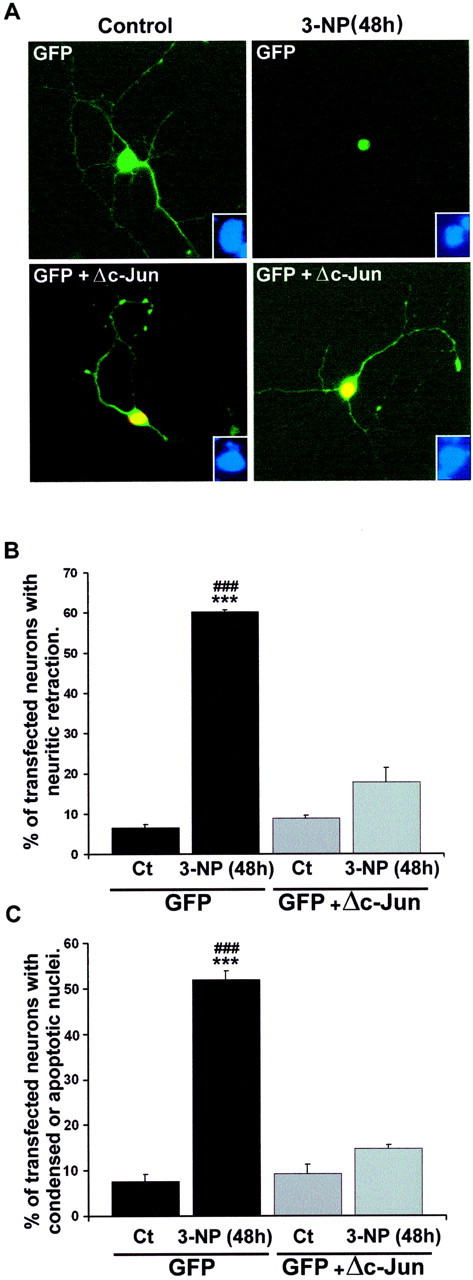
A dominant negative form of c-Jun protects neurons from apoptosis induced by 3-NP. Primary striatal neurons were transfected with GFP alone or in combination with a dominant negative form of c-Jun (Δc-Jun, lacking the first 169 amino acids). A, Transfected neurons were visualized by GFP (top panels, green). In cotransfected neurons (bottom panels), detection of Δc-Jun expression was performed using an antibody that recognizes the FLAG epitope. The coexpression of GFP and Δc-Jun is shown in yellow. Note the protection from 3-NP-induced neuritic retraction and nuclear condensation in cells cotransfected with GFP and Δc-Jun.Insets, Corresponding Hoechst staining in these neurons.B, Quantification of neurite retraction from transfected cells. C, Quantification of transfected neurons having condensed or fragmented nuclei. Data are representative of three independent experiments (for each experiment, ∼500 transfected neurons were analyzed). Statistical analysis ***p< 0.001 when comparing controls (Ct) with3-NP treatment; ###p < 0.001 when comparing 3-NP-treated neurons transfected with GFP alone or withGFP+Δc-Jun (Scheffé test).
In control conditions, neurons transfected with GFP alone or in combination with Δc-Jun showed a very long neuritic extension and a normal cell body (Fig. 9A). After 48 hr of 3-NP treatment, 60% of neurons transfected with GFP alone exhibited a strong retraction of neurites and shrinkage of cell bodies (F(3,8) = 185.246; p< 0.0001; Fig. 9A,B). In contrast, only 17% of neurons coexpressing GFP and Δc-Jun showed a neuritic retraction after 3-NP treatment (Fig. 9A,B). Similar protective effects were found by Hoechst staining, because no fragmented nuclei were found in Δc-Jun-transfected neurons treated with 3-NP (F(3,8)=356.710; p < 0.0001; Fig. 9C). No significant differences in either neuritic or nuclear characteristics of Δc-Jun-transfected neurons were found between control and 3-NP-treated cells at 48 hr (Fig.9A–C). Thus, activation of c-Jun plays a critical role in 3-NP-induced striatal neurodegeneration.
DISCUSSION
We provide here the first demonstration that chronic infusion of 3-NP in rats activates the JNK pathway specifically in the striatum. This activation was further illustrated in vitro in primary striatal cultures, and in this model system, the dominant negative c-Jun blocked striatal death induced by 3-NP. We therefore propose that 3-NP-induced striatal death is controlled at least in part by a genetic program involving the activation of the JNK pathway.
Chronic infusion of 3-NP induces JNK activation and c-Jun expression in the striatum
In vivo, chronic infusion of 3-NP produces neurological deficits such as motor dysfunction. These alterations are primarily attributable to striatal degeneration in the lateral part, a region functionally connected by cortical motor afferents (Brown, 1992;Sgambato et al., 1997). Activation of JNK occurred at neurological scores of 5–6 in a strict spatial register with striatal degeneration. Although we found less phospho-JNK- than TUNEL-positive cells, we must consider that phosphorylation of JNK is a transient phenomenon, highly controlled by specific phosphatases. Thus, it remains possible that the peak time point for detection of phospho-JNK in such a chronic model is temporally restricted and thereby less detectable compared with TUNEL.
JNK activation is classically linked to transcriptional regulation that occurs early in the apoptotic process. Consistent with this, we found upregulation of c-Jun phosphorylation as well as its expression in the dorsolateral striatum. Transcriptional regulation of c-Jun is under the control of an activator protein-1 (AP1)/12-O-tetradecanoylphorbol 13-acetate response element site located in its promoter. Of interest, this AP1 site is activated by an heterodimeric complex formed by c-Jun itself and activating transcription factor-2, and phosphorylation of c-Jun by activated JNK strongly potentiates the transcriptional activity of this complex. This was demonstrated recently in an elegant study using deficient mice for JNK3, in which kainate-induced c-Jun phosphorylation and AP1 activity were strongly reduced in the hippocampus (Yang et al., 1997).
Possible partners for selective activation of JNK/c-Jun in the striatum
Despite the widespread effect of 3-NP, i.e., inhibition of SDH activity in various brain areas (Brouillet et al., 1998; Dautry et al., 2000), JNK activation was restricted to the striatum. Different intracellular and extracellular events can be responsible for JNK activation in neuronal cells. One of these events is production of reactive oxygen species and nitrogen species by 3-NP (Schulz et al., 1995). In line with this, administration of the free radical scavengers coenzyme Q10 and N-acetylcysteine or the nitric oxide synthase inhibitor 7-nitroindazole exerts neuroprotective effects on 3-NP-treated rats (Schulz et al., 1995; Matthews et al., 1998; La Fontaine et al., 2000). Lesions induced by malonate, another selective inhibitor of SDH, are also attenuated by free radical scavengers (Schulz et al., 1996). Although reactive oxygen and nitrogen species are necessary and sufficient to activate JNK in different model systems, it is highly probable that other partners are necessary to account for JNK activation in vivo in the chronic 3-NP model.
The striatum is highly innervated by glutamatergic afferents incoming from the cerebral cortex, and a cortical lesion strongly impairs 3-NP-induced striatal degeneration (Beal et al., 1993). In striatal neurons, defects in energy metabolism produced by 3-NP may lead to membrane depolarization and may produce glutamate toxicity via the removal of the Mg2+ block from NMDA receptors (Beal et al., 1993). Glutamate is known to induce JNK activation on primary striatal cultures (Schwarzschild et al., 1997) as well as on striatal slices (Vanhoutte et al., 1999). However, the action of glutamate cannot solely explain striatal activation of JNK to systemically administered 3-NP, because glutamate is not found exclusively in the striatum but also in regions of the brain that are not sensitive to 3-NP such as the cerebral cortex (Cotman et al., 1987).
One must keep in mind that the striatum is the major input structure for dopaminergic afferences (Björklund and Lindvall, 1984). Dopamine released from nigrostriatal terminals has neurotoxic effects when directly injected into the striatum (Filloux and Townsend, 1993). Furthermore, amphetamine, which increases striatal dopamine release, potentiates 3-NP toxicity in acute or chronic models of 3-NP administration (Bowyer et al., 1996; Reynolds et al., 1998). Conversely, denervation of the nigrostriatal pathway by 6-hydroxydopamine reduces the lesion induced by chronic 3-NP in vivo (Reynolds et al., 1998). At high doses (500 μm), dopamine is able to induce JNK activation in 293 cells, and blocking JNK activation by overexpression of a dominant negative version of the kinase responsible for its phosphorylation [dominant negative stress-activated protein kinase/Erk kinase 1 (SEK1)] reverses dopamine-induced apoptosis (Luo et al., 1998). These authors also showed, in primary striatal cultures, that overexpression of dominant negative c-Jun is able to block apoptosis induced by dopamine. Because dopamine can produce reactive oxygen species (ROS) intracellularly via auto-oxidation processes (Luo and Roth, 2000), a tentative explanation is that combined effects of dopamine and 3-NP on ROS production could account for JNK activation that we observed in the striatum. The decline of oxidative stress defense mechanisms during aging could be responsible for the higher vulnerability of the aged brain to the toxicity of reactive oxygen species (for review, see Luo and Roth, 2000). This might explain why 3-NP is much more toxic in older compared with young adult animals (Brouillet et al., 1993) and primates (Brouillet et al., 1995).
Role of the JNK/c-Jun module in 3-NP-induced striatal degeneration
To further investigate the specific role of the JNK pathway in 3-NP-induced striatal degeneration, we used primary striatal cultures treated with 3-NP (1 mm) and found activation of the JNK pathway with much more rapid kinetics than in vivo. Thus, high doses of 3-NP alone are able to activate JNK in an isolated context. Using the same dose of 3-NP as in the present study, Pang and Geddes (1997) showed 50% of hippocampal cell loss after 48 hr of treatment, which corresponded to both necrotic and apoptotic features. Although the rapid necrotic cell death was blocked by the NMDA receptor antagonist MK801, apoptosis was not attenuated by this compound but instead prevented by a protein synthesis inhibitor, cycloheximide. These data indicated that 3-NP-induced apoptosis required activation of a genetic program. In this way, it is now generally admitted that although many components of the apoptotic machinery preexist in a latent form, de novo gene expression is required to initiate apoptosis in neurons. Alterations in gene expression are triggered by specific intracellular pathways that control the phosphorylation state of nuclear transcription factors. Indeed, we found nuclear translocation of activated JNK as well as hyperphosphorylation of the transcription factor c-Jun on 3-NP treatment.
Overexpression of a truncated version of c-Jun, lacking the first 169 amino acids and thereby phosphorylation sites for JNK, protected striatal neurons from apoptosis. By controlling the phosphorylation state of c-Jun, the JNK pathway controls the heterodimeric transcription factor AP1. Distinct genes are known to be regulated according to the specific composition of this complex (Kaminska et al., 2000). One possible target gene of the JNK/c-Jun module could be Fas ligand (FasL), as proposed recently from different model systems of neuronal apoptosis (Herdegen et al., 1998; Le-Niculescu et al., 1999; Martin-Villalba et al., 1999; Morishima et al., 2001). By binding to its cognate receptor and activating caspase 8 in a paracrine or autocrine manner, FasL is an important mediator of death machinery. More recently a proapoptotic Bcl-2 family member, Bim, which is strongly induced in sympathetic neurons on NGF withdrawal, was shown to be under the transcriptional control of the JNK/c-Jun module (Putcha et al., 2001; Whitfield et al., 2001).
Can JNK/c-Jun activation account for striatal degeneration in Huntington's disease?
One potential role of mutated huntingtin (Htt) is to alter transcription levels of antiapoptotic proteins. In this way, mutated Htt toxicity is attributable to its accumulation in the nucleus, and this may involve cleavage by caspase 3 (Cattaneo et al., 2001). Locally, the truncated region of Htt (the N-terminal region containing a polyglutamine tract) acts as a transcriptional repressor by interacting with the tumor suppressor p53 and sequestrating the cAMP response element-binding protein (CREB)-binding protein (Steffan et al., 2000; Nucifora et al.; 2001). By interfering with the functions of the transcription factor CREB, one important mediator of cell survival, mutated Htt could be involved in downregulation of the antiapoptotic bcl-2 (Riccio et al., 1999) and brain-derived neurotrophic factor (BDNF; Tao et al., 1998) genes. In this regard, it is interesting to note that BDNF mRNA and protein levels are modified in the cerebral cortex in HD patients, as well as in a mouse transgenic model of HD (Zuccato et al., 2001).
Besides downregulation of antiapoptotic genes, our data raise the interesting possibility that overexpression of proapoptotic genes could also be involved in HD, via activation of the JNK/c-Jun module. Given the defective mitochondrial energy production and increased levels of free radicals observed in HD patients (Beal, 1996; Sharp and Ross, 1996), we propose that JNK activation could occur in the striatum via mechanisms similar to those observed in 3-NP experimental models. Alternatively, recent evidence indicates that mutated Htt can activate JNK in a hippocampal neuronal cell line via a mechanism involving mixed lineage kinase 2 (Liu, 1998; Liu et al., 2000). In these studies, overexpression of dominant negative SEK1 blocked apoptosis induced by mutated Htt (Liu, 1998). Analyzing whether and how JNK/c-Jun is activated in HD could elucidate some fundamental molecular aspects of striatal neurodegeneration in this disease.
Footnotes
This work was supported by Commissariat à l'Energie Atomique and Centre National de la Recherche Scientifique (CNRS) (E.B.), University Pierre et Marie Curie, CNRS, and European Commission Grant BMH4-CT-97-2215 (M.J.B. and J.C.). M.G. was supported by the Ministère de la Recherche Médicale. We thank M. Yaniv for the Δc-Jun plasmid.
Correspondence should be addressed to Jocelyne Caboche, Neuronal Signaling and Gene Regulation, Centre National de la Recherche Scientifique/University Pierre et Marie Curie, Unité Mixte de Recherche 7102, 9 quai Saint Bernard, 75005 Paris, France. E-mail:Jocelyne.Caboche@snv.jussieu.fr.
REFERENCES
- 1.Angel P, Hattori K, Smeal T, Karin M. The jun proto-oncogene is positively autoregulated by its product, Jun/AP-1. Cell. 1988;55:875–885. doi: 10.1016/0092-8674(88)90143-2. [DOI] [PubMed] [Google Scholar]
- 2.Beal MF. Mitochondria, free radicals, and neurodegeneration. Curr Opin Neurobiol. 1996;6:661–666. doi: 10.1016/s0959-4388(96)80100-0. [DOI] [PubMed] [Google Scholar]
- 3.Beal MF. Energetics in the pathogenesis of neurodegenerative diseases. Trends Neurosci. 2000;23:298–304. doi: 10.1016/s0166-2236(00)01584-8. [DOI] [PubMed] [Google Scholar]
- 4.Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT. Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid. J Neurosci. 1993;13:4181–4192. doi: 10.1523/JNEUROSCI.13-10-04181.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Behrens A, Sibilia M, Wagner EF. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat Genet. 1999;21:326–329. doi: 10.1038/6854. [DOI] [PubMed] [Google Scholar]
- 6.Björklund A, Lindvall O. Dopamine-containing systems in the CNS. In: Björklund A, Hökfelt T, editors. Classical transmitter in the CNS. Elsevier; Amsterdam: 1984. pp. 55–74. [Google Scholar]
- 7.Bowyer JF, Clausing P, Schmued L, Davies DL, Binienda Z, Newport GD, Scallet AC, Slikker W., Jr Parenterally administered 3-nitropropionic acid and amphetamine can combine to produce damage to terminals and cell bodies in the striatum. Brain Res. 1996;712:221–229. doi: 10.1016/0006-8993(95)01417-9. [DOI] [PubMed] [Google Scholar]
- 8.Brouillet E, Jenkins BG, Hyman BT, Ferrante RJ, Kowall NW, Srivastava R, Roy DS, Rosen BR, Beal MF. Age-dependent vulnerability of the striatum to the mitochondrial toxin 3-nitropropionic acid. J Neurochem. 1993;60:356–359. doi: 10.1111/j.1471-4159.1993.tb05859.x. [DOI] [PubMed] [Google Scholar]
- 9.Brouillet E, Hantraye P, Ferrante RJ, Dolan R, Leroy-Willig A, Kowall NW, Beal MF. Chronic mitochondrial energy impairment produces selective striatal degeneration and abnormal choreiform movements in primates. Proc Natl Acad Sci USA. 1995;92:7105–7109. doi: 10.1073/pnas.92.15.7105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brouillet E, Guyot MC, Mittoux V, Altairac S, Conde F, Palfi S, Hantraye P. Partial inhibition of brain succinate dehydrogenase by 3-nitropropionic acid is sufficient to initiate striatal degeneration in rat. J Neurochem. 1998;70:794–805. doi: 10.1046/j.1471-4159.1998.70020794.x. [DOI] [PubMed] [Google Scholar]
- 11.Brouillet E, Conde F, Beal MF, Hantraye P. Replicating Huntington's disease phenotype in experimental animals. Prog Neurobiol. 1999;59:427–468. doi: 10.1016/s0301-0082(99)00005-2. [DOI] [PubMed] [Google Scholar]
- 12.Brown LL. Somatotopic organization in rat striatum: evidence for a combinational map. Proc Natl Acad Sci USA. 1992;89:7403–7407. doi: 10.1073/pnas.89.16.7403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Browne SE, Bowling AC, MacGarvey U, Baik MJ, Berger SC, Muqit MM, Bird ED, Beal MF. Oxidative damage and metabolic dysfunction in Huntington's disease: selective vulnerability of the basal ganglia. Ann Neurol. 1997;41:646–653. doi: 10.1002/ana.410410514. [DOI] [PubMed] [Google Scholar]
- 14.Cattaneo E, Rigamonti D, Goffredo D, Zuccato C, Squitieri F, Sipione S. Loss of normal huntingtin function: new developments in Huntington's disease research. Trends Neurosci. 2001;24:182–188. doi: 10.1016/s0166-2236(00)01721-5. [DOI] [PubMed] [Google Scholar]
- 15.Cotman CW, Monaghan DT, Ottersen OP, Storm-Mathisen J. Anatomical organization of excitatory amino acid receptors and their pathways. Trends Neurosci. 1987;10:273–280. [Google Scholar]
- 16.Dautry C, Vaufrey F, Brouillet E, Bizat N, Henry PG, Conde F, Bloch G, Hantraye P. Early N-acetylaspartate depletion is a marker of neuronal dysfunction in rats, primates chronically treated with the mitochondrial toxin 3-nitropropionic acid. J Cereb Blood Flow Metab. 2000;20:789–799. doi: 10.1097/00004647-200005000-00005. [DOI] [PubMed] [Google Scholar]
- 17.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 18.Estus S, Zaks WJ, Freeman RS, Gruda M, Bravo R, Johnson EM., Jr Altered gene expression in neurons during programmed cell death: identification of c-jun as necessary for neuronal apoptosis. J Cell Biol. 1994;127:1717–1727. doi: 10.1083/jcb.127.6.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Filloux F, Townsend JJ. Pre- and postsynaptic neurotoxic effects of dopamine demonstrated by intrastriatal injection. Exp Neurol. 1993;119:79–88. doi: 10.1006/exnr.1993.1008. [DOI] [PubMed] [Google Scholar]
- 20.Greene JG, Sheu SS, Gross RA, Greenamyre JT. 3-Nitropropionic acid exacerbates N-methyl-d-aspartate toxicity in striatal culture by multiple mechanisms. Neuroscience. 1998;84:503–510. doi: 10.1016/s0306-4522(97)00389-8. [DOI] [PubMed] [Google Scholar]
- 21.Gu M, Gash MT, Mann VM, Javoy-Agid F, Cooper JM, Schapira AH. Mitochondrial defect in Huntington's disease caudate nucleus. Ann Neurol. 1996;39:385–389. doi: 10.1002/ana.410390317. [DOI] [PubMed] [Google Scholar]
- 22.Ham J, Babij C, Whitfield J, Pfarr CM, Lallemand D, Yaniv M, Rubin LL. A c-Jun dominant negative mutant protects sympathetic neurons against programmed cell death. Neuron. 1995;14:927–939. doi: 10.1016/0896-6273(95)90331-3. [DOI] [PubMed] [Google Scholar]
- 23.Herdegen T, Skene P, Bähr M. The c-Jun transcription factor-bipotential mediator of neuronal death, survival and regeneration. Trends Neurosci. 1997;20:227–231. doi: 10.1016/s0166-2236(96)01000-4. [DOI] [PubMed] [Google Scholar]
- 24.Herdegen T, Claret FX, Kallunki T, Martin-Villalba A, Winter C, Hunter T, Karin M. Lasting N-terminal phosphorylation of c-Jun and activation of c-Jun N-terminal kinases after neuronal injury. J Neurosci. 1998;18:5124–5135. doi: 10.1523/JNEUROSCI.18-14-05124.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- 26.Jenkins BG, Koroshetz WJ, Beal MF, Rosen BR. Evidence for impairment of energy metabolism in vivo in Huntington's disease using localized 1H NMR spectroscopy. Neurology. 1993;43:2689–2695. doi: 10.1212/wnl.43.12.2689. [DOI] [PubMed] [Google Scholar]
- 27.Jenkins BG, Rosas HD, Chen YC, Makabe T, Myers R, MacDonald M, Rosen BR, Beal MF, Koroshetz WJ. 1H NMR spectroscopy studies of Huntington's disease: correlations with CAG repeat numbers. Neurology. 1998;50:1357–1365. doi: 10.1212/wnl.50.5.1357. [DOI] [PubMed] [Google Scholar]
- 28.Kaminska B, Pyrzynska B, Ciechomska I, Wisniewska M. Modulation of the composition of AP-1 complex and its impact on transcriptional activity. Acta Neurobiol Exp. 2000;60:395–402. doi: 10.55782/ane-2000-1358. [DOI] [PubMed] [Google Scholar]
- 29.La Fontaine MA, Geddes JW, Banks A, Butterfield DA. Effect of exogenous and endogenous antioxidants on 3-nitropionic acid-induced in vivo oxidative stress and striatal lesions: insights into Huntington's disease. J Neurochem. 2000;75:1709–1715. doi: 10.1046/j.1471-4159.2000.0751709.x. [DOI] [PubMed] [Google Scholar]
- 30.Le-Niculescu H, Bonfoco E, Kasuya Y, Claret FX, Green DR, Karin M. Withdrawal of survival factors results in activation of the JNK pathway in neuronal cells leading to Fas ligand induction and cell death. Mol Cell Biol. 1999;19:751–763. doi: 10.1128/mcb.19.1.751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lipton SA, Rosenberg PA. Excitatory amino acids as a final common pathway for neurologic disorders. N Engl J Med. 1994;330:613–622. doi: 10.1056/NEJM199403033300907. [DOI] [PubMed] [Google Scholar]
- 32.Liu YF. Expression of polyglutamine-expanded Huntingtin activates the SEK1-JNK pathway and induces apoptosis in a hippocampal neuronal cell line. J Biol Chem. 1998;273:28873–28877. doi: 10.1074/jbc.273.44.28873. [DOI] [PubMed] [Google Scholar]
- 33.Liu YF, Dorow D, Marshall J. Activation of MLK2-mediated signaling cascades by polyglutamine-expanded huntingtin. J Biol Chem. 2000;275:19035–19040. doi: 10.1074/jbc.C000180200. [DOI] [PubMed] [Google Scholar]
- 34.Luo Y, Roth GS. The roles of dopamine oxidative stress and dopamine receptor signaling in aging and age-related neurodegeneration. Antioxid Redox Signal. 2000;2:449–460. doi: 10.1089/15230860050192224. [DOI] [PubMed] [Google Scholar]
- 35.Luo Y, Umegaki H, Wang X, Abe R, Roth GS. Dopamine induces apoptosis through an oxidation-involved SAPK/JNK activation pathway. J Biol Chem. 1998;273:3756–3764. doi: 10.1074/jbc.273.6.3756. [DOI] [PubMed] [Google Scholar]
- 36.Martin-Villalba A, Herr I, Jeremias I, Hahne M, Brandt R, Vogel J, Schenkel J, Herdegen T, Debatin KM. CD95 ligand (Fas-L/APO-1L) and tumor necrosis factor-related apoptosis-inducing ligand mediate ischemia-induced apoptosis in neurons. J Neurosci. 1999;19:3809–3817. doi: 10.1523/JNEUROSCI.19-10-03809.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matthews RT, Yang L, Browne S, Baik M, Beal MF. Coenzyme Q10 administration increases brain mitochondrial concentrations and exerts neuroprotective effects. Proc Natl Acad Sci USA. 1998;95:8892–8897. doi: 10.1073/pnas.95.15.8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morishima Y, Gotoh Y, Zieg J, Barrett T, Takano H, Flavell R, Davis RJ, Shirasaki Y, Greenberg ME. Beta-amyloid induces neuronal apoptosis via a mechanism that involves the c-Jun N-terminal kinase pathway and the induction of Fas ligand. J Neurosci. 2001;21:7551–7560. doi: 10.1523/JNEUROSCI.21-19-07551.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nucifora FC, Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA. Interference by huntingtin and atrophin-1 with CBP-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- 40.Ouary S, Bizat N, Altairac S, Menetrat H, Mittoux V, Conde F, Hantraye P, Brouillet E. Major strain differences in response to chronic systemic administration of the mitochondrial toxin 3-nitropropionic acid in rats: implications for neuroprotection studies. Neuroscience. 2000;97:521–530. doi: 10.1016/s0306-4522(00)00020-8. [DOI] [PubMed] [Google Scholar]
- 41.Pang Z, Geddes JW. Mechanisms of cell death induced by the mitochondrial toxin 3-nitropropionic acid: acute excitotoxic necrosis and delayed apoptosis. J Neurosci. 1997;17:3064–3073. doi: 10.1523/JNEUROSCI.17-09-03064.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Putcha GV, Moulder KL, Golden JP, Bouillet P, Adams JA, Strasser A, Johnson EM. Induction of BIM, a proapoptotic BH3-only BCL-2 family member, is critical for neuronal apoptosis. Neuron. 2001;29:615–628. doi: 10.1016/s0896-6273(01)00238-0. [DOI] [PubMed] [Google Scholar]
- 43.Reynolds DS, Carter RJ, Morton AJ. Dopamine modulates the susceptibility of striatal neurons to 3-nitropropionic acid in the rat model of Huntington's disease. J Neurosci. 1998;18:10116–10127. doi: 10.1523/JNEUROSCI.18-23-10116.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD. Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science. 1999;286:2358–2361. doi: 10.1126/science.286.5448.2358. [DOI] [PubMed] [Google Scholar]
- 45.Sawa A, Wiegand GW, Cooper J, Margolis RL, Sharp AH, Lawler JF, Jr, Greenamyre JT, Snyder SH, Ross CA. Increased apoptosis of Huntington disease lymphoblasts associated with repeat length-dependent mitochondrial depolarization. Nat Med. 1999;5:1194–1198. doi: 10.1038/13518. [DOI] [PubMed] [Google Scholar]
- 46.Schulz JB, Matthews RT, Beal MF. Role of nitric oxide in neurodegenerative diseases. Curr Opin Neurol. 1995;8:480–486. doi: 10.1097/00019052-199512000-00016. [DOI] [PubMed] [Google Scholar]
- 47.Schulz JB, Matthews RT, Henshaw DR, Beal MF. Neuroprotective strategies for treatment of lesions produced by mitochondrial toxins: implications for neurodegenerative diseases. Neuroscience. 1996;71:1043–1048. doi: 10.1016/0306-4522(95)00527-7. [DOI] [PubMed] [Google Scholar]
- 48.Schwarzschild MA, Cole RL, Hyman SE. Glutamate, but not dopamine, stimulates stress-activated protein kinase and AP-1-mediated transcription in striatal neurons. J Neurosci. 1997;17:3455–3466. doi: 10.1523/JNEUROSCI.17-10-03455.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sgambato V, Abo V, Rogard M, Besson MJ, Deniau JM. Effect of electrical stimulation of the cerebral cortex on the expression of the Fos protein in the basal ganglia. Neuroscience. 1997;81:93–112. doi: 10.1016/s0306-4522(97)00179-6. [DOI] [PubMed] [Google Scholar]
- 50.Sgambato V, Pages C, Rogard M, Besson MJ, Caboche J. Extracellular signal-regulated kinase (ERK) controls immediate early gene induction on corticostriatal stimulation. J Neurosci. 1998;18:8814–8825. doi: 10.1523/JNEUROSCI.18-21-08814.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sharp AH, Ross CA. Neurobiology of Huntington's disease. Neurobiol Dis. 1996;3:3–15. doi: 10.1006/nbdi.1996.0002. [DOI] [PubMed] [Google Scholar]
- 52.Steffan JS, Kazantsev A, Spasic-Boskovic O, Greenwald M, Zhu YZ, Gohler H, Wanker EE, Bates GP, Housman DE, Thompson LM. The Huntington's disease protein interacts with p53, CREB-binding protein, represses transcription. Proc Natl Acad Sci USA. 2000;97:6763–6768. doi: 10.1073/pnas.100110097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tao X, Finkbeiner S, Arnold DB, Shaywitz AJ, Greenberg ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron. 1998;20:709–726. doi: 10.1016/s0896-6273(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 54.Vanhoutte P, Barnier JV, Guibert B, Pages C, Besson MJ, Hipskind RA, Caboche J. Glutamate induces phosphorylation of Elk-1 and CREB, along with c-fos activation, via an extracellular signal-regulated kinase-dependent pathway in brain slices. Mol Cell Biol. 1999;19:136–146. doi: 10.1128/mcb.19.1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Whitfield J, Neame SJ, Paquet L, Bernard O, Ham J. Dominant-negative c-Jun promotes neuronal survival by reducing BIM expression and inhibiting mitochondrial cytochrome c release. Neuron. 2001;29:629–643. doi: 10.1016/s0896-6273(01)00239-2. [DOI] [PubMed] [Google Scholar]
- 56.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 57.Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- 58.Zeevalk GD, Derr-Yellin E, Nicklas WJ. NMDA receptor involvement in toxicity to dopamine neurons in vitro caused by the succinate dehydrogenase inhibitor 3-nitropropionic acid. J Neurochem. 1995;64:455–458. doi: 10.1046/j.1471-4159.1995.64010455.x. [DOI] [PubMed] [Google Scholar]
- 59.Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin-mediated BDNF gene transcription in Huntington's disease. Science. 2001;293:493–498. doi: 10.1126/science.1059581. [DOI] [PubMed] [Google Scholar]