

Abstract
The extracellular protease cascade of tissue plasminogen activator (tPA) and plasminogen has been implicated in neuronal plasticity and degeneration. We show here that unstimulated expression of tPA in the mouse hippocampus is concentrated in the mossy fiber pathway, with little or no expression within the perforant path, the Schaffer collaterals, or neuronal cell bodies. tPA protein is also expressed in vascular endothelial cells throughout the brain parenchyma. Four hours after excitotoxic injury, tPA protein is transiently induced within CA1 pyramidal neurons. The induced CA1 tPA is localized to neurons that survive the injury and is enzymatically active. Within the mossy fiber pathway, injury resulted in decreased tPA protein. In contrast, mossy fiber tPA activity displayed a biphasic character: transient increase at 8 hr, then a decrease by 24 hr after injury. Analysis of plasminogen activator inhibitor-1 (PAI-1) expression showed that PAI-1 antigen is upregulated by 24 hr and could account for the tPA activity downregulation seen at this time point. Plasminogen immunohistochemistry suggested an increase within the mossy fiber pathway after injury. Finally, hippocampal tPA expression among various mammalian species was strikingly different. These results indicate a complex control of tPA protein and enzymatic activity in the hippocampus that may help regulate neuronal plasticity.
Keywords: tissue plasminogen activator (tPA), hippocampus, plasminogen activator inhibitor 1 (PAI-1), mossy fiber pathway, excitotoxic injury, protease, mouse, rat, human, hamster, gerbil, cat, synaptic plasticity
Extracellular proteases are expressed by neurons in the CNS (Sappino et al., 1993; Chen et al., 1995; Nishibori et al., 1995; Backstrom et al., 1996; Gschwend et al., 1997; Scarisbrick et al., 2001), and their function can vary from potentiating synaptic transmission (Hoffman et al., 1998; Gingrich et al., 2000) to structural alterations associated with long-lasting forms of synaptic plasticity (Baranes et al., 1998; Neuhoff et al., 1999). Evidence shows that extracellular proteases are also capable of mediating regulatory components of learning and memory (Shiosaka and Yoshida, 2000).
Protease expression is balanced by proteolytic inhibitors (Festoff et al., 1996; Osterwalder et al., 1996; Hastings et al., 1997; Jaworski and Fager, 2000; Hino et al., 2001; Kato et al., 2001). Given the neuronal roles of extracellular proteases and their inhibitors (Shiosaka and Yoshida, 2000), knowledge of their expression and regulation is critical to an understanding of neuronal communication, learning and memory, and certain instances of neuronal death. This article focuses on one proteolytic axis implicated in all the events described above, the plasminogen activator (PA)–plasmin system.
Tissue plasminogen activator (tPA) is a serine protease that catalyzes the activation of plasminogen to plasmin. Previous experiments suggest the following model for tPA expression in adult neurons: tPA is synthesized under basal conditions and is stored in vesicles (Baranes et al., 1996; Gualandris et al., 1996; Parmer et al., 1997; Lochner et al., 1998). On depolarization, tPA is released into the extracellular space (Gualandris et al., 1996; Parmer et al., 1997), and tPA mRNA is upregulated (Qian et al., 1993; Carroll et al., 1994). Plasminogen, a serine protease zymogen, is present in most extracellular fluids and can be recruited by cells requiring proteolysis through the expression of a PA. This proteolytic cascade is countered by the expression of protease inhibitors, including plasminogen activator inhibitor 1 (PAI-1), and neuroserpin. Within the nervous system, the PA–plasmin axis has been implicated in neuronal growth cone penetration (Krystosek and Seeds 1981), Wallerian degeneration and neuronal regeneration (Bignami et al., 1982; Salles et al., 1990), migration of cerebellar granule neurons during development (Friedman and Seeds, 1995), long-term potentiation (Frey et al., 1996; Huang et al., 1996; Baranes et al., 1998; Madani et al., 1999), long-term depression (Calabresi et al., 2000), NMDA receptor-mediated signaling (Nicole et al., 2001), synaptic remodeling (Baranes et al., 1998; Neuhoff et al., 1999), and various pathological situations such as excitotoxic injury (Tsirka et al., 1995; Chen and Strickland, 1997; Wang et al., 1998) and peripheral nerve injury (Akassoglou et al., 2000). Additionally, overexpression of tPA enhances long-term potentiation (LTP) and improves performance in the Morris water maze (Madani et al., 1999).
To better understand the localization and regulation of the PA–plasmin system within the CNS, we have identified several antibodies that have allowed us to localize the expression of tPA, PAI-1, and plasminogen in the hippocampus after excitotoxic injury. Our results suggest a more restricted expression of tPA within in the CNS than previously recognized and a complex regulatory pathway that includes control both of tPA expression and activity.
MATERIALS AND METHODS
Reagents and animals. Antibodies used were as follows: rabbit anti-human tPA antisera (Waller and Schleuning, 1985; or Molecular Innovations, catalog #ASHTPA), sheep anti-mouse PAI-1 (American Diagnostica, catalog #1040), sheep anti-rat plasminogen antisera (a kind gift from E. Reich, State University of New York, Stony Brook, NY), and neuroserpin antisera (a gift from D. Lawrence, American Red Cross, Rockville, MD). Wild-type animals were C57BL/6J. tPA-deficient animals (tPA−/−; Carmeliet et al., 1994) and were back-crossed to C57BL/6J mice for nine generations. PAI-1-deficient animals (PAI-1−/−) were from the Jackson Laboratory (Bar Harbor, ME) and were back-crossed to C57BL/6J mice for seven generations. Plasminogen-deficient animals (plg−/−; Bugge et al., 1995) were of mixed genetic background, 129 and C57BL/6 (a gift from J. Degen, Children's Hospital Medical Center, Cincinnati, OH). Other brains were obtained as follows: Mongolian gerbil brains (a gift from F. J. Antonawich, State University of New York, Stony Brook, NY), hamster brain (a gift from L. P. Morin, State University of New York, Stony Brook, NY), cat brain (a gift from I. Solomon, State University of New York, Stony Brook, NY), and human hippocampus brain sections from six individuals (a gift from D. W. Dickson, Mayo Clinic, Jacksonville, FL).
Immunohistochemistry. Mouse, rat, and gerbil brain tissue was obtained from ice-cold PBS-perfused animals. All tissue was frozen in optimal cutting temperature compound under powdered dry ice, sectioned to 14 μm, and stored at −70°C until analysis. Before use, sections were allowed to air dry and were fixed in 4% paraformaldehyde in PBS for 20 min. After washing, sections were blocked in PBS containing; 10% sera from species of the secondary antibody, 1% BSA, 0.5% Triton X-100, and 0.5% Tergitol (NP-40) for at least 30 min. Primary antibody in fresh blocking buffer was then applied at the appropriate dilution [tPA (Waller and Schleuning, 1985), 1:500; tPA (Molecular Innovations), 1:1500; PAI-1, 1:200; plasminogen, 1:500; and neuroserpin 1:2000] and incubated overnight at 4°C in a humidified chamber. Immediately before use, the tPA antibodies were preabsorbed for 30 min at room temperature with an acetone brain powder generated from the brains of tPA-deficient animals (Harlow and Lane, 1988). The brain powder was pelleted by centrifugation, and the supernatant was applied to the sections as described. For the tPA preabsorption experiment, tPA-Sepharose (Andrade-Gordon and Strickland, 1990) was added to the primary antibody-brain powder mixture in a >50-fold protein excess and incubated for 30 min at room temperature, and the supernatant was used.
In situ zymography. Zymography to detect tPA was performed essentially as described by Sappino et al. (1993) using 14 μm fresh-frozen sections on microscope slides, covered with a coverslip, and allowed to develop at room temperature in a humidified chamber. Zymographies were developed for 8–20 hr. Zymography to detect plasminogen was performed as above with the substitution of 0.1 μg of tPA for plasminogen in the casein mix, and development times were extended to 48–72 hr. For the plasminogen zymography, excess tPA had to be added to the matrix to visualize plasminogen activity. The plasminogen activity was specific, because lytic zones were restricted to tissue regions, and no activity was detected in the absence of tPA.
In situ hybridization. In situ hybridizations were performed using a modified version of the method of Hebert et al. (1991). Fresh-frozen brain sections were fixed with 4% paraformaldehyde in PBS for 20 min at room temperature and washed with 0.1 m DEPC-treated phosphate buffer, pH 7.4. Slides were incubated sequentially in 0.2 m HCl for 10 min, 0.5% acetic anhydride in 0.1 m Tris, pH 8.0, for 10 min, and finally 14 μg/ml proteinase K in Tris-buffered EDTA for 10 min, with several phosphate buffer washes between each step. Sections were dehydrated serially through ethanol and finally washed with chloroform and allowed to air dry. Digoxygenin-labeled (Roche Molecular Biochemicals, Indianapolis, IN) RNA probes were synthesized as per the manufacturer's recommendations using T3 or T7 RNA polymerase. Colorimetric development was performed with an anti-digoxygenin primary antibody and a peroxidase-labeled secondary antibody using diaminobenzidine as the substrate. Three different tPA antisense probes were used: a 5′ probe encompassing the 5′-terminal 500 bp, an internal probe encompassing the end of the coding region and the 5′ portion of the 3′ untranslated region (UTR;Sappino et al., 1993), and a 3′ probe encompassing the 3′-most 500 bp all within the 3′ UTR. In our experience, we found that using the antisense probe on tPA−/− brain tissue was a superior control to tPA sense transcripts for hybridization.
Stereotaxic injections. Intrahippocampal injections in mice were performed unilaterally as described previously (Tsirka et al., 1997). Kainic acid (Tocris, Ballwan, MO; 0.3–1.0 nmol/μl) was resuspended in sterile PBS and then filtered through a 0.2 μm filter before loading the glass capillary injection needle. Rat stereotaxic injections were performed on 300 gm Sprague Dawley rats as described previously (Lee et al., 1997).
RESULTS
tPA is abundantly expressed in the mossy fiber pathway
Expression and localization of tPA protein were visualized using a rabbit polyclonal antibody (Waller and Schleuning, 1985). This antibody was raised against human tPA purified from HeLa cells. Within the hippocampus, high-level tPA expression was seen primarily in two cell types: first, a large punctate staining within the neuritic layer comprising the mossy fiber pathway (Fig.1A–D); and second, a slightly more uniform staining within morphologically identified blood vessels (Fig. 1D,arrows). No specific detectable neuronal cell body staining was visualized in any of the hippocampal cell layers (CA1, CA3, and dentate gyrus) under basal conditions. Several lines of evidence indicate that the antibody is specifically staining tPA. First, the antibody was preabsorbed with recombinant human tPA (Activase; a gift from Genentech, San Francisco, CA) conjugated to Sepharose (Andrade-Gordon and Strickland, 1990). Antibody preabsorbtion completely removed the observed mossy fiber and blood vessel staining (Fig. 1E,F). A second control was to use the antibody on brain sections from animals genetically deficient for tPA (Fig. 1G, tPA−/−). This control revealed an essentially indistinguishable staining pattern as that seen with the wild-type control sections using preabsorbed antibody (Fig. 1,E vs G). Third, all the tPA staining patterns detailed within this article were reproduced using a second independently derived polyclonal antibody (Molecular Innovations; data not shown). Finally, the mossy fiber and vascular staining observed in these controls correlated almost perfectly with the tPA distribution pattern seen by in situ zymography, an assay for tPA enzymatic activity (Sappino et al., 1993; Fig. 1H). These data, taken together, strongly suggest that the residual staining pattern seen in Figure 1, E and G, is background staining. This background staining included the cell bodies of neurons in the CA2 area (Fig. 1E–G, arrowheads).
Fig. 1.
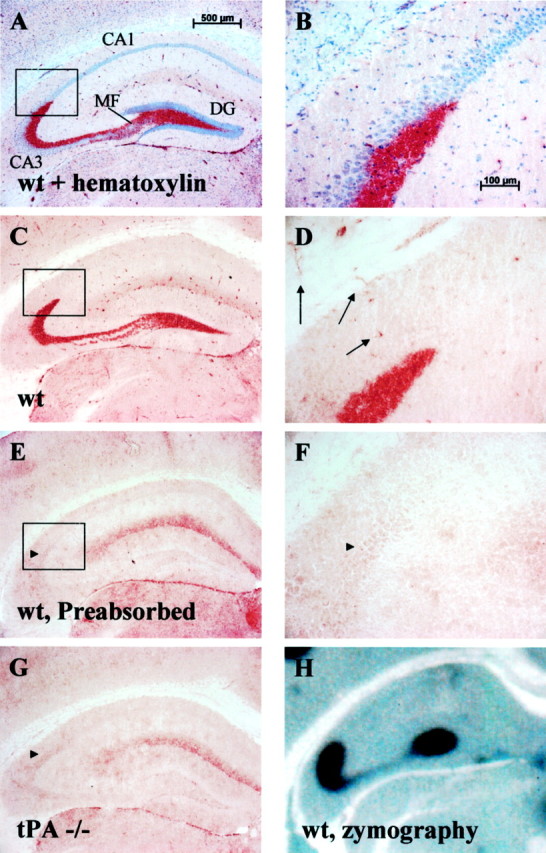
tPA is abundantly expressed in the mossy fiber pathway. A, B, tPA immunohistochemistry on wild-type (wt) sections shows robust tPA staining (red) in the hippocampal mossy fiber pathway. Sections are counter-stained with hematoxylin to visualize the neuronal cell body layers of the hippocampus. DG, Dentate gyrus;MF, mossy fiber pathway; n = 20 animals. C, D, tPA immunostain without the hematoxylin counterstain showing lack of significant staining within the neuronal cell bodies (compare high-magnification B withD). Arrows in D highlight vascular endothelial cell staining that is seen throughout the brain.E, F, tPA immunohistochemistry on wild-type sections after preabsorbing the antibody (Figure legend continues.) (Figure legend continued.) with tPA as described in Materials and Methods. Notice the loss of mossy fiber and vascular staining but residual background parenchymal stain;n = 7 animals. G, tPA immunohistochemistry on tPA-deficient (tPA−/−) brain sections. Notice a similar level of background parenchymal staining compared with E; n = 10 animals.H, Zymographic analysis of tPA activity (dark zone of lysis against a milky background) closely parallels staining seen by immunohistochemistry;n = 20 animals. Activity is plasminogen-dependent (data not shown). Boxes in A,C, and E show the region magnified inB, D, and F.Arrowheads in E–G highlight background CA2 cell body staining.
Outside of the hippocampus, robust tPA staining could be observed in vascular tissue throughout the brain (Fig. 1D,arrows) and in the meninges as well as in the neuropil of the central nucleus, the bed nucleus, and the hypothalamus (data not shown). We did not find robust staining within microglia. The reasons for the discrepancy between this lack of staining and previous results that found microglial expression of tPA (Tsirka et al., 1997) are not known.
tPA is upregulated in CA1 neurons after excitotoxic injury
Because we could not find significant tPA staining in the CA1 region in basal conditions, we examined tPA expression after excitotoxic injury. After unilateral kainic acid (KA) injection, tPA antigen was upregulated in CA1 neuronal cell bodies, consistent with its characterization as an immediate early gene (Qian et al., 1993). The pattern of tPA CA1 upregulation was somewhat variable in terms of both localization and timing. The variability is most likely attributable to very precise requirements of experimental parameters. In general, tPA upregulation was seen in the ipsilateral or injected hippocampus in neuronal cell bodies comprising the border of neuronal degeneration, as evidenced by staining adjacent sections with Nissl stain (Fig. 2A,B,arrows). Dying neurons, which are shrunken with pyknotic nuclei, are easily identified by Nissl stain (Fig.2B). In many experiments, with excessive degeneration in the ipsilateral hippocampus, a strong induction of tPA could be seen in the contralateral or uninjected CA1 neuronal cell bodies (Fig.2C). This upregulated CA1 tPA was active, as measured byin situ zymography (Fig. 2D).
Fig. 2.
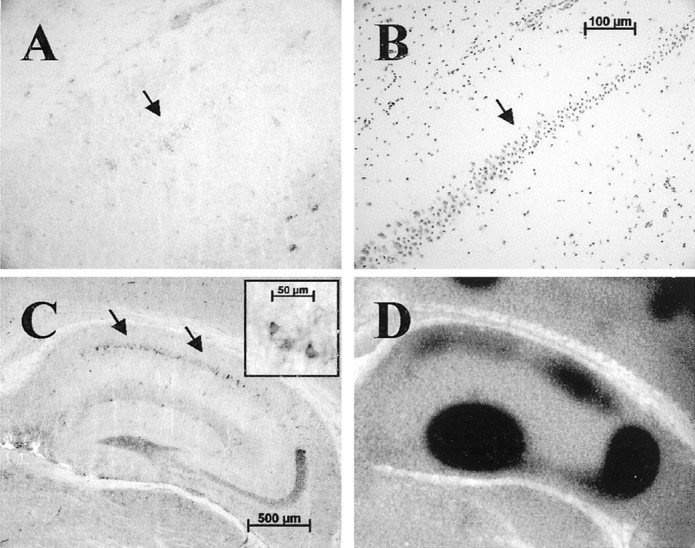
tPA is upregulated in CA1 neurons after excitotoxic injury. A, After a weak excitotoxic injury (between 0.3 and 0.6 nmol of KA in a 300 nl volume, depending on lot potency), at 8 hr after injury tPA protein is upregulated in neuronal cell bodies within the CA1 layer (arrow);n = 18 animals, with 14 showing some level of staining. These neurons occupy the border of surviving and degenerating neurons as seen in the adjacent section (B) stained with Nissl (arrow). Degenerating neurons are visualized as having pyknotic nuclei versus the surviving neurons that appear normal. C, After a robust injury (between 0.9 and 1.2 nmol of KA in a 300 nl volume, depending on lot potency), at 8 hr after injury tPA protein is strongly upregulated within some neuronal cell bodies of the contralateral uninjected hippocampus (arrows); n = 16 animals, with 8 showing significant staining. Inset, Magnification of several pyramidal neurons within the CA1 showing cytoplasmic tPA accumulation. Note that not all CA1 neurons upregulate tPA.D, This upregulated protein is active, as measured by the zymographic analysis. Contralateral CA1 tPA expression was seen when treated with sufficient KA to destroy ∼80% of ipsilateral hippocampus. In two animals, we could detect both ipsilateral and contralateral CA1 staining.
A time course revealed that the tPA induction was transient. On average, tPA protein staining could be detected by 2.5 hr, peaking between 5 and 8 hr and returning to baseline by 24–48 hr after injection (Fig. 3). Despite the variability in tPA induction noted above, in all cases, tPA staining was barely detectable by 24–48 hr (Fig. 3E). Subcutaneous administration of KA also produced tPA upregulation in the CA1 (data not shown).
Fig. 3.
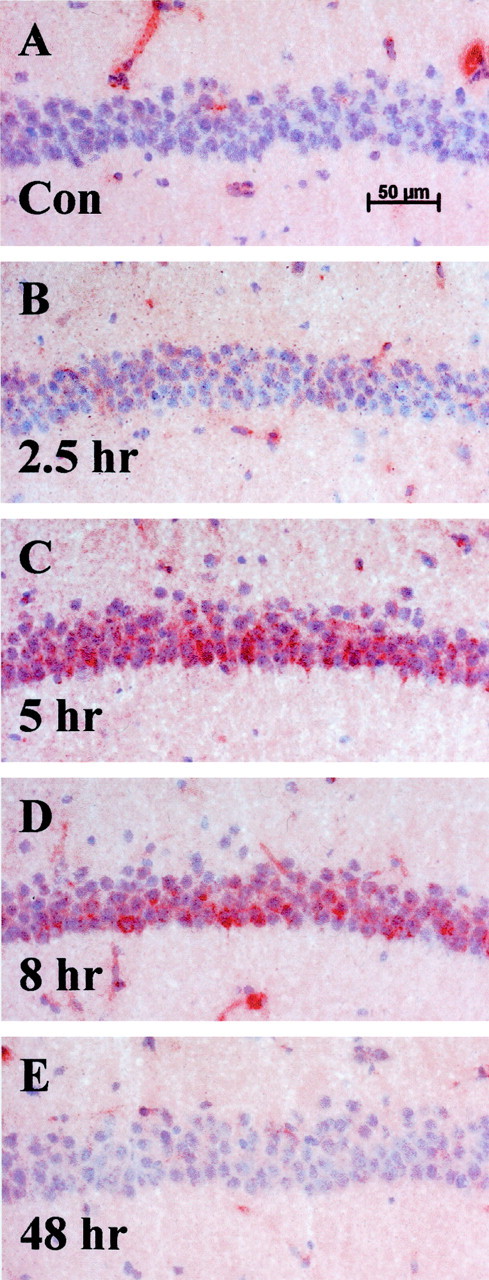
Time course of tPA induction in CA1 neurons after injury. Results show transiently upregulated neuronal cell body tPA in CA1 pyramidal neurons after injury in the contralateral uninjected hippocampus after a robust excitotoxic lesion. Sections were counterstained with hematoxylin to visualize the neuronal cell layer. Notice the lack of pyknotic nuclei and neuronal degeneration.A, No significant tPA staining is seen in the CA1 layer of control animals; n = 20 animals.B, CA1 neuronal cell layer 2.5 hr after lesion;n = 4 animals. C, tPA staining is induced 5 hr after injury (red staining within neuronal cell layer); n = 4 animals. D, tPA staining seen at 8 hr after injury; n = 8 animals.E, Forty-eight hours after injury, tPA is no longer detectable within the cell layer, yet the neurons still appear healthy;n = 4 animals. Additional time points assayed were 1 and 24 hr (data not shown).
To determine whether tPA protein upregulation was attributable to increased tPA mRNA, in situ hybridization was performed. These analyses revealed no detectable tPA mRNA staining under basal conditions in any region of the brain (Fig.4A) even after long development periods for the in situ detection (>3 weeks). Our actin and laminin controls for mRNA detection robustly developed overnight (data not shown), indicating appropriate hybridization sensitivity. Eight hours after KA injection, however, when a strong upregulation of CA1 tPA antigen was detected, we could easily detect a strong induction of tPA mRNA in all pyramidal layers within a 3 d development period (Fig. 4B). Two lines of evidence indicate that the analysis is specific for tPA mRNA: (1) results were consistent using three different tPA mRNA probes (see Materials and Methods); and (2) tPA hybridization on brain sections from tPA-deficient animals was negative (Fig. 4C).
Fig. 4.
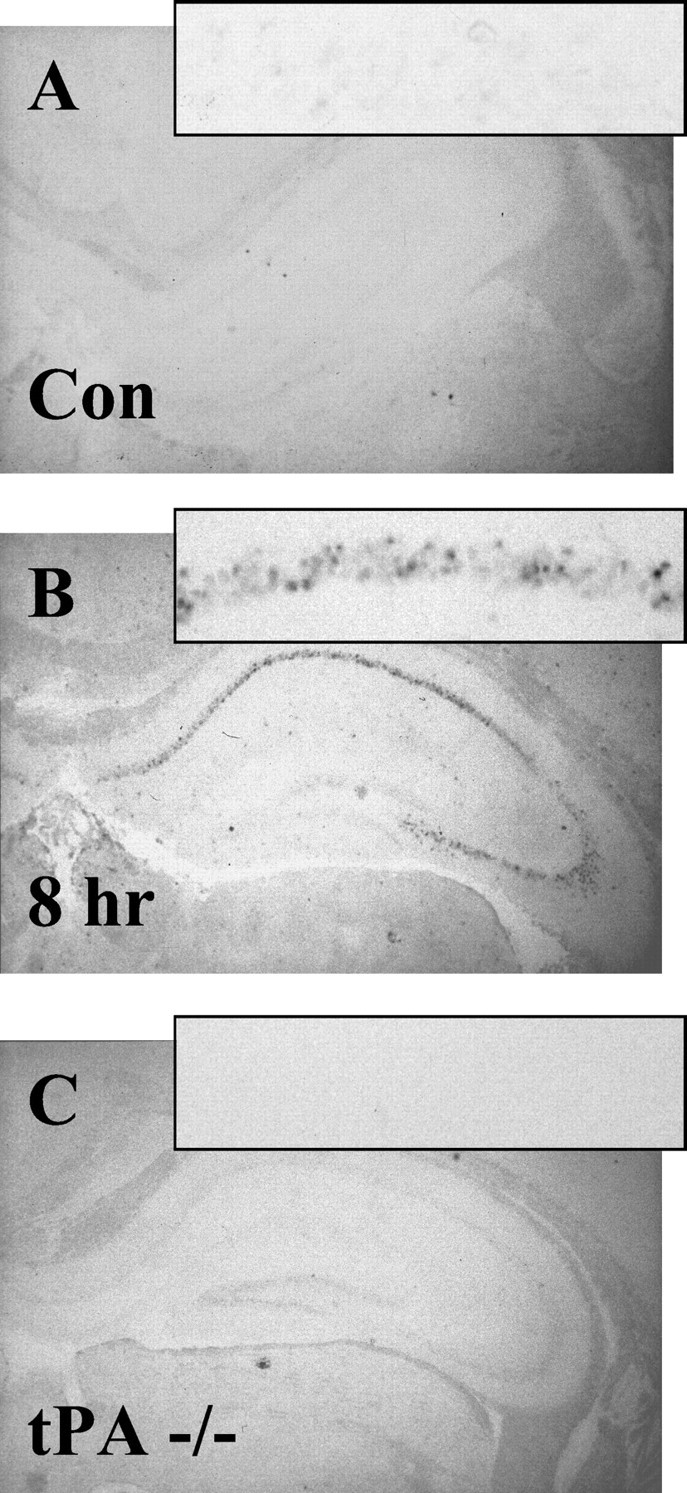
tPA mRNA is strongly upregulated after excitotoxic injury in the contralateral uninjected CA1 pyramidal neurons byin situ hybridization. tPA mRNA was not detectable in control animals (n = 10; A) or tPA−/− animals (n = 10; C). tPA mRNA was only detectable after injury in wild-type animals (n = 5; B).
Modulation of mossy fiber tPA activity by protease inhibitors
In wild-type samples (data not shown) and control PBS-injected samples, tPA protein staining (Fig.5A) and activity levels (Fig.5B) were equivalent in the ipsilateral (open arrows) and contralateral (closed arrows) hippocampus. By 8 hr after unilateral KA injection into CA1 neurons, we observed a consistent decrease in tPA protein staining in the ipsilateral mossy fiber pathway compared with the contralateral side (Fig, 5C, compare open, closed arrows). Surprisingly, tPA activity was transiently elevated in the injected side at this time point (Fig. 5D, open vs closed arrows). This increased activity coincides with the decrease in antigen staining, suggesting the downregulation of protease inhibitor activity between 0 and 8 hr after injection. At 24 hr after injury, tPA immunostaining (Fig. 5E) was the same as that observed at 8 hr, yet the level of tPA activity decreased to barely detectable levels for both sides of the hippocampus (Fig. 5, compare F,open, closed arrows, with B,open, closed arrows). This decrease in activity at 24 hr occurs despite the normal level of tPA protein staining in the contralateral control hippocampus (Fig. 5E, closed arrow) and the decreased but detectable level of tPA antigen in the ipsilateral hippocampus. This observation suggests that between 8 and 24 hr after injury, a protease inhibitor is upregulated within the hippocampus. PBS control injections showed no alteration in tPA protein staining or activity at either 8 or 24 hr after injection (8 hr, data not shown; 24 hr, Fig. 5A,B). Together, our evidence indicates a biphasic inhibitor profile: transient downregulation of protease inhibitor(s) by 8 hr with upregulation by 24 hr.
Fig. 5.
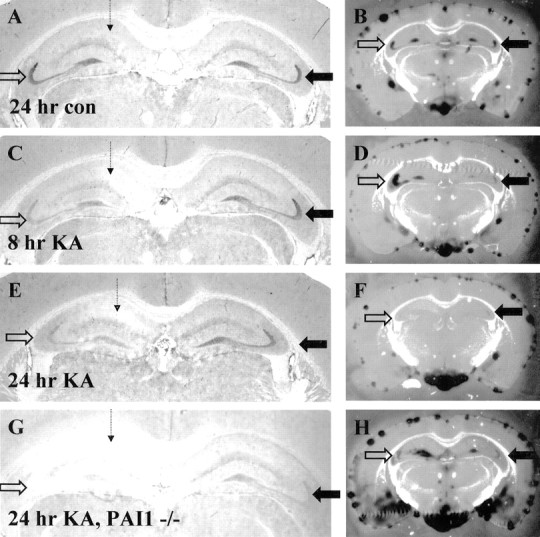
Modulation of protease inhibitor activity in the hippocampus after excitotoxic injury. The dashed arrowrepresents the site of excitotoxin injection. A, C, E, G, tPA protein staining; B, D, F, H, tPA activity zymographies performed on sections adjacent to those displayed for protein stains. A, B, Control PBS-injected wild type brain harvested 24 hr after injection; n = 6 animals. C, D, KA-injected brain harvested 8 hr after injection; n = 6 animals. E, F, KA-injected brain harvested 24 hr after injection;n = 6 animals. High-level upregulation of CA1 tPA protein was not seen in this experiment (see Fig. 2 legend). G, H, KA-injected brain from a PAI-1−/− animal harvested 24 hr after injection; n = 4 animals. All zymographies for each replicate shown were performed at the same time and incubated identically for the same time. The zymographies shown include the meningeal layer, which typically displays significant amounts of tPA activity, as an internal control to allow comparison of assay development time. The PAI-1 tissue showed slightly higher levels of tPA activity through out the tissue than wild type (H). Activities were all plasminogen-dependent. Open arrows indicate injected or ipsilateral hippocampus; closed arrows indicate uninjected or contralateral hippocampus.
We analyzed whether PAI-1, a specific tPA inhibitor, could account for the biphasic nature of the inhibitory activity by assessing tPA regulation after excitotoxic injury in the hippocampus of PAI-1-deficient animals. There were no obvious differences in KA-induced neuronal death between the wild-type and PAI-1−/− animals. Our analysis revealed that under basal conditions and at 8 hr after injury, both the tPA protein staining and the tPA activity profile in PAI-1-deficient tissue were the same as those seen in wild-type brain sections (data not shown). These data suggest that the increased tPA activity in the injected hippocampus at 8 hr was not attributable to downregulation of PAI-1. However, in the PAI-1−/− animals at 24 hr after injury, there was no detectable decrease in tPA activity (Fig.5H), in sharp contrast to the wild-type animals. These data suggest that the inhibition of tPA activity seen at 24 hr is at least partially attributable to upregulation of PAI-1 and are consistent with a published report on the upregulation of PAI-1 mRNA after KA stimulation (Masos and Miskin, 1997).
To further analyze the expression of PAI-1, we screened for the presence of PAI-1 antigen using a polyclonal antibody raised in sheep (Fig. 6) in wild-type animals. Neither under basal conditions nor at 8 hr after injury could we detect significant specific PAI-1 protein staining in the brain (Fig.6A,B) compared with control PAI-1−/− tissue (basal, Fig. 6D; 8 hr, data not shown). However, PAI-1 antigen was upregulated significantly at 24 hr after injury (Fig.6C) compared with the PAI-1−/− control tissue (Fig.6D). The distribution of PAI-1 antigen was greatest in the stratum oriens of the CA3 region as well as in the hilus of the dentate gyrus of the injected side. The uninjected hippocampus also showed an upregulation of PAI-1 antigen, but it was decreased in magnitude compared with the injected side (data not shown).
Fig. 6.
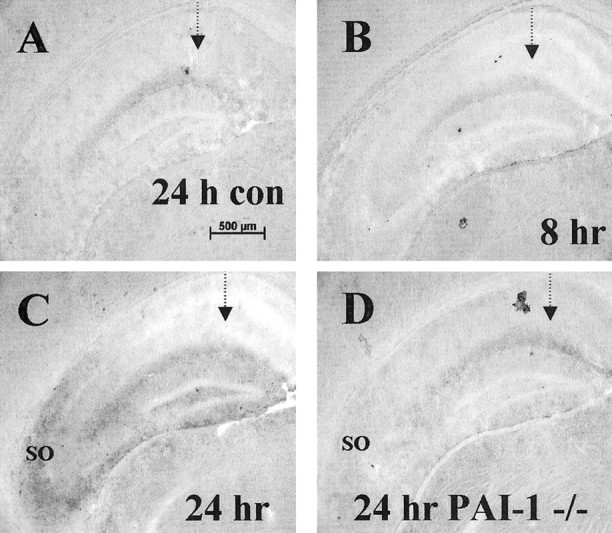
PAI-1 upregulation in the hippocampus after excitotoxic injury. Ipsilateral injected hippocampus was immunostained for PAI-1. The dashed arrow represents the site of excitotoxin injection. A, Control PBS-injected wild-type brain harvested 24 hr after injection; n = 6 animals. B, KA-injected brain harvested 8 hr after injection; n = 6 animals. C, KA-injected brain harvested 24 hr after injection (SO, stratum oriens); n = 6 animals. D, KA-injected brain from a PAI-1−/− animal harvested 24 hr after injection; n = 4 animals.
In an attempt to identify the presumptive proteolytic inhibitor whose activity is downregulated in the ipsilateral hippocampus at 8 hr after injury, we immunostained for neuroserpin, a serine protease inhibitor known to be expressed in hippocampal neurons. Under basal conditions, neuroserpin could be detected mostly in neuronal cell bodies, with no detectable staining within the mossy fiber pathway. At 8 hr after injury, there was a slight increase in staining within CA1 neurons of the injected hippocampus, and at later time points, staining was lost in the degenerating cell bodies. These data suggest that neuroserpin is not modulating the observed tPA activity upregulation seen (data not shown).
It is important to note that protease activities visualized in brain sections reflect the balance between proteases and inhibitory activity. Local proteolysis could be uncontrolled in pathological situations because of discrete compartmentalization. Sectioning and zymographic analyses could allow two localizations separated in vivo to intermingle through physical disruption. Additionally, it was possible that the increase in tPA activity seen at 8 hr after injection was attributable to release of vesicularly localized tPA. To test this hypothesis, we performed the in situ zymographies in the presence of 0.1 and 0.5% Triton X100 or saponin to promote vesicular tPA liberation. These treatments yielded a slightly more diffuse appearance to the zones of lysis but did not alter the lytic zone localizations or the overall level of activity (data not shown), indicating that the increase in tPA activity seen was not attributable to vesicular release.
Plasminogen expression after excitotoxic injury
Plasminogen is present at high concentrations in the blood and in the extracellular space of most tissues. However, little is known about the concentration of plasminogen in the basal neuropil and how that might change after injury. We used a sheep polyclonal antibody against rat plasminogen to determine the expression profile of plasminogen after injury. We could detect little to no plasminogen in wild-type brain tissue (PBS-perfused) when compared with immunostains of Plgn−/− brain tissue (PBS-perfused) controls (data not shown). The antibody showed a significant amount of “background” neuronal cell body staining that was also seen in the plasminogen-deficient control tissue. However, after excitotoxic injury, we detected a large increase in plasminogen staining within the mossy fiber pathway (Fig.7, open arrows) and notably in the hilus of the dentate gyrus. Additionally, we observed a variably shaped diffuse region localizing to the CA1–CA3 areas (Fig. 7). This staining correlated well with the pattern obtained for plasminogen activity using zymography (Fig. 7C,D).
Fig. 7.
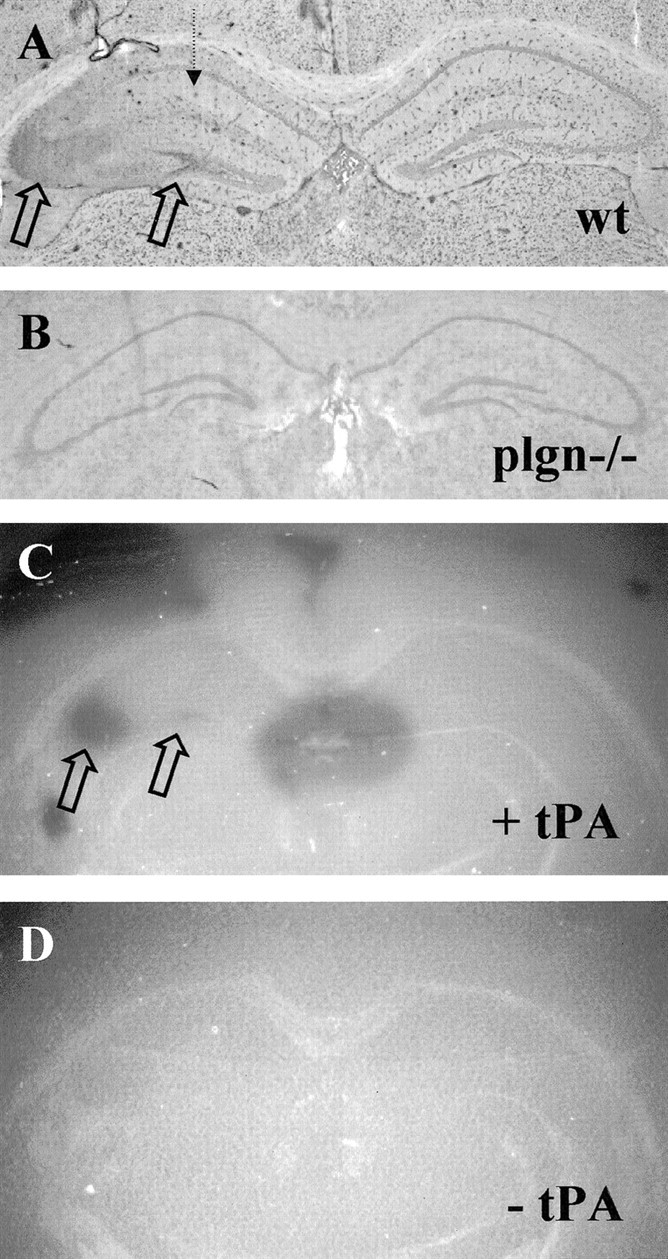
Expression of plasminogen in the hippocampus after excitotoxic injury. The dashed arrow represents the site of excitotoxin injection. A, Plasminogen immunostain on KA-injected wild-type brain section 8 hr after injection. Open arrows highlight areas of intense staining;n = 6 animals. B, Plasminogen immunostain on plg−/− brain section; n = 3 animals. C, Plasminogen zymography of wild-type brain section adjacent to section shown in A indicating dark lytic zones of plasminogen activity (open arrows).D, Control zymography with tPA omitted from the mixture.
Because this observed plasminogen staining could originate from a vascular source due to blood–brain barrier breakdown or from local protein synthesis, we analyzed vascular leakage by staining for IgG in the neuropil, the traditional marker for blood–brain barrier breakdown (data not shown). Using this assay, we could not exclude the possibility that the detected plasminogen was derived from vascular leakage, because the plasminogen staining pattern was always a subset of the visualized IgG staining (data not shown).
Basal high-level tPA mossy fiber expression varies among species
To determine whether our observed staining pattern of tPA expression in the hippocampus was consistent among different species, we first stained adult (300 gm) Sprague Dawley rat coronal brain sections using the tPA antibody to determine the expression of tPA in rat hippocampus. Surprisingly, the rat mossy fiber pathway does not express significant levels of tPA under basal conditions (Fig.8A). The antibody used cross-reacts with rat antigen, as evidenced by the robust staining in the vascular endothelium (Fig. 8A, open arrows). The staining was also determined to be specific by immunodepletion using tPA-Sepharose (data not shown). This absence of basal mossy fiber tPA staining was corroborated by the lack of mossy fiber tPA activity, as measured by in situ zymography (Fig.8B). A previous report had shown that tPA activity was upregulated in the rat hippocampus by KA injection into the ventricle in a pattern reminiscent of mossy fiber expression (Nagai et al., 1999). In our hands, stereotaxic KA injection produced seizure activity and resulted in the appearance of tPA protein staining within a variable portion of the mossy fiber pathway (Fig. 8C). This staining was also associated with activity via the zymography assay (data not shown). These results suggest that mouse and rat mossy fiber pathways regulate tPA expression differently.
Fig. 8.
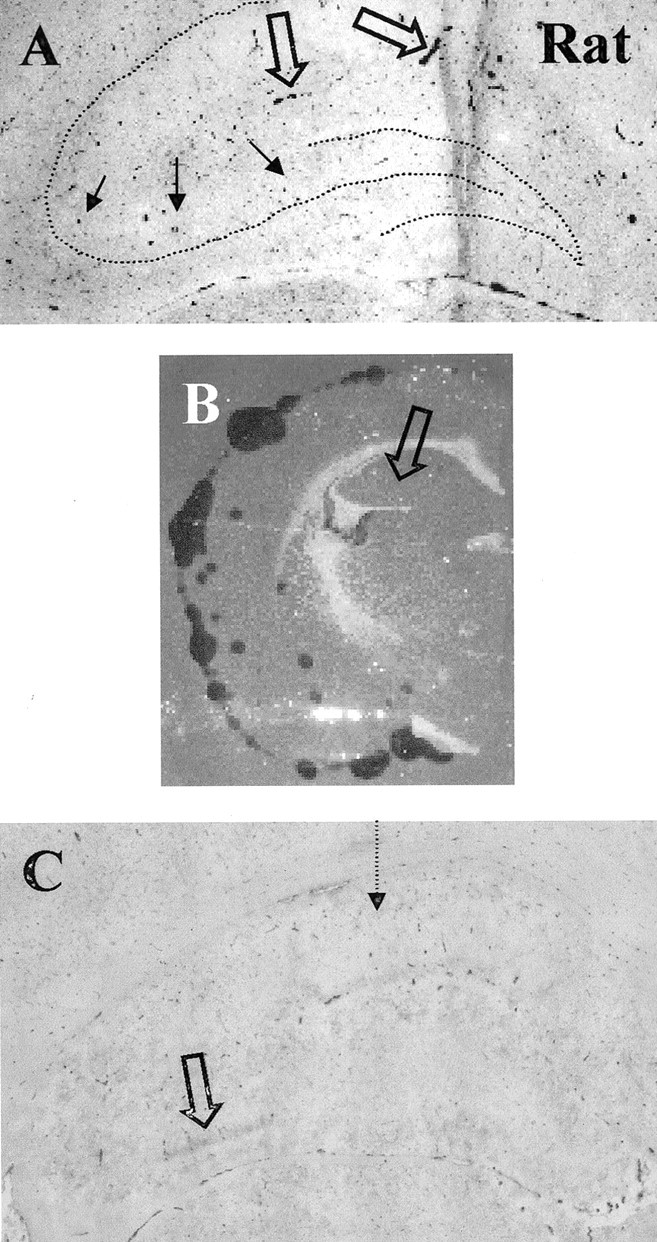
Rat mossy fiber pathway is devoid of significant tPA staining but is upregulated after excitotoxic injury.A, tPA protein is seen primarily in vascular endothelial cells. The dotted line shows the position of hippocampal neuronal cell layers. Filled arrows point to the mossy fiber pathway, where no significant tPA staining is detected.Open arrows point to tPA-stained vascular endothelium;n = 6 animals. B, In situ zymography assay showing tPA activity and lack of any detectable activity in the mossy fiber pathway. C, tPA protein staining is upregulated (open arrow) in a portion of the mossy fiber pathway 8 hr after stereotaxic KA injection into the CA1 region (dotted arrow);n = 8 animals, with 5 showing some level of tPA upregulation.
We then examined a variety of other species to analyze the expression of tPA. Gerbil and hamster brains both expressed high basal levels of tPA in the mossy fiber pathway, whereas cat and human brains did not (data not shown). These differences in species staining led us to test a variety of mouse strains to see whether there were differences in hippocampal tPA staining among lines of mice. All strains tested basally expressed tPA specifically in the mossy fiber pathway.
DISCUSSION
Our results demonstrate that tPA protein and activity are concentrated in the mossy fiber pathway within the mouse hippocampus. This localization is essentially identical to the activity localization reported previously (Sappino et al., 1993). However, numerous reports have localized tPA mRNA to all neuronal layers within the hippocampus (Qian et al., 1993; Sappino et al., 1993; Carroll et al., 1994; Tsirka et al., 1997). One possible explanation for this discrepancy was that our antibody would not recognize tPA in an inactive conformation or bound to inhibitors. There are three lines of evidence that combined strongly argue against this possibility. First, we are using a polyclonal antibody that should recognize multiple epitopes on tPA. Second, we have identified a second independently generated polyclonal antibody that yields an identical staining pattern (data not shown; see Materials and Methods). Finally, we observed robust tPA staining in sections that did not exhibit activity because of a high level of the tPA inhibitor PAI-1 (see Figs. 5, E vs F, 6C).
We therefore reasoned that the tPA mRNA in the CA1 may be under translational control, in which the mRNA would be continually present but only translated after significant activity or injury, providing rapid protease production. In oogenesis, tPA mRNA is under translational control (Huarte et al., 1987), and translational control by a similar molecular mechanism has recently been proposed for calcium/calmodulin-dependent kinase II in neurons (Wu et al., 1998). We performed in situ hybridization experiments on control and KA-injected hippocampus to determine the relative levels of mRNA after injury. Our results clearly show upregulation of tPA mRNA coincident with tPA protein production (see Fig. 4). Although our results are consistent with transcriptional regulation, we cannot rule out translational control of any presynthesized CA1 mRNA. We do not have an explanation for our inability to detect tPA mRNA in wild-type animals under basal conditions.
Previous work has shown CA1 hippocampal neurons to be extremely sensitive to tPA-mediated excitotoxin-induced degeneration (Tsirka et al., 1995). Because we find that the CA1 region is essentially devoid of detectable tPA protein and activity, we initially hypothesized that the upregulation of tPA protein was required for neurodegeneration. However, our observations lead us to suspect that this is not the case. First, we looked for tPA expression at several time points before observable neurodegeneration but well into immediate early gene expression (as early as 1 hr after injury) and could never see tPA expression in CA1 neurons destined for degeneration. Second, CA1 neurons that upregulate tPA seem to survive the excitotoxic injury. For example, in the injected hippocampus, tPA-expressing (upregulated) neurons lie at the border of the neurodegenerating region (Fig.2A,B) and look healthy compared with adjacent degenerating neurons. Additionally, when tPA was upregulated in contralateral CA1 neurons, the neurons looked healthy and never showed phenotypic signs of neurodegeneration.
So why are CA1 pyramidal neurons most sensitive to tPA-mediated excitotoxin-induced degeneration? There are several possible explanations. First, tPA might be expressed below the level of detection, and it is this low-level tPA that is crucial for CA1 neuronal degeneration. Second, cell death is likely to be attributable to a confluence of many factors, because we know that tPA overexpression alone is not sufficient to kill neurons (Tsirka et al., 1996; Madani et al., 1999). It is possible that different situations exist for individual neurons, such that neurons upregulating tPA synthesis are only subjected to cell death in the presence of secondary factor(s) (e.g., KA exposure and other cellular interactions). This combinatorial regulation is suggested by the upregulation of tPA only in specific subsets of neurons within the CA regions (see Fig.2A,C). Finally, the presence of mossy fiber tPA may be required for CA1 neuronal death, possibly through feedback potentiation of electrical activity. Therefore, loss of mossy fiber tPA could short circuit enhanced electrical activity in CA1 neurons.
Chen et al. (1997) showed that tPA and plasmin act through cleavage of laminin within CA1 to lead to neuronal degeneration in response to excitotoxic injury. How do we reconcile the potential lack of tPA within CA1 with these results? Chen et al. (1997) noted that their results did not prove that the loss of laminin was a direct effect of local plasmin. Therefore, high-level tPA expression within the mossy fiber pathway may be required for synthesis, activation, or release of a second protease(s) within CA1 that acts on laminin. A potentially interesting candidate would be neuropsin, a serine protease that is expressed primarily in the pyramidal neurons within the hippocampus (Chen et al., 1995).
Why is tPA so highly expressed under basal conditions in the mossy fiber pathway? Several studies suggest that a major role of tPA in neurons is synaptic plasticity (Frey et al., 1996; Huang et al., 1996;Baranes et al., 1998; Madani et al., 1999). The mossy fiber pathway is anatomically unique among synaptic connections within the hippocampus, being characterized by extremely large axon terminals contacting multiple synaptic spines. Additionally, the mossy fiber pathway is characterized electrophysiologically by NMDA-independent LTP. High-level tPA expression could be required for the structural remodeling necessary within these types of synaptic terminals and may confer specific signaling properties to these synapses. For example, the robust presence of proteolytic potential could allow for the observed NMDA-independent character of mossy fiber LTP.
If protease expression is required for structural modifications during synaptic plasticity, a releasable “resident” protease may be required at all synaptic terminals to allow appropriate extracellular matrix modifications. There may be a variety of proteases with different inhibition characteristics, target protein specificities, and downstream signaling capabilities that participate in different synapse types, e.g., tPA and plasminogen in mossy fibers leading to CA3 neurons and neuropsin in Schaffer collaterals leading to CA1 neurons. It is quite possible that variations in neuronal protease expression alter circuitry as well as learning and memory characteristics. Interestingly the lack of significant tPA expression in the rat mossy fiber pathway may confer different electrophysiological, structural, or behavioral characteristics on mossy fibers between rats and mice. Additionally, under basal conditions, rat mossy fiber terminals might express high levels of a protease other than tPA.
Neuronal extracellular proteolytic activity when improperly released within a pathological setting may lead to excessive matrix modification and subsequent neuronal death. This is exemplified by experiments showing that loss of tPA or plasminogen conferred neuronal protection after excitotoxic injury (Tsirka et al., 1995) and stroke (Wang et al., 1998). Several other proteins have been identified whose absence confers resistance to excitotoxin-induced neuronal degeneration: jnk3 (Yang et al., 1997), preprotachykinin A (Liu et al., 1999), glutathione peroxidase (Jiang et al., 2000), and spinophilin (Feng et al., 2000). It is not clear that all of these players lie in the same pathway for neurodegeneration, but loss of any of these components provides some degree of neuroprotection. It is possible that normal physiological functions, including learning and memory, are also mediated through interactions between some or all of these players. The interrelationships among proteases, their inhibitors, and matrix molecules may help explain the relationship between normal neuronal functions and neuropathological states.
Footnotes
This work was supported by National Institutes of Health Grants NS35704 and NS38472. We thank Katerina Akassoglou, Sarah Baker, Stephanie Bury, Zu-Lin Chen, Duane Day, Dennis Dickson, Andy Lee, Rime Mahdavi, Charles Ouimet, Ed Reich, Irene Solomon, Andre Stutz, Stella Tsirka, and the Strickland laboratory for helpful discussions and reagents. We are grateful to Robert Pawlak for very helpful comments on this manuscript.
Correspondence should be addressed to Dr. Fernando Sallés, Cogent Neuroscience, Inc., 4321 Medical Park Drive, Durham, NC 27704. E-mail:fsalles@cogentneuroscience.com.
REFERENCES
- 1.Akassoglou K, Kombrinck KW, Degen JL, Strickland S. Tissue plasminogen activator-mediated fibrinolysis protects against axonal degeneration and demyelination after sciatic nerve injury. J Cell Biol. 2000;149:1157–1166. doi: 10.1083/jcb.149.5.1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Andrade-Gordon P, Strickland S. Fractionation of heparin by chromatography on a tissue plasminogen activator-Sepharose column. Proc Natl Acad Sci USA. 1990;87:1865–1869. doi: 10.1073/pnas.87.5.1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Backstrom JR, Lim GP, Cullen MJ, Tökés ZA. Matrix metalloproteinase-9 (MMP-9) is synthesized in neurons of the human hippocampus and is capable of degrading the amyloid-β peptide (1–40). J Neurosci. 1996;16:7910–7919. doi: 10.1523/JNEUROSCI.16-24-07910.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baranes D, López-García JC, Chen M, Bailey CH, Kandel ER. Reconstitution of the hippocampal mossy fiber and associational-commissural pathways in a novel dissociated cell culture system. Proc Natl Acad Sci USA. 1996;93:4706–4711. doi: 10.1073/pnas.93.10.4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baranes D, Lederfein D, Huang YY, Chen M, Bailey CH, Kandel ER. Tissue plasminogen activator contributes to the late phase of LTP and to synaptic growth in the hippocampal mossy fiber pathway. Neuron. 1998;21:813–825. doi: 10.1016/s0896-6273(00)80597-8. [DOI] [PubMed] [Google Scholar]
- 6.Bignami A, Cella G, Chi NH. Plasminogen actiators in rat neural tissues during development and in wallerian degeneration. Acta Neuropathol (Berl) 1982;58:224–228. doi: 10.1007/BF00690805. [DOI] [PubMed] [Google Scholar]
- 7.Bugge TH, Flick MJ, Daugherty CC, Degen JL. Plasminogen deficiency causes severe thrombosis but is compatible with development and reproduction. Genes Dev. 1995;9:794–807. doi: 10.1101/gad.9.7.794. [DOI] [PubMed] [Google Scholar]
- 8.Calabresi P, Napolitano M, Centonze D, Marfia GA, Gubellini P, Teule MA, Berretta N, Bernardi G, Frati L, Tolu M, Gulino A. Tissue plasminogen activator controls multiple forms of synaptic plasticity and memory. Eur J Neurosci. 2000;12:1002–1012. doi: 10.1046/j.1460-9568.2000.00991.x. [DOI] [PubMed] [Google Scholar]
- 9.Carmeliet P, Schoonjans L, Kieckens L, Ream B, Degen J, Bronson R, De Vos R, van den Oord JJ, Collen D, Mulligan RC. Physiological consequences of loss of plasminogen activator gene function in mice. Nature. 1994;368:419–424. doi: 10.1038/368419a0. [DOI] [PubMed] [Google Scholar]
- 10.Carroll PM, Tsirka SE, Richards WG, Frohman MA, Strickland S. The mouse tissue plasminogen activator gene 5′ flanking region directs appropriate expression in development and a seizure-enhanced response in the CNS. Development. 1994;120:3173–3183. doi: 10.1242/dev.120.11.3173. [DOI] [PubMed] [Google Scholar]
- 11.Chen Z-L, Strickland S. Neuronal death in the hippocampus is promoted by plasmin-catalyzed degradation of laminin. Cell. 1997;91:917–925. doi: 10.1016/s0092-8674(00)80483-3. [DOI] [PubMed] [Google Scholar]
- 12.Chen Z-L, Yoshida S, Kato K, Momota Y, Suzuki J, Tanaka T, Ito J, Nishino H, Aimoto S, Kiyama H, Shiosaka S. Expression and activity-dependent changes of a novel limbic-serine protease gene in the hippocampus. J Neurosci. 1995;15:5088–5097. doi: 10.1523/JNEUROSCI.15-07-05088.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feng J, Yan Z, Ferreira A, Tomizawa K, Liauw JA, Zhuo M, Allen PB, Ouimet CC, Greengard P. Spinophilin regulates the formation and function of dendritic spines. Proc Natl Acad Sci USA. 2000;97:9287–9292. doi: 10.1073/pnas.97.16.9287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Festoff BW, Nelson PG, Brenneman DE. Prevention of activity-dependent neuronal death: vasoactive intestinal polypeptide stimulates astrocytes to secrete the thrombin-inhibiting neurotrophic serpin, protease nexin I. J Neurobiol. 1996;30:255–266. doi: 10.1002/(SICI)1097-4695(199606)30:2<255::AID-NEU7>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 15.Frey U, Müller M, Kuhl D. A different form of long-lasting potentiation revealed in tissue plasminogen activator mutant mice. J Neurosci. 1996;16:2057–2063. doi: 10.1523/JNEUROSCI.16-06-02057.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Friedman GC, Seeds NW. Tissue plasminogen activator mRNA expression in granule neurons coincides with their migration in the developing cerebellum. J Comp Neurol. 1995;360:658–670. doi: 10.1002/cne.903600410. [DOI] [PubMed] [Google Scholar]
- 17.Gingrich MB, Junge CE, Lyuboslavsky P, Traynelis SF. Potentiation of NMDA receptor function by the serine protease thrombin. J Neurosci. 2000;20:4582–4595. doi: 10.1523/JNEUROSCI.20-12-04582.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gschwend TP, Krueger SR, Kozlov SV, Wolfer DP, Sonderegger P. Neurotrypsin, a novel multidomain serine protease expressed in the nervous system. Mol Cell Neurosci. 1997;9:207–219. doi: 10.1006/mcne.1997.0616. [DOI] [PubMed] [Google Scholar]
- 19.Gualandris A, Jones TE, Strickland S, Tsirka SE. Membrane depolarization induces calcium-dependent secretion of tissue plasminogen activator. J Neurosci. 1996;16:2220–2225. doi: 10.1523/JNEUROSCI.16-07-02220.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harlow E, Lane D. Antibodies: a laboratory manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1988. [Google Scholar]
- 21.Hastings GA, Coleman TA, Haudenschild CC, Stefansson S, Smith EP, Barthlow R, Cherry S, Sandkvist M, Lawrence DA. Neuroserpin, a brain-associated inhibitor of tissue plasminogen activator is localized primarily in neurons. Implications for the regulation of motor learning and neuronal survival. J Biol Chem. 1997;272:33062–33067. doi: 10.1074/jbc.272.52.33062. [DOI] [PubMed] [Google Scholar]
- 22.Hebert JM, Boyle M, Martin GR. mRNA localization studies suggest that murine FGF-5 plays a role in gastrulation. Development. 1991;112:407–415. doi: 10.1242/dev.112.2.407. [DOI] [PubMed] [Google Scholar]
- 23.Hino H, Akiyama H, Iseki E, Kato M, Kondo H, Ikeda K, Kosaka K. Immunohistochemical localization of plasminogen activator inhibitor-1 in rat, human brain tissues. Neurosci Lett. 2001;297:105–108. doi: 10.1016/s0304-3940(00)01679-7. [DOI] [PubMed] [Google Scholar]
- 24.Hoffman KB, Larson J, Bahr BA, Lynch G. Activation of NMDA receptors stimulates extracellular proteolysis of cell adhesion molecules in hippocampus. Brain Res. 1998;811:152–155. doi: 10.1016/s0006-8993(98)00907-x. [DOI] [PubMed] [Google Scholar]
- 25.Huang Y-Y, Bach ME, Lipp H-P, Zhuo M, Wolfer DP, Hawkins RD, Schoonjans L, Kandel ER, Godfraind J-M, Mulligan R, Collen D, Carmeliet P. Mice lacking the gene encoding tissue-type plasminogen activator show a selective interference with late-phase long-term potentiation in both Schaffer collateral and mossy fiber pathways. Proc Natl Acad Sci USA. 1996;93:8699–8704. doi: 10.1073/pnas.93.16.8699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huarte J, Belin D, Vassalli A, Strickland S, Vassalli J-D. Meiotic maturation of mouse oocytes triggers the translation and polyadenylation of dormant tissue-type plasminogen activator mRNA. Genes Dev. 1987;1:1201–1211. doi: 10.1101/gad.1.10.1201. [DOI] [PubMed] [Google Scholar]
- 27.Jaworski DM, Fager N. Regulation of tissue inhibitor of metalloproteinase-3 (Timp-3) mRNA expression during rat CNS development. J Neurosci Res. 2000;61:396–408. doi: 10.1002/1097-4547(20000815)61:4<396::AID-JNR6>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 28.Jiang D, Akopian G, Ho YS, Walsh JP, Andersen JK. Chronic brain oxidation in a glutathione peroxidase knockout mouse model results in increased resistance to induced epileptic seizures. Exp Neurol. 2000;164:257–268. doi: 10.1006/exnr.2000.7431. [DOI] [PubMed] [Google Scholar]
- 29.Kato K, Kishi T, Kamachi T, Akisada M, Oka T, Midorikawa R, Takio K, Dohmae N, Bird P, Sun J, Scott F, Miyake Y, Yamamoto K, Machida A, Tanaka T, Matsumoto K, Shibata M, Shiosaka S. Serine proteinase inhibitor 3 and murinoglobulin I are potent inhibitors of neuropsin in adult mouse brain. J Biol Chem. 2001;276:14562–14571. doi: 10.1074/jbc.M010725200. [DOI] [PubMed] [Google Scholar]
- 30.Krystosek A, Seeds NW. Plasminogen activator release at the neuronal growth cone. Science. 1981;213:1532–1534. doi: 10.1126/science.7197054. [DOI] [PubMed] [Google Scholar]
- 31.Lee S, Williamson J, Lothman EW, Szele FG, Chesselet MF, Von Hagen S, Sapolsky RM, Mattson MP, Christakos S. Early induction of mRNA for calbindin-D28k and BDNF but not NT-3 in rat hippocampus after kainic acid treatment. Brain Res Mol Brain Res. 1997;47:183–194. doi: 10.1016/s0169-328x(97)00043-0. [DOI] [PubMed] [Google Scholar]
- 32.Liu H, Cao Y, Basbaum AI, Mazarati AM, Sankar R, Wasterlain CG. Resistance to excitotoxin-induced seizures and neuronal death in mice lacking the preprotachykinin A gene. Proc Natl Acad Sci USA. 1999;96:12096–12101. doi: 10.1073/pnas.96.21.12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lochner JE, Kingma M, Kuhn S, Meliza CD, Cutler B, Scalettar BA. Real-time imaging of the axonal transport of granules containing a tissue plasminogen activator/green fluorescent protein hybrid. Mol Biol Cell. 1998;9:2463–2476. doi: 10.1091/mbc.9.9.2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Madani R, Hulo S, Toni N, Madani H, Stelmer T, Muller D, Vassalli J-D. Enhanced hippocampal long-term potentiation and learning by increased neuronal expression of tissue-type plasminogen activator in transgenic mice. EMBO J. 1999;18:3007–3012. doi: 10.1093/emboj/18.11.3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Masos T, Miskin R. mRNAs encoding urokinase-type plasminogen activator and plasminogen activator inhibitor-1 are elevated in the mouse brain following kainate-mediated excitation. Brain Res Mol Brain Res. 1997;47:157–169. doi: 10.1016/s0169-328x(97)00040-5. [DOI] [PubMed] [Google Scholar]
- 36.Nagai N, Urano T, Endo A, Takahashi H, Takada Y, Takada A. Neuronal degeneration and a decrease in laminin-like immunoreactivity is associated with elevated tissue-type plasminogen activator in the rat hippocampus after kainic acid injection. Neurosci Res. 1999;33:147–154. doi: 10.1016/s0168-0102(98)00125-4. [DOI] [PubMed] [Google Scholar]
- 37.Neuhoff H, Roeper J, Schweizer M. Activity-dependent formation of perforated synapses in cultured hippocampal neurons. Eur J Neurosci. 1999;11:4241–4250. doi: 10.1046/j.1460-9568.1999.00856.x. [DOI] [PubMed] [Google Scholar]
- 38.Nicole O, Docagne F, Ali C, Margaill I, Carmeliet P, MacKenzie ET, Vivien D, Buisson A. The proteolytic activity of tissue-plasminogen activator enhances NMDA receptor-mediated signaling. Nat Med. 2001;7:59–64. doi: 10.1038/83358. [DOI] [PubMed] [Google Scholar]
- 39.Nishibori M, Ohtsuka A, Kawabata M, Nakaya N, Murakami T, Saeki K. A novel serpin-like protein, B-43, exists in both neurons and astrocytes: an immunohistochemical study in the parietal region of the bovine brain. Neurosci Lett. 1995;200:125–128. doi: 10.1016/0304-3940(95)12095-l. [DOI] [PubMed] [Google Scholar]
- 40.Osterwalder T, Contartese J, Stoeckli ET, Kuhn TB, Sonderegger P. Neuroserpin, an axonally secreted serine protease inhibitor. EMBO J. 1996;15:2944–2953. [PMC free article] [PubMed] [Google Scholar]
- 41.Parmer RJ, Mahata M, Mahata S, Sebald MT, O'Connor DT, Miles LA. Tissue plasminogen activator (t-PA) is targeted to the regulated secretory pathway. Catecholamine storage vesicles as a reservoir for the rapid release of t-PA. J Biol Chem. 1997;272:1976–1982. doi: 10.1074/jbc.272.3.1976. [DOI] [PubMed] [Google Scholar]
- 42.Qian Z, Gilbert ME, Colicos MA, Kandel ER, Kuhl D. Tissue-plasminogen activator is induced as an immediate-early gene during seizure, kindling, and long-term potentiation. Nature. 1993;361:453–457. doi: 10.1038/361453a0. [DOI] [PubMed] [Google Scholar]
- 43.Salles FJ, Schechter N, Strickland S. A plasminogen activator is induced during goldfish optic nerve regeneration. EMBO J. 1990;9:2471–2477. doi: 10.1002/j.1460-2075.1990.tb07425.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sappino A-P, Madani R, Huarte J, Belin D, Kiss JZ, Wohlwend A, Vassalli J-D. Extracellular proteolysis in the adult murine brain. J Clin Invest. 1993;92:679–685. doi: 10.1172/JCI116637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Scarisbrick I, Isackson P, Ciric B, Windebank A, Rodriguez M. MSP, a trypsin-like serine protease, is abundantly expressed in the human nervous system. J Comp Neurol. 2001;431:347–361. [PubMed] [Google Scholar]
- 46.Shiosaka S, Yoshida S. Synaptic microenvironments—structural plasticity, adhesion molecules, proteases, their inhibitors. Neurosci Res. 2000;37:85–89. doi: 10.1016/s0168-0102(00)00115-2. [DOI] [PubMed] [Google Scholar]
- 47.Tsirka SE, Gualandris A, Amaral DG, Strickland S. Excitotoxin-induced neuronal degeneration and seizure are mediated by tissue plasminogen activator. Nature. 1995;377:340–344. doi: 10.1038/377340a0. [DOI] [PubMed] [Google Scholar]
- 48.Tsirka SE, Rogove AD, Strickland S. Neuronal cell death and tPA. Nature. 1996;384:123–124. doi: 10.1038/384123b0. [DOI] [PubMed] [Google Scholar]
- 49.Tsirka SE, Rogove AD, Bugge TH, Degen JL, Strickland S. An extracellular proteolytic cascade promotes neuronal degeneration in the mouse hippocampus. J Neurosci. 1997;17:543–552. doi: 10.1523/JNEUROSCI.17-02-00543.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Waller EK, Schleuning WD. Induction of fibrinolytic activity in HeLa cells by phorbol myristate acetate. Tissue-type plasminogen activator antigen and mRNA augmentation require intermediate protein biosynthesis. J Biol Chem. 1985;260:6354–6360. [PubMed] [Google Scholar]
- 51.Wang YF, Tsirka SE, Strickland S, Stieg PE, Soriano SG, Lipton SA. Tissue plasminogen activator (tPA) increases neuronal damage after focal cerebral ischemia in wild-type and tPA-deficient mice. Nat Med. 1998;4:228–231. doi: 10.1038/nm0298-228. [DOI] [PubMed] [Google Scholar]
- 52.Wu L, Wells D, Tay J, Mendis D, Abbott M-A, Barnitt A, Quinlan E, Heynen A, Fallon JR, Richter JD. CPEB-mediated cytoplasmic polyadenylation and the regulation of experience-dependent translation of α−CAMKII mRNA at synapses. Neuron. 1998;21:1129–1139. doi: 10.1016/s0896-6273(00)80630-3. [DOI] [PubMed] [Google Scholar]
- 53.Yang DD, Kuan CY, Whitmarsh AJ, Rincon M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]