

Abstract
The proper formation of neuromuscular synapses requires ongoing synaptic activity that is translated into complex structural changes to produce functional synapses. One mechanism by which activity could be converted into these structural changes is through the regulated expression of specific synaptic regulatory factors. Here we demonstrate that blocking synaptic activity with curare reduces synaptic neuregulin expression in a dose-dependent manner yet has little effect on synaptic agrin or a muscle-derived heparan sulfate proteoglycan. These changes are associated with a fourfold increase in number and a twofold reduction in average size of synaptic acetylcholine receptor clusters that appears to be caused by excessive axonal sprouting with the formation of new, smaller acetylcholine receptor clusters. Activity blockade also leads to threefold reductions in brain-derived neurotrophic factor and neurotrophin 3 expression in muscle without appreciably changing the expression of these same factors in spinal cord. Adding back these or other neurotrophic factors restores synaptic neuregulin expression and maintains normal end plate band architecture in the presence of activity blockade. The expression of neuregulin protein at synapses is independent of spinal cord and muscle neuregulin mRNA levels, suggesting that neuregulin accumulation at synapses is independent of transcription. These findings suggest a local, positive feedback loop between synaptic regulatory factors that translates activity into structural changes at neuromuscular synapses.
Keywords: neuregulin, neuromuscular, synapse, neurotrophin, BDNF, activity, GDNF, NT-3
During synaptogenesis, an excess number of synaptic connections are reduced to a smaller number of functional connections dependent on their degree of activity; those connections that are more active become more stable, and those that are less active deteriorate. One of the best studied synapses is this regard is that between a motor neuron and a muscle cell or the neuromuscular junction (NMJ; Sanes and Lichtman, 1999). In addition to many presynaptic and postsynaptic proteins important for acetylcholine signal transduction, a growing list of regulatory factors have been discovered that may provide the necessary modulatory signals to aid in this selection process.
In the anterograde direction, motor neurons promote the accumulation of acetylcholine receptors (AChRs) at NMJs through at least two regulatory factors, including a specific form of agrin that leads to the clustering of preexisting AChRs (Ruegg and Bixby, 1998) and AChR-inducing activity (ARIA), which is made in motor neurons, transported down axons (Loeb et al., 1999), and released and concentrated in the synaptic basal lamina (Burgess et al., 1995;Goodearl et al., 1995; Loeb and Fischbach, 1995; Loeb et al., 1998,1999; Meier et al., 1998; Wang et al., 2000). There it induces the synthesis of new AChRs necessary to maintain proper synaptic function (Sandrock et al., 1997). ARIA is derived from the NRG-1 gene and belongs to a large family of alternatively spliced growth and differentiation factors now called neuregulins (NRGs; Fischbach and Rosen, 1997).
A number of retrograde regulatory factors are produced in muscle; however, their individual effects on NMJ development are less clear. Brain-derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), NT-4, and glial cell line-derived neurotrophic factor (GDNF) are expressed in muscle during synaptic development (Funakoshi et al., 1995; Moore et al., 1996; Sanchez et al., 1996) and transported to motor neuron cell bodies (Koliatsos et al., 1993; Leitner et al., 1999; Watson et al., 1999). They support the survival of motor neurons (Henderson et al., 1993, 1994; Koliatsos et al., 1993; Moore et al., 1996; Sanchez et al., 1996; Alcantara et al., 1997) and, for BDNF and NT-4, promote synaptic differentiation (Gonzalez et al., 1999).
Electrical activity is critical in orchestrating the normal formation of neuromuscular synapses. Blocking muscle activity with curare prevents motor neuron cell death, the pruning of early synaptic connections, and the loss of extrajunctional AChRs (Chang et al., 1975;Burden, 1977; Oppenheim et al., 1989; Hory-Lee and Frank, 1995). One way that activity may be translated into synaptic structure is through the regulated expression of these regulatory factors. For example, NT-4 expression in postnatal rat muscle increases with activity (Funakoshi et al., 1995), and the neurotrophic factors BDNF, NT-3, NT-4, and GDNF rapidly and selectively increase NRG in cultured motor neurons, raising the possibility that this may be part of a positive feedback loop (Loeb and Fischbach, 1997).
To determine the relationship among synaptic activity, neurotrophic factors, and synaptic structure, we developed an in vivoexperimental system in which we measure regulatory factor expression on both sides of the synapse in the presence and absence of normal synaptic activity. Despite early embryonic expression of NRG in chick motor neuron axons at embryonic day 3 (E3), NRG is not detectable in the synaptic basal lamina until E16 during synapse elimination (Burden, 1977; Loeb et al., 1999). This late accumulation is concurrent with a concentration of basal lamina heparan sulfate proteoglycans (HSPGs), including agrin (Fallon and Gelfman, 1989; Loeb et al., 1999), that bind to the heparin-binding domain of NRG (Loeb and Fischbach, 1995;Meier et al., 1998) and thus focally enrich it at synapses. Using this system, we demonstrate that synaptic NRG can be regulated by an interplay between synaptic activity and neurotrophic factors. Activity blockade with curare significantly reduces synaptic NRG accumulation, reduces muscle neurotrophic factor expression, and produces a dramatic change in end plate band architecture. Both NRG accumulation and normal end plate band architecture were restored by adding back exogenous neurotrophic factors. The accumulation of NRG was independent of transcription, suggesting that local cues at individual synaptic contacts may contain all of the signaling information necessary to release NRG.
MATERIALS AND METHODS
Reagents and antibodies. All pharmacologic agents were purchased from Sigma (St. Louis, MO) unless stated otherwise. Rhodamine and Bodipy-labeled α-bungarotoxin were from Molecular Probes (Eugene, OR); secondary antibodies coupled with either Cy2 or Cy3 were from Amersham Biosciences (Piscataway, NJ). Affinity-purified polyclonal antiserum against the extracellular domain of NRG (183N) was prepared as described previously (Loeb et al., 1999). Monoclonal antibodies against SV2 were a generous gift from Kathy Buckley (Harvard Medical School, Boston, MA), and those against agrin (6D2) were a generous gift from Willi Halfter (University of Pittsburgh, Pittsburgh, PA). Mouse monoclonal IgG supernatant against neurofilaments (RT-97) was obtained from the Developmental Studies Hybridoma Bank at the University of Iowa (Iowa City, IA) under contract N01-HD-7-3263 from the National Institute of Child Health and Human Development. BDNF, NT-3, and GDNF were generous gifts from Amgen (Thousand Oaks, CA) and were diluted in 0.2% BSA and PBS before use.
Eggs and in ovo treatments. Fertilized chicken eggs were obtained from Michigan State University Poultry Farms (East Lansing, MI) and incubated at 37°C in a Kuhl (Flemington, NJ) rocking humidified incubator at 50% humidity. Addition of chemical agents and neurotrophic factors was made by opening the egg on the top and tearing a small hole through the air sac without damaging underlying blood vessels. The eggs were sealed after each treatment using transparent plastic packing tape. At indicated times, tissues were processed for total RNA or immunohistochemistry as described below.
Activity blockade. Stock solutions ofd-tubocurarine were prepared in sterile saline at 20 mg/ml. Aliquots containing the indicated total amounts were added daily together with or without neurotrophic factors. The total amount of liquid added each day for a given experiment was the same for all eggs. Activity blockade was assessed daily before and after the addition of curare by noting the absence of any movements over a 2 min period. The paralytic concentration of curare used produced complete paralysis in most embryos at all times examined by these measurements. No attempts were made to quantify numbers of movements in partially paralyzed animals, as described recently by Oppenheim et al. (2000).
Curare concentrations were determined empirically and from previous literature requiring 3 mg/d for complete paralysis and 0.3 mg/d for partial activity blockade (Hory-Lee and Frank, 1995). Similarly, 50 μg/d α-bungarotoxin was required for paralysis.
Immunohistochemistry and quantitative analysis of synapses.Anterior latisimus dorsi (ALD) muscles were exposed by removing the skin, and a small piece of Kimwipe soaked in fresh 4% paraformaldehyde in PBS was placed on top of the muscles for 40 min to fix the musclein situ to maintain normal muscle shape. Overfixation or underfixation led to a reduction in NRG staining with 183N antisera. This was followed by overnight washing twice in cold PBS and then placing in 30% sucrose before preparing 20 μm frozen sections on Superfrost slides (Fisher Scientific, Pittsburgh, PA) on a cryostat. Sections were washed in PBS for 20 min, followed by blocking with 10% normal goat serum in PBS for 30 min. Antibody solutions prepared in the same blocking solution were added in a humidified chamber as described previously (Loeb et al., 1999). The following dilutions were used: 183N at 1:100, SV2 at 1:10, RT-97 (neurofilament) at 1:10, 6D2 at 1:1, goat anti-mouse Cy3 or Cy2 at 1:500, and goat anti-rabbit Cy3 or Cy2 at 1:500. For each experiment and figure, all sections were derived from the same experiment and stained in parallel and repeated at least three times.
Digital images were obtained on a Nikon Eclipse 600 epifluorescent microscope using rhodamine or FITC filters, unless stated otherwise, with a Princeton Instruments Micromax 5 MHz cooled CCD camera. Quantitation of synaptic number and size were performed using MetamorphSoftware (Universal Imaging, West Chester, PA). To count the number and average size of synapses per 10,000 μm2, nonsaturated images of ALD muscle stained with α-bungarotoxin were captured digitally for the same time. To normalize for staining intensity differences from slide to slide, the threshold of each image was adjusted to account for the relative background intensity of the muscle so that only synapses were selected as objects. For each image, the total intensity (sum of each pixel intensity within a region) and total area measurements of ∼40 synapses were selected that could be seen in their entirety. This result yielded an average synapse size and intensity for these whole synapses. This value was used to measure the total number of synapses an image by dividing the total intensity of signal greater than the thresholded value by the average intensity for a single synapse. Finally, the total muscle surface area was measured, and the ratio of total number of synapses per 10,000 μm2 was thus calculated and found to represent best the number of synapses obtained by counting manually. This process was repeated for at least three images for each condition analyzed and expressed as an average ± 1 SD. Statistical significance was defined as p < 0.05 using a two-tailed t test assuming unequal variances.
Real-time quantitative PCR. We measured mRNA levels of BDNF, NT-3, and GDNF using reverse transcription (RT)-PCR. Total RNA was isolated by homogenization in Ultraspect (Biotecx Labs, Houston, TX) and prepared according to the manufacturer's instructions. RNA samples used for RT-PCR were cleaned up further and treated with DNase using RNeasy columns purchased from Qiagen (Valencia, CA) and quantified using the Ribogreen method of Molecular Probes. We used a real-time kinetic analysis for PCR products using the SYBR green system by Applied Biosystems (Foster City, CA) on an Applied Biosystems Prism 7700 sequence detection system in quadruplicate using samples from multiple animals. On chick muscle, this system gives linear measurements over three orders of magnitude for each of the primer sets and for chick glyceraldehyde-3-phosphate dehydrogenase (GAPDH). To adjust for RNA/cDNA concentration differences, each sample (in quadruplicate) was normalized to the rate of chicken GAPDH amplicon synthesis from the same RNA sample. Minus RT reactions (adding the same amount of RNA with RT treatment) were performed routinely in parallel to be certain that our samples were not contaminated with genomic DNA. The following primer pairs were selected using Applied Biosystems Primer-Express software: GDNF chick-172 forward (F), ATGCCAGAGGATTACCCAGATC; GDNF chick-317 reverse (R), TCTACGTTTGTGGCTGCACTTT; BDNF chick-233F, AGCCCAGTGAGGAAAACAAGG; BDNF chick-363R, CATGTTTGCAGCATCCAGGT; NT-3 chick-431F, CACCACTGTACCTCACAGAGGATT; NT-3 chick-578R, GATGATTTGTCCGTGACCCATA; GAPDH chick-445F, TGATGGGTGTCAACCATGAGA; and GAPDH chick-591R, TGGCATGGA CAGTGGTCATAA.
Northern blot analysis and quantitation. Northern blots for NRG using a full-length proARIA-1 2.3 kb probe (Falls et al., 1993) were performed as described previously and normalized to GAPDH (Loeb and Fischbach, 1997). Data were quantified either using a PhosphorImager (Amersham Biosciences) or on nonsaturated x-ray films using Metamorph image analysis software (Universal Imaging).
RESULTS
We examined the effects of nonparalytic (low-dose) and paralytic (high-dose) concentrations of the AChR antagonist curare, applied daily from E14 through E18, on synaptic structure of the polyinnervated ALD muscle. Dose-dependent changes in synaptic structure were observed both presynaptically, stained for SV2, and postsynaptically, stained with α-bungarotoxin (Fig.1A), as described previously (Srihari and Vrbova, 1978; Oppenheim et al., 1989). On low-power magnification, there was a marked disorganization of end plate band architecture that increased with curare dose. Where ordered, linear arrays of synapses depicting the course of the nerve across the muscle were clearly seen in the control muscle, synapses in muscles treated with high-dose curare were distributed haphazardly, making it almost impossible to predict the course of the nerve. The low-dose curare showed a pattern in between the control and high-dose curare-treated muscle. Higher-magnification views (Fig.1A, insets), together with neurofilament double-labeling studies (shown in Fig. 6A), suggest that this disorganization with activity blockade is attributable to increased branching and sprouting of terminal axons. The higher-magnification views also reveal that activity blockade produces more numerous AChR clusters of variable size, most of which are associated with nerve endings. Although there may also be some AChR clusters not associated with nerve endings, most of these clusters appear to have been produced from the sprouting of distal axons to produce new synapses. Quantitation of the number of synapses per unit area and the average size of AChR clusters using digital morphometric analysis revealed a fourfold increase in the total number of synapses with a corresponding twofold reduction in average size of the AChR clusters with the high dose of curare (Fig. 1B,C). Although there was some atrophy in muscle thickness seen with only with the high curare concentration, the total surface area of sections taken through the muscle was not appreciably reduced and cannot fully account for the changes seen.
Fig. 1.
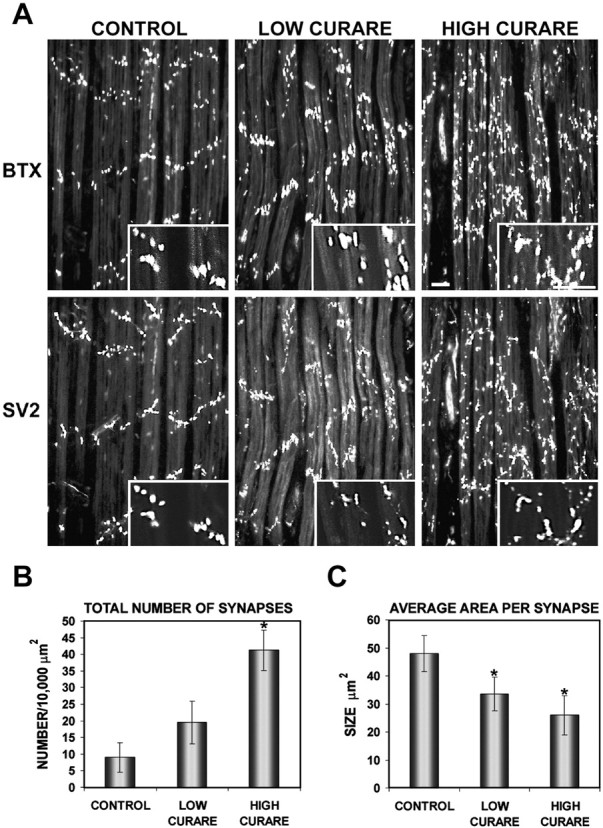
Activity blockade with curare results in a disorganized end plate band architecture with an increase in number and a decrease in average synaptic size. A, Chick embryos were treated daily with saline (CONTROL), a nonparalytic concentration (0.3 mg/d) of d-tubocurarine (LOW CURARE), or a paralytic dose (3 mg/d) ofd-tubocurarine (HIGH CURARE) for 4 d from E14 to E18. ALD muscles at E18 were double-labeled with rhodamine α-bungarotoxin (BTX), which labels AChRs, and SV2, which labels synaptic vesicles in the nerve terminals.Insets, Higher-magnification views of AChR clusters and nerve terminals showing that with curare treatment, a mixture of AChR clusters of variable size is seen, and most of these are associated with nerve terminals. Scale bar, 50 μm. Quantitation of the number (B) and size (C) of postsynaptic AChRs stained with α-bungarotoxin revealed a fourfold to fivefold increase in the number of postsynaptic AChR clusters and a twofold reduction in the average size of the AChR clusters. *Significantly different from the control; p = 0.0018 (B, HIGH CURARE), 0.048 (C, LOW CURARE), and 0.016 (C, HIGH CURARE).
Fig. 6.
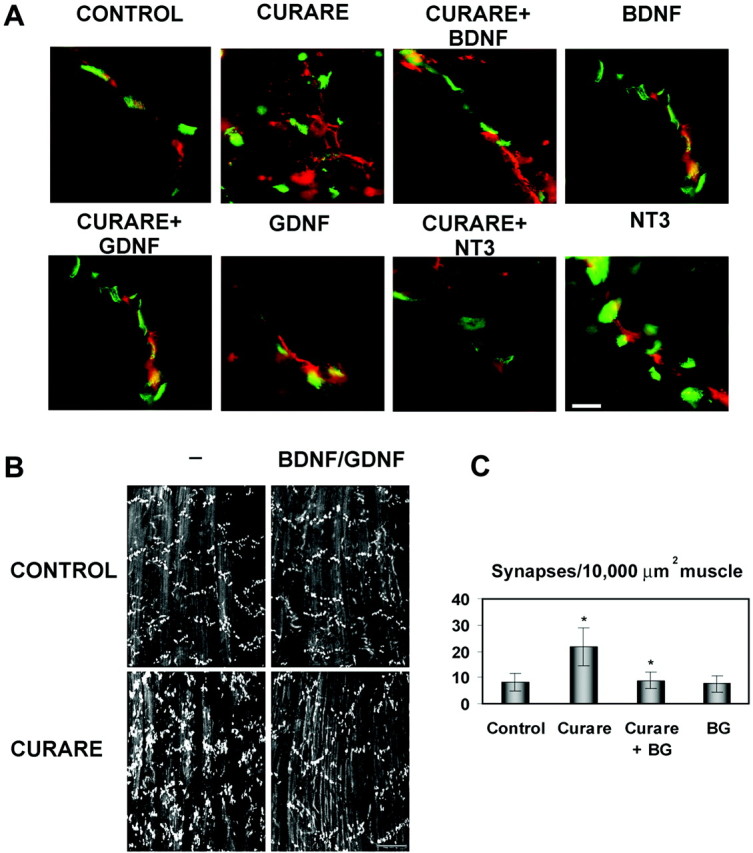
Neurotrophic factors maintain normal end plate band architecture in the face of activity blockade. A, High-magnification views of synapses treated with saline (CONTROL), high-dose curare (CURARE), or high-dose curare plus BDNF, NT-3, or GDNF (1 μg/d) double-labeled with α-bungarotoxin (green) and neurofilaments and synaptic terminals (red) using a mixture of RT-97 and SV2 antibodies. This reveals that the increased number of smaller synapses seen with curare is attributable to excessive sprouting of the terminal axons and that this sprouting is suppressed by the coaddition of BDNF, NT-3, or GDNF. Scale bar, 10 μm. B, Examination of AChR patterns in ALD with α-bungarotoxin at lower magnification reveals that BDNF (2 ng/ml) plus GDNF (20 ng/ml) in the presence of paralytic doses of curare helps maintain a normal architecture. These factors given without curare had no effects. Scale bar, 100 μm. C, Quantitation of the number of AChR clusters per 10,000 μm2 revealed increased numbers with curare alone (as in Fig. 1) that normalized to control levels with BDNF and GDNF (BG). The density with curare treatment was significantly increased from controls (p= 0.010) and the curare with BDNF and GDNF treatment was significantly lower than with curare alone (p = 0.014).
As a first step toward determining the molecular mediators of this change in synaptic organization, we monitored synaptic NRG expression in these muscles (Fig. 2, top panel). With increasing amounts of curare, a dose-dependent reduction in synaptic NRG protein expression was seen so that, at paralytic concentrations of curare, little to no NRG could be detected at synapses. A paralytic dose of another nondepolarizing antagonist, α-bungarotoxin, also reduced the accumulation of NRG at synapses (data not shown). To be certain that the reduction in NRG expression was not attributable to changes in the synaptic basal lamina that might reduce NRG-binding sites, we labeled adjacent muscle sections with antibodies against two synaptic HSPGs: agrin and a muscle-specific HSPG (Bayne et al., 1984), shown in Figure 2, bottom four panels. Each of these was double-labeled with α-bungarotoxin to colocalize HSPG staining to synaptic regions, because these proteoglycans are expressed both synaptically and extrasynaptically. These results show that both HSPGs remain concentrated at synapses with activity blockade and, in fact, often showed a more intense pattern of staining at higher curare concentrations. The same result for agrin was observed using a different antibody, 5B1 (data not shown; Fallon and Gelfman, 1989).
Fig. 2.
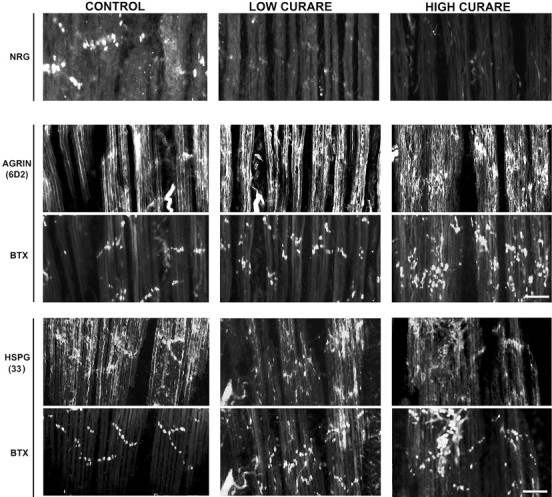
Activity blockade blocks synaptic NRG expression in a dose-dependent manner but has little effect on synaptic HSPGs. NRG immunoreactivity (top panel) at neuromuscular synapses was assessed with affinity-purified 183N antisera on the same ALD muscle sections shown in Figure 1 treated with saline or low or high doses of curare. A dose-dependent reduction in NRG immunoreactivity was noted so that, at paralytic doses of curare, little to no NRG immunoreactivity was detected. Agrin immunoreactivity at neuromuscular synapses was measured using the monoclonal antibody 6D2 double-labeled with Bodipy-α-bungarotoxin (BTX;middle two panels), and a muscle HSPG antibody (33) was double-labeled with Bodipy-α-bungarotoxin (bottom two panels). Scale bars, 50 μm.
The effect of activity blockade on synaptic NRG accumulation was examined further and is shown in Figure3. In this experiment, replicate embryos were treated with either paralytic doses of curare or saline as a control from E14 to E17. Some embryos were analyzed at E17, showing that those treated with curare had no NRG at synapses, but those with the saline control had low but detectable levels of synaptic NRG. On subsequent days from E17 to E19, animals were either further treated with the same agents or reversed so that some initially treated with curare now were switched to saline and those in saline were switched to curare. In each of these conditions, activity blockade was confirmed each day by noting the absence or presence of movements of the curare-treated embryos. Embryos with continued treatment with curare showed no detectable NRG immunoreactivity at synapses, and those treated with saline from E14 to E19 showed an intense pattern of NRG staining, indicating additional NRG accumulation at synapses from E17 to E19. The embryos initially treated with saline and then curare showed no additional increase in NRG staining, suggesting that no additional NRG accumulation occurred after the addition of curare. The embryos first treated with curare and then saline showed that with reversal of the activity blockade, NRG accumulation at synapses resumed. These results demonstrate that NRG accumulation at synapses is closely coupled to synaptic activity and can resume once activity blockade is discontinued, but once released into the synaptic cleft, NRG remains concentrated there through interactions with the synaptic basal lamina.
Fig. 3.

Synaptic NRG expression resumes after reversal of activity blockade. Chicken embryos were treated with either paralytic concentrations of curare (3 mg/d) or the same volume of saline from E14 to E17. Some of the embryos were examined on E17, showing that activity blockade blocked the appearance of NRG immunoreactivity compared with untreated embryos that expressed submaximal, synaptic NRG levels. All muscle sections were double-labeled at the same time with 183N antibodies against NRG and α-bungarotoxin (BTX) for localization of synapses. Each group was either continued with the same treatment or switched to the opposite treatment from E17 to E19. Those that were in saline for the entire period from E14 to E19 had maximal NRG staining, whereas curare treatment from E17 to E19 prevented any additional increase in NRG staining. Those animals maintained in curare for the entire period had no NRG staining at synapses, whereas those switched from curare to saline recovered their ability to release NRG at synapses. Scale bar, 20 μm.
We have shown previously that the neurotrophic factors BDNF, NT-3, and GDNF, which are expressed in muscle during synaptogenesis, regulate NRG mRNA and protein expression in motor neurons in vitro (Loeb and Fischbach, 1997). Because they may have similar effects in vivo, we investigated here whether these neurotrophic factors are also regulated by electrical activity. If so, we might predict that blocking synaptic activity postsynaptically would result in a reduction of muscle neurotrophic factor expression that, in turn, may lead to a reduction of presynaptic NRG expression in motor neurons. Consistently, using real-time, quantitative RT-PCR on ALD muscle, we found threefold reductions in BDNF and NT-3 mRNA levels in curare-treated muscles compared with saline-treated controls (Fig.4). GDNF mRNA levels were unchanged or modestly increased. Spinal cords from the same animals showed no changes in mRNA levels of these same three factors, suggesting that activity blockade with curare selectively regulates these neurotrophic factors in muscle but not in the spinal cord containing motor neurons.
Fig. 4.

Activity blockade results in altered neurotrophic factor expression in muscle but not in spinal cord. Using real-time RT-PCR methods normalized to chicken GAPDH, both BDNF and NT-3 mRNAs were reduced by approximately threefold with paralytic concentrations of curare from E14 to E18 relative to saline-treated ALD muscle. In contrast, GDNF expression increased slightly in muscle. None of these neurotrophic factors was changed in spinal cords from the same animals, suggesting that curare is working postsynaptically to induce its effects on these factors. Each point represents the average ± 1 SD from RT-PCR measurements from three independent animals, and each RT-PCR from each animal was performed in quadruplicate.
Because activity blockade with curare results in a reduction of neurotrophic factor mRNA in muscle, we would predict that adding back neurotrophic factors in the presence of curare would restore the accumulation of NRG in the synaptic basal lamina. This in fact was seen and is shown in Figure 5, where curare treatment alone blocked NRG staining at synapses, but the addition of 20 ng/ml BDNF, NT-3, or GDNF with curare restored synaptic NRG expression. This effect could also be seen with 10-fold lower concentrations of BDNF or with combinations of BDNF and GDNF (data not shown). Giving these same neurotrophic factors alone had no clear effects on synaptic NRG (Fig. 5).
Fig. 5.

Neurotrophic factors restore synaptic NRG expression in the presence of activity blockade. Chick embryos were treated daily with saline (CONTROL), a paralytic dose of curare (3 mg/d; CURARE) or the same dose of curare with 1 μg (∼20 ng/ml) of BDNF, NT-3, or GDNF for 4 d from E14 to E18. ALD muscles at E18 were double-labeled for NRG (183N) and acetylcholine receptors (α-bungarotoxin; BTX) to localize synapses. Although the curare alone blocked NRG expression, each of these neurotrophic factors restored NRG expression in the presence of activity blockade. The neurotrophic factors alone had no effect. Scale bar, 20 μm.
Not only did these trophic factors restore NRG expression, but they also preserved synaptic structure (Fig.6). High-power views of synapses double-labeled for AChRs and terminal axons demonstrated that curare treatment promotes massive axon terminal sprouting that was prevented by BDNF, NT-3, or GDNF (Fig. 6A). When combined together or given alone, BDNF and GDNF treatment with curare produced AChR cluster densities and an end plate band architecture indistinguishable from controls (Fig. 6B,C). BDNF and GDNF alone, without activity blockade, had no effect on synaptic organization. Taken together, these results suggest that activity-dependent expression of neurotrophic factors in muscle may be needed both for the release of NRG at synapses and to maintain normal end plate band architecture, perhaps by preventing the unregulated sprouting of new axon terminals.
Activity-dependent accumulation of NRG into the synaptic cleft could be mediated by transcriptional or post-transcriptional processes. To see the contribution of transcriptional control mechanisms on the regulation of NRG expression at synapses, we measured NRG mRNA levels by Northern blot analysis in spinal cord and muscle with activity blockade with curare with or without added neurotrophic factors (Fig.7A,B). We have found that most NRG mRNA and protein in spinal cord is in motor neurons and that there is a close correlation between NRG mRNA and protein levels during development (Loeb et al., 1999). Despite the absence of NRG accumulation at synapses treated with curare, we found a dose-dependent increase in NRG mRNA in spinal cord and no change in the lower levels of NRG mRNA expressed in muscle. α-Bungarotoxin had similar stimulatory effects on NRG in spinal cord. This suggests that, while synaptic activity appears to regulate NRG mRNA expression in spinal cord but not in muscle, the mechanism that translates synaptic activity into NRG concentration at synapses is independent of transcription. Addition of BDNF and GDNF, with or without curare, produced an even greater, fivefold to eightfold, increase in NRG mRNA in spinal cord (Fig. 7B). This confirms previous in vitroobservations, now in vivo, that neurotrophic factors increase NRG mRNA levels (Loeb and Fischbach, 1997) and raises the possibility that neurotrophic factors have additional effects on post-transcriptional processes that lead to the accumulation of NRG at synapses.
Fig. 7.

NRG mRNA levels are increased in spinal cord with activity blockade and neurotrophic factors but are unchanged in muscle.A, Total RNA from lumbar spinal cords and gastrocnemius muscles taken from the embryos described previously were analyzed for NRG mRNA by Northern blot analysis using a full-length pro-NRG probe revealing two transcripts. Note that considerably less NRG mRNA is expressed in muscle compared with spinal cord. Although there were significant increases in NRG mRNA for both low and high doses of curare, no differences were noted in muscle. Positions of the ribosomal RNA bands are indicated by arrows on theleft. B, The top, 7 kb transcript shown in A together with data from similar Northern blots were quantified and normalized to the level of chicken GAPDH mRNA (shown in the bottom of A) and summarized as the mean ± SD for spinal cord samples on theleft and gastrocnemius and ALD muscles on theright. Although curare alone produced a twofold induction of NRG mRNA levels, BDNF and GDNF alone or together with curare produced a fivefold to eightfold induction of NRG mRNA. No difference was noted between gastrocnemius and ALD muscles. The numbers of animals in each condition were as follows: Control,n = 32; Low Curare,n = 8; High Curare,n = 13; α-Bungarotoxin,n = 2; BDNF/GDNF +Curare, n = 6;BDNF/GDNF, n = 2; Control Gas, n = 2; Low Curare Gas,n = 3; High Curare Gas,n = 5; Control ALD,n = 4; and High Curare ALD,n = 2.
DISCUSSION
Activity-dependent accumulation of NRG at NMJs
In this report we have examined how activity at chick neuromuscular synapses may be translated into changes in synaptic structure and organization through an interplay between presynaptic and postsynaptic regulatory factors. We chose to look at a specific developmental stage when synapse elimination occurs, because at this time we are able to make direct assessments of NRG accumulation at synapses together with measurements of both NRG and neurotrophic factor mRNA expression and to correlate these with synaptic structure. Our results are consistent with a model in which acetylcholine-induced depolarization of muscle results in increased levels of muscle neurotrophic factors that, in turn, promote both the synthesis and release of NRG from motor neurons. NRG then acts to increase the local synthesis of AChRs that helps maintain synaptic function (Sandrock et al., 1997).
The profound changes observed in synaptic number, size, and organization with activity blockade are a testament to the importance of normal synaptic activity during development and may result from an imbalance of normal neurotrophic factor signaling. Paralytic doses of curare (or α-bungarotoxin) produced a disordered array of synapses with approximately fourfold more synapses per square micrometer of muscle that were of variable size, on average, half the size of those in untreated muscle. Nonparalytic doses of curare produced an intermediate response. These changes appear to be attributable to excessive new sprouting of axons. In other studies, axonal sprouting has been observed with activity blockade or denervation and may represent a general response to perturbations in neurotrophic factor expression (Duchen and Strich, 1968; Srihari and Vrbova, 1978; Brown et al., 1981; Holland and Brown, 1981; Zhao and Nonet, 2000). Consistently, increased proximal nerve branching is observed with the overexpression or underexpression of synaptic regulatory factors, including NRGs, neurotrophins, and agrin (Funakoshi et al., 1995;DeChiara et al., 1996; Gautam et al., 1996; Riethmacher et al., 1997;Nguyen et al., 1998; Gonzalez et al., 1999; Morris et al., 1999; Lin et al., 2000; Wolpowitz et al., 2000). Finally, during the period of synapse elimination, activity blockade prevents the loss of multiply innervated end plates that may also contribute to this hyperinnervated appearance (Srihari and Vrbova, 1978; Colman and Lichtman, 1993).
In our experimental system, activity blockade greatly reduced the normal deposition of NRG in the synaptic basal lamina. However, agrin expression at synapses was preserved if not increased. These effects on NRG and agrin correlate with changes in synaptic size and number observed: the increase in the number of acetylcholine clusters is associated with agrin expression at many new synaptic sites, yet the smaller size of each of these clusters could be attributable to a lack of NRG. Consistently, mice partially deficient in NRG had a reduced density in synaptic AChRs and a corresponding reduction in neuromuscular transmission (Sandrock et al., 1997). These results lend further support for complimentary roles of agrin and NRG in concentrating synaptic AChRs, in which agrin is critical for forming new AChR clusters, NRG may build up the density and size of these clusters on the basis of their level of activity.
The accumulation of synaptic NRG in the synaptic basal lamina is a unique feature of this developmental stage that enabled us to monitor synaptic NRG expression as a function of activity. This likely results through interactions between the NRG heparin-binding, Ig-like domain and HSPGs in the synaptic cleft (Goodearl et al., 1995; Jo et al., 1995; Loeb and Fischbach, 1995; Loeb et al., 1999). We have shown that NRG–HSPG interactions promote sustained erbB receptor phosphorylation required to induce AChR synthesis (Li and Loeb, 2001). Agrin also binds heparin and is itself an HSPG present in the basal lamina (Tsen et al., 1995) that can bind directly to the heparin-binding domain of NRG (Meier et al., 1998). In fact, it is only one of several synaptic HSPGs made by both muscle and nerve (Anderson et al., 1984; Bayne et al., 1984; Sanes et al., 1986; Sanes, 1989; Peng et al., 1999). A critical observation here is that the absence of NRG at NMJs with activity blockade does not appear to be attributable to a lack of HSPGs, because both agrin and another, muscle-derived HSPG are expressed at NMJs at the same or higher levels than controls. Therefore, our results imply that the absence of NRG staining is in fact attributable to a failure of mechanisms that release NRG into the synaptic cleft rather than an absence of NRG binding sites.
When expressed ectopically in muscle, neural agrin is capable of inducing clusters of AChRs with associated increases in local AChR transcription (Meier et al., 1998). There is some evidence to suggest that these ectopic AChR clusters are associated with NRG immunoreactivity, raising the possibility that muscle-derived NRG may also be recruited to NMJs.
Presynaptic versus postsynaptic effects of activity on NRG and neurotrophic factor expression
One advantage of studying synaptogenesis at peripheral NMJs is the relative ease of differentiating presynaptic from postsynaptic events. Our observations here that synaptic activity blockade selectively changes neurotrophic factor expression in muscle but not in spinal cord and changes NRG expression in spinal cord but not in muscle help define the directionality of activity-dependent neurotrophic factor signaling at NMJs. Consistently, neurotrophic factors increase both NRG mRNA and protein release from cultured motor neurons (Loeb and Fischbach, 1997). However, the exact sites of action and source of each of these factors are not without ambiguity. Although NRG is most highly expressed in spinal motor neurons by both in situ hybridization and immunostaining (Loeb et al., 1999), significantly lower levels are expressed in muscle (Ng et al., 1997) (Fig. 7). Thus, the preponderance of evidence suggests that synaptic NRG is released from motor nerve endings in an activity-dependent manner; however, contributions of NRG from muscle are possible.
Similarly, although the primary sites of action of curare and α-bungarotoxin are postsynaptic, it has been suggested that curare-induced motor neuron survival may be mediated through presynaptic, neuronal nicotinic AChRs (Hory-Lee and Frank, 1995). Arguing against this possibility, Oppenheim et al. (2000) found that programmed cell death could be prevented only with antagonists that block muscle AChRs and that the previous study did not adequately assess the extent of muscle paralysis. Although we suspect that the primary actions of curare and α-bungarotoxin are postsynaptic, we cannot rule out additional, presynaptic effects.
Finally, a growing number of target-derived, “survival” factors for motor neurons have been identified, including BDNF, NT-3, NT-4, GDNF, cardiotrophin-1, and hepatocyte growth factor/scatter factor (Ebens et al., 1996; Oppenheim, 1996). It is becoming evident that these factors are expressed at different stages of development and have both redundant and distinct activities on motor neuron survival. Understanding how this multiplicity of target-derived factors functions in motor neuron programmed cell death and in synaptogenesis is far from simple. Here we found that activity blockade with curare produced threefold reductions in BDNF and NT-3 mRNA levels and a slight increase in GDNF mRNA; however, all three of these factors were able to rescue the disorganizing effects of curare when given systemically. Funakoshi et al. (1995) found similar activity-dependent changes in NT-4 mRNA expression, but when applied locally in muscle, NT-4 promoted sprouting. A key question is whether changes in neurotrophic factor mRNA levels indeed predict how much of each protein is released at synapses. In contrast to the above findings, Gonzalez et al. (1999)argued that anterograde signaling of neurotrophic factors, from nerve to muscle, was responsible for changes in AChR clustering in mice with defective trkB signaling. Although there is some additional evidence for trkB signaling in cultured muscle (Wells et al., 1999), most other studies support a retrograde supply of neurotrophic factors from muscle to nerve (Koliatsos et al., 1993; Leitner et al., 1999; Watson et al., 1999).
Local cues at individual synapses
The lack of synaptic NRG accumulation despite an increase in spinal cord NRG mRNA with activity blockade suggests that there is no clear relationship between NRG transcription and its release at synapses. On the other hand, we found that both spinal cord NRG mRNA and synaptic NRG protein increased in animals given exogenous neurotrophic factors, consistent with previous in vitroobservations (Loeb and Fischbach, 1997). Even so, it is difficult to imagine how transcriptional control mechanisms, which require contact with the cell nucleus, could differentially regulate the stability of one of many individual synaptic connections made by a single motor neuron.
Taken together, our results are consistent with a mechanism whereby activity-dependent regulation of postsynaptic neurotrophic factors promotes the release of presynaptic NRG from individual motor nerve terminals that is independent of motor neuron transcription. Previous work has shown that NRG is first synthesized as a transmembrane precursor that is proteolytically cleaved and released in response to activation of protein kinase C (Burgess et al., 1995; Loeb et al., 1998). Through similar signaling pathways, target-derived neurotrophic factors may stimulate NRG release at individual synapses based on the degree of activity of a particular connection. This would form a local feedback loop in which the directed release of neurotrophic factors promotes the growth of connections through the reciprocal release of NRG that, in turn, promotes AChR densities at more active synapses.
Footnotes
This work was supported by the Children's Research Center of Michigan (J.A.L.), by National Science Foundation Grant IBN-0092623 (J.A.L.), by National Institutes of Health (NIH) Grants NS01659 (J.A.L.) and NS18458 (G.D.F.), and by an Osserman/Sosin/McClure fellowship from the Myasthenia Gravis Foundation of America (A.H.). We thank Janet Robbins and Janelle Novak for expert technical assistance and the Wayne State University Applied Genomics Facility, where the quantitative RT-PCR studies were performed.
Correspondence should be addressed to Dr. Jeffrey Loeb, Department of Neurology and Center for Molecular Medicine and Genetics, Elliman 3217, 421 East Canfield Avenue, Detroit, MI 48201. E-mail:jloeb@med.wayne.edu.
REFERENCES
- 1.Alcantara S, Frisen J, del Rio JA, Soriano E, Barbacid M, Silos-Santiago I. TrkB signaling is required for postnatal survival of CNS neurons and protects hippocampal and motor neurons from axotomy-induced cell death. J Neurosci. 1997;17:3623–3633. doi: 10.1523/JNEUROSCI.17-10-03623.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anderson MJ, Klier FG, Tanguay KE. Acetylcholine receptor aggregation parallels the deposition of a basal lamina proteoglycan during development of the neuromuscular junction. J Cell Biol. 1984;99:1769–1784. doi: 10.1083/jcb.99.5.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bayne EK, Anderson MJ, Fambrough DM. Extracellular matrix organization in developing muscle: correlation with acetylcholine receptor aggregates. J Cell Biol. 1984;99:1486–1501. doi: 10.1083/jcb.99.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brown MC, Holland RL, Hopkins WG. Motor nerve sprouting. Annu Rev Neurosci. 1981;4:17–42. doi: 10.1146/annurev.ne.04.030181.000313. [DOI] [PubMed] [Google Scholar]
- 5.Burden S. Development of the neuromuscular junction in the chick embryo: the number, distribution, and stability of acetylcholine receptors. Dev Biol. 1977;57:317–329. doi: 10.1016/0012-1606(77)90218-4. [DOI] [PubMed] [Google Scholar]
- 6.Burgess TL, Ross SL, Qian YX, Brankow D, Hu S. Biosynthetic processing of neu differentiation factor. Glycosylation trafficking, and regulated cleavage from the cell surface. J Biol Chem. 1995;270:19188–19196. doi: 10.1074/jbc.270.32.19188. [DOI] [PubMed] [Google Scholar]
- 7.Chang CC, Chuang ST, Huang MC. Effects of chronic treatment with various neuromuscular blocking agents on the number and distribution of acetylcholine receptors in the rat diaphragm. J Physiol (Lond) 1975;250:161–173. doi: 10.1113/jphysiol.1975.sp011047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colman H, Lichtman JW. Interactions between nerve and muscle: synapse elimination at the developing neuromuscular junction. Dev Biol. 1993;156:1–10. doi: 10.1006/dbio.1993.1054. [DOI] [PubMed] [Google Scholar]
- 9.DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, Kinetz E, Compton DL, Rojas E, Park JS, Smith C, DiStefano PS, Glass DJ, Burden SJ, Yancopoulos GD. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell. 1996;85:501–512. doi: 10.1016/s0092-8674(00)81251-9. [DOI] [PubMed] [Google Scholar]
- 10.Duchen LW, Strich SJ. The effects of botulinum toxin on the pattern of innervation of skeletal muscle in the mouse. Q J Exp Physiol Cogn Med Sci. 1968;53:84–89. doi: 10.1113/expphysiol.1968.sp001948. [DOI] [PubMed] [Google Scholar]
- 11.Ebens A, Brose K, Leonardo ED, Hanson MG, Jr, Bladt F, Birchmeier C, Barres BA, Tessier-Lavigne M. Hepatocyte growth factor/scatter factor is an axonal chemoattractant and a neurotrophic factor for spinal motor neurons. Neuron. 1996;17:1157–1172. doi: 10.1016/s0896-6273(00)80247-0. [DOI] [PubMed] [Google Scholar]
- 12.Fallon JR, Gelfman CE. Agrin-related molecules are concentrated at acetylcholine receptor clusters in normal and aneural developing muscle. J Cell Biol. 1989;108:1527–1535. doi: 10.1083/jcb.108.4.1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Falls DL, Rosen KM, Corfas G, Lane WS, Fischbach GD. ARIA, a protein that stimulates acetylcholine receptor synthesis, is a member of the neu ligand family. Cell. 1993;72:801–815. doi: 10.1016/0092-8674(93)90407-h. [DOI] [PubMed] [Google Scholar]
- 14.Fischbach GD, Rosen KM. ARIA: a neuromuscular junction neuregulin. Annu Rev Neurosci. 1997;20:429–458. doi: 10.1146/annurev.neuro.20.1.429. [DOI] [PubMed] [Google Scholar]
- 15.Funakoshi H, Belluardo N, Arenas E, Yamamoto Y, Casabona A, Persson H, Ibanez CF. Muscle-derived neurotrophin-4 as an activity-dependent trophic signal for adult motor neurons. Science. 1995;268:1495–1499. doi: 10.1126/science.7770776. [DOI] [PubMed] [Google Scholar]
- 16.Gautam M, Noakes PG, Moscoso L, Rupp F, Scheller RH, Merlie JP, Sanes JR. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell. 1996;85:525–535. doi: 10.1016/s0092-8674(00)81253-2. [DOI] [PubMed] [Google Scholar]
- 17.Gonzalez M, Ruggiero FP, Chang Q, Shi YJ, Rich MM, Kraner S, Balice-Gordon RJ. Disruption of Trkb-mediated signaling induces disassembly of postsynaptic receptor clusters at neuromuscular junctions. Neuron. 1999;24:567–583. doi: 10.1016/s0896-6273(00)81113-7. [DOI] [PubMed] [Google Scholar]
- 18.Goodearl AD, Yee AG, Sandrock AW, Jr, Corfas G, Fischbach GD. ARIA is concentrated in the synaptic basal lamina of the developing chick neuromuscular junction. J Cell Biol. 1995;130:1423–1434. doi: 10.1083/jcb.130.6.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henderson CE, Camu W, Mettling C, Gouin A, Poulsen K, Karihaloo M, Rullamas J, Evans T, McMahon SB, Armanini MP, Berkemeier L, Phillips HS, Rosenthal A. Neurotrophins promote motor neuron survival and are present in embryonic limb bud. Nature. 1993;363:266–270. doi: 10.1038/363266a0. [DOI] [PubMed] [Google Scholar]
- 20. Henderson CE, Phillips HS, Pollock RA, Davies AM, Lemeulle C, Armanini M, Simmons L, Moffet B, Vandlen RA, Simpson LC, Moffet B, Vandlen RA, Koliatsos VE, Rosenthal A. GDNF: a potent survival factor for motoneurons present in peripheral nerve and muscle. Science 266 1994. 1062 1064 [erratum (1995) 267:777] [DOI] [PubMed] [Google Scholar]
- 21.Holland RL, Brown MC. Nerve growth in botulinum toxin poisoned muscles. Neuroscience. 1981;6:1167–1179. doi: 10.1016/0306-4522(81)90081-6. [DOI] [PubMed] [Google Scholar]
- 22.Hory-Lee F, Frank E. The nicotinic blocking agents d-tubocurare and alpha-bungarotoxin save motoneurons from naturally occurring death in the absence of neuromuscular blockade. J Neurosci. 1995;15:6453–6460. doi: 10.1523/JNEUROSCI.15-10-06453.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jo SA, Zhu X, Marchionni MA, Burden SJ. Neuregulins are concentrated at nerve-muscle synapses and activate ACh-receptor gene expression. Nature. 1995;373:158–161. doi: 10.1038/373158a0. [DOI] [PubMed] [Google Scholar]
- 24.Koliatsos VE, Clatterbuck RE, Winslow JW, Cayouette MH, Price DL. Evidence that brain-derived neurotrophic factor is a trophic factor for motor neurons in vivo. Neuron. 1993;10:359–367. doi: 10.1016/0896-6273(93)90326-m. [DOI] [PubMed] [Google Scholar]
- 25.Leitner ML, Molliver DC, Osborne PA, Vejsada R, Golden JP, Lampe PA, Kato AC, Milbrandt J, Johnson EM. Analysis of the retrograde transport of glial cell line-derived neurotrophic factor (GDNF), neurturin, and persephin suggests that in vivo signaling for the GDNF family is GFRα coreceptor-specific. J Neurosci. 1999;19:9322–9331. doi: 10.1523/JNEUROSCI.19-21-09322.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li Q, Loeb JA. Neuregulin-HSPG interactions produce sustained ErbB receptor activation required for the induction of AChRs in muscle. J Biol Chem. 2001;276:38068–38075. doi: 10.1074/jbc.M104485200. [DOI] [PubMed] [Google Scholar]
- 27.Lin W, Sanchez HB, Deerinck T, Morris JK, Ellisman M, Lee KF. Aberrant development of motor axons and neuromuscular synapses in erbB2-deficient mice. Proc Natl Acad Sci USA. 2000;97:1299–1304. doi: 10.1073/pnas.97.3.1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Loeb JA, Fischbach GD. ARIA can be released from extracellular matrix through cleavage of a heparin-binding domain. J Cell Biol. 1995;130:127–135. doi: 10.1083/jcb.130.1.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Loeb JA, Fischbach GD. Neurotrophic factors increase neuregulin expression in embryonic ventral spinal cord neurons. J Neurosci. 1997;17:1416–1424. doi: 10.1523/JNEUROSCI.17-04-01416.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loeb JA, Susanto ET, Fischbach GD. The Neuregulin precursor proARIA is processed to ARIA after expression on the cell surface by a protein kinase C-enhanced mechanism. Mol Cell Neurosci. 1998;11:77–91. doi: 10.1006/mcne.1998.0676. [DOI] [PubMed] [Google Scholar]
- 31.Loeb JA, Khurana TS, Robbins JT, Yee AG, Fischbach GD. Expression patterns of transmembrane and released forms of neuregulin during spinal cord and neuromuscular synapse development. Development. 1999;126:781–791. doi: 10.1242/dev.126.4.781. [DOI] [PubMed] [Google Scholar]
- 32.Meier T, Masciulli F, Moore C, Schoumacher F, Eppenberger U, Denzer AJ, Jones G, Brenner HR. Agrin can mediate acetylcholine receptor gene expression in muscle by aggregation of muscle-derived neuregulins. J Cell Biol. 1998;141:715–726. doi: 10.1083/jcb.141.3.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moore MW, Klein RD, Farinas I, Sauer H, Armanini M, Phillips H, Reichardt LF, Ryan AM, Carver-Moore K, Rosenthal A. Renal and neuronal abnormalities in mice lacking GDNF. Nature. 1996;382:76–79. doi: 10.1038/382076a0. [DOI] [PubMed] [Google Scholar]
- 34.Morris JK, Lin W, Hauser C, Marchuk Y, Getman D, Lee KF. Rescue of the cardiac defect in ErbB2 mutant mice reveals essential roles of ErbB2 in peripheral nervous system development. Neuron. 1999;23:273–283. doi: 10.1016/s0896-6273(00)80779-5. [DOI] [PubMed] [Google Scholar]
- 35.Ng YP, Pun S, Yang JF, Ip NY, Tsim KW. Chick muscle expresses various ARIA isoforms: regulation during development, denervation, and regeneration. Mol Cell Neurosci. 1997;9:132–143. doi: 10.1006/mcne.1997.0613. [DOI] [PubMed] [Google Scholar]
- 36.Nguyen QT, Parsadanian AS, Snider WD, Lichtman JW. Hyperinnervation of neuromuscular junctions caused by GDNF overexpression in muscle. Science. 1998;279:1725–1729. doi: 10.1126/science.279.5357.1725. [DOI] [PubMed] [Google Scholar]
- 37.Oppenheim RW. Neurotrophic survival molecules for motoneurons: an embarrassment of riches. Neuron. 1996;17:195–197. doi: 10.1016/s0896-6273(00)80151-8. [DOI] [PubMed] [Google Scholar]
- 38.Oppenheim RW, Bursztajn S, Prevette D. Cell death of motoneurons in the chick embryo spinal cord. XI. Acetylcholine receptors and synaptogenesis in skeletal muscle following the reduction of motoneuron death by neuromuscular blockade. Development. 1989;107:331–341. doi: 10.1242/dev.107.2.331. [DOI] [PubMed] [Google Scholar]
- 39.Oppenheim RW, Prevette D, D'Costa A, Wang S, Houenou LJ, McIntosh JM. Reduction of neuromuscular activity is required for the rescue of motoneurons from naturally occurring cell death by nicotinic-blocking agents. J Neurosci. 2000;20:6117–6124. doi: 10.1523/JNEUROSCI.20-16-06117.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peng HB, Xie H, Rossi SG, Rotundo RL. Acetylcholinesterase clustering at the neuromuscular junction involves perlecan and dystroglycan. J Cell Biol. 1999;145:911–921. doi: 10.1083/jcb.145.4.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Riethmacher D, Sonnenberg-Riethmacher E, Brinkmann V, Yamaai T, Lewin GR, Birchmeier C. Severe neuropathies in mice with targeted mutations in the ErbB3 receptor. Nature. 1997;389:725–730. doi: 10.1038/39593. [DOI] [PubMed] [Google Scholar]
- 42.Ruegg MA, Bixby JL. Agrin orchestrates synaptic differentiation at the vertebrate neuromuscular junction. Trends Neurosci. 1998;21:22–27. doi: 10.1016/s0166-2236(97)01154-5. [DOI] [PubMed] [Google Scholar]
- 43.Sanchez MP, Silos-Santiago I, Frisen J, He B, Lira SA, Barbacid M. Renal agenesis and the absence of enteric neurons in mice lacking GDNF. Nature. 1996;382:70–73. doi: 10.1038/382070a0. [DOI] [PubMed] [Google Scholar]
- 44.Sandrock AW, Jr, Dryer SE, Rosen KM, Gozani SN, Kramer R, Theill LE, Fischbach GD. Maintenance of acetylcholine receptor number by neuregulins at the neuromuscular junction in vivo. Science. 1997;276:599–603. doi: 10.1126/science.276.5312.599. [DOI] [PubMed] [Google Scholar]
- 45.Sanes JR. Extracellular matrix molecules that influence neural development. Annu Rev Neurosci. 1989;12:491–516. doi: 10.1146/annurev.ne.12.030189.002423. [DOI] [PubMed] [Google Scholar]
- 46.Sanes JR, Lichtman JW. Development of the vertebrate neuromuscular junction. Annu Rev Neurosci. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- 47.Sanes JR, Schachner M, Covault J. Expression of several adhesive macromolecules (N-CAM, L1, J1, NILE, uvomorulin, laminin, fibronectin, and a heparan sulfate proteoglycan) in embryonic, adult, and denervated adult skeletal muscle. J Cell Biol. 1986;102:420–431. doi: 10.1083/jcb.102.2.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Srihari T, Vrbova G. The role of muscle activity in the differentiation of neuromuscular junctions in slow and fast chick muscles. J Neurocytol. 1978;7:529–540. doi: 10.1007/BF01260887. [DOI] [PubMed] [Google Scholar]
- 49.Tsen G, Halfter W, Kroger S, Cole GJ. Agrin is a heparan sulfate proteoglycan. J Biol Chem. 1995;270:3392–3399. doi: 10.1074/jbc.270.7.3392. [DOI] [PubMed] [Google Scholar]
- 50.Wang JY, Miller SJ, Falls DL. The N-terminal region of neuregulin isoforms determines the accumulation of cell surface and released neuregulin ectodomain. J Biol Chem. 2000;275:2841–2851. doi: 10.1074/jbc.M005700200. [DOI] [PubMed] [Google Scholar]
- 51.Watson FL, Heerssen HM, Moheban DB, Lin MZ, Sauvageot CM, Bhattacharyya A, Pomeroy SL, Segal RA. Rapid nuclear responses to target-derived neurotrophins require retrograde transport of ligand-receptor complex. J Neurosci. 1999;19:7889–7900. doi: 10.1523/JNEUROSCI.19-18-07889.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wells DG, McKechnie BA, Kelkar S, Fallon JR. Neurotrophins regulate agrin-induced postsynaptic differentiation. Proc Natl Acad Sci USA. 1999;96:1112–1117. doi: 10.1073/pnas.96.3.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wolpowitz D, Mason TB, Dietrich P, Mendelsohn M, Talmage DA, Role LW. Cysteine-rich domain isoforms of the neuregulin-1 gene are required for maintenance of peripheral synapses. Neuron. 2000;25:79–91. doi: 10.1016/s0896-6273(00)80873-9. [DOI] [PubMed] [Google Scholar]
- 54.Zhao H, Nonet ML. A retrograde signal is involved in activity-dependent remodeling at a C. elegans neuromuscular junction. Development. 2000;127:1253–1266. doi: 10.1242/dev.127.6.1253. [DOI] [PubMed] [Google Scholar]