

Abstract
NMDA and AMPA receptors (NMDARs and AMPARs) are colocalized at most excitatory synapses in the CNS. Consequently, both receptor types are activated by a single quantum of transmitter and contribute to miniature and evoked EPSCs. However, in amphibian retina, miniature EPSCs in ganglion cell layer neurons are mediated solely by AMPARs, although both NMDARs and AMPARs are activated during evoked EPSCs. One explanation for this discrepancy is that NMDARs are located outside of the synaptic cleft and are activated only when extrasynaptic glutamate levels increase during coincident release from multiple synapses. Alternatively, NMDARs may be segregated at synapses that either are not spontaneously active or yield miniature EPSCs that are too small to detect. In this study, we examined excitatory, glutamatergic synaptic inputs to neurons in the ganglion cell layer of acute slices of rat retina. EPSCs, elicited by electrically stimulating presynaptic bipolar cells, exhibited both NMDAR- and AMPAR-mediated components. However, spontaneous EPSCs exhibited only an AMPAR-mediated component. The effects of low-affinity, competitive receptor antagonists indicated that NMDARs encounter less glutamate than AMPARs during an evoked synaptic response. Reducing glutamate uptake or changing the probability of release preferentially affected the NMDAR component in evoked EPSCs; reducing uptake revealed an NMDAR component in spontaneous EPSCs. These results indicate that NMDARs are located extrasynaptically and that glutamate transporters prevent NMDAR activation by a transmitter released from a single vesicle and limit their activation during evoked responses.
Keywords: rat, retina, ganglion cell, low-affinity antagonist, glutamate transporter, spillover
At most excitatory synapses in the CNS, AMPA receptors (AMPARs) and NMDA receptors (NMDARs) are colocalized in the postsynaptic membrane (Bekkers and Stevens, 1989;McBain and Dingledine, 1992; Silver et al., 1992). Consequently, both receptor types usually contribute to evoked EPSCs and to miniature EPSCs, the postsynaptic response to a single quantum of transmitter.
In the retina, ganglion cells receive excitatory and inhibitory synaptic input from bipolar cells and amacrine cells at synapses in the inner plexiform layer (IPL). Ganglion cells integrate and translate this input into patterns of action potentials that are propagated along their axons in the optic nerve to targets in the lateral geniculate nucleus and superior colliculus. Cells in the ganglion cell layer (GLCs) (Matsui et al., 1998) of numerous species express both NMDARs and AMPARs (Aizenman et al., 1988; Mittman et al., 1990; Cohen et al., 1994). Accordingly, EPSCs evoked by either light or electrical stimulation exhibit NMDAR and AMPAR components (Mittman et al., 1990;Diamond and Copenhagen, 1993; Lukasiewicz and Roeder, 1995; Matsui et al., 1998; Higgs and Lukasiewicz, 1999; Matsui et al., 1999). However, in acute slices of amphibian retina spontaneous EPSCs (sEPSCs) lack an NMDAR-mediated component (Taylor et al., 1995; Matsui et al., 1998). Two mechanisms have been proposed to explain this result: first, that NMDARs are located extrasynaptically and are activated only by the concomitant release of many vesicles (Matsui et al., 1998; Higgs and Lukasiewicz, 1999); second, that NMDA and AMPA receptors are expressed separately at different synapses and that only the AMPAR-mediated sEPSCs are detectable (Taylor et al., 1995). Physiological data from amphibian retina supports the first possibility (Matsui et al., 1998), but the punctate expression and colocalization of NMDARs and the postsynaptic density protein PSD-95 in the IPL of rat retina (Fletcher et al., 2000) support the second idea, although the identity of the postsynaptic, immunopositive neurons was not established in the latter study.
We examined this issue in the rat retina by recording evoked and spontaneous EPSCs from GLCs in acute slices. Consistent with reports in amphibian retina, we find that electrically evoked EPSCs are mediated by both AMPARs and NMDARs, whereas sEPSCs are mediated solely by AMPARs. Using low-affinity competitive antagonists of either receptor type, we show that AMPARs are exposed to more synaptically released glutamate than are NMDARs during an evoked response. In addition, reducing glutamate uptake or changing release probability affects the amplitudes of NMDAR EPSCs to a greater extent than those of AMPAR EPSCs. Finally, reducing glutamate uptake causes an NMDAR-mediated component to emerge in the sEPSCs. Taken together, these results suggest that NMDARs on GLCs are located outside of excitatory synapses and are activated only when multiple release events increase extrasynaptic glutamate levels sufficiently. These results suggest that glutamate transporters regulate NMDAR activation and, subsequently, the manner in which GLCs integrate synaptic input.
MATERIALS AND METHODS
Slice preparation and solutions. Retinal slices were prepared from Sprague Dawley rats (17–22 d) in accordance with the National Institute of Neurological Disorders and Stroke Animal Care and Use Committee guidelines. Both eyes were removed and immersed in oxygenated extracellular solution at room temperature. Extracellular solution contained (in mm): 119 NaCl, 2.5 KCl, 1.3 MgCl2, 2.5 CaCl2, 26.2 NaHCO3, 1 NaH2PO4, 20 glucose, 2 Na pyruvate, and 4 Na lactate, bubbled with 95% O2and 5% CO2. The cornea, iris, lens, and vitreous were removed from one eye with scissors. The retina was mechanically detached from the eyecup and immersed in 2% agarose (low-gelling temperature, type VII; Sigma, St. Louis, MO) and cut into 200-μm-thick slices on a vibratome (Leica, Nussloch, Germany). Slices were prepared and stored in oxygenated extracellular solution; they were transferred one at a time to the recording chamber, in which picrotoxin (100 μm) and strychnine (10 μm) were added to oxygenated extracellular solution to block inhibitory synaptic transmission. For outside-out patch experiments, the NaHCO3 in the extracellular solution was replaced with 20 mmHEPES. In magnesium-free solutions, MgCl2 was replaced with CaCl2. The patch pipette solution contained (in mm): 120 Cs methanesulfonate, 10 EGTA, 20 HEPES, 2 MgATP, and 0.2 NaGTP. All solutions were adjusted to pH 7.4 with NaOH or CsOH and adjusted to 290–300 mOsm with sucrose. Reagents were obtained from Sigma, except forl-glutamate,l-2-amino-5-phosphonopentaenoic acid (l-AP-5), andd,l-threo-β-benzyloxyaspartate (TBOA), which were obtained from Tocris Cookson (Ballwin, MO).
Solution delivery. The recording chamber was superfused constantly at a low rate (1 ml/min) with control extracellular solution. During outside-out patch recordings, control and test solutions were delivered simultaneously through theta glass tubing (Warner Instruments, Hamden, CT) pulled to a tip width of 100 μm per barrel. The solution flow created a sharp interface between solutions delivered through neighboring barrels. Solution changes were made by moving the tubing rapidly with a piezoelectric bimorph (Piezo Systems, Cambridge, MA), such that the solution interface traversed the width of the patch pipette tip, enabling brief (1–2 msec) applications ofl-glutamate.
Electrophysiology. All recordings were made from GLCs with an Axopatch 1D amplifier (Axon Instruments, Foster City, CA) in voltage-clamp mode. Patch electrodes (#0010 glass; World Precision Instruments, Sarasota, FL) had tip resistances of 4–5 MΩ when filled with internal solution. Access resistance was 10–20 MΩ; it was monitored continuously and not compensated. Data acquisition and analysis were performed with custom macros written in IgorPro (WaveMetrics, Lake Oswego, OR). Data were filtered at 5 kHz and sampled at 10 kHz. Responses were elicited with a stainless steel bipolar electrode (Frederick Haer, Bowdoinham, ME), positioned in the outer plexiform layer or the distal part of the inner nuclear layer. Because large stimulus currents often elicited longer-lasting, multiphasic responses, the stimulus strength was adjusted such that the AMPAR EPSC decayed in a relatively rapid, monotonic manner (Fig.1).
Fig. 1.

Electrically evoked EPSCs in rat GCLs are shown.A, Infrared differential interference contrast image of a rat retinal slice. A bipolar stimulating electrode is positioned in the outer plexiform layer (OPL; one pole visible,left). A ganglion cell layer (GCL) was patched and filled with Lucifer yellow (right); the fluorescence image has been superimposed. The axonal process just below the soma (arrow) indicates that this cell is probably a ganglion cell. INL, Inner nuclear layer.B, Evoked EPSCs (holding potential, −80 mV) were blocked reversibly by the calcium channel blocker CdCl2 (20 μm). C, EPSCs (−80 mV) were blocked by the non-NMDA receptor antagonist DNQX (10 μm).D, EPSCs were blocked by GYKI (25 μm), an AMPAR antagonist.
Outside-out patches were obtained by slowly withdrawing the patch pipette after establishing a whole-cell recording. Becausel-glutamate-evoked currents were very small in conventional outside-out patches, gentle suction was applied during withdrawal to obtain nucleated patches.
All experiments were performed at room temperature (21–23°C). Unless otherwise indicated, all data are expressed as means ± SD;p values indicate paired t tests, andp < 0.05 was considered significant.
RESULTS
Electrically evoked synaptic responses in GLC neurons
Whole-cell voltage clamp recordings were made from GLCs in acute slices of rat retina (Fig. 1A). Electrical stimuli (10–20 μA, 600 μsec) were delivered through a stainless steel bipolar stimulating electrode positioned in the outer plexiform layer (Fig. 1A). When the cell was voltage clamped at −80 mV, stimulation elicited inward currents that were reduced either by the Ca2+ channel blocker Cd2+ (20 μm; 87 ± 10% block; n = 7) (Fig.1B), 6,7-dinitroquinoxaline-2,3-dione (DNQX, 10 μm; 96 ± 2% block; n = 6) (Fig. 1C) or 1-[4-aminophenyl]-4-methyl-7,8-methylenedioxy-5H-2,3-benzodiazepine (GYKI 52466, 25 μm; 94 ± 6% block;n = 4) (Fig. 1D), indicating that the responses reflected primarily synaptic activation of AMPARs.
In a subset of cells, the charge transfer (Q) during both evoked EPSCs and sEPSCs was measured to estimate the quantal content of the evoked response. The quantal content (Qevoked/QsEPSC) was quite variable across cells (16 ± 13; n = 7). This approach requires that sEPSCs reflect the postsynaptic response to a single quantum, rather than multivesicular release, which has been demonstrated at some synapses (Tong and Jahr, 1994; Auger et al., 1998;Wadiche and Jahr, 2001). QsEPSC was unaffected by changes in pr (see Fig.6A,B), consistent with the sEPSCs being uniquantal. However, it remains possible that multivesicular release occurs during an evoked response, which could lead to an underestimate of the quantal content with this method.
Fig. 6.
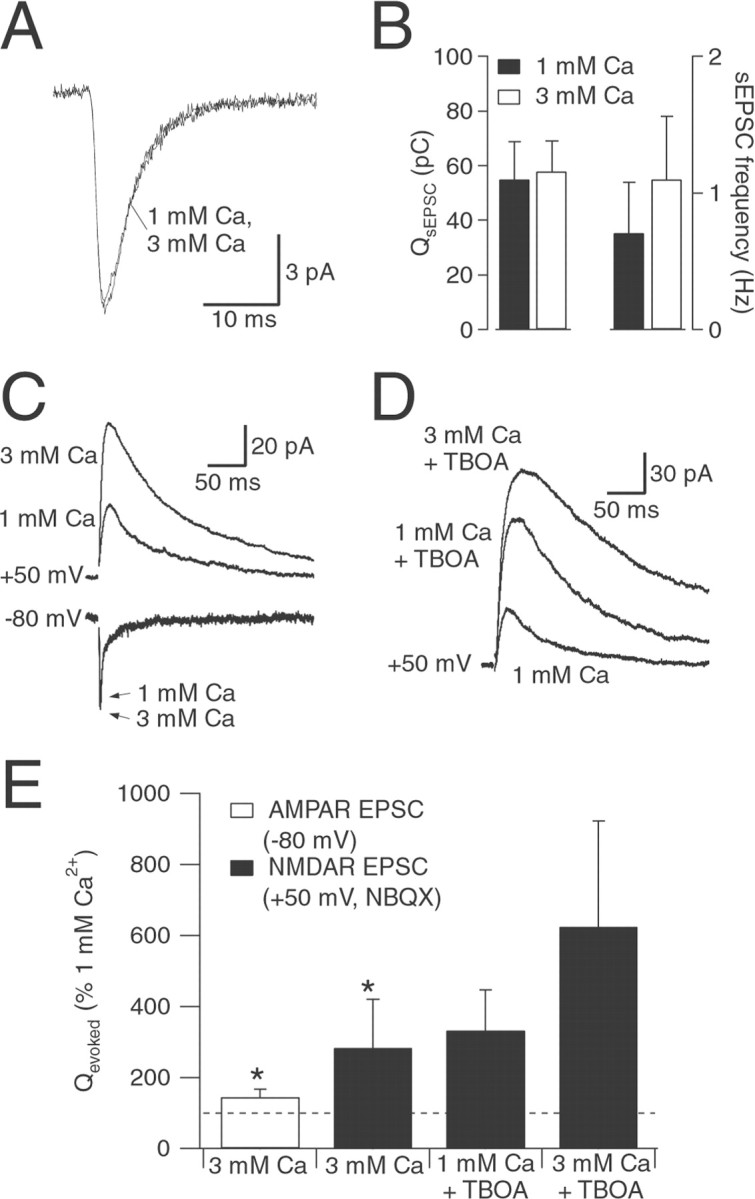
Changing prpreferentially affects NMDAR EPSC. A, Average sEPSCs from one cell in 1 and 3 mm[Ca2+]o. B, Changing [Ca2+]o had no significant effect onQspont (n = 5) but increased sEPSC frequency (frequency in 3 mm[Ca2+]o = 187 ± 101% of frequency in 1 mm[Ca2+]o; n= 5; p = 0.05). C, AMPAR EPSCs (holding potential, −80 mV) and NMDAR EPSCs (holding potential, +50 mV; 5 μm NBQX) in superfusion solution containing either 1 or 3 mm [Ca2+]o.D, Effects on NMDAR EPSCs (holding potential, +50 mV; 5 μm NBQX) of changing [Ca2+]o in the presence of TBOA (10 μm). E, Summary of effects of changing [Ca2+]o in the absence (n = 4) and the presence (n = 4) of TBOA (10 μm). Experiments with TBOA were performed in a separate set of cells. Asterisks indicate a statistically significant difference compared with control (p < 0.05).
Electrically evoked EPSCs exhibited faster time courses than light-evoked EPSCs that were recorded in mammalian ganglion cells previously (Cohen, 2000), presumably because the bipolar cell depolarization elicited by electrical stimulation has a faster time course than that elicited by light (cf. Higgs and Lukasiewicz, 1999;Berntson and Taylor, 2000). Although the GCL in rat retina contains both ganglion cells and displaced amacrine cells (Perry, 1981), no systematic differences in EPSC characteristics distinguished the cell types. Consequently, the data from all cells have been pooled; the postsynaptic neurons are referred to as GLCs (Matsui et al., 1998). Generally, larger cells that were not immediately adjacent to the IPL were selected for recording; when a subset of recorded cells was filled with Lucifer yellow, 9 of 10 cells exhibited a visible axonal process, indicative of ganglion cells (Fig. 1A).
Evoked responses exhibit AMPAR and NMDAR components
Synaptic, glutamatergic excitation of GLCs in amphibian retinal slices is mediated by both AMPARs and NMDARs (Mittman et al., 1990;Diamond and Copenhagen, 1993; Lukasiewicz and Roeder, 1995; Lukasiewicz et al., 1997; Matsui et al., 1998). However, in rat ganglion cells grown in culture, evoked synaptic responses are mediated solely by AMPARs, even though the cells express functional NMDARs (Taschenberger et al., 1995). To determine whether this discrepancy is attributable to a difference in species or in preparation, we looked in rat retinal slices to see whether evoked EPSCs in GLCs exhibit an NMDAR component. NMDARs are mostly blocked at negative potentials by external magnesium ions, but this blockade is primarily relieved at positive potentials (Mayer et al., 1984; Nowak et al., 1984). Accordingly, when the postsynaptic membrane was clamped at positive potentials, the evoked EPSC decayed much more slowly (Fig.2A), indicating the presence of an NMDAR component (Hestrin et al., 1990a; Mittman et al., 1990). The kinetic differences between the two components allowed them to be examined simultaneously: the early component of the EPSC exhibited a linear, ohmic conductance, typical of AMPARs, and the late component exhibited a J-shaped current–voltage relationship, indicative of NMDARs (Mayer et al., 1984; Nowak et al., 1984) (Fig.2B).
Fig. 2.

Electrically evoked EPSCs exhibit AMPAR and NMDAR components. A, EPSCs recorded at holding potentials (in millivolts) are indicated at left.Dashed lines indicate the early (circles) and late (squares) time points at which EPSC amplitudes were measured in B. Stimulus artifacts have been removed for clarity. B, The early component of the EPSC (circles) exhibited an ohmic conductance typical of the AMPAR, whereas the late component (squares) exhibited the J-shaped conductance signature of the NMDAR (Mayer et al., 1984;Nowak et al., 1984). Similar results were observed in seven cells.C, The AMPAR antagonist NBQX (5 μm) blocked the entire EPSC at −80 mV and the early component at +40 mV. Similar results were observed in six cells. D, The NMDAR antagonist CPP exerted little effect at −80 mV and blocked a slow component at +40 mV. Similar results were observed in six cells.
The AMPAR and NMDAR components could also be distinguished pharmacologically. As shown in Figure 1, at −80 mV the response was abolished by AMPAR antagonists, including DNQX or 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide (NBQX; 5 μm) (Fig. 2C). In contrast, the NMDAR antagonist (RS)-3-(2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (CPP; 5 μm) had little effect on the EPSC at −80 mV (Fig. 2D). At +40 mV, NBQX blocked a fast component of the EPSC (Fig. 2C), whereas CPP blocked the slow component (Fig. 2D).
Spontaneous EPSCs exhibit only an AMPAR component
At hippocampal and cerebellar synapses, evoked EPSCs are also mediated by NMDARs and AMPARs (Hestrin et al., 1990a; Silver et al., 1992). Moreover, miniature EPSCs also exhibit NMDAR and AMPAR components at these synapses, indicating that the two receptor types are colocalized in the postsynaptic membrane (Bekkers and Stevens, 1989; McBain and Dingledine, 1992; Silver et al., 1992). In contrast, sEPSCs in amphibian (Taylor et al., 1995; Matsui et al., 1998) and mammalian (Taschenberger et al., 1995; Tian et al., 1998) GLCs do not exhibit an NMDAR component, suggesting that NMDARs may be expressed either extrasynaptically or at synapses different from those at which AMPARs are expressed. We addressed this question in rat retinal slices by recording sEPSCs in GLCs (Fig. 3). The sodium channel blocker tetrodotoxin (TTX; 1 μm) did not affect the frequency, amplitude, or waveform of spontaneous events recorded in GLCs (data not shown); therefore, the experiments described here were performed in the absence of TTX, and the events are referred to as sEPSCs.
Fig. 3.

Spontaneous EPSCs do not exhibit an NMDAR component. A, EPSCs evoked in control solution at holding potentials of −80 and +40 mV. B, The charge transferred during EPSCs (Qevoked) at +40 mV was significantly greater than at −80 mV. C, Representative recordings showing spontaneous activity at −80 and +50 mV. Inset, Average sEPSC at −80 mV (inward trace, average of 101 events from one cell) and +50 mV (outward trace, average of 65 events from the same cell). D, Average charge transfer (Qspont) at −80 and +50 mV (n = 5). Bar on rightindicates data at +50 mV scaled to reflect an 80 mV driving force.E, Effect on EPSCs of superfusing the slice with nominally Mg-free extracellular solution. F, Comparison of Qevoked in control and nominally Mg-free solution. G, Representative recordings of spontaneous activity (−80 mV) in control solution, nominally Mg-free solution, and Mg-free solution plus 10 μm DNQX. Inset, Average sEPSCs from one cell in control (n = 291 events) and nominally Mg-free solution (n = 284 events). H, Qspont in control solution and in nominally Mg-free solution (n = 7).Asterisks indicate a statistically significant difference compared with control (p < 0.05).
As shown above, evoked EPSCs exhibited a prominent NMDAR component at positive potentials (Figs. 2, 3A). Because of the prolonged time course of the NMDAR conductance, the charge transferred during an EPSC (Qevoked) clamped at +40 mV was significantly greater than at −80 mV, despite the reduced driving force (Qevoked at +40 mV = 711 ± 656% of Qevoked at −80 mV; n = 7; p = 0.003) (Fig.3B). However, the average charge transferred during sEPSCs (Qspont) recorded at +50 mV, when scaled to account for the difference in driving force, was not significantly different from that during theQspont recorded at −80 mV [Qspont (+50, scaled) = 89 ± 27% of Qspont at −80 mV;n = 5; p = 0.49] (Fig.3D).
When magnesium was removed from the extracellular solution, an NMDAR component was detected in the evoked response at −80 mV (Fig.3E), which led to a significant increase inQevoked (411 ± 253% of control;n = 6; p = 0.01) (Fig.3F). This did not appear to result from any change in the probability of release, because no effects were observed in the evoked EPSC at +40 mV or in the AMPAR component of the EPSC at −80 mV (Fig. 3E). At −80 mV, removing external magnesium did not affect Qspont (104 ± 5% of control; n = 7; p = 0.22) (Fig.3G,H), and sEPSCs were abolished by 10 μm DNQX (Fig. 3G). The lack of an NMDAR component in the sEPSCs may reflect the depletion of endogenous glycine, a coagonist of the NMDAR (Johnson and Ascher, 1987), from the slice. To control for this possibility, sEPSCs were also recorded in the absence of magnesium and the presence of 10 μmd-serine, a nontransported NMDAR glycine site agonist, with similar results [Qspont(d-serine, 0 Mg2+) = 115 ± 26% of control (0 Mg2+); n = 5;p = 0.27] (data not shown). These results indicate that sEPSCs in rat GLCs are mediated solely by AMPARs, as has been reported for other species (Taylor et al., 1995; Matsui et al., 1998;Tian et al., 1998).
NMDARs encounter a lower transmitter concentration during a synaptic event
The experiments illustrated in Figure 3 suggest that NMDARs on rat GLCs may be located either extrasynaptically or in synapses separate from AMPARs. If NMDARs were located extrasynaptically, they would be likely to encounter a lower glutamate concentration during a synaptic event than AMPARs located within the synaptic cleft, closer to the site of glutamate release. To test this prediction, we examined the actions of low-affinity competitive antagonists on synaptic responses and on receptor-mediated currents in outside-out, nucleated patches excised from GLC somata. The efficacy of γ-d-glutamylglycine (γ-DGG; 500 μm), a low-affinity AMPAR antagonist (Watkins and Olverman, 1987), and l-AP-5 (200 μm), a low-affinity NMDAR antagonist (Watkins and Olverman, 1987), was calibrated by measuring their effects on patch currents elicited by brief (2 msec) applications of 1 mml-glutamate (see Materials and Methods) (Fig.4A,C). In one group of patches, AMPARs were pharmacologically isolated by including 5 μm CPP in all solutions, whereas in another group NMDARs were isolated by including 5 μmNBQX. The duration of glutamate application was monitored by measuring open-tip currents at the end of each experiment (Fig.4A,C) and was similar for both groups.
Fig. 4.

NMDARs encounter less synaptically released glutamate than AMPARs. A, AMPAR responses (NMDARs blocked with 5 μm CPP) in outside-out patches to brief pulses (1–2 msec) of l-glutamate (1 mm) in control solution and in the continuous presence of γ-DGG (500 μm; holding potential, −80 mV). Top trace, Open-tip current across open electrode, indicating the speed of solution exchange across the pipette tip. B, Evoked AMPAR EPSCs (5 μm CPP) recorded in control solution and in the presence of γ-DGG (500 μm; holding potential, −80 mV). Stimulus artifacts have been removed for clarity.C, As in A, except that NMDARs were isolated (5 μm NBQX), and the effects ofl-AP-5 (200 μm) were tested (holding potential, +50 mV). D, Evoked NMDAR EPSCs (5 μm NBQX) recorded in control solution and in the presence of l-AP-5 (200 μm; holding potential, +50 mV). Inset, EPSC recorded from the same neuron at −80 mV. Calibration: 25 pA, 30 msec. E, Effects of 500 μm γ-DGG on patch currents (n = 6) and evoked EPSCs (n = 6). F, Effects of 200 μml-AP-5 on patch currents (n = 6) and evoked EPSCs (n = 5). The asterisk indicates a statistically significant difference compared with control (p < 0.05).
γ-DGG (500 μm) reversibly reduced AMPAR responses in excised patches (peak amplitude in γ-DGG was 22 ± 10% of control; n = 6) (Fig. 4A,E). This degree of blockade was comparable to the antagonism of γ-DGG against AMPAR EPSCs (peak amplitude in γ-DGG was 21 ± 9% of control; n = 6; p = 0.83, unpaired t test between patch and EPSC data) (Fig.4B,E), suggesting that AMPARs encountered comparable amounts of glutamate in the patch and synaptic responses. In contrast, the effect of l-AP-5 on NMDAR patch currents (peak amplitude in l-AP-5 was 67 ± 8% of control; n = 6) (Fig. 4C,F) was significantly weaker than its effect on NMDAR EPSCs (peak amplitude inl-AP-5 was 33 ± 14% of control;n = 6; p = 0.001, unpaired ttest between patch and EPSC data) (Fig. 4D,F), suggesting that NMDARs encountered less glutamate during the synaptic responses than during the patch responses. Although the fragility of the patches pulled from GLCs precluded a direct comparison of the two antagonists in the same patch, these results indicate that AMPARs encounter more glutamate than NMDARs during a synaptic response.
Blocking glutamate uptake preferentially enhances the NMDAR component of the EPSC
The results presented thus far are consistent with a scenario in which NMDARs on GLC dendrites are located outside the synaptic cleft. Glutamate uptake plays a crucial role in clearing synaptically released glutamate at many synapses (Otis et al., 1996; Diamond and Jahr, 1997;Higgs and Lukasiewicz, 1999; Carter and Regehr, 2000). Despite the presence of glutamate transporters in neuronal membranes (Rothstein et al., 1994), including those of rat bipolar and ganglion cells (Rauen et al., 1996), most glutamate uptake in the inner retina appears to occur at glial (Müller cell) membranes (Rauen et al., 1998), beyond the immediate vicinity of the postsynaptic density. Thus, uptake may limit activation of receptors outside the synaptic cleft (or in neighboring, inactive clefts) more than it would affect receptor activation within an active synapse. Therefore, one would predict that the activation of extrasynaptic NMDARs would be limited more than that of synaptic AMPARs by glutamate uptake.
To test this prediction, we examined the effects of TBOA (Shimamoto et al., 1998) on evoked EPSCs. TBOA is a competitive, nonsubstrate antagonist of glutamate transporters that does not interact with NMDARs (Jabaudon et al., 1999). TBOA (10 μm) did not affect the charge transferred during AMPAR EPSCs (Qevoked = 107 ± 12% of control; n = 5; p = 0.2) (Fig.5A,C). In contrast, TBOA potentiated and prolonged NMDAR EPSCs (Qevoked = 365 ± 145% of control; n = 5; p = 0.02) (Fig.5B,C). Therefore, blocking transporters preferentially enhanced the NMDAR component of the EPSC, which is consistent with there being an extrasynaptic location for the NMDARs.
Fig. 5.
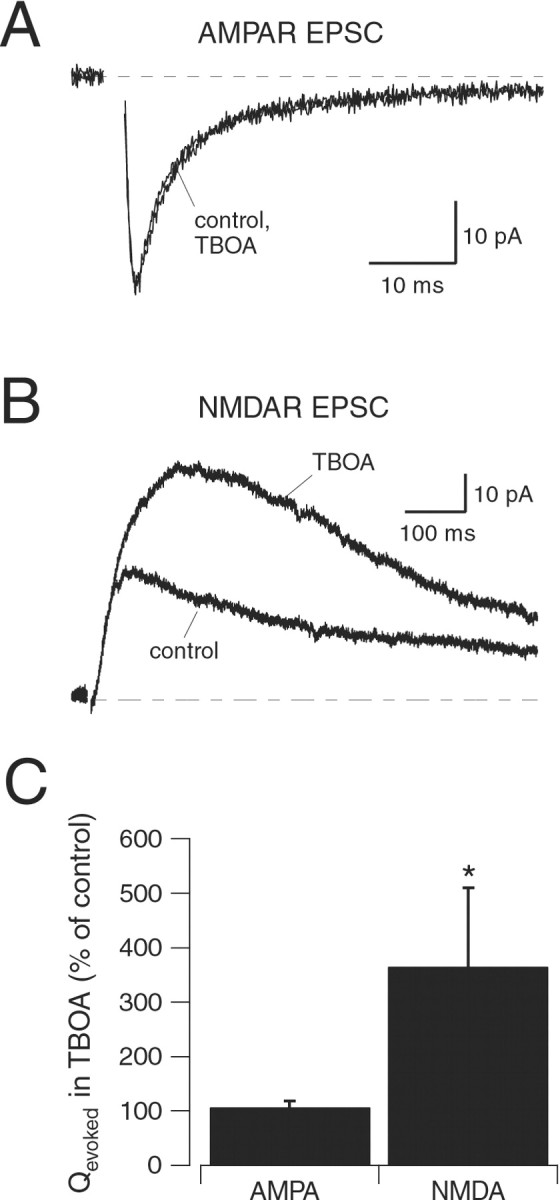
Glutamate transporters limit synaptic activation of NMDARs. A, Evoked AMPAR EPSCs recorded in control (5 μm CPP) and in the presence of 10 μm TBOA (holding potential, −80 mV). B, Evoked NMDAR EPSCs recorded in control (5 μm NBQX) and in the presence of 10 μm TBOA (holding potential, +40 mV). C, Effects of TBOA on Qevoked of AMPAR EPSCs (n = 5) and NMDAR EPSCs (n = 5). The asterisk indicates a statistically significant difference compared with control (p < 0.05).
Changing release probability preferentially affects NMDAR activation
NMDAR activation in GLCs appears to require coincident release of multiple quanta. During an evoked response, glutamate released from multiple vesicles at different synapses, or from within the same synapse, may accumulate in the extrasynaptic space to levels sufficient to activate extrasynaptic NMDARs. NMDARs could even be activated by transmitter released at a synapse made on a different postsynaptic cell. One might predict from this model that changing the probability of release (pr) would change the extrasynaptic accumulation of glutamate and affect the NMDAR EPSC, perhaps more than the AMPAR EPSC. This would stand in contrast to other central synapses, where changing praffects NMDAR and AMPAR EPSCs to a similar extent (Perkel and Nicoll, 1993; Tong and Jahr, 1994).
pr was manipulated by varying [Ca2+]o from 1 to 3 mm([Mg2+]o was maintained at 1.3 mm), which caused a nearly threefold increase in the charge transferred during the NMDAR EPSC (284 ± 136% increase; n = 4) (Fig.6C,E). The AMPAR EPSC was increased to a lesser extent (145 ± 22% increase;n = 4) (Fig. 6C,E). The relatively small effect of changing pr on the AMPAR EPSC suggests that pr may be quite high in 1 mm[Ca2+]o, perhaps close to 1. If AMPARs were occupied to a significant extent during release of a single vesicle, as suggested previously (Clements et al., 1992; Silver et al., 1996; Liu et al., 1999; Wadiche and Jahr, 2001), then multivesicular release at the same synapse would cause a relatively small increase in AMPAR activation.
Changing [Ca2+]ofrom 1 to 3 mm also enhanced the NMDAR EPSC in the presence of TBOA (181 ± 38%; n = 4) (Fig.6D,E), although to a somewhat lesser extent than in the absence of TBOA. Whereas the results varied from cell to cell, the trend was consistent with the idea that blocking transporters allows more extrasynaptic NMDARs to be occupied during an EPSC in the “lowpr” condition (1 mm[Ca2+]o), resulting in less potentiation when pris increased.
Reducing uptake reveals an NMDAR component in sEPSCs
Because glutamate uptake appeared to limit NMDAR activation during evoked, multiquantal responses, we tested whether the same was true during spontaneous (likely monoquantal) activity by examining the effects of TBOA on sEPSCs (Fig. 7). TBOA (10 μm) did not affect the sEPSC charge transfer in normal (1.3 mm[Mg2+]o) solution (Qspont = 99 ± 18% of control;n = 5; p = 0.77) (Fig. 7A, top left), consistent with analogous experiments in amphibian GLCs (Higgs and Lukasiewicz, 1999; Matsui et al., 1999). However, when Mg2+ was removed from the superfusion solution in the continued presence of TBOA a slow component emerged, significantly increasing Qspont(210 ± 66% of TBOA alone; n = 5;p = 0.02) (Fig. 7A, top left). This slow component was mediated by NMDARs because it was blocked by 5 μm CPP (Qspontreduced to 116 ± 21% of TBOA alone; n = 5;p = 0.2) (Fig. 7A, top left). Subtraction of the average sEPSC waveform in the presence of CPP from that in TBOA/0 Mg2+ revealed a CPP-sensitive component with a slow rise and decay (Fig. 7A, bottom left).
Fig. 7.

Blocking transporters reveals an NMDAR component in sEPSCs. A, Top, Average sEPSCs recorded from one GLC at −80 mV in control extracellular solution (1.3 Mg2+, 62 events), control solution plus 10 μm TBOA (83 events), 0 Mg2+extracellular solution plus 10 μm TBOA (128 events), and 0 Mg2+ extracellular solution plus 10 μm TBOA plus 5 μm CPP (109 events).Left, Average of all events. Center,Average of smallest 20% of events. Right, Average of largest 20% of events. Average sEPSCs in all four conditions are superimposed. A, Bottom, Subtraction of average traces in TBOA/0 Mg2+/CPP from average traces in TBOA/0 Mg2+. B, Summary of the effects of external magnesium, TBOA, and CPP on sEPSCs (n = 5 cells). Asterisks indicate a statistically significant difference compared with control (p < 0.05).
Interestingly, the NMDAR component in the TBOA–0 Mg2+ condition was most apparent in the largest sEPSCs. No significant effect of removing Mg2+ was observed in the smallest 20% of events (Qspont = 108 ± 30% of TBOA alone; n = 5; p = 0.6) (Fig.7A, center), whereas a marked effect was observed in the largest 20% of events (Qspont = 183 ± 63% of TBOA alone; n = 5;p = 0.04) (Fig. 7A, right). These results suggest that extrasynaptic NMDARs would be activated by glutamate released from a single vesicle were it not for high-affinity glutamate uptake, particularly during the quantal events that generate the largest postsynaptic responses.
DISCUSSION
The experiments presented here indicate that NMDARs on GLCs are activated only during evoked responses, when multiple release events occur. Glutamate released from a single quantum is not, by itself, sufficient to activate NMDARs, apparently because glutamate uptake limits transmitter access to the NMDARs. Thus, in contrast to most central synapses, in which glutamate transporters regulate the synaptic activation of NMDARs only moderately (Hestrin et al., 1990b; Sarantis et al., 1993; Asztely et al., 1997; cf. Overstreet et al., 1999), transporters in the inner retina appear to play a critical role in limiting NMDAR activation by synaptically released glutamate.
Extrasynaptic NMDA receptors
These results in rat retinas are consistent with previous work in amphibian GLCs that showed that sEPSCs are mediated solely by AMPARs, despite the presence of an NMDAR component in the evoked EPSC (Taylor et al., 1995; Matsui et al., 1998). Taylor et al. (1995) suggested that NMDARs may be segregated from AMPARs at different synapses; Matsui et al. (1998) proposed that NMDARs are located extrasynaptically. The data presented here are interpreted most easily in the context of the second scenario: NMDARs were shown to encounter less glutamate during a synaptic response (Fig. 4); reducing transport with TBOA or increasingpr preferentially enhanced the NMDAR component of the EPSC (Figs. 5, 6); and TBOA caused an NMDAR component to emerge in sEPSCs (Fig. 7). Taken together, these results suggest that NMDARs are located at some distance from the site of release.
However, most of our results can also be interpreted in terms of the segregated-receptor hypothesis (Taylor et al., 1995). This scenario is potentially consistent with immunohistochemical evidence from rat retina that NMDARs colocalize with PSD-95 at postsynaptic densities in the IPL (Fletcher et al., 2000), although the NMDAR-immunopositive dendrites were not identified in that study and could have originated from amacrine cells in the inner nuclear layer. If NMDARs were segregated at synapses into which less glutamate was released during a synaptic response, perhaps attributable to differences in release machinery (Choi et al., 2000), NMDAR sEPSCs could be difficult to detect, and low-affinity antagonists would block the evoked NMDAR EPSC to a relatively greater extent than the AMPAR EPSC (Fig. 4). Blocking transporters could encourage spillover between neighboring synapses, preferentially enhancing the NMDAR component of the evoked EPSC (Fig.5) (Asztely et al., 1997). However, TBOA would cause an NMDAR component to emerge in the sEPSCs (Fig. 7) only if substantial spillover consistently occurred between synaptic contacts made on the same postsynaptic neuron. At “dyad” synapses in the IPL, bipolar cell synaptic terminals are apposed to two postsynaptic elements that only very rarely, if ever, arise from the same ganglion cell (Dowling, 1987). Even if they did, it seems unlikely, given the small dimensions of dyad synapses (∼200 nm diameter) (Koulen et al., 1998; Fletcher et al., 2000), that even a high density of glutamate transporters could so sharply partition the transmitter concentration within the synapse. Given these considerations, the results presented here are most consistent with an extrasynaptic location of NMDARs on GLC dendrites.
In rat retinal ganglion cells grown in cell culture, neither sEPSCs nor EPSCs exhibit an NMDAR component, even though the cells exhibit functional NMDARs (Taschenberger et al., 1995). Whereas it is possible that culture conditions would, for some reason, favor AMPAR synapses over NMDAR synapses [although this does not appear to be the case in the hippocampus (Bekkers and Stevens, 1989; Gomperts et al., 1998)], it seems more likely that synaptically released glutamate, on reaching the perimeter of the cleft, would be diluted by the large extracellular volume of the culture media before reaching extrasynaptic NMDARs.
Transporters limit glutamate receptor activation
Decreasing glutamate transport with TBOA enhanced the NMDAR EPSC but did not affect the AMPAR EPSC. This is in contrast to previous reports in amphibian retinas that showed that the glutamate transport inhibitorl-trans-pyrrolidine-2,4-dicarboxylic acid (PDC) prolonged AMPAR EPSCs evoked by electrical or light stimulation (Higgs and Lukasiewicz, 1999; Matsui et al., 1999). In both of these previous studies, blocking transporters enhanced primarily a slow component of the evoked EPSC that was not evident in the responses reported here. The discrepancy is likely attributable to differences in stimulus strength; when we used stronger stimuli, we also observed a slow component in the EPSC, similar to that reported in amphibians (Higgs and Lukasiewicz, 1999; Matsui et al., 1999), that appeared to be attributable to the spillover of glutamate onto AMPARs in other synapses. It was enhanced by TBOA and blocked to a greater extent than the fast component by γ-DGG (data not shown). We purposely limited stimulus intensity to examine the differences between the NMDAR and AMPAR components of the EPSC, although the effect of PDC on the light-evoked AMPAR EPSC (Higgs and Lukasiewicz, 1999; Matsui et al., 1999) strongly suggests that glutamate transporters limit AMPAR activation in a physiologically meaningful manner.
When glutamate uptake was reduced with TBOA, an NMDAR component emerged in sEPSCs, but only in larger events (Fig. 7). One possible explanation for this result is that larger sEPSCs may reflect activity at larger synapses that express more AMPARs within the synaptic cleft and NMDARs extrasynaptically. In contrast, smaller synapses would express fewer AMPARs and perhaps no extrasynaptic NMDARs. Alternatively, larger sEPSCs may reflect the release of more glutamate, which could activate a larger fraction of synaptic AMPARs and, with transporters inhibited, might succeed in activating extrasynaptic NMDARs. [A wide range of transmitter concentrations would be achieved if larger sEPSCs reflected spontaneous multivesicular release. However, sEPSC amplitude was insensitive to changes in [Ca2+]o (Fig.6A,B), a manipulation that affects the incidence of multivesicular release (Tong and Jahr, 1994; Auger et al., 1998;Wadiche and Jahr, 2001), suggesting that sEPSCs reflect postsynaptic responses to single quanta. Such a range of transmitter concentrations, then, would have to arise from variations in vesicular transmitter content (Frerking et al., 1995; Liu et al., 1999).] Additional experiments are required to distinguish between these presynaptic and postsynaptic possibilities.
Possible physiological roles for extrasynaptic NMDARs
Functional NMDARs are expressed by retinal ganglion cells in numerous species, but a specific role for NMDARs in ganglion cell synaptic processing remains unclear. Changing [Ca2+]o:[Mg2+]ofrom 2.5:1.3 to 3.8:0 caused little change in the EPSC amplitude (Fig.3E), suggesting that pr at bipolar cell terminals may be maximal under control conditions (2.5 mm[Ca2+]o, 1.3 mm[Mg2+]o). Increasing [Ca2+]o:[Mg2+]ofrom 1:1.3 to 3:1.3 enhanced NMDAR EPSCs (indicating a modulation ofpr) but had relatively little effect on the AMPAR EPSC (Fig. 6). This may mean thatpr in 1 mm[Ca2+]o is nearly 1; if synaptic receptors were significantly occupied under these conditions, then increasing pr further would cause a subproportional change in receptor activation. Perhaps their extrasynaptic location allows NMDARs to avoid saturation and accurately reflect increases in pr>1, a range over which synaptic receptors may be relatively insensitive. It is possible that NMDARs may not be activated under low-light conditions but may play a role in boosting the synaptic response to stronger light stimuli. Although NMDARs on salamander ganglion cells appear to mediate a similar fraction of the response to weak and strong light stimulation (Diamond and Copenhagen, 1995), recent work in mouse amacrine cells suggests that postsynaptic NMDAR activation during a light response may depend on bipolar cell terminalpr, which is regulated by feedback inhibition from amacrine cells (Matsui et al., 2001).
Footnotes
This work was supported by the National Institute of Neurological Disorders and Stroke Intramural Research Program. We thank Matthew Higgs and Joshua Singer for critically reading this manuscript.
Correspondence should be addressed to Dr. Jeffrey S. Diamond, National Institutes of Health, National Institute of Neurological Disorders and Stroke, Synaptic Physiology Unit, Building 36, Room 2C09, 36 Convent Drive, Bethesda, MD 20892-4066. E-mail: diamondj@ninds.nih.gov.
REFERENCES
- 1.Aizenman E, Frosch MP, Lipton SA. Responses mediated by excitatory amino acid receptors in solitary retinal ganglion cells from rat. J Physiol (Lond) 1988;396:75–91. doi: 10.1113/jphysiol.1988.sp016951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Asztely F, Erdemli G, Kullmann DM. Extrasynaptic glutamate spillover in the hippocampus: dependence on temperature and the role of active glutamate uptake. Neuron. 1997;18:281–293. doi: 10.1016/s0896-6273(00)80268-8. [DOI] [PubMed] [Google Scholar]
- 3.Auger C, Kondo S, Marty A. Multivesicular release at single functional synaptic sites in cerebellar stellate and basket cells. J Neurosci. 1998;18:4532–4547. doi: 10.1523/JNEUROSCI.18-12-04532.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bekkers JM, Stevens CF. NMDA and non-NMDA receptors are co-localized at individual excitatory synapses in cultured rat hippocampus. Nature. 1989;341:230–233. doi: 10.1038/341230a0. [DOI] [PubMed] [Google Scholar]
- 5.Berntson A, Taylor WR. Response characteristics and receptive field widths of on-bipolar cells in the mouse retina. J Physiol (Lond) 2000;524:879–889. doi: 10.1111/j.1469-7793.2000.00879.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carter AG, Regehr WG. Prolonged synaptic currents and glutamate spillover at the parallel fiber to stellate cell synapse. J Neurosci. 2000;20:4423–4434. doi: 10.1523/JNEUROSCI.20-12-04423.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choi S, Klingauf J, Tsien RW. Postfusional regulation of cleft glutamate concentration during LTP at “silent synapses.”. Nat Neurosci. 2000;3:330–336. doi: 10.1038/73895. [DOI] [PubMed] [Google Scholar]
- 8.Clements JD, Lester RA, Tong G, Jahr CE, Westbrook GL. The time course of glutamate in the synaptic cleft. Science. 1992;258:1498–1501. doi: 10.1126/science.1359647. [DOI] [PubMed] [Google Scholar]
- 9.Cohen ED. Light-evoked excitatory synaptic currents of X-type retinal ganglion cells. J Neurophysiol. 2000;83:3217–3229. doi: 10.1152/jn.2000.83.6.3217. [DOI] [PubMed] [Google Scholar]
- 10.Cohen ED, Zhou ZJ, Fain GL. Ligand-gated currents of alpha and beta ganglion cells in the cat retinal slice. J Neurophysiol. 1994;72:1260–1269. doi: 10.1152/jn.1994.72.3.1260. [DOI] [PubMed] [Google Scholar]
- 11.Diamond JS, Copenhagen DR. The contribution of NMDA and non-NMDA receptors to the light-evoked input-output characteristics of retinal ganglion cells. Neuron. 1993;11:725–738. doi: 10.1016/0896-6273(93)90082-3. [DOI] [PubMed] [Google Scholar]
- 12.Diamond JS, Copenhagen DR. The relationship between light-evoked synaptic excitation and spiking behaviour of salamander retinal ganglion cells. J Physiol (Lond) 1995;487:711–725. doi: 10.1113/jphysiol.1995.sp020912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Diamond JS, Jahr CE. Transporters buffer synaptically released glutamate on a submillisecond time scale. J Neurosci. 1997;17:4672–4687. doi: 10.1523/JNEUROSCI.17-12-04672.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dowling JE. The retina: an approachable part of the brain. Belknap, Harvard UP; Cambridge, MA: 1987. [Google Scholar]
- 15.Fletcher EL, Hack I, Brandstatter JH, Wassle H. Synaptic localization of NMDA receptor subunits in the rat retina. J Comp Neurol. 2000;420:98–112. [PubMed] [Google Scholar]
- 16.Frerking M, Borges S, Wilson M. Variation in GABA mini amplitude is the consequence of variation in transmitter concentration. Neuron. 1995;15:885–895. doi: 10.1016/0896-6273(95)90179-5. [DOI] [PubMed] [Google Scholar]
- 17.Gomperts SN, Rao A, Craig AM, Malenka RC, Nicoll RA. Postsynaptically silent synapses in single neuron cultures. Neuron. 1998;21:1443–1451. doi: 10.1016/s0896-6273(00)80662-5. [DOI] [PubMed] [Google Scholar]
- 18.Hestrin S, Nicoll RA, Perkel DJ, Sah P. Analysis of excitatory synaptic action in pyramidal cells using whole-cell recording from rat hippocampal slices. J Physiol (Lond) 1990a;422:203–225. doi: 10.1113/jphysiol.1990.sp017980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hestrin S, Sah P, Nicoll RA. Mechanisms generating the time course of dual component excitatory synaptic currents recorded in hippocampal slices. Neuron. 1990b;5:247–253. doi: 10.1016/0896-6273(90)90162-9. [DOI] [PubMed] [Google Scholar]
- 20.Higgs MH, Lukasiewicz PD. Glutamate uptake limits synaptic excitation of retinal ganglion cells. J Neurosci. 1999;19:3691–3700. doi: 10.1523/JNEUROSCI.19-10-03691.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jabaudon D, Shimamoto K, Yasuda-Kamatani Y, Scanziani M, Gahwiler BH, Gerber U. Inhibition of uptake unmasks rapid extracellular turnover of glutamate of nonvesicular origin. Proc Natl Acad Sci USA. 1999;96:8733–8738. doi: 10.1073/pnas.96.15.8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson JW, Ascher P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature. 1987;325:529–531. doi: 10.1038/325529a0. [DOI] [PubMed] [Google Scholar]
- 23.Koulen P, Fletcher EL, Craven SE, Bredt DS, Wassle H. Immunocytochemical localization of the postsynaptic density protein PSD- 95 in the mammalian retina. J Neurosci. 1998;18:10136–10149. doi: 10.1523/JNEUROSCI.18-23-10136.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu G, Choi S, Tsien RW. Variability of neurotransmitter concentration and nonsaturation of postsynaptic AMPA receptors at synapses in hippocampal cultures and slices. Neuron. 1999;22:395–409. doi: 10.1016/s0896-6273(00)81099-5. [DOI] [PubMed] [Google Scholar]
- 25.Lukasiewicz PD, Roeder RC. Evidence for glycine modulation of excitatory synaptic inputs to retinal ganglion cells. J Neurosci. 1995;15:4592–4601. doi: 10.1523/JNEUROSCI.15-06-04592.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lukasiewicz PD, Wilson JA, Lawrence JE. AMPA-preferring receptors mediate excitatory synaptic inputs to retinal ganglion cells. J Neurophysiol. 1997;77:57–64. doi: 10.1152/jn.1997.77.1.57. [DOI] [PubMed] [Google Scholar]
- 27.Matsui K, Hosoi N, Tachibana M. Excitatory synaptic transmission in the inner retina: paired recordings of bipolar cells and neurons of the ganglion cell layer. J Neurosci. 1998;18:4500–4510. doi: 10.1523/JNEUROSCI.18-12-04500.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsui K, Hosoi N, Tachibana M. Active role of glutamate uptake in the synaptic transmission from retinal nonspiking neurons. J Neurosci. 1999;19:6755–6766. doi: 10.1523/JNEUROSCI.19-16-06755.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsui K, Hasegawa J, Tachibana M. Modulation of excitatory synaptic transmission by GABA(C) receptor-mediated feedback in the mouse inner retina. J Neurophysiol. 2001;86:2285–2298. doi: 10.1152/jn.2001.86.5.2285. [DOI] [PubMed] [Google Scholar]
- 30.Mayer ML, Westbrook GL, Guthrie PB. Voltage-dependent block by Mg2+ of NMDA responses in spinal cord neurones. Nature. 1984;309:261–263. doi: 10.1038/309261a0. [DOI] [PubMed] [Google Scholar]
- 31.McBain C, Dingledine R. Dual-component miniature excitatory synaptic currents in rat hippocampal CA3 pyramidal neurons. J Neurophysiol. 1992;68:16–27. doi: 10.1152/jn.1992.68.1.16. [DOI] [PubMed] [Google Scholar]
- 32.Mittman S, Taylor WR, Copenhagen DR. Concomitant activation of two types of glutamate receptor mediates excitation of salamander retinal ganglion cells. J Physiol (Lond) 1990;428:175–197. doi: 10.1113/jphysiol.1990.sp018206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- 34.Otis TS, Wu YC, Trussell LO. Delayed clearance of transmitter and the role of glutamate transporters at synapses with multiple release sites. J Neurosci. 1996;16:1634–1644. doi: 10.1523/JNEUROSCI.16-05-01634.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Overstreet LS, Kinney GA, Liu YB, Billups D, Slater NT. Glutamate transporters contribute to the time course of synaptic transmission in cerebellar granule cells. J Neurosci. 1999;19:9663–9673. doi: 10.1523/JNEUROSCI.19-21-09663.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Perkel DJ, Nicoll RA. Evidence for all-or-none regulation of neurotransmitter release: implications for long-term potentiation. J Physiol (Lond) 1993;471:481–500. doi: 10.1113/jphysiol.1993.sp019911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perry VH. Evidence for an amacrine cell system in the ganglion cell layer of the rat retina. Neuroscience. 1981;6:931–944. doi: 10.1016/0306-4522(81)90174-3. [DOI] [PubMed] [Google Scholar]
- 38.Rauen T, Rothstein JD, Wassle H. Differential expression of three glutamate transporter subtypes in the rat retina. Cell Tissue Res. 1996;286:325–336. doi: 10.1007/s004410050702. [DOI] [PubMed] [Google Scholar]
- 39.Rauen T, Taylor WR, Kuhlbrodt K, Wiessner M. High-affinity glutamate transporters in the rat retina: a major role of the glial glutamate transporter GLAST-1 in transmitter clearance. Cell Tissue Res. 1998;291:19–31. doi: 10.1007/s004410050976. [DOI] [PubMed] [Google Scholar]
- 40.Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 41.Sarantis M, Ballerini L, Miller B, Silver RA, Edwards M, Attwell D. Glutamate uptake from the synaptic cleft does not shape the decay of the non-NMDA component of the synaptic current. Neuron. 1993;11:541–549. doi: 10.1016/0896-6273(93)90158-n. [DOI] [PubMed] [Google Scholar]
- 42.Shimamoto K, Lebrun B, Yasuda-Kamatani Y, Sakaitani M, Shigeri Y, Yumoto N, Nakajima T. dl-Threo-β-benzyloxyaspartate, a potent blocker of excitatory amino acid transporters. Mol Pharmacol. 1998;53:195–201. doi: 10.1124/mol.53.2.195. [DOI] [PubMed] [Google Scholar]
- 43.Silver RA, Traynelis SF, Cull-Candy SG. Rapid-time-course miniature and evoked excitatory currents at cerebellar synapses in situ. Nature. 1992;355:163–166. doi: 10.1038/355163a0. [DOI] [PubMed] [Google Scholar]
- 44.Silver RA, Cull-Candy SG, Takahashi T. Non-NMDA glutamate receptor occupancy and open probability at a rat cerebellar synapse with single and multiple release sites. J Physiol (Lond) 1996;494:231–250. doi: 10.1113/jphysiol.1996.sp021487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Taschenberger H, Engert F, Grantyn R. Synaptic current kinetics in a solely AMPA-receptor-operated glutamatergic synapse formed by rat retinal ganglion neurons. J Neurophysiol. 1995;74:1123–1136. doi: 10.1152/jn.1995.74.3.1123. [DOI] [PubMed] [Google Scholar]
- 46.Taylor WR, Chen E, Copenhagen DR. Characterization of spontaneous excitatory synaptic currents in salamander retinal ganglion cells. J Physiol (Lond) 1995;486:207–221. doi: 10.1113/jphysiol.1995.sp020803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tian N, Hwang TN, Copenhagen DR. Analysis of excitatory and inhibitory spontaneous synaptic activity in mouse retinal ganglion cells. J Neurophysiol. 1998;80:1327–1340. doi: 10.1152/jn.1998.80.3.1327. [DOI] [PubMed] [Google Scholar]
- 48.Tong G, Jahr CE. Multivesicular release from excitatory synapses of cultured hippocampal neurons. Neuron. 1994;12:51–59. doi: 10.1016/0896-6273(94)90151-1. [DOI] [PubMed] [Google Scholar]
- 49.Wadiche JI, Jahr CE. Multivesicular release at climbing fiber-Purkinje cell synapses. Neuron. 2001;32:301–313. doi: 10.1016/s0896-6273(01)00488-3. [DOI] [PubMed] [Google Scholar]
- 50.Watkins J, Olverman H. Agonists and antagonists for excitatory amino acid receptors. Trends Neurosci. 1987;10:265–272. [Google Scholar]