

Abstract
Methamphetamine (MA), a widely used drug of abuse, produces oxidative damage of nigrostriatal dopaminergic terminals. We examined the effect of subtype-selective ligands of metabotropic glutamate (mGlu) receptors on MA neurotoxicity in mice. MA (5 mg/kg, i.p.; injected three times, every 2 hr) induced, 5 d later, a substantial degeneration of striatal dopaminergic terminals associated with reactive gliosis. MA toxicity was primarily attenuated by the coinjection of the noncompetitive mGlu5 receptor antagonists 2-methyl-6-(phenylethynyl)pyridine and (E)-2-methyl-6-styrylpyridine both at 10 mg/kg, i.p.). In contrast, the mGlu1 receptor antagonist 7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylate ethyl ester (10 mg/kg, i.p.), and the mGlu2/3 receptor agonist (−)-2-oxa-4-aminocyclo[3.1.0]hexane-4,6-dicarboxylic acid (1 mg/kg, i.p.), failed to affect MA toxicity. mGlu5 receptor antagonists reduced the production of reactive oxygen species but did not reduce the acute stimulation of dopamine release induced by MA both in striatal synaptosomes and in the striatum of freely moving mice. We conclude that endogenous activation of mGlu5 receptors enables the development of MA neurotoxicity and that mGlu5 receptor antagonists are neuroprotective without interfering with the primary mechanism of action of MA.
Keywords: neuroprotection, methamphetamine toxicity, mGlu5 antagonists, microdialysis, striatal dopaminergic terminals, reactive oxygen species
During the past 10 years, the use of methamphetamine (MA) as a cheap, accessible, recreational drug has increased rapidly in both Europe and Western countries (Albertson et al., 1999). MA, also known as “speed,” “crank,” “smoke,” or “crystal ice,” damages nigrostriatal dopaminergic neurons in mice (Wagner et al., 1980) and induces, in chronic users, neuropathological lesions of the basal ganglia reminiscent of those occurring in Parkinson's disease (Wilson et al., 1996). MA is transported into dopaminergic terminals where it increases the amount of extravesicular dopamine (DA). DA is initially released, but then accumulates inside the terminal because MA inhibits the DA transporter (DAT), the vesicular monoamine transporter-2, and monoamine oxidase activity (for review, see Fleckenstein et al., 2000). Formation of reactive oxygen species by DA oxidation is classically implicated in the pathophysiology of MA neurotoxicity (O'Dell et al., 1991; Cubells et al., 1994; Yamamoto and Zhu, 1998; LaVoie and Hastings, 1999). However, a growing body of evidence indicates that glutamate plays a permissive role in the development of MA toxicity. MA enhances glutamate release (Nash and Yamamoto, 1992; Abekawa et al., 1994;Stephans and Yamamoto 1994), and NMDA-receptor antagonists are protective against MA neurotoxicity (Sonsalla et al., 1989, 1998). This raises the exciting possibility that glutamate receptor blockers can be used to prevent the degeneration of nigrostriatal dopaminergic fibers in MA abusers. Unfortunately, NMDA receptor antagonists are not suitable for long-term treatments because they impair fast excitatory synaptic transmission, causing sedation, ataxia, and deficits in learning and memory. In addition, some of these antagonists induce drug addiction and are endowed with intrinsic toxicity (for reviewed, seeNicoletti et al., 1996). Metabotropic glutamate receptor type 5 (mGlu5) receptors are coexpressed with, and functionally coupled to, NMDA receptors. Activation of mGlu5 receptors enhances NMDA responses in neurons (Pisani et al., 1997; Jia et al., 1998; Ugolini et al., 1999;Awad et al., 2000; Dohert et al., 2000; Salt and Binns, 2000) and contributes to the development of excitotoxic neuronal death (Bruno et al., 2000). Pharmacological blockade of mGlu5 receptors is expected to produce neuroprotection without the undesirable side effects typical of NMDA or AMPA receptor antagonists because mGlu5 receptors “modulate” rather than “mediate” excitatory synaptic transmission (for review, see Nicoletti et al., 1996). The advent of potent, subtype-selective, and centrally available mGlu5 receptor antagonists allows the study of mGlu5 receptors in in vivomodels of neurodegeneration. We report that mGlu5 receptor antagonists are highly protective against neurodegeneration induced by MA in mice. This effect is associated with a reduced formation of reactive oxygen species and does not depend on the interaction of these drugs with the dopamine transporter.
MATERIALS AND METHODS
Materials. Standard solutions of DA and metabolites were purchased from Sigma (St. Louis, MO). MA HCl was purchased from Sigma. 2,3-Dihydroxybenzoic acid (2,3-DHBA) was purchased from Sigma-Aldrich Chemical (Steinheim, Germany). 2-Methyl-6-(phenylethynyl)pyridine (MPEP), (E)-2-methyl-6-styrylpyridine (SIB-1893), and 6-methyl-2-(phenylazo)pyridin-3-ol (SIB-1757), and 7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylate ethyl ester (CPCCOEt) were purchased from Tocris Cookson (Bristol, UK). (-)-2-Oxa-4-aminocyclo[3.1.0]hexane-4,6-dicarboxylic acid (LY379268) was kindly provided by Eli Lilly & Company (Indianapolis, IN).
Animals. Male C57/6N black mice (Charles River, Calco, Como, Italy), 10 weeks old, weighing 22–24 gm, were kept under environmentally controlled conditions with food and water ad libitum. Because MA toxicity depends on the aggregation of the animals (Wagner et al., 1981), mice were housed 10 per cage in small cages (38 × 22 cm wide and 15 cm high). Experiments were performed following the Guidelines for Animal Care and Use of the National Institutes of Health.
Experimental design. To measure the effects induced by systemic administration of mGlu receptor ligands on MA toxicity, mice were divided into groups of 10 animals. Groups were treated intraperitoneally with CPCCOEt (10 mg/kg), MPEP (10 mg/kg), SIB-1893 (10 mg/kg), or LY379268 (1 mg/kg) 30 min before receiving each injection of MA. These doses of CPCCOEt, MPEP, SIB-1893, and LY379268 are reported to induce central effects when injected systemically (Monn et al., 1999; Chapman et al., 2000; Catania et al., 2001). MA was injected three times, with a 2 hr interval, at the dose of 5 mg/kg, i.p. We have selected this dose to produce a consistent degeneration of striatal dopaminergic terminals (Fornai et al., 1999). Control groups were injected with saline ± mGlu receptor ligands. Five days after the administration of MA or saline, mice were killed by decapitation, and the brains were dissected for the detection of striatal monoamine levels and for the immunohistochemical analysis of tyrosine hydroxylase (TH), DAT, and glial fibrillary acidic protein (GFAP) in the corpus striatum and substantia nigra.
Monoamine assay. The striatum was sonicated in 0.6 ml of ice-cold 0.1 m perchloric acid. Fifty microliters of the homogenates were used for protein determination (Lowry et al., 1951). The remaining aliquots were centrifuged at 8000 ×g for 10 min, and 20 μl of the supernatants was injected into an HPLC equipped with an autosampler 507 (Beckman Instruments, Fullerton, CA), a programmable solvent module 126 (Beckman), an analytical C18 reverse-phase column kept at 30°C ]Ultrasphere ODS 5 μm, 80 Å pore, 250 × 4.6 mm (Beckman)], and a Coulochem II electrochemical detector (ESA, Inc., Chelmsford, MA). The holding potentials were set at +350 and −350 mV for the detection of DA, 3,4-dihydroxyphenylacetic acid (DOPAC), and homovanillic acid (HVA). The mobile phase consisted of 80 mm sodium phosphate, 40 mm citric acid, 0.4 mm EDTA, 3 mm1-heptansulfonic acid, and 12.5% methanol, brought to pH 2.75 with phosphoric acid (run under isocratic conditions, at 1 ml/min).
TH, DAT, and GFAP immunostaining. Brains were immediately frozen and stored at −80°C. Ten micrometer sections were incubated overnight with primary antibodies and then for 1 hr with secondary antibodies. 3,3′-Diaminobenzidine (DAB) immunostaining was used for the detection of TH and DAT (ABC elite kit, Vector Laboratories, Burlingame, CA). The primary antibodies were monoclonal mouse antibodies against TH (1:2000; Sigma-Aldrich, Milan, Italy) and monoclonal rat antibodies against DAT (1:1000; Chemicon, Temecula, CA). Secondary antibodies used were biotinylated horse anti-mouse IgG or biotinylated rabbit anti-rat IgG (1:200; Vector). TH and DAT immunoreactivity were quantified by measuring the relative optical densities of the dorsal striatum and the substantia nigra in the stained sections using a computer-based microdensitometer (NIH Image Software, Bethesda, MD). GFAP labeling was performed by immunofluorescence. The primary antibody was monoclonal mouse antibody against GFAP (1:400; Sigma-Aldrich). Secondary antibodies were fluorescein isothiocyanate-conjugated horse anti-mouse IgG (1:100; Vector). Control staining was performed without the primary antibodies.
Microdialysis in freely moving mice. Male C57/6N Black mice, 10 weeks old, were used to measure the release of DA in the striatum of freely moving mice by microdialysis. Mice weighing 24–26 gm were implanted with microdialysis intracerebral guides (CMA/7 Guide Cannula,CMA/Microdialysis, Stockholm, Sweden) under ketamine (100 mg/kg, i.p.) + xylazine (10 mg/kg, i.p.) anesthesia, in a Kopf stereotaxic frame. The site of implantation was the left striatum [coordinates: 0.6 mm anterior to the bregma, 1.7 mm lateral to the midline, 3.5–5.5 mm ventral from the surface of skull, according to the atlas of Franklin and Paxinos (1997)]. After surgery, mice were housed in separate cages in a temperature-controlled environment on a 12 hr light/dark cycle, with ad libitum access to water and food, and allowed to recover for 4 d before the experiment. On the evening before the experiment, a probe was inserted into the intracerebral guide, after a dummy was removed, and mice were transferred to a plastic bowl cage with a moving arm (CMA/120 System for Freely-Moving Animals,CMA/Microdialysis), with ad libitum access to water and food. Concentric vertical microdialysis probes 2 mm long and 0.24 mm in outer diameter having a cuprophane membrane with a molecular cutoff of 6000 Da (CMA/7 Microdialysis Probe, CMA/Microdialysis) were used. The probes were perfused continuously with artificial CSF at a flow rate of 1.5 μl/min, using a microinjection pump (Bioanalytical System, West Lafayette, IN). The ACSF contained (in mm): 150 NaCl, 3 KCl, 1.7 CaCl2, and 0.9 MgCl2. This solution was not buffered, and the pH was typically 6.5. On the following morning, 30 μl (20 min) of consecutive perfusate sample fractions were continuously collected by a fraction collector (CMA/142 Microfraction Collector,CMA/Microdialysis). After four sample fractions, which were used to determine the basal levels of monoamines, control mice received a single injection of MA (5 mg/kg, i.p.), and sample fractions were collected for the following 2 hr. Animals injected systemically with MPEP (20 min before MA) or locally perfused with MPEP (100 μm), SIB-1757 (100 μm), or CPCCOEt (100 μm) received a single injection of MA (5 mg/kg, i.p.), and sample fractions were collected for the following 2 hr. In another set of animals, formation of reactive oxygen species was examined by monitoring 2,3-DHBA, a product of the reaction of salicylate (5 mm, added to artificial CSF with hydroxyl radicals. Analysis of DA and 2,3-DHBA was performed by HPLC with electrochemical detection, as described above.
Measurement of body temperature. To test whether group-I mGlu receptor antagonists produced hypothermia, we measured body temperature in control mice and in mice injected with MA. Mice were treated with MA (5 mg/kg; three times, 2 hr apart) and MPEP, SIB-1893, SIB-1757, or CPCCOEt (all at 10 mg/kg), including saline, were injected 20 min before each MA or saline injection. Body temperature was measured at 60, 180, and 300 min after saline or MA injection, i.e., 60 min after each injection of MA or saline. Measurements were performed at constant daytime intervals starting at 9:00 A.M. to avoid the influence of circadian variations. Core temperature was measured by inserting a thermometer probe lubricated with oil at least 3 cm into the rectum of the mice. To prevent sudden movements occurring especially in MA-treated mice, animals were gently handled with a wool glove while their tail was moved to allow the probe insertion. This was done to prevent the effects of restrain stress on body temperature. When the attempt to insert probe was not successful (i.e., sudden movements of the animal or the need to restrain the mouse), the animal was excluded from the groups. Baseline temperature did not vary after saline injection.
Release of [3H]DA from striatal synaptosomes. For the preparation of striatal synaptosomes, the brains were quickly removed and the striatum was dissected at 4°C. Synaptosomal fractions were prepared as described by Gray and Whittaker (1962). Synaptosomal pellets were suspended in 0.32 mglucose (8 mg of protein/ml). The suspension was diluted 1:10 with Krebs'–Ringer's medium (130 mm NaCl, 3 mmMgSO4, 2.5 mmNa2HPO4, 1 mmascorbic acid, and 20 mm Tris buffer, pH 7.5) and preincubated for 10 min at 37°C. Synaptosomes were then labeled with 0.1 μmdihydroxy-phenyl-ethylamine,3,4-[7-3H] ([3H]DA) (specific activity 36 Ci/mmol; NEN-DuPont) for 10 min at 37°C. After a 1:10 dilution with Krebs'–Ringer's medium, pH 7.5, containing 10 mmglucose, aliquots of the suspension were placed on Millipore filters lying at the bottom of parallel superfusion chambers thermostatically maintained at 37°C (Cerrito et al., 1993). The following drugs were added to the perfusate: MA (0.1 μm),d-amphetamine (0.1 μm), MPEP (5 μm), SIB-1757 (5 μm), or SIB-1893 (5 μm). The amount of [3H]DA present in each fraction and that extracted from synaptosomes at the end of the perfusion were separated from labeled metabolites on Biorex-70 columns (Smith et al., 1975), and the radioactivity present as [3H]DA was counted by scintillation spectrometry. [3H]DA release was expressed as percentage of the total [3H]DA.
RESULTS
mGlu5 receptor antagonists protect striatal dopaminergic terminals against MA toxicity
MA treatment (5 mg/kg, i.p.; injected three times at 2 hr intervals) led to >80% reduction in striatal DA levels, 5 d after the last injection (Fig.1A). This reduction was attenuated, but not abolished, in mice injected with the mGlu5 receptor antagonists MPEP or SIB-1893 (10 mg/kg, i.p.), which did not affect striatal DA levels on their own (Fig. 1A). Neither the mGlu2/3 receptor agonist, LY379268 (1 mg/kg, i.p.), nor the mGlu1 receptor antagonist, CPCCOEt (10 mg/kg, i.p.), had any effect on striatal DA levels in mice injected with MA (Fig.1A). MPEP and SIB-1893 also attenuated the reduction in striatal DOPAC and HVA levels induced by MA (Fig.1B,C). MA treatment also led to a substantial reduction in striatal TH and DAT immunoreactivity, associated with an increase in GFAP immunoreactivity, as already reported by others (Eisch et al., 1992; Pu and Vorhees, 1993) (Fig.2). Under our experimental conditions, no significant changes in TH or DAT immunoreactivity were observed in the substantia nigra (data not shown). The effects of MA on striatal TH, DAT, or GFAP immunoreactivity were markedly reduced in mice systemically injected with either MPEP or SIB-1893 (Fig. 2).
Fig. 1.
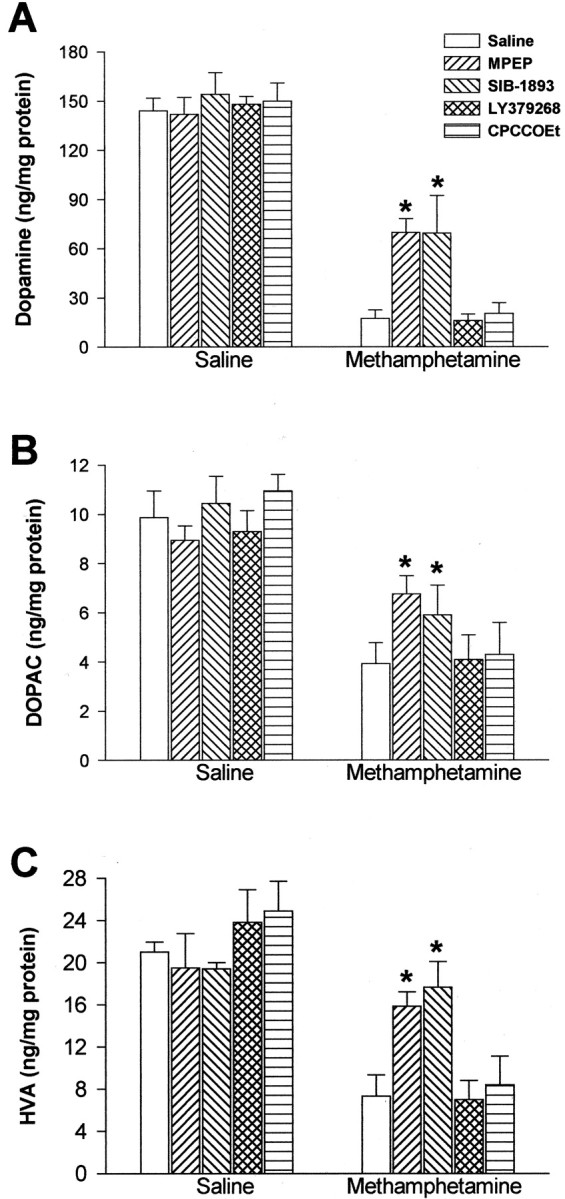
Striatal DA (A), DOPAC (B), and HVA (C) levels in mice injected with saline or MA (5 mg/kg, i.p.; 3 times, 2 hr apart) alone or in combination with MPEP (10 mg/kg), SIB-1893 (10 mg/kg), LY379268 (1 mg/kg), and CPCCOEt (10 mg/kg). Values are mean ± SEM of 8–10 determinations. *p < 0.05 (one-way ANOVA + Dunnett's test for post hoc analysis), if compared with the respective “saline” values.
Fig. 2.
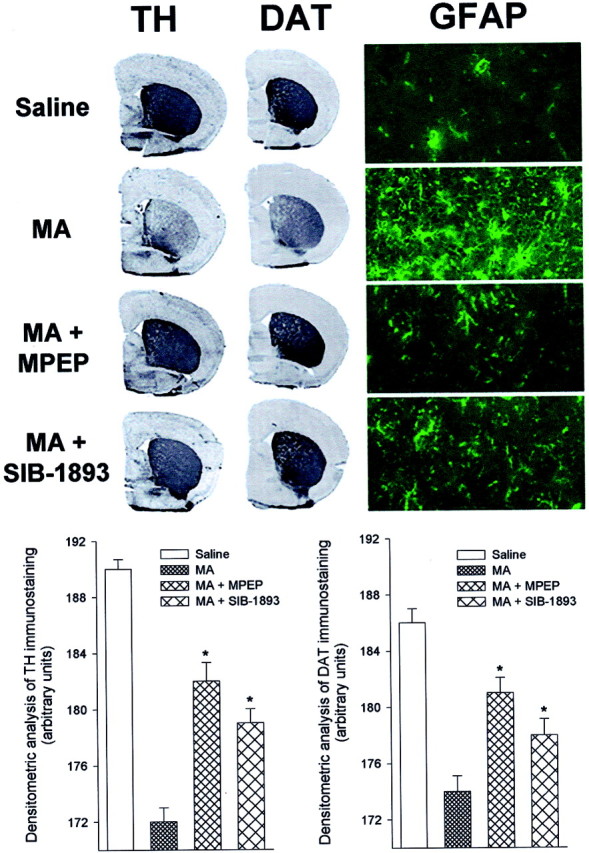
Immunohistochemical analysis of TH, DAT, and GFAP in the corpus striatum of mice injected with MA alone or in combination with MPEP or SIB-1893 (Fig. 1). Densitometric analysis of striatal TH or DAT immunostaining has been performed on comparable sections from five to six mice per group. *p < 0.05 (one-way ANOVA + Dunnett's test) versus MA alone.
mGlu5 receptor antagonists reduce the production of reactive oxygen species induced by MA in the striatum of freely moving mice
Using in vivo microdialysis, we measured the production of reactive oxygen species by monitoring the levels of 2,3-DHBA generated by the reaction of salicylic acid with hydroxyl radicals. A single injection of MA (5 mg/kg, i.p.) led to a rapid increase in the formation of 2,3-DHBA that lasted for at least 2 hr. This increase was almost abolished in mice systemically injected with MPEP (10 mg/kg, i.p.) (Fig. 3). A similar reduction was observed when MPEP or SIB-1757, but not CPCCOEt, was added to the perfusate (Fig. 3). All mGlu receptor antagonists were added to the perfusate at a concentration of 100 μm to yield ∼10 μm in the tissue. These tissular concentrations should produce maximal or near-to-maximal inhibition of mGlu5 or mGlu1 receptors (Schoepp et al., 1999).
Fig. 3.

Assessment of hydroxyl radical formation in the striatum of freely moving mice injected intraperitoneally with MA (A) and locally perfused with CPCCOEt (B), SIB-1757 (C), or MPEP (D) (all at 100 μm), since the beginning of the perfusion, or injected with MPEP (E) (10 mg/kg, i.p.) 20 min before MA. Thevertical arrows indicate the time of injection of the drugs. Values are means ± SEM of five determinations. *p < 0.05, if compared with the respective time points obtained in rats injected locally (B,C, F(5.32) = 14.83;D, F(5.32) = 27.82) or systemically (E, F(5.32)= 28.88) with saline (dotted line) (two-way ANOVA + Dunnett's test). #p < 0.05, if compared with the respective values at 20 min (A) (one-way ANOVA + Dunnett's test).
Effect of mGlu5 receptor antagonists on MA-induced dopamine release
We examined the stimulation of striatal DA release by MA both by microdialysis and in synaptosomal preparations. In freely moving mice, a single injection of MA (5 mg/kg, i.p.) induced a rapid and long-lasting increase in extracellular DA, although the extent of the increase (approximately twofold above basal) was lower than that reported by El Daly et al. (2000) with MA in mice (Fig.4). In mice injected with MPEP (10 mg/kg, i.p.) or locally infused with MPEP or SIB-1757 (both at 100 μm in the perfusate), MA stimulated DA release to the same extent as in control mice, although the stimulation of release was delayed in mice treated with MPEP. CPCCOEt had no effect on MA-stimulated DA release (Fig. 4).
Fig. 4.

Microdialysis study of extracellular striatal DA levels in mice injected intraperitoneally with MA (A) and locally perfused with CPCCOEt (B), SIB-1757 (C), or MPEP (D) (all at 100 μm), since the beginning of the perfusion, or injected with MPEP (E) (10 mg/kg, i.p.) 20 min before MA. Thevertical arrows indicate the time of injection of the drugs. Values are means ± SEM of five determinations. *p < 0.05 (one-way ANOVA + Dunnett's test) if compared with the respective values at 20 min.
We also examined the effect of mGlu5 receptor antagonists on the basal or MA-evoked release of [3H]DA in striatal synaptosomes. Addition of MA or d-amphetamine to the perfusion chamber increased [3H]DA release from striatal synaptosomes. This increase was time dependent, reaching a peak after 3–4 min, and was abolished by the DAT inhibitor nomifensine. None of the mGlu5 receptor antagonists reduced the stimulation of [3H]DA release induced by MA or d-amphetamine, indicating that these drugs do not compete with MA or d-amphetamine for the transport into dopaminergic terminals. MPEP, but not SIB-1757 or SIB-1893, produced a slight increase in the basal [3H]DA release, which was additive to the larger increase produced by MA ord-amphetamine (Fig.5A,B).
Fig. 5.

Effect of MPEP, SIB-1757, or SIB-1893 (all at 5 μm) on [3H]DA release stimulated by 0.1 μmd-amphetamine (A) or 0.1 μm MA (B) in striatal synaptosomes. We have also used nomifensine (5 μm) to validate the model. Thearrows indicate the time of addition ofd-amphetamine or MA. mGlu5 receptor antagonists or nomifensine were added to the perfusate 20 min befored-amphetamine or MA. Values are means ± SEM of six to eight determinations. SIB-1757 and SIB-1893 had no effect on [3H]DA release on their own.
Systemic injection of mGlu5 receptor antagonists does not affect MA-induced hyperthermia in mice
Knowing that hypothermia contributes to the protective effects of NMDA receptor antagonists or other drugs against MA neurotoxicity (Ali et al., 1994; Bowyer et al., 1994; Miller and O'Callaghan, 1994;Albers and Sonsalla, 1995), we measured the effect of MPEP, SIB-1757, SIB-1893, or CPCCOEt (all at 10 mg/kg, i.p.) on core body temperature in control mice and in mice receiving injections of MA (5 mg/kg, i.p.; three times, 2 hr apart). MA induced an increase of ∼2°C in body temperature, which was detected after 60, 180, and 300 min. None of the mGlu receptor antagonists reduced MA-induced hyperthermia or had any effect on body temperature in control mice (Table1).
Table 1.
Group-I mGlu receptor antagonists fail to affect body temperature in control and MA-treated mice
| Treatments | Body temperature (°C) | ||
|---|---|---|---|
| 60 min | 180 min | 300 min | |
| Saline | 37.6 ± 0.09 | 37.2 ± 0.15 | 37.2 ± 0.11 |
| MA | 39.3 ± 0.17 | 39.3 ± 0.18 | 39.2 ± 0.16 |
| MPEP | 37.5 ± 0.25 | 37.2 ± 0.03 | 37.1 ± 0.20 |
| SIB-1893 | 37.4 ± 0.26 | 36.9 ± 0.58 | 37.1 ± 0.28 |
| SIB-1757 | 37.4 ± 0.15 | 37.1 ± 0.13 | 37.3 ± 0.17 |
| CPCCOEt | 37.6 ± 0.21 | 38.1 ± 0.9 | 37.2 ± 0.15 |
| MA + MPEP | 39.2 ± 0.09 | 39.2 ± 0.13 | 39.4 ± 0.12 |
| MA + SIB-1893 | 39.4 ± 0.10 | 39.6 ± 0.09 | 39.2 ± 0.19 |
| MA + SIB-1757 | 39.5 ± 0.68 | 39.4 ± 0.21 | 39.2 ± 0.21 |
| MA + CPCCOEt | 39.3 ± 0.16 | 39.5 ± 0.13 | 39.3 ± 0.18 |
Body temperature was measured after three repeated injections of methamphetamine, 5 mg/kg, 2 hr apart. Effects of each compound (all at 10 mg/kg) on body temperature were measured 1 hr after each methamphetamine injection. Values are mean ± SEM of four animals for each time point.
DISCUSSION
In the last few years, MA abuse has become a widespread phenomenon, and treatment of MA neurotoxicity is emerging as a compelling clinical goal. Mice were treated with MA using an experimental paradigm that produces substantial neurotoxicity (5 mg/kg, i.p.; three injections at 2 hr intervals). This treatment led to a massive degeneration of striatal dopaminergic terminals, associated with reactive gliosis. Both effects were attenuated by systemically injected MPEP and SIB-1893, which both behave as potent and selective noncompetitive mGlu5 receptor antagonists. In contrast, inhibition of mGlu1 receptors with CPCCOEt or activation of mGlu2/3 receptors with LY379268 did not affect MA toxicity. MPEP, SIB-1893, or their congener SIB-1757 did not reduce the stimulation of DA release induced by MA in synaptosomes or in the striatum of freely moving animals, excluding the fact that mGlu5 receptor antagonists limit the access of MA into dopaminergic terminals. In addition, the effect of mGlu5 receptor antagonist was not secondary to a reduction in body temperature, as claimed previously for the NMDA receptor antagonist MK-801 (Ali et al., 1994; Bowyer et al., 1994; Miller and O'Callaghan, 1994;Albers and Sonsalla, 1995).
The slight stimulation of basal DA release produced by MPEP in synaptosomal preparations was not observed with SIB-1757 or SIB-1893 and therefore cannot be related to the recent observation that mGlu5 receptors negatively modulate DAT activity (Page et al., 2001). The significance of this effect, which was not observed in microdialysis studies, is obscure at present. Because mGlu5 receptor antagonists did not reduce MA-stimulated DA release, neuroprotection cannot be reconducted to changes in the amount of extravesicular DA available as a substrate for oxidative processes. A possible scenario is that the glutamate released in response to MA (Nash and Yamamoto, 1992; Abekawa et al., 1994; Stephans and Yamamoto, 1994) activates mGlu5 receptors, thereby triggering a chain of intracellular reactions that contribute to damage dopaminergic terminals. Interestingly, mGlu5 receptors are physically linked to the NR2 subunit of NMDA receptors through a chain of anchoring proteins including PSD-95, GKAP, SHANK, and Homer (Tu et al., 1999). Activation of mGlu5 receptors enhances NMDA currents (Ugolini et al., 1999; Awad et al., 2000; Salt and Binns, 2000), whereas activation of NMDA receptors potentiates mGlu5 receptor responses by inhibiting receptor desensitization (Alagarsamy et al., 1999). Neuroprotection observed with MPEP, SIB-1893, or SIB-1757 against NMDA toxicity in neuronal cultures (Bruno et al., 2000) suggests that the functional partnership between NMDA and mGlu5 receptors is necessary for the development of excitotoxic death. Because both NMDA (Sonsalla et al., 1989, 1998) and mGlu5 (present data) receptor antagonists are protective against MA toxicity, we conclude that endogenous activation of both receptors is required for the induction of damage. Activation of mGlu5 receptors generates oscillatory increases in intracellular Ca2+, as a result of inositoltrisphosphate formation and protein kinase C activation (Kawabata et al., 1996). A sustained activity of Ca2+-dependent enzymes is classically implicated in the development of neuronal damage through various mechanisms, which include the formation of reactive oxygen species (Choi, 1994). This chain of events may readily contribute to the overall production of reactive oxygen species induced by MA and, therefore, to the oxidative damage of striatal dopaminergic terminals (De Vito and Wagner, 1989). Accordingly, both MPEP (injected systemically or infused locally) and SIB-1757 (infused locally) substantially reduced the formation of hydroxyl radicals in the striatum of freely moving mice injected with MA. CPCCOEt, which was not neuroprotective, failed to affect MA-induced hydroxyl radicals formation, supporting a role for radical oxygen species in nigrostriatal damage. However, it is noteworthy that mGlu5 receptor antagonists (particularly MPEP) were more effective in reducing MA-induced hydroxyl radical formation than the depletion in striatal DA, DAT, TH, or GFAP observed in MA-treated mice. This suggests that oxidative damage contributes to, but is not the only mechanism responsible for, MA neurotoxicity.
MA may damage striatal dopaminergic terminals by increasing the formation of reactive oxygen species via two distinct mechanisms: (1) auto oxidation of extravesicular DA and (2) activation of mGlu5 and NMDA receptors by the endogenous glutamate. We speculate that activation of Ca2+-dependent enzymes with ensuing formation of radical oxygen species that follows the stimulation of NMDA and mGlu5 receptors combines with autoxidation of DA to reach the threshold of neuronal death. This would explain why mGlu5 receptor antagonists are protective without reducing MA-stimulated DA release. A series of studies suggest that mGlu5 receptors are present on striatal dopaminergic terminals (Page et al., 2001; Yu et al., 2001), thus providing a possible substrate for this hypothesis.
mGlu5 receptors may represent a novel target for drugs aimed at preventing the development of MA neurotoxicity in drug abusers. As opposed to DAT inhibitors (which prevent the access of MA into dopaminergic terminals), mGlu5 receptor antagonists do not limit the enhanced release of DA and therefore should not interfere with the rewarding properties of MA. This should make mGlu5 receptor antagonists more “acceptable” by MA abusers than DAT inhibitors or other drugs that interfere with dopaminergic transmission.
Footnotes
Correspondence should be addressed to Dr. Ferdinando Nicoletti, Department of Human Physiology and Pharmacology, Piazzale Aldo Moro, 5, 00185 Rome, Italy. E-mail: nicoletti@neuromed.it.
REFERENCES
- 1.Abekawa T, Ohmori T, Koyama T. Effects of repeated administration of a high dose of methamphetamine on dopamine and glutamate release in rat striatum and nucleus accumbens. Brain Res. 1994;643:276–281. doi: 10.1016/0006-8993(94)90033-7. [DOI] [PubMed] [Google Scholar]
- 2.Alagarsamy S, Marino MJ, Rouse ST, Gereau IVRW, Heinemann SF, Conn PJ. Activation of NMDA receptors reverses desensitization of mGluR5 in native and recombinant systems. Nat Neurosci. 1999;2:234–240. doi: 10.1038/6338. [DOI] [PubMed] [Google Scholar]
- 3.Albers DS, Sonsalla PK. Methamphetamine-induced hyperthermia and dopaminergic neurotoxicity in mice: pharmacological profile of protective and nonprotective agents. J Pharmacol Exp Ther. 1995;275:1104–1114. [PubMed] [Google Scholar]
- 4.Albertson TE, Derlet RW, Van Hoozen BE. Methamphetamine and the expanding complications of amphetamines. West J Med. 1999;170:214–219. [PMC free article] [PubMed] [Google Scholar]
- 5.Ali SF, Newport GD, Holson RR, Slikker W, Jr, Bowyer JF. Low environmental temperature or pharmacological agents that produce hyperthermia decrease methamphetamine neurotoxicity in mice. Brain Res. 1994;658:33–38. doi: 10.1016/s0006-8993(09)90007-5. [DOI] [PubMed] [Google Scholar]
- 6.Awad H, Hubert GW, Smith Y, Levey AI, Conn PJ. Activation of metabotropic glutamate receptor 5 has direct excitatory effects and potentiates NMDA receptor currents in neurons of the subthalamic nucleus. J Neurosci. 2000;20:7871–7879. doi: 10.1523/JNEUROSCI.20-21-07871.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bowyer JF, Davies DL, Schmued L, Broening HW, Newport GD, Slikker W, Jr, Holson RR. Further studies of the role of hyperthermia in methamphetamine neurotoxicity. J Pharmacol Exp Ther. 1994;268:1571–1580. [PubMed] [Google Scholar]
- 8.Bruno V, Ksiazek I, Battaglia G, Lukic S, Leonhardt T, Sauer D, Gasparini F, Kuhn R, Nicoletti F, Flor PJ. Selective blockade of metabotropic glutamate receptor subtype 5 is neuroprotective. Neuropharmacology. 2000;39:2223–2230. doi: 10.1016/s0028-3908(00)00079-4. [DOI] [PubMed] [Google Scholar]
- 9.Catania MV, Bellomo M, Di Giorgi-Gerevini V, Seminara G, Giuffrida R, Romeo R, De Blasi A, Nicoletti F. Endogenous activation of group-I metabotropic glutamate receptors is required for differentiation and survival of cerebellar Purkinje cells. J Neurosci. 2001;21:7664–7673. doi: 10.1523/JNEUROSCI.21-19-07664.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cerrito F, Aloisi G, Arminio P. Age-related changes in rat brain monoamines release: peculiarity of dopamine release. J Neurosci Res. 1993;34:363–370. doi: 10.1002/jnr.490340314. [DOI] [PubMed] [Google Scholar]
- 11.Chapman AG, Nanan K, Williams M, Meldrum BS. Anticonvulsant activity of two metabotropic glutamate group I antagonists selective for the mGlu5 receptor: 2-methyl-6-(phenylethynyl)-pyridine (MPEP), and (E)-6-methyl-2-styryl-pyridine (SIB-1893). Neuropharmacology. 2000;39:1567–1574. doi: 10.1016/s0028-3908(99)00242-7. [DOI] [PubMed] [Google Scholar]
- 12.Choi DW. Calcium and excitotoxic neuronal injury. Ann NY Acad Sci. 1994;747:162–171. doi: 10.1111/j.1749-6632.1994.tb44407.x. [DOI] [PubMed] [Google Scholar]
- 13.Cubells JF, Rayport S, Rajendran G, Sulzer D. Methamphetamine neurotoxicity involves vacuolation of endocytic organelles and dopamine-dependent intracellular oxidative stress. J Neurosci. 1994;14:2260–2271. doi: 10.1523/JNEUROSCI.14-04-02260.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Vito MJ, Wagner GC. Methamphetamine-induced neuronal damage: a possible role for free radicals. Neuropharmacology. 1989;28:1145–1150. doi: 10.1016/0028-3908(89)90130-5. [DOI] [PubMed] [Google Scholar]
- 15.Eisch AJ, Gaffney M, Weihmuller FB, O'Dell SJ, Marshall JF. Striatal subregions are differentially vulnerable to the neurotoxic effects of methamphetamine. Brain Res. 1992;598:321–326. doi: 10.1016/0006-8993(92)90201-j. [DOI] [PubMed] [Google Scholar]
- 16.El Daly E, Chefer V, Sardill S, Shippenberg TS. Modulation of the neurotoxic effects of methamphetamine by the selective kappa-opioid receptor agonist U69593. J Neurochem. 2000;74:1553–1562. doi: 10.1046/j.1471-4159.2000.0741553.x. [DOI] [PubMed] [Google Scholar]
- 17.Fleckenstein AE, Gibb JW, Hanson GR. Differential effects of stimulants on monoaminergic transporters: pharmacological consequences and implications for neurotoxicity. Eur J Pharmacol. 2000;406:1–13. doi: 10.1016/s0014-2999(00)00639-7. [DOI] [PubMed] [Google Scholar]
- 18.Fornai F, Giorgi FS, Alessandrì MG, Giusiani M, Corsini GU. Effects of pre-treatment with DSP-4 on methamphetamine-induced striatal dopamine loss and pharmacokinetics. J Neurochem. 1999;72:777–784. doi: 10.1046/j.1471-4159.1999.0720777.x. [DOI] [PubMed] [Google Scholar]
- 19.Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Academic; London: 1997. [Google Scholar]
- 20.Gray EG, Whittaker VP. The isolation of nerve endings from rat brain: an electron microscope study of cell fragments derived by homogenization and centrifugation. J Anat. 1962;96:79–87. [PMC free article] [PubMed] [Google Scholar]
- 21.Jia Z, Lu Y, Henderson J, Taverna F, Romano C, Abramow-Newerly W, Wojtowicz JM, Roder J. Selective abolition of the NMDA component of long-term potentiation in mice lacking mGluR5. Learn Mem. 1998;5:331–343. [PMC free article] [PubMed] [Google Scholar]
- 22.Kawabata S, Tsutsumi R, Kohara A, Yamaguchi T, Naknishi S, Okada M. Control of calcium oscillations by phosphorylation of metabotropic glutamate receptors. Nature. 1996;383:89–92. doi: 10.1038/383089a0. [DOI] [PubMed] [Google Scholar]
- 23.LaVoie MJ, Hastings TG. Dopamine quinone formation and protein modification associated with the striatal neurotoxicity of methamphetamine: evidence against a role for extracellular dopamine. J Neurosci. 1999;19:1484–1491. doi: 10.1523/JNEUROSCI.19-04-01484.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 25.Miller DB, O'Callaghan JP. Environment-, drug- and stress-induced alterations in body temperature affects the neurotoxicity of substituted amphetamines in the C57BL/6J mouse. J Pharmacol Exp Ther. 1994;270:752–760. [PubMed] [Google Scholar]
- 26.Monn JA, Valli MJ, Massey SM, Hansen MM, Kress TJ, Wepsiec JP, Harkness AR, Grutsch JL, Jr, Wright RA, Johnson BG, Andis SL, Kingston A, Tomlinson R, Lewis R, Griffey KR, Tizzano JP, Schoepp DD. Synthesis, pharmacological characterization, and molecular modelling of heterobicyclic amino acids related to (+)-2-aminobicyclo[3.1.0] hexane-2,6-dicarboxylic acid (LY354740): identification of two new potent, selective, and systemically active agonists for group II metabotropic glutamate receptors. J Med Chem. 1999;42:1027–1040. doi: 10.1021/jm980616n. [DOI] [PubMed] [Google Scholar]
- 27.Nash JF, Yamamoto BK. Methamphetamine neurotoxicity and striatal glutamate release: comparison to 3,4-methylenedioxymethamphetamine. Brain Res. 1992;581:237–243. doi: 10.1016/0006-8993(92)90713-j. [DOI] [PubMed] [Google Scholar]
- 28.Nicoletti F, Bruno V, Copani A, Casabona G, Knöpfel T. Metabotropic glutamate receptors: a new target for the therapy of neurodegenerative disorders? Trends Neurosci. 1996;1:267–271. doi: 10.1016/S0166-2236(96)20019-0. [DOI] [PubMed] [Google Scholar]
- 29.O'Dell SJ, Weihmuller FB, Marshall JF. Multiple methamphetamine injections induce marked increases in extracellular striatal dopamine which correlate with subsequent neurotoxicity. Brain Res. 1991;564:256–260. doi: 10.1016/0006-8993(91)91461-9. [DOI] [PubMed] [Google Scholar]
- 30.Page G, Peters M, Najimi GM, Maloteaux JM, Hermans E. Modulation of the neuronal dopamine transporter activity metabotropic glutamate receptors mGluR5 in rat striatal synaptosomes through phosphorylation mediated processes. J Neurochem. 2001;76:1282–1290. doi: 10.1046/j.1471-4159.2001.00179.x. [DOI] [PubMed] [Google Scholar]
- 31.Pisani A, Calabresi P, Centonze D, Bernardi G. Enhancement of NMDA responses by group-I metabotropic glutamate receptor activation in striatal neurones. Br J Pharmacol. 1997;120:1007–1014. doi: 10.1038/sj.bjp.0700999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pu C, Vorhees CV. Developmental dissociation of methamphetamine-induced depletion of dopaminergic terminals and astrocytes reaction in rat striatum. Brain Res. 1993;72:325–328. doi: 10.1016/0165-3806(93)90201-k. [DOI] [PubMed] [Google Scholar]
- 33.Salt TE, Binns KE. Contributions of mGlu1 and mGlu5 receptors to interactions with N-methyl-d-aspartate receptor-mediated responses and nociceptive sensory responses of rat thalamic neurons. Neuroscience. 2000;100:375–380. doi: 10.1016/s0306-4522(00)00265-7. [DOI] [PubMed] [Google Scholar]
- 34.Schoepp DD, Jane DE, Monn AJ. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- 35.Smith JE, Lane JD, Shea PA, McBride WJ, Aprison MH. A method for concurrent measurement of picomole quantities of acetylcholine, choline, dopamine, norepinephrine, serotonin, 5-hydroxytryptophan, 5-hydroxyindolacetic acid, tryptophan, tyrosine, glycine, aspartate, glutamate, alanine and gamma-aminobutyric acid in single tissue from different areas of rat central nervous system. Anal Biochem. 1975;63:149–169. doi: 10.1016/0003-2697(75)90417-0. [DOI] [PubMed] [Google Scholar]
- 36.Sonsalla PK, Nicklas WJ, Heikkila RE. Role for excitatory amino acids in methamphetamine-induced nigrostriatal dopaminergic toxicity. Science. 1989;243:398–400. doi: 10.1126/science.2563176. [DOI] [PubMed] [Google Scholar]
- 37.Sonsalla PK, Albers DS, Zeevalk GD. Role of glutamate in neurodegeneration in dopamine neurons in several animal models of parkinsonism. Amino Acids. 1998;14:69–74. doi: 10.1007/BF01345245. [DOI] [PubMed] [Google Scholar]
- 38.Stephans SE, Yamamoto BK. Methamphetamine-induced neurotoxicity: roles for glutamate and dopamine efflux. Synapse. 1994;17:203–209. doi: 10.1002/syn.890170310. [DOI] [PubMed] [Google Scholar]
- 39.Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakman P, Doan A, Aakalu VK, Lanahan AA, Sheng M, Worley PF. Coupling of mGluR/Homer and PSD-95 complexes by the Shank family of postsynaptic density proteins. Neuron. 1999;23:583–592. doi: 10.1016/s0896-6273(00)80810-7. [DOI] [PubMed] [Google Scholar]
- 40.Ugolini A, Corsi M, Bordi F. Potentiation of NMDA and AMPA responses by the specific mGluR5 agonist CHPG in spinal cord motoneurons. Neuropharmacology. 1999;38:1569–1576. doi: 10.1016/s0028-3908(99)00095-7. [DOI] [PubMed] [Google Scholar]
- 41.Wagner GC, Ricaurte GA, Seiden LS, Schuster CR, Miller RJ, Westley J. Long-lasting depletion of striatal dopamine and loss of dopamine uptake sites following repeated administration of methamphetamine. Brain Res. 1980;181:151–160. doi: 10.1016/0006-8993(80)91265-2. [DOI] [PubMed] [Google Scholar]
- 42.Wagner GC, Lucot JB, Schuster CR, Seiden LS. The ontogeny of aggregation-enhanced toxicity. Psychopharmacology. 1981;75:92–93. doi: 10.1007/BF00433509. [DOI] [PubMed] [Google Scholar]
- 43.Wilson JM, Kalasinsky KS, Levey AI, Bergeron C, Reiber G, Anthony RM, Schmunk GA, Shannak K, Haycock JW, Kish SJ. Striatal dopamine nerve terminal markers in human, chronic methamphetamine users. Nat Med. 1996;2:699–703. doi: 10.1038/nm0696-699. [DOI] [PubMed] [Google Scholar]
- 44.Yamamoto BK, Zhu W. The effects of methamphetamine on the production of free radicals and oxidative stress. J Pharmacol Exp Ther. 1998;287:107–114. [PubMed] [Google Scholar]
- 45.Yu TS, Wang SD, Liu JC, Yin HS. Changes in the gene expression of GABA(A) receptor alpha1 and alpha2 subunits and metabotropic glutamate receptor 5 in the basal ganglia of the rats with unilateral 6-hydroxydopamine lesion and embryonic mesencephalic grafts. Exp Neurol. 2001;168:231–241. doi: 10.1006/exnr.2000.7590. [DOI] [PubMed] [Google Scholar]